Introduction
Incidence rates of hyperuricemia have increased
during the past decade, with a prevalence of 13.3% in mainland
China (1). Hyperuricemia or a
high-normal level of serum uric acid (UA) have been demonstrated to
be independent risk factors for the initiation and prognosis of
chronic kidney disease (2,3),
which can be attenuated through urate-reducing therapy (1,4,5).
UA, as monosodium crystals or soluble (S) UA, induces
tubulointerstitial inflammation (6-8).
However, the mechanism by which SUA triggers renal tubular
inflammation and how this process is regulated are poorly
understood. Serum UA is useful for predicting (9) and screening the incidence of
metabolic disorders (10).
Additionally, type 2 diabetic patients with hyperuricemia,
typically associated with insulin resistance (IR), exhibit an
increased incidence of renal calculus compared with patients
without hyperuricemia (11).
Formation of UA nephrolithiasis decreases the improvement of IR in
mice with type 2 diabetes and metabolic syndrome (12). These data suggest that IR may
facilitate hyperuricemia-induced development of nephrolithiasis.
Thus, the present study evaluates whether IR increases the
vulnerability of renal tubules to SUA-elicited inflammation.
Adiponectin (APN) is an adipokine primarily derived
from adipocytes (13-15). APN is generally recognized as an
insulin sensitizer and hypoadiponectinemia caused by genetic and
environmental factors impairs insulin sensitivity, leading to
diabetes and metabolic syndrome (16). Under conditions of IR,
transcription of APN receptor 1 (AdipoR1) (17) and the activation of adenosine
monophosphate-activated protein kinase (AMPK) (16) are abolished, decreasing the
response sensitivity of APN (16). Over the past two decades, APN has
been identified as a potent anti-inflammatory mediator (18-22) via receptor-dependent mechanisms
(23). Various studies indicated
that APN is expressed in and acts protectively in renal podocytes
(18,24,25), mesangial cells (22,26) and tubular epithelial cells
(25,27) in humans and rodents (19,28). It was demonstrated that APN
knockout (KO) worsens the severity of kidney structural damage,
increases the infiltration of macrophages and upregulates the
intrarenal production of proinflammatory factors in subtotal
nephrectomized and diabetic mice, whereas the overexpression of APN
ameliorated these disorders (22,28). Conversely, few studies argued that
APN serves as a proinflammatory factor in the renal tubular cell
line HK-2 upon stimulation with lipopolysaccharide (LPS) (29) and in acute kidney injury induced
by ischemia-reperfusion (30).
Therefore, further research is required to clarify the association
between APN and renal inflammation. Furthermore, whether and how
APN modifies renal tubular inflammation induced by SUA and whether
IR-associated abnormal APN signaling facilitates renal tubular
injury have not yet been determined.
Here, it was hypothesized that APN conferred
resistance in renal tubular inflammation following SUA exposure.
Effects of APN and its signaling mechanism in SUA-stimulated human
proximal renal tubular epithelial cells (PTECs) with
loss-of-function experiment were performed to validate this
hypothesis. The present study suggested that APN protects against
SUA-induced renal tubular inflammatory responses via the
AdipoR1/AMPK signaling pathway.
Materials and methods
Materials and reagents
PTECs (cat. no. 4100) and epithelial cell medium
(ECM; cat. no. 4101) were obtained from ScienCell Research
Laboratories, Inc. (San Diego, CA, USA). BioXtra UA was purchased
from Sigma-Aldrich (cat. no. U0881; Merck KGaA, Darmstadt,
Germany). Primary antibodies against APN (cat. no. Ab22554;
1:1,000) AdipoR1 (cat. no. Ab126611; 1:2,000), NACHT, leucine rich
repeat and pyrin domain-containing protein 3 (NLRP3; cat. no.
Ab109314; 1:1,000) and tumor necrosis factor (TNF) α (cat. no.
Ab9635; 1:2,000) were from Abcam (Cambridge, UK). Primary
antibodies against AMPKα (cat. no. 2532; 1:1,000) and
phosphorylated (p) AMPKα-Thr172 (cat. no. 2535; 1:1,000) were
provided by Cell Signaling Technology, Inc. (Danvers, MA, USA). The
anti-GAPDH antibody (cat. no. 10094-T52; 1:10,000) and the
horseradish peroxidase (HRP)-conjugated goat anti-mouse (cat. no.
SSA007; 1:1,000) and anti-rabbit IgG (cat. no. SSA004; 1:1,000)
secondary antibodies were from Sino Biological, Inc. (Shanghai,
China). High sensitivity ELISA kits for interleukin (IL)-1β (cat.
no. BMS224HS) and TNF-α (cat. no. BMS223HS) were from eBioScience
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
lentivirus-mediated short hairpin (sh) RNA against AdipoR1, the
scramble-shRNA and Polybrene were from Shanghai GeneChem Co., Ltd.
(Shanghai, China). Recombinant human globular (g) APN (cat. no.
450-21) was supplied by PeproTech, Inc. (Rocky Hill, NJ, USA).
Compound C (cat. no. US1171260), a specific AMPK inhibitor, was
from Merck KGaA.
SUA preparation
SUA was freshly prepared prior to each experiment as
previously described (31).
Briefly, BioXtra UA was dissolved in 1 M NaOH to a final
concentration of 50 mg/ml. The solution was filtered (pore size,
0.22 µm) and tested for mycoplasma and endotoxin. Polarizing
microscopy (magnification, ×200) was used to check for crystals in
the SUA solution for the duration of the experiments.
Cell culture
PTECs were cultured at 37°C in a humidified
atmosphere with 5% CO2 and incubated in ECM, which
consisted of basal medium, 2% fetal bovine serum albumin, 1%
epithelial cell growth supplement and 1% penicillin/streptomycin
solution. Cells passaged 4-7 times were used in the following
experiments as previously described (27).
Cell viability
Growth-arrested PTECs were seeded in 96-well plates
at 2.5×104 cells/well. Cells were exposed to SUA at
increasing concentrations (0, 25, 50, 100 and 200 µg/ml) for
24, 48 and 72 h. A commercial MTT assay kit from Amresco, LLC
(Solon, OH, USA) was used to assess the viability of PTECs
following SUA exposure, as previously described (8). In brief, PTECs were incubated with
20 µl MTT at 37°C for 4 h, followed by an addition of 150
µl dimethyl sulfoxide. Cell viability is represented as the
percentage change in the absorbance measured at 570 nm compared
with untreated cells.
Transfection with AdipoR1-shRNA
AdipoR1-shRNA was designed to target the following
sequence: 5′-CAA AGC TGA AGA AGA GCA A-3′. The negative control
scrambled sequence was: 5′-TTC TCC GAA CGT GTC ACG T-3′. The
uniqueness of these sequences was confirmed using the GenBank/EBI
database (https://www.ncbi.nlm.nih.gov/nucleotide/ and
https://www.ebi.ac.uk/ena). Reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blot assays were performed to evaluate the efficiency of
AdipoR1 silencing. Next, 2×105 PTECs were transfected
with 70 µl AdipoR1-shRNA lentivirus (3×108
transducing U/ml) at a multiplicity of infection = 50 for 24 h
prior to incubation for 48 h with 100 µg/ml SUA. Polybrene
(5 µl/ml) was used to facilitate transfection reactions.
RNA extraction and gene expression levels
of APN and AdipoR1
Growth-arrested PTECs (5×105) were
exposed to 100 µg/ml SUA for 4 h. Total RNA was extracted
using TRIzol reagent from Takara Bio, Inc. (Otsu, Japan). cDNA
synthesis and RT were performed as previously described (8). Briefly, extracted RNA was reverse
transcribed into cDNA at 42°C for 50 min and 85°C for 5 min using a
PrimeScript™ RT Master Mix kit (Takara Bio, Inc.). qPCR reactions
(total volume, 25 µl) were conducted in duplicate using a
SYBR Premix Ex Taq™ kit (Takara Bio, Inc.) and the Fast Real-Time
PCR System 7300 (Applied Biosystems; Thermo Fisher Scientific,
Inc.). Each reaction was performed at 95°C for 10 min, followed by
40 cycles of 95°C for 10 sec and 60°C for 30 sec, then 95°C for 15
sec and 60°C for 1 min, and 95°C for 15 sec and 60°C for 15 sec.
Primer pairs for amplifying APN (forward, 5′-CTA TGA TGG CTC CAC
TGG TA-3′ and reverse, 5′-GAG CAT AGC CTT GTC CTT CT-3′; product,
124 bp), AdipoR1 (forward, 5′-CGG TGG AAC TGG CTG AAC TG-3′
and reverse, 5′-CCG CAC CTC CTC CTC TTC TT-3′; product, 125 bp) and
GAPDH (forward, 5′-ATG GGG AAG GTG AAG GTC G-3′ and reverse, 5′-GGG
GTC ATT GAT GGC AAC AAT A-3′; product, 107 bp) were provided by
Invitrogen (Thermo Fisher Scientific, Inc.). The relative levels of
the target mRNAs were normalized and expressed using the
2−ΔΔCq method as described (8,32).
Protein expression levels in cell
lysates
PTECs were lysed with lysis buffer containing a
protease inhibitor cocktail (Sigma-Aldrich; Merck KGaA). Protein
concentrations were determined by bicinchoninic acid protein assays
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Immunoblot
analyses were conducted as previously described (8). Equal amounts of protein (10
µg) were loaded and separated on 11% SDS-PAGE gels, followed
by transfer to polyvinylidene fluoride membranes. Following 30 min
incubation with blocking buffer (ZLI 9027; ZSGB-BIO; OriGene
Technologies, Inc., Beijing, China), composed of 5% bovine serum
albumin in TBS with 0.05% Tween-20, membranes were probed with
antibodies against NLRP3, APN, AdipoR1, AMPKα, pAMPKα, TNFα and
GAPDH at 4°C overnight. Immune complexes were visualized following
incubation with HRP-conjugated anti-mouse or anti-rabbit secondary
antibodies for 1 h at room temperature. Immunoreactive bands were
detected using enhanced chemiluminescence (Amersham; GE Healthcare,
Chicago, IL, USA). Signals were quantified using the Tanon-4500 gel
imaging system with GIS ID analysis software v4.1.5 (Tanon Science
and Technology Co., Ltd., Shanghai, China).
APN, IL-1β and TNFα levels in cell
supernatants
Growth- arrested PTECs (5×105) were
incubated with 100 µg/ml SUA for 48 h as previously
described (8), with or without
pretreatment with AdipoR1-shRNA lentivirus for 24 h or 10 mM
compound C for 90 min Cell supernatants were collected. APN levels
were measured at 450 nm with a high sensitivity (<60 pg/ml)
ELISA kit (cat. no. EK0595; Boster Biological Technology,
Pleasanton, CA, USA). The standard provided with the kit is a 30
kDa APN protein, representing a full-length APN. IL-1β and TNFα
were quantified using commercial ELISA kits at 450 nm. The
sensitivity of the TNFα assay was 0.13 pg/ml and its intra- and
interassay coefficients of variation were 8.5 and 9.8%,
respectively. The sensitivity and intra- and interassay
coefficients of variation for the IL-1β assay were 0.05 pg/ml, 6.7
and 8.1%, respectively.
Statistical analysis
Statistical analyses were performed using SPSS 21.0
software (IBM Corp., Armonk, NY, USA). The results are expressed as
the mean ± standard errors of the mean. Comparisons between two
groups were analyzed by a two-tailed Student's t-test.
Comparisons among three or more groups were analyzed by one-way
analysis of variance followed by Tukey's post hoc test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Effect of SUA on cultured human PTEC
viability
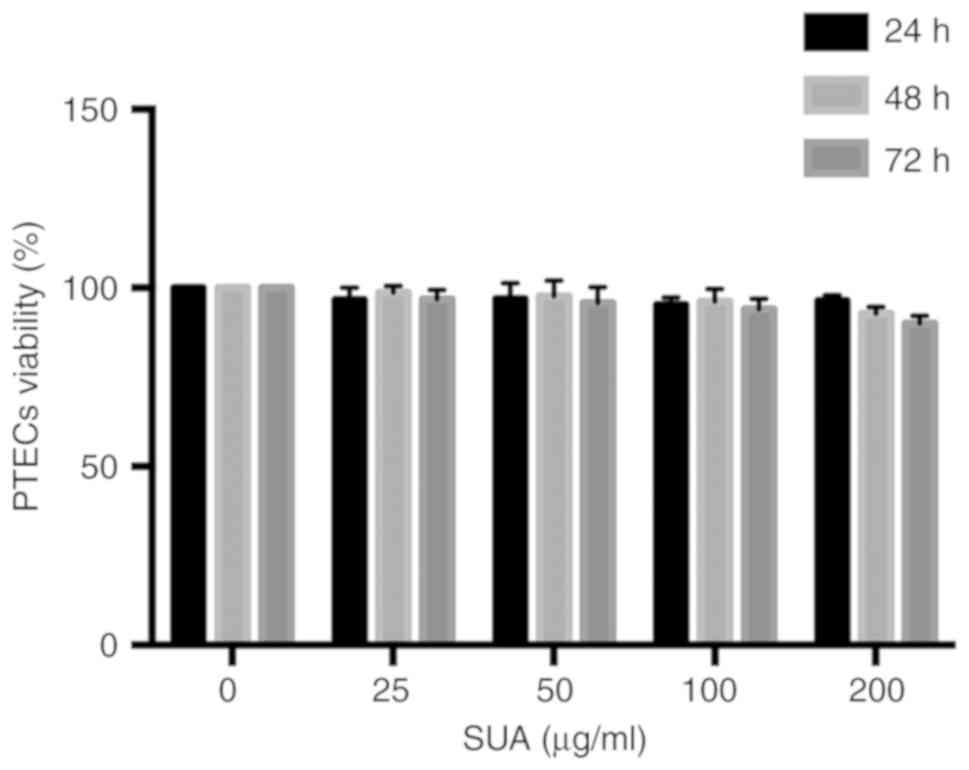
To determine whether SUA impaired cell viability,
increasing doses of SUA (25-200 µg/ml) were added to PTECs
for varying time periods (24-72 h). Cell viability was assessed
using an MTT assay and expressed as the percentage change in the
absorbance relative to untreated PTECs. Fig. 1 revealed that SUA administration
did not significantly affect cell viability at any of the indicated
time points (P>0.05). A dose of 100 µg/ml SUA,
corresponding to a common level of UA in hyperuricemia in humans,
was employed in the following experiment (33).
Inflammatory responses are elevated in
SUA-treated PTECs
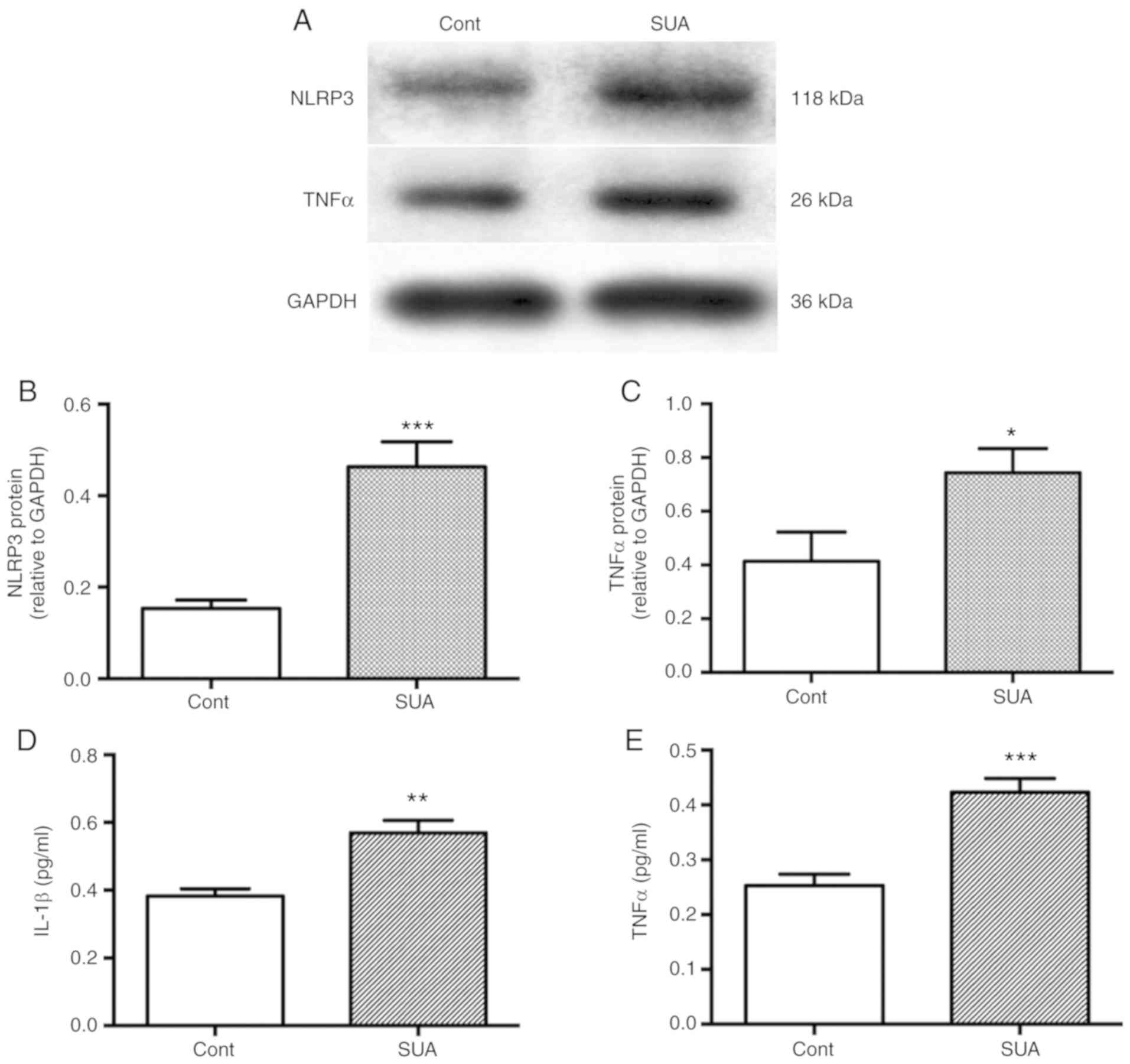
The impact of SUA on inflammatory responses was
evaluated in cultured PTECs. PTECs were incubated in the presence
of absence of 100 µg/ml SUA for 48 h. SUA treatment
significantly enhanced the protein synthesis of NLRP3 and TNFα in
PTECs compared with the untreated control (P<0.001 and
P<0.05, respectively; Fig.
2A–C). SUA further significantly promoted the release of IL-1β
(P<0.01; Fig. 2D) and TNFα
from PTECs into the serum compared with the untreated control
(P<0.001; Fig. 2E).
APN expression is increased in
SUA-treated PTECs
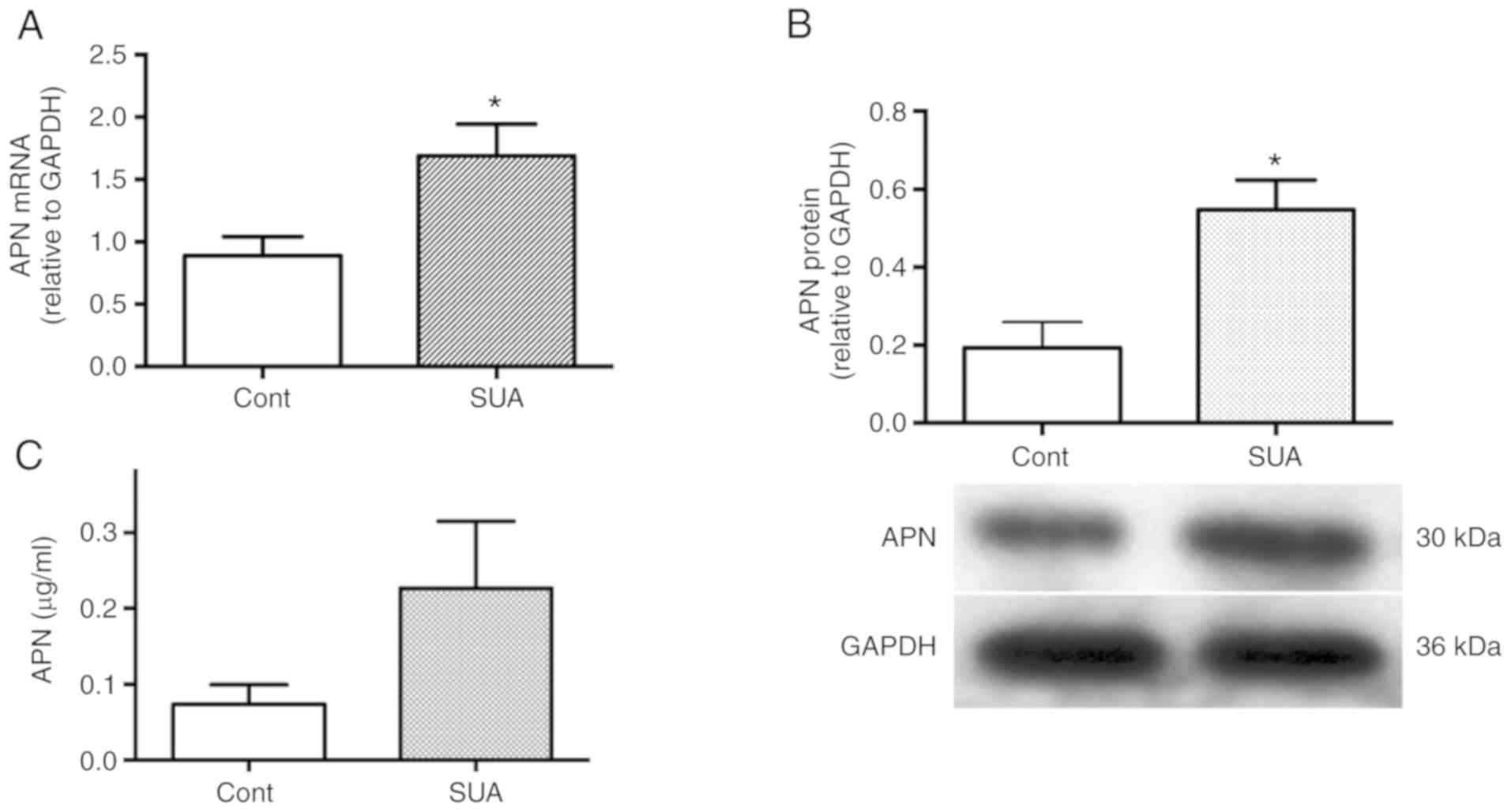
Effects of SUA on the expression of APN in cultured
PTECs were established. PTECs were incubated in the presence or
absence of 100 µg/ml SUA for indicated time periods. SUA
treatment significantly increased APN mRNA expression following a
4-h incubation and protein expression following a 48-h incubation
(P<0.05; Fig. 3A and B).
APN release was increased in SUA-treated PTECs
compared with the untreated control (0.23±0.09 vs. 0.07±0.03
µg/ml; P=0.16; Fig. 3C).
To analyze whether APN treatment attenuates SUA-induced
inflammation responses in PTECs, cells were cultured in the
presence or absence of 2.5 µg/ml gAPN for 6 h prior to
incubation with 100 µg/ml SUA for 48 h. Exogenous gAPN
inhibited the SUA-induced elevation of cellular and secreted
protein levels (Fig. 4). TNFα
levels were significantly decreased in gAPN-pretreated cells
compared with the SUA-treated cells as determined by western
blotting and ELISA analysis (P<0.001; Fig. 4A, C and E). gAPN administration
further significantly decreased IL-1β levels in the supernatant of
PTECs incubated with SUA (P<0.01; Fig. 4D). No significant changes in NLRP3
protein expression were observed in SUA-treated cells pretreated
with APN (P>0.05; Fig. 4A and
B). The data indicated that APN may inhibit SUA-induced
inflammation in vitro and may aid in restoring the balance
between anti- and proinflammatory environments.
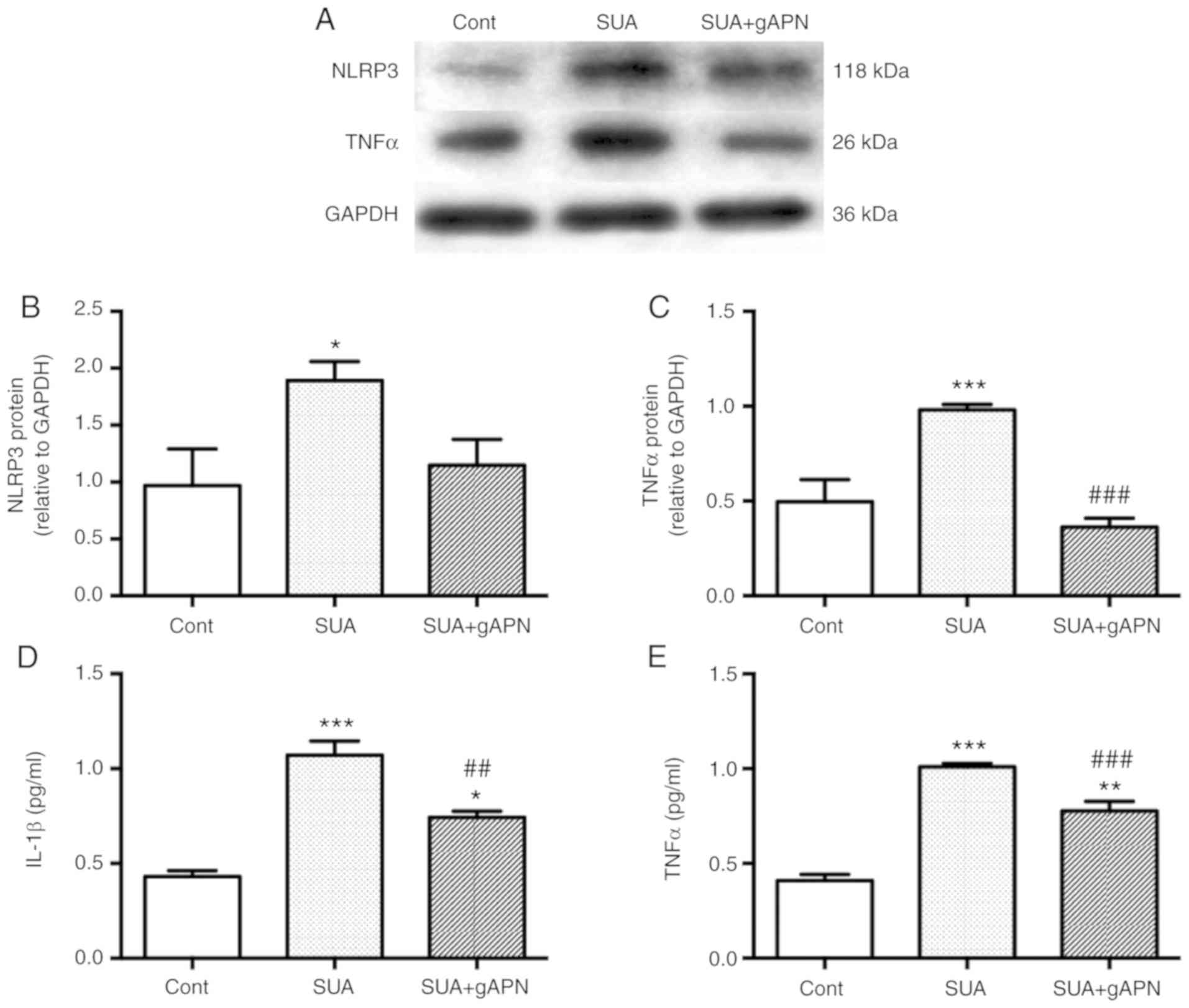 | Figure 4APN supplement alleviates
SUA-triggered inflammatory responses in PTECs. PTECs were
pretreated gAPN (0 or 2.5 µg/ml) for 6 h prior to incubation
with SUA (100 µg/ml) for 48 h (n=3). (A) Western blot images
and quantified protein expression of (B) NLRP3 and (C) TNFα. Levels
of (D) IL-1β and (E) TNFα in cell supernatants measured using
ELISA. Data are representative of three independent experiments.
*P<0.05, **P<0.01 and
***P<0.001 vs. Cont; ##P<0.01 and
###P<0.001 vs. SUA. APN, adiponectin; g,
globular; PTEC, proximal renal tubular epithelial cell; SUA,
soluble uric acid; NLRP3, NACHT, leucine rich repeat and pyrin
domain-containing protein 3; TNF, tumor necrosis factor; IL,
interleukin; Cont, untreated cells. |
APN activates the AdipoR1/AMPK signaling
pathway in PTECs
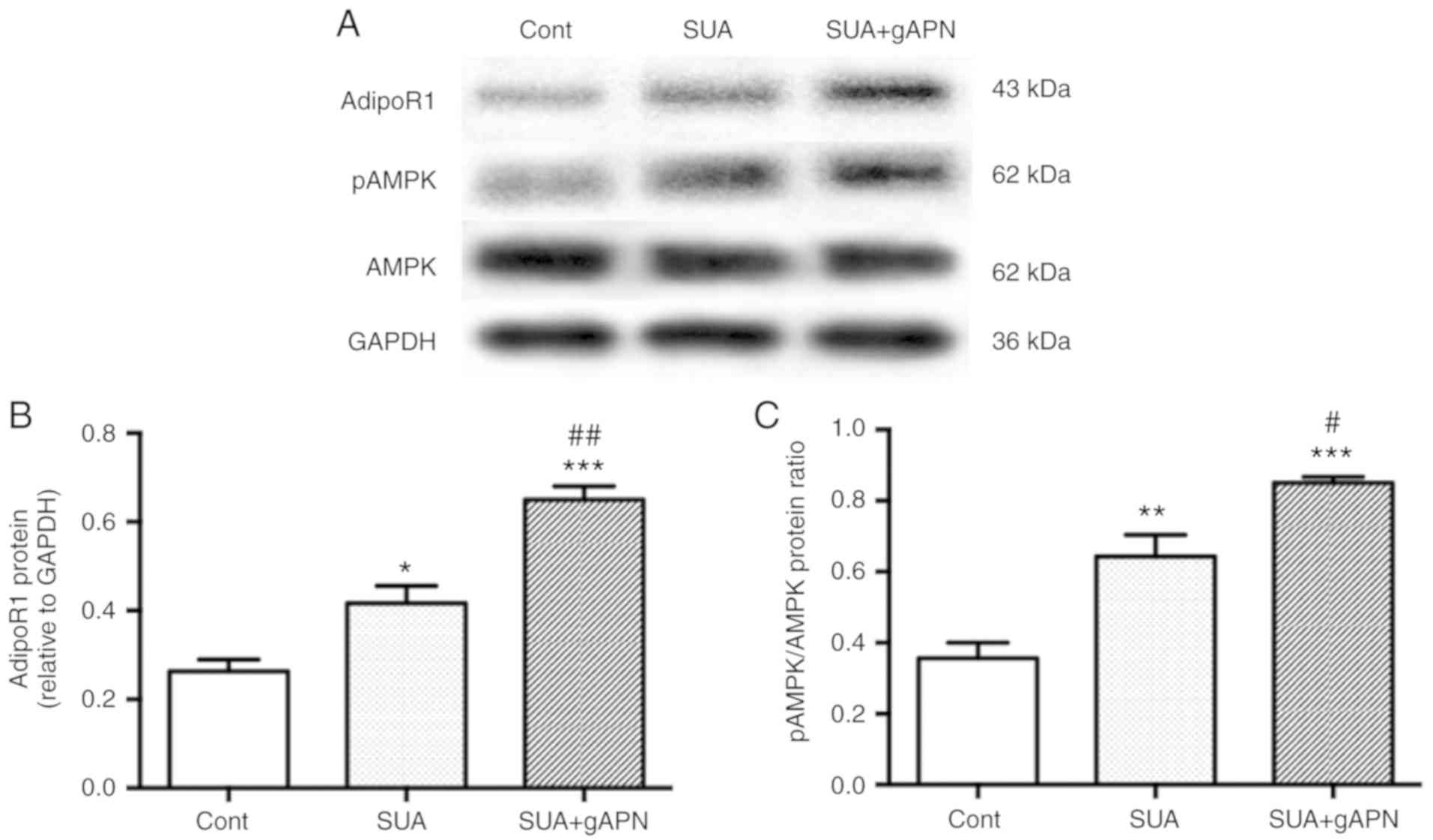
To investigate whether the AdipoR1/AMPK signaling
pathway is involved in the anti-inflammatory mechanism of gAPN,
PTECs were treated with 2.5 µg/ml gAPN for 6 h followed by
incubation with 100 µg/ml SUA for 48 h. AdipoR1 protein
expression and AMPK phosphorylation at threonine-172 were assessed.
The results suggested that SUA significantly increased AdipoR1
protein expression and AMPK phosphorylation in PTECs compared with
the untreated control (P<0.05 and P<0.01, respectively;
Fig. 5). gAPN treatment further
significantly increased AdipoR1 protein and AMPK phosphorylation
levels compared with the SUA-treated cells (P<0.01 and
P<0.05, respectively).
AdipoR1 knockdown amplifies SUA-induced
inflammatory responses in PTECs
To clarify whether AdipoR1 is involved in the
anti-inflammatory effects of APN, PTECs were subjected to 24 h
transfection with AdipoR1-shRNA or scramble-shRNA prior to SUA
treatment (100 µg/ml) for 48 h. AdipoR1-shRNA transfection
significantly decreased AdipoR1 mRNA and protein levels compared
with the untreated control by 32.33 and 61.45% (P<0.05 and
P<0.001, respectively; Fig. 6A and
B). No significant difference between the scramble-shRNA and
the AdipoR1-shRNA was detected at mRNA level (P>0.05); however,
protein levels were significantly different (P<0.001).
AdipoR1-shRNA transfection markedly increased NLRP3 protein levels
compared with the SUA-treated cells (P>0.05; Fig. 6C and D). AdipoR1 knockdown
significantly increased synthesis and secretion of TNFα (P<0.05
and P<0.001, respectively; Fig.
6C, E and G) and increased IL-1β secretion from cultured PTECs
compared with the SUA-treated cells (P<0.01; Fig. 6F). The data indicated that AdipoR1
knockdown further promoted inflammatory responses induced by SUA in
PTECs.
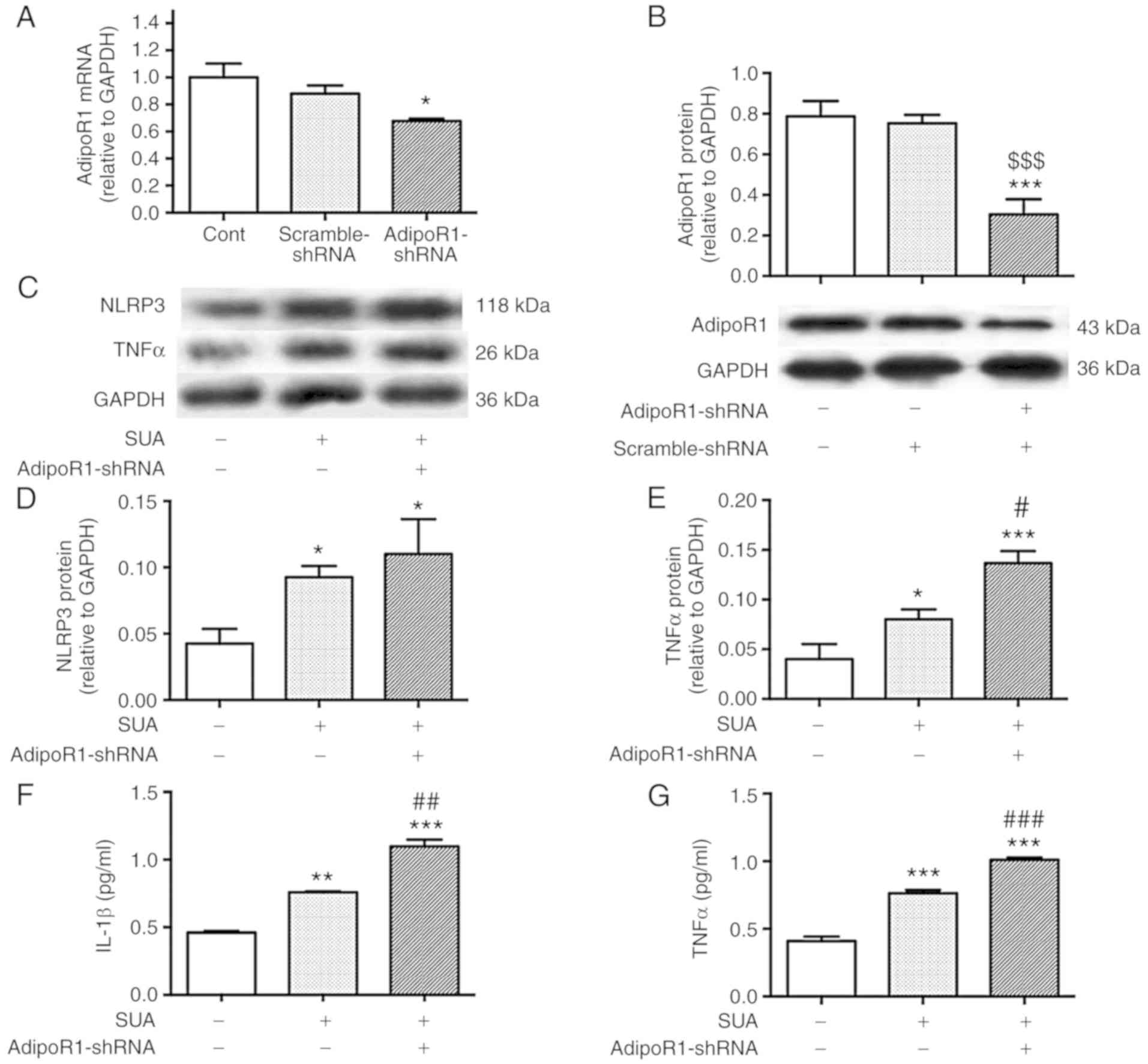 | Figure 6AdipoR1 knockdown promotes
inflammation in PTECs induced by SUA. PTECs were transfected with
AdipoR1-shRNA or scramble-shRNA prior to treatment with SUA (100
µg/ml) for 48 h (n=3). AdipoR1 (A) mRNA and (B) protein
expression in shRNA transfected cells. (C) Western blot images and
quantified protein expression of (D) NLRP3 and (E) TNFα. Levels of
(F) IL-1β and (G) TNFα in cell supernatants measured using ELISA.
Data are representative of three independent experiments.
*P<0.05, **P<0.01 and
***P<0.001 vs. Cont; #P<0.05,
##P<0.01 and ###P<0.001 vs. SUA;
$$$P<0.001 vs. scramble-shRNA. APN, adiponectin;
PTEC, proximal renal tubular epithelial cell; SUA, soluble uric
acid; NLRP3, NACHT, leucine rich repeat and pyrin domain-containing
protein 3; TNF, tumor necrosis factor; IL, interleukin; Cont,
untreated cells; sh, short hairpin; AdipoR1, APN receptor 1. |
AMPK inhibitor promotes SUA-induced TNFα
secretion from PTECs
To test whether AMPK signaling was associated with
APN-induced resistance to SUA-induced inflammation, PTECs were
subjected to incubation with compound C (10 mM), a specific AMPK
inhibitor, for 90 min followed by exposure to SUA (100
µg/ml) for 48 h. Compound C administration further promoted
the SUA-induced inflammatory responses (Fig. 7). Protein expression of NLRP3 and
TNFα was slightly increased by the inhibitor treatment compared
with the SUA-treated cells (P>0.05; Fig. 7A–C). Secretion of IL-1β was
further markedly increased by the inhibitor treatment (P>0.05;
Fig. 7D), while TNFα secretion
exhibited a significant increase in the compound C treated cells
compared with the SUA-treated cells (P<0.05; Fig. 7E). The data indicated that an AMPK
inhibitor partially increased the proinflammatory reaction induced
by SUA in cultured PTECs.
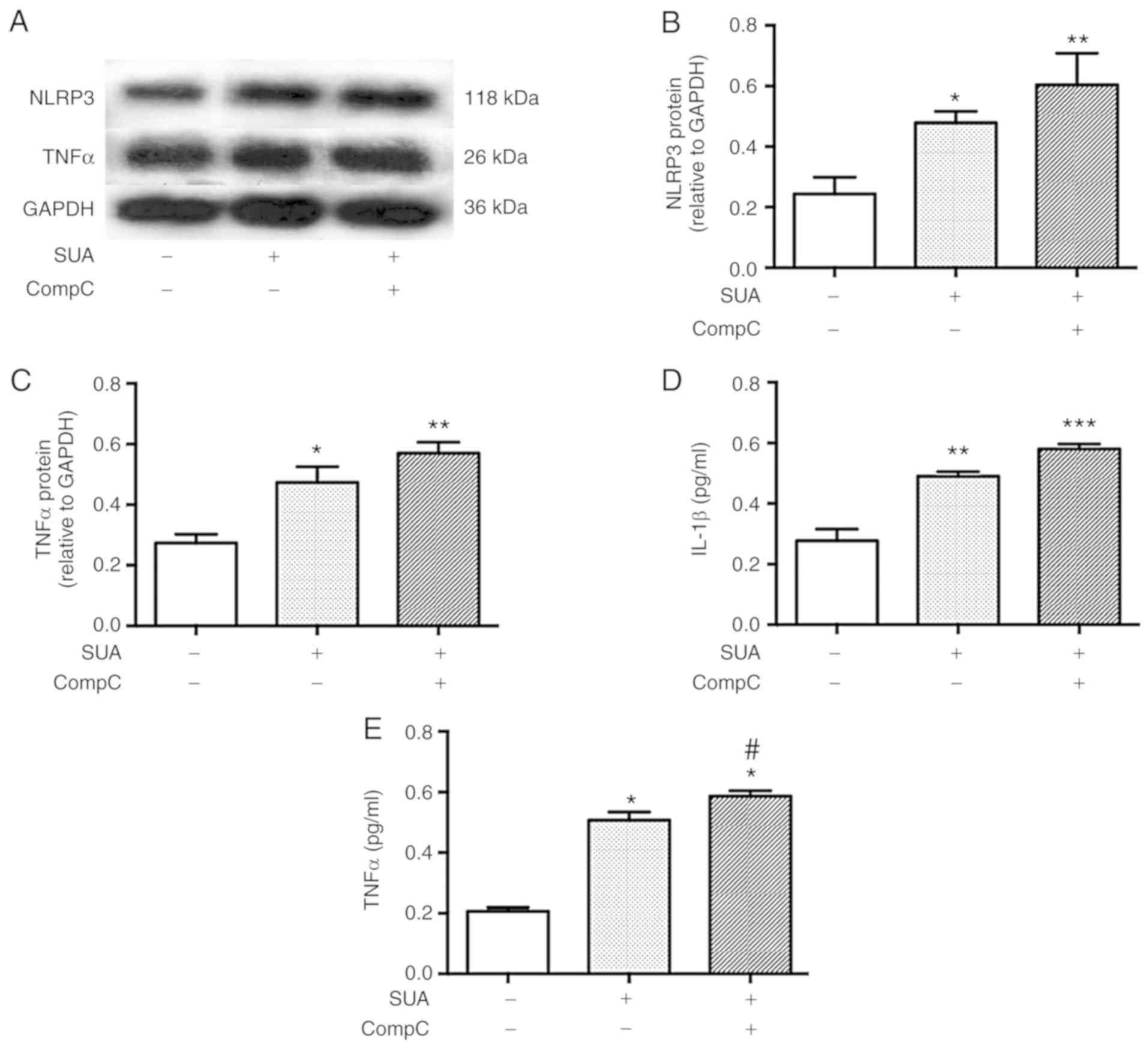 | Figure 7AMPK inhibitor aggravates SUA-induced
inflammation in PTECs. PTECs were preincubated with compound C (10
µM) for 90 min prior to stimulation with SUA (100
µg/ml) for 48 h (n=3). (A) Western blot images and
quantified protein expression of (B) NLRP3 and (C) TNFα. Levels of
(D) IL-1β and (E) TNFα in cell supernatants measured using ELISA.
Data are representative of three independent experiments.
*P<0.05, **P<0.01 and
***P<0.001 vs. Cont; and #P<0.05
vs. SUA. APN, adiponectin; PTEC, proximal renal tubular
epithelial cell; SUA, soluble uric acid; NLRP3, NACHT, leucine rich
repeat and pyrin domain-containing protein 3; TNF, tumor necrosis
factor; IL, interleukin; Cont, untreated cells; sh, short hairpin;
AdipoR1, APN receptor 1; CompC, compound C; AMPK, adenosine
monophosphate-activated protein kinases. |
Discussion
The present study demonstrated that adiponectin
(APN) induced a resistance to SUA-triggered inflammation in PTECs.
APN supplementation suppressed SUA-induced inflammatory mediators
partially via the AdipoR1/AMPK signaling pathway. Both AdipoR1
knockdown and AMPK inhibition intensified the SUA-induced release
of proinflammatory cytokines. To the best of our knowledge, this
report is the first report describing regulatory function of APN in
renal tubular injury in the context of SUA stimulation. The
findings of the present study may assist in clarifying the
pathogenesis of SUA-associated renal disease.
Previous studies have revealed that SUA triggers
renal inflammation (8,34) by activating the NLRP3/IL-1β
signaling pathway (8) and
amplifying TNFα mRNA expression (7) as a result of nuclear factor (NF)-κB
activation (35,36). However, the mechanism by which
tubular inflammation may be inhibited is poorly understood. In the
present study, it was observed that APN supplementation alleviated
the production and release of TNFα in SUA-treated PTECs, similar to
the findings described for cardiomyocytes, adipocytes (37) and macrophages (38,39). APN was reported to decrease TNFα
production in myocytes under ischemia-reperfusion or LPS exposure
(36,37,39) and APN deficiency increases TNFα
levels in ischemic-reperfused heart tissue (37). Accumulating evidence has
demonstrated that APN exerts renoprotective effects through
inhibiting NF-κB activity in high glucose- or TNFα-treated renal
mesangial cells (28) and
alleviating NF-κB-associated inflammatory responses induced by
angiotensin II (27). In
contrast, APN depletion exacerbates inflammatory damage and
upregulates the intrarenal levels of proinflammatory factors in
diabetic (28) and subtotal
nephrectomized mice (25). TNFα
stimulates APN expression, which in turn increases IL-8 release via
AdipoR1, in lung epithelial cells (40). These findings indicate that TNFα
induces an APN-AdipoR1-dependent proinflammatory effect in lungs
(40). Conversely,
numerous studies have indicated that APN exerts anti-inflammatory
effects via suppression of TNFα synthesis and induction of
anti-inflammatory cytokines (41-43). These observations are consistent
with the findings of the present study suggesting that gAPN
suppressed the release of TNFα and IL-1β from SUA-treated PTECs.
Unlike results presented by Miller et al (40), TNFα and APN levels were
simultaneously increased in SUA-treated PTECs compared with the
untreated control. The regulatory effect of APN on inflammatory
factors may depend on the targeted cell type, dose and the APN
stimulator. It is hypothesized that SUA-stimulated TNFα amplifies
APN expression via a positive feedback loop and overexpressed APN
reacts to alleviate inflammation. Direct evidence of TNFα-induced
APN expression in SUA-treated renal cells requires further
elucidation.
IL-1β induces proinflammatory activity (44). Maturation of IL-1β is controlled
by the NF-κB-dependent production and the NLRP3
inflammasome-dependent proteolytic processing of pro-IL-1β
(45). Notably, it was observed
that gAPN administration inhibited IL-1β release without
significantly influencing NLRP3 production. IL-1β may be an adaptor
molecule of the NLRP3 inflammasome complex, similar to
apoptosis-associated speck-like protein containing a caspase
recruitment domain or caspase-1, instead of an NLRP3-controled
maturation of IL-1β in SUA-exposed PTECs. Current findings
indicated a potent anti-inflammatory function of APN in SUA-exposed
PTECs.
Cellular APN secreted from HK-2 cells (<2 ng/ml)
(19) and PTECs (70 ng/ml;
present study) was decreased compared with plasma APN from healthy
volunteers (1.9 to 17.0 mg/ml) (46). Findings of the present study
demonstrated that SUA increased inflammatory cytokine expression in
the presence of endogenous APN in PTECs, similar to the
lipopolysaccharide-induced NF-κB upregulation in HK-2 cells
observed previously (29).
Anti-inflammatory effects of APN were determined in the present
study for gAPN supplementation at a dose of 2.5 µg/ml; this
dose is increased compared with the endogenous APN concentration
determined previously (<2 ng/ml) (19). Findings suggested a
dose-associated biological effect of APN and clarify why a moderate
amount of endogenous APN, 70 ng/ml in the present study, did not
inhibit SUA-induced inflammation. These comparisons may be regarded
with caution due to the data originating from various cell systems
with dissimilar APN doses and treatment durations. Future
experiments may investigate whether PTECs produce elastases
involved in cleaving APN and generating its globular domain at the
amino-terminal collagenous domain as suggested previously (47). Further research is required to
analyze whether UA further affects these processes.
AdipoR1 and AdipoR2 are well-known APN receptors,
which have been detected in renal tubular cells (19,48). AdipoR1 levels are increased
compared with AdipoR2 (48),
suggesting a superior function association with APN. A previous
study on 3T3-L1 preadipocytes demonstrated distinctly increased
mRNA levels of AdipoR1 compared with AdipoR2 (49). AdipoR1 exhibits a high affinity
for gAPN (23) and contributes to
the phosphorylation and activation of AMPK (23,50,51). In the present study effects of
gAPN treatment on AdipoR1 expression were evaluated. gAPN
upregulated AdipoR1 protein expression while reducing synthesis and
secretion of inflammatory mediators in SUA-treated PTECs. These
data indicated that AdipoR1 may be involved in the resistance
conferred by APN to SUA-induced renal tubular inflammation and may
advice the potential use of AdipoR1 in urate nephropathy.
AMPK, a sensitive energy sensor, participates in
various pathophysiological processes and mediates beneficial
actions of APN (18,20,22,37). It was demonstrated that AMPK
activity is decreased in the hearts of APN-KO mice (37). AMPK deficiency further abrogated
antiapoptotic activities of APN in cardiomyocytes following
hypoxia-reoxygenation (37). The
current research demonstrated that gAPN administration limited
inflammatory responses and enhanced AMPK phosphorylation in
SUA-treated PTECs, suggesting a positive involvement of AMPK in
APN-mediated anti-inflammatory processes. Taken together, these
findings imply that AMPK may mediate renoprotective effects of APN
(52-54). Future studies are required to
investigate the effect of gAPN-AMPK inhibitor co-treatment on PTECs
to determine whether treatment with an AMPK-inhibitor abolishes
APN-induced inhibition of inflammation following SUA treatment. A
gene KO procedure may be performed to support conclusions drawn
from reports of AMPK-independent effects of compound C in various
cells (55,56). The low efficiency of shRNA
transfections in the present study may be the reason for the
inability of the AdipoR1-shRNA to fully suppress AMPK activity. The
presence of AdipoR2 (19,48) may provide an additional
explanation for minor changes in AMPK phosphorylation.
IR is essential in hyperuricemia-induced renal
tubular nephrolithiasis (11,12). Furthermore, abnormalities in APN
signaling and IR affect each other. Hypoadiponectinemia contributes
to diabetes and metabolic syndrome (16), which are characterized by IR.
Conversely, IR impairs AdipoR1 transcription (17) and AMPK activation, decreasing the
response sensitivity of APN (16). APN/AdipoR1/AMPK signaling pathway
disruptions may partially describe physiological conditions
associated with IR. Therefore, the present study mimicked IR by
treatment with AdipoR1-shRNA and AMPK inhibitor, which augmented
the release of inflammatory cytokines from SUA-treated PTECs. The
results suggested that abnormal APN signaling and IR may increase
the vulnerability of PTECs to SUA-induced inflammation. However,
the present study was a preliminary in vitro study and did
not provide direct data on IR. Future investigations using a
hyperuricemic model may evaluate the extent of IR and potential
AMPK targets in AdipoOR1 and AMPK KO cells. Additionally,
interventions in a hyperuricemic animal model with and without IR
may be performed to elucidate whether alleviating IR ameliorates
UA-induced renal tubular inflammatory injury.
Several limitations still exist in the present
study. AdipoR1 signaling differentially modulated protein
expression of TNFα, IL-1β and NLRP3. This raises the question
whether other signaling pathways are involved in the regulation of
TNFα and whether alternative receptors for APN, including AdipoR2
or T-cadherin (57), are involved
in the regulation of NLRP3 expression. There was a non-significant
difference at AdipoR1 mRNA levels between the scramble- and the
AdipoR1-shRNA. This may be partially associated with off-target
effects. Further study will focus on modifying the shRNA design to
improve the knockdown efficiency. Furthermore, the present study
did not explore if alternative components of the mature NLRP3
inflammasome were involved in anti-inflammatory effects exerted by
APN. Further study will address this question using RT-qPCR and
western blot analysis. Additionally, control experiments studying
PTECs pretreated with APN in the absence of SUA were not performed
and therefore, direct effects of APN on AMPK or inflammatory
factors, as previously described, were not evaluated here (27,39). The underlying mechanism by which
APN is upregulated in SUA-treated PTECs remains to be
elucidated.
In summary, the present study indicated that APN
exerted protective effects against SUA-induced inflammation in
PTECs at least partially via the AdipoR1/AMPK signaling pathway
(Fig. 8).
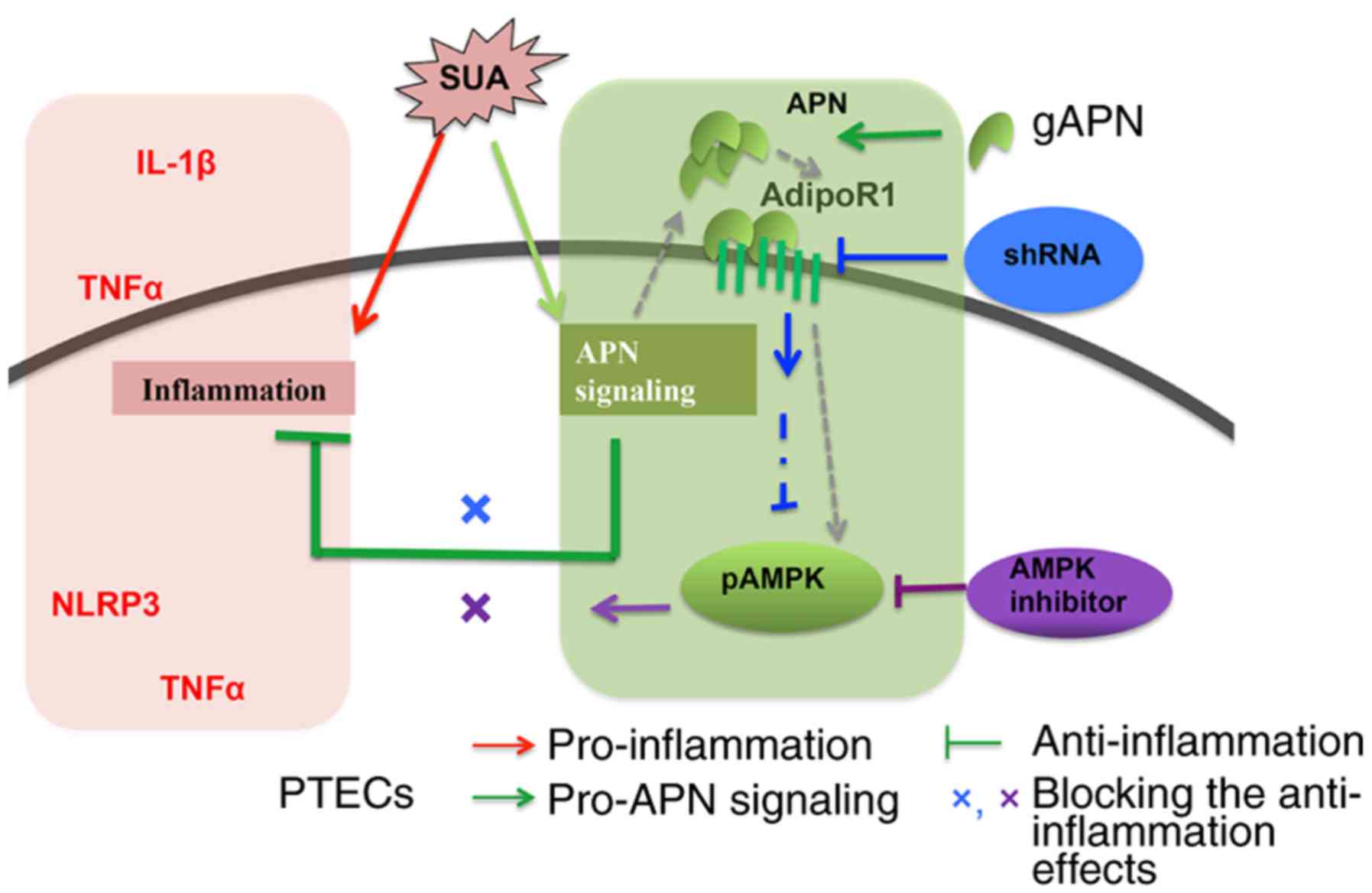 | Figure 8Proposed model for APN inhibition of
inflammatory responses in PTECs induced by SUA. SUA evokes
inflammatory responses and induces protein expression of NLRP3 and
TNFα, and the release of TNFα and IL-1β. APN, upregulated by SUA,
inhibits expression of inflammatory factors and promotes AdipoR1
synthesis and AMPK activation. AdipoR1 knockdown and AMPK
inhibition increase proinflammatory mediators. APN, adiponectin; g,
globular; AdipoR1, ANP receptor 1; PTEC, proximal renal tubular
epithelial cell; SUA, soluble uric acid; NLRP3, NACHT, leucine rich
repeat and pyrin domain-containing protein 3; TNF, tumor necrosis
factor; IL, interleukin; g, globular; AMPK, adenosine
monophosphate-activated protein kinases; p, phosphorylated; sh,
short hairpin. |
Funding
The present study was supported by the Shanghai
Municipal Commission of Health and Family Planning (grant no.
201840271), the Key Developing Disciplines (grant no. 2015ZB0501),
and the Jinshan Science and Technology Committee of Shanghai (grant
no. 2016-3-02).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ZY and QY conceived and designed the experiments. QY
and JX performed the experiments. CF and XZ analyzed the data. QY,
JZ and ZZ interpreted data and prepared the manuscript. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Liu R, Han C, Wu D, Xia X, Gu J, Guan H,
Shan Z and Teng W: Prevalence of hyperuricemia and gout in mainland
China from 2000 to 2014: A systematic review and meta-analysis.
BioMed Res Int. 2015:7628202015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mohandas R and Johnson RJ: Uric acid
levels increase risk for new-onset kidney disease. J Am Soc
Nephrol. 19:2251–2253. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ficociello LH, Rosolowsky ET, Niewczas MA,
Maselli NJ, Weinberg JM, Aschengrau A, Eckfeldt JH, Stanton RC,
Galecki AT, Doria A, et al: High-normal serum uric acid increases
risk of early progressive renal function loss in type 1 diabetes:
Results of a 6-year follow-up. Diabetes Care. 33:1337–1343. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang H, Wei Y, Kong X and Xu D: Effects of
urate-lowering therapy in hyperuricemia on slowing the progression
of renal function: A meta-analysis. J Ren Nutr. 23:389–396. 2013.
View Article : Google Scholar
|
|
5
|
Ito S, Naritomi H, Ogihara T, Shimada K,
Shimamoto K, Tanaka H and Yoshiike N: Impact of serum uric acid on
renal function and cardiovascular events in hypertensive patients
treated with losartan. Hypertens Res. 35:867–873. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Johnson RJ, Nakagawa T, Jalal D,
Sanchez-Lozada LG, Kang DH and Ritz E: Uric acid and chronic kidney
disease: Which is chasing which? Nephrol Dial Transplant.
28:2221–2228. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou Y, Fang L, Jiang L, Wen P, Cao H, He
W, Dai C and Yang J: Uric acid induces renal inflammation via
activating tubular NF-κB signaling pathway. PLoS One. 7:e397382012.
View Article : Google Scholar
|
|
8
|
Xiao J, Zhang XL, Fu C, Han R, Chen W, Lu
Y and Ye Z: Soluble uric acid increases NALP3 inflammasome and
interleukin-1β expression in human primary renal proximal tubule
epithelial cells through the Toll-like receptor 4-mediated pathway.
Int J Mol Med. 35:1347–1354. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mangge H, Zelzer S, Puerstner P, Schnedl
WJ, Reeves G, Postolache TT and Weghuber D: Uric acid best predicts
metabolically unhealthy obesity with increased cardiovascular risk
in youth and adults. Obesity. 21:E71–E77. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Z, Que S, Zhou L and Zheng S:
Dose-response relationship of serum uric acid with metabolic
syndrome and non-alcoholic fatty liver disease incidence: A
meta-analysis of prospective studies. Sci Rep. 5:143252015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Daudon M, Traxer O, Conort P, Lacour B and
Jungers P: Type 2 diabetes increases the risk for uric acid stones.
J Am Soc Nephrol. 17:2026–2033. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Iba A, Kohjimoto Y, Mori T, Kuramoto T,
Nishizawa S, Fujii R, Nanpo Y, Matsumura N, Shintani Y, Inagaki T,
et al: Insulin resistance increases the risk of urinary stone
formation in a rat model of metabolic syndrome. BJU Int.
106:1550–1554. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Scherer PE, Williams S, Fogliano M,
Baldini G and Lodish HF: A novel serum protein similar to C1q,
produced exclusively in adipocytes. J Biol Chem. 270:26746–26749.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu E, Liang P and Spiegelman BM: AdipoQ is
a novel adipose-specific gene dysregulated in obesity. J Biol Chem.
271:10697–10703. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maeda K, Okubo K, Shimomura I, Funahashi
T, Matsuzawa Y and Matsubara K: cDNA cloning and expression of a
novel adipose specific collagen-like factor, apM1 (AdiPose Most
abundant Gene transcript 1). Biochem Biophys Res Commun.
221:286–289. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kadowaki T, Yamauchi T, Kubota N, Hara K,
Ueki K and Tobe K: Adiponectin and adiponectin receptors in insulin
resistance, diabetes, and the metabolic syndrome. J Clin Invest.
116:1784–1792. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun X, He J, Mao C, Han R, Wang Z, Liu Y
and Chen Y: Negative regulation of adiponectin receptor 1 promoter
by insulin via a repressive nuclear inhibitory protein element.
FEBS Lett. 582:3401–3407. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sharma K, Ramachandrarao S, Qiu G, Usui
HK, Zhu Y, Dunn SR, Ouedraogo R, Hough K, McCue P, Chan L, et al:
Adiponectin regulates albuminuria and podocyte function in mice. J
Clin Invest. 118:1645–1656. 2008.PubMed/NCBI
|
|
19
|
Perri A, Vizza D, Lofaro D, Gigliotti P,
Leone F, Brunelli E, Malivindi R, De Amicis F, Romeo F, De Stefano
R, et al: Adiponectin is expressed and secreted by renal tubular
epithelial cells. J Nephrol. 26:1049–1054. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cammisotto PG and Bendayan M: Adiponectin
stimulates phosphorylation of AMP-activated protein kinase alpha in
renal glomeruli. J Mol Histol. 39:579–584. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Park HS, Lim JH, Kim MY, Kim Y, Hong YA,
Choi SR, Chung S, Kim HW, Choi BS, Kim YS, et al: Resveratrol
increases AdipoR1 and AdipoR2 expression in type 2 diabetic
nephropathy. J Translat Med. 14:1762016. View Article : Google Scholar
|
|
22
|
Tan M, Tang G and Rui H: Adiponectin
attenuates Ang II- induced TGFβ1 production in human mesangial
cells via an AMPK-dependent pathway. Biotechnol Appl Biochem.
62:848–854. 2015. View Article : Google Scholar
|
|
23
|
Yamauchi T, Kamon J, Ito Y, Tsuchida A,
Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et
al: Cloning of adiponectin receptors that mediate antidiabetic
metabolic effects. Nature. 423:762–769. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rutkowski JM, Wang ZV, Park AS, Zhang J,
Zhang D, Hu MC, Moe OW, Susztak K and Scherer PE: Adiponectin
promotes functional recovery after podocyte ablation. J Am Soc
Nephrol. 24:268–282. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ohashi K, Iwatani H, Kihara S, Nakagawa Y,
Komura N, Fujita K, Maeda N, Nishida M, Katsube F, Shimomura I, et
al: Exacerbation of albuminuria and renal fibrosis in subtotal
renal ablation model of adiponectin-knockout mice. Arterioscler
Thromb Vasc Biol. 27:1910–1917. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Su YX, Deng HC, Zhang MX, Long J and Peng
ZG: Adiponectin inhibits PDGF-induced mesangial cell proliferation:
Regulation of mammalian target of rapamycin-mediated survival
pathway by adenosine 5-monophosphate-activated protein kinase. Horm
Metab Res. 44:21–27. 2012. View Article : Google Scholar
|
|
27
|
Fang F, Liu GC, Kim C, Yassa R, Zhou J and
Scholey JW: Adiponectin attenuates angiotensin II-induced oxidative
stress in renal tubular cells through AMPK and cAMP-Epac signal
transduction pathways. Am J Physiol Renal Physiol. 304:F1366–F1374.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fang F, Bae EH, Hu A, Liu GC, Zhou X,
Williams V, Maksimowski N, Lu C, Konvalinka A, John R, et al:
Deletion of the gene for adiponectin accelerates diabetic
nephropathy in the Ins2+/C96Y mouse. Diabetologia.
58:1668–1678. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Perri A, Vizza D, Lupinacci S, Toteda G,
De Amicis F, Leone F, Gigliotti P, Lofaro D, La Russa A and
Bonofiglio R: Adiponectin secreted by tubular renal cells during
LPS exposure worsens the cellular inflammatory damage. J Nephrol.
29:185–194. 2016. View Article : Google Scholar
|
|
30
|
Jin X, Chen J, Hu Z, Chan L and Wang Y:
Genetic deficiency of adiponectin protects against acute kidney
injury. Kidney Int. 83:604–614. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Webb R, Jeffries M and Sawalha AH: Uric
acid directly promotes human T-cell activation. Am J Med Sci.
337:23–27. 2009. View Article : Google Scholar
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
33
|
Yang Q, Fu C, Xiao J and Ye Z: Uric acid
upregulates the adiponectinadiponectin receptor 1 pathway in renal
proximal tubule epithelial cells. Mol Med Rep. 17:3545–3554.
2018.PubMed/NCBI
|
|
34
|
Vilaysane A, Chun J, Seamone ME, Wang W,
Chin R, Hirota S, Li Y, Clark SA, Tschopp J, Trpkov K, et al: The
NLRP3 inflammasome promotes renal inflammation and contributes to
CKD. J Am Soc Nephrol. 21:1732–1744. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Han HJ, Lim MJ, Lee YJ, Lee JH, Yang IS
and Taub M: Uric acid inhibits renal proximal tubule cell
proliferation via at least two signaling pathways involving PKC,
MAPK, cPLA2, and NF-kappaB. Am J Physiol Renal Physiol.
292:F373–F381. 2007. View Article : Google Scholar
|
|
36
|
Yang Z, Xiaohua W, Lei J, Ruoyun T,
Mingxia X, Weichun H, Li F, Ping W and Junwei Y: Uric acid
increases fibronectin synthesis through upregulation of lysyl
oxidase expression in rat renal tubular epithelial cells. Am J
Physiol Renal Physiol. 299:F336–346. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shibata R, Sato K, Pimentel DR, Takemura
Y, Kihara S, Ohashi K, Funahashi T, Ouchi N and Walsh K:
Adiponectin protects against myocardial ischemia-reperfusion injury
through AMPK- and COX-2-dependent mechanisms. Nat Med.
11:1096–1103. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wulster-Radcliffe MC, Ajuwon KM, Wang J,
Christian JA and Spurlock ME: Adiponectin differentially regulates
cytokines in porcine macrophages. Biochem Biophys Res Commun.
316:924–929. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ouchi N and Walsh K: Adiponectin as an
anti-inflammatory factor. Clin Chim Acta. 380:24–30. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Miller M, Cho JY, Pham A, Ramsdell J and
Broide DH: Adiponectin and functional adiponectin receptor 1 are
expressed by airway epithelial cells in chronic obstructive
pulmonary disease. J Immunol. 182:684–691. 2009. View Article : Google Scholar
|
|
41
|
Whitehead JP, Richards AA, Hickman IJ,
Macdonald GA and Prins JB: Adiponectin - a key adipokine in the
metabolic syndrome. Diabetes Obes Metab. 8:264–280. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tilg H and Moschen AR: Adipocytokines:
Mediators linking adipose tissue, inflammation and immunity. Nat
Rev Immunol. 6:772–783. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Moore KW, de Waal Malefyt R, Coffman RL
and O'Garra A: Interleukin-10 and the interleukin-10 receptor. Annu
Rev Immunol. 19:683–765. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zielinski CE, Mele F, Aschenbrenner D,
Jarrossay D, Ronchi F, Gattorno M, Monticelli S, Lanzavecchia A and
Sallusto F: Pathogen-induced human TH17 cells produce IFN-gamma or
IL-10 and are regulated by IL-1β. Nature. 484:514–518. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bauernfeind FG, Horvath G, Stutz A,
Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks
BG, Fitzgerald KA, et al: Cutting edge: NF-kappaB activating
pattern recognition and cytokine receptors license NLRP3
inflammasome activation by regulating NLRP3 expression. J Immunol.
183:787–791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Arita Y, Kihara S, Ouchi N, Takahashi M,
Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K,
et al: Paradoxical decrease of an adipose-specific protein,
adiponectin, in obesity. Biochem Biophys Res Commun. 257:79–83.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Waki H, Yamauchi T, Kamon J, Kita S, Ito
Y, Hada Y, Uchida S, Tsuchida A, Takekawa S and Kadowaki T:
Generation of globular fragment of adiponectin by leukocyte
elastase secreted by monocytic cell line THP-1. Endocrinology.
146:790–796. 2005. View Article : Google Scholar
|
|
48
|
Shen YY, Hughes JT, Charlesworth JA, Kelly
JJ and Peake PW: Adiponectin is present in the urine in its native
conforation, and specifically reduces the secretion of MCP-1 by
proximal tubular cells. Nephrology. 13:405–410. 2008. View Article : Google Scholar
|
|
49
|
Lazra Y, Falach A, Frenkel L, Rozenberg K,
Sampson S and Rosenzweig T: Autocrine/paracrine function of
globular adiponectin: Inhibition of lipid metabolism and
inflammatory response in 3T3-L1 adipocytes. J Cell Biochem.
116:754–766. 2015. View Article : Google Scholar
|
|
50
|
Iwabu M, Yamauchi T, Okada-Iwabu M, Sato
K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata
M, et al: Adiponectin and AdipoR1 regulate PGC-1α and mitochondria
by Ca2+ and AMPK/SIRT1. Nature. 464:1313–1319. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhou L, Deepa SS, Etzler JC, Ryu J, Mao X,
Fang Q, Liu DD, Torres JM, Jia W, Lechleiter JD, et al: Adiponectin
activates AMP-activated protein kinase in muscle cells via
APPL1/LKB1-dependent and phospholipase
C/Ca2+/Ca2+/calmodulin-dependent protein
kinase kinase-dependent pathways. J Biol Chem. 284:22426–22435.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang Y, Viollet B, Terkeltaub R and
Liu-Bryan R: AMP-activated protein kinase suppresses urate
crystal-induced inflammation and transduces colchicine effects in
macrophages. Ann Rheum Dis. 75:286–294. 2016. View Article : Google Scholar
|
|
53
|
Liu Y, Palanivel R, Rai E, Park M, Gabor
TV, Scheid MP, Xu A and Sweeney G: Adiponectin stimulates autophagy
and reduces oxidative stress to enhance insulin sensitivity during
high-fat diet feeding in mice. Diabetes. 64:36–48. 2015. View Article : Google Scholar
|
|
54
|
Gao H, Fall T, van Dam RM, Flyvbjerg A,
Zethelius B, Ingelsson E and Hägg S: Evidence of a causal
relationship between adiponectin levels and insulin sensitivity: A
mendelian randomization study. Diabetes. 62:1338–1344. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Vucicevic L, Misirkic M, Janjetovic K,
Vilimanovich U, Sudar E, Isenovic E, Prica M, Harhaji-Trajkovic L,
Kravic-Stevovic T, Bumbasirevic V, et al: Compound C induces
protective autophagy in cancer cells through AMPK
inhibition-independent blockade of Akt/mTOR pathway. Autophagy.
7:40–50. 2011. View Article : Google Scholar
|
|
56
|
Kudo TA, Kanetaka H, Mizuno K, Ryu Y,
Miyamoto Y, Nunome S, Zhang Y, Kano M, Shimizu Y and Hayashi H:
Dorsomorphin stimulates neurite outgrowth in PC12 cells via
activation of a protein kinase A-dependent MEK-ERK1/2 signaling
pathway. Genes Cells. 16:1121–1132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Takeuchi T, Adachi Y, Ohtsuki Y and
Furihata M: Adiponectin receptors, with special focus on the role
of the third receptor, T-cadherin, in vascular disease. Med Mol
Morphol. 40:115–120. 2007. View Article : Google Scholar : PubMed/NCBI
|