Introduction
The farnesoid X receptor (FXR) is a bile
acid-activated nuclear receptor, which regulates bile acid
synthesis and transport and the metabolism of cholesterol, the bile
acid precursor (1-3). In mouse models, the activation of
FXR attenuates the development of atherosclerosis and reduces
plasma total cholesterol (TC) and high-density lipoprotein
cholesterol (HDL-C). FXR is expressed at high levels in the liver,
intestine and adrenal glands. FXR forms a heterodimer with retinoid
X receptor (RXR) to modulate the expression of target genes by
binding to DNA sequences referred to as FXR response elements
(FXREs). The most common DNA-binding motif bound by FXR is the
highly conserved inverted repeat separated by 1 nucleotide (IR-1:
G/AGGTGA A TAACCT) (4-8).
Scavenger receptor class B type I (SR-BI) is a
physiologically relevant HDL receptor and serves distinct roles in
plasma HDL metabolism and transhepatic cholesterol efflux in
preclinical animal models and in humans (9-12).
Previous studies have shown that the activation of FXR by its
natural ligand, chenodeoxycholic acid (4), or synthetic ligands, including
obeticholic acid (6E-CDCA; INT-747;OCA;OCALIVA™) (13), which is approved by the Food and
Drug Administration for treating primary biliary cholangitis,
increases the hepatic expression of SR-BI, which has been
considered the primary mechanism responsible for FXR-mediated
reductions in plasma HDL-C (14).
Of note, different regulatory mechanisms for the FXR-mediated
induced gene expression of SR-BI have been suggested. One study
reported that the activation of FXR by synthetic ligand GW4064
increased the protein levels of the hepatocyte nuclear factor 4α
(HNFα), transcription factor, which led to transcriptional
activation of the SR-BI gene via HNF4α-binding sequences embedded
in the promoter region and intronic sequences of the murine SR-BI
gene (15). Two other studies
identified functional FXR binding sites (IR-1s) in the first intron
of the mouse SR-BI gene, which mediate the FXR-induced expression
of SR-BI in mice (4,16). Furthermore, it was reported that
treating HepG2 cells with GW4064 increased the mRNA levels of
SR-BI. This effect was linked to the binding of FXR to a putative
FXRE site (DR8) in the promoter region of the human SR-BI gene
(17).
Our previous investigations conducted in hamster
models have shown that the activation of FXR by obeticholic acid
(OCA) increases the hepatic expression of SR-BI and accelerates the
removal of circulating HDL-C with increased fecal cholesterol
excretion in hamsters fed a high fat and high cholesterol diet
(18). It was observed that, in
hamsters fed a normal chow diet, OCA treatment induced the
expression of small heterodimer partner (SHP) and other typical
FXR-target genes; however, it did not increase the expression of
SR-BI nor enhance transhepatic cholesterol output. These previous
findings suggest that dietary cholesterol may be involved in
OCA-induced SR-BI transcription in hamsters.
Liver X receptors (LXRs) are ligand-activated
receptors that act as cholesterol sensors (19). LXRs form an obligate heterodimer
with RXR, and the heterodimer binds to LXR response elements
(LXREs) in LXR target genes. LXREs consist of direct repeats of the
consensus half-site sequence AGGTCA separated by four nucleotides
(DR4) (6,20). In mice, the activation of LXRs by
endogenous ligands of oxidized cholesterol derivatives and
pharmaceutical agonists, including GW3965, leads to induction of
genes involved in reverse cholesterol transport and mobilization of
cholesterol, including the ATP binding cassette (ABC) transporter
genes Abca1, Abcg1, Abcg5, Abcg8, and genes involved in bile acid
synthesis, including Cyp7A1 (21,22). However, the mRNA levels of Sr-b1
in the liver were not upregulated by the LXR agonist in chow-fed
mice (23), nor in normolipidemic
hamsters (18).
In the present study, the involvement of LXR in the
OCA-induced upregulated hepatic expression of SR-BI was examined.
By conducting genomic sequence analysis and direct DNA binding
assays, a highly conserved regulatory region was identified in the
first intron of the hamster SR-BI gene, which contains a functional
FXR response element IR-1 motif and an LXRE site separated by 57
base pairs (bp). Luciferase reporter gene activity assays
demonstrated the functional involvement of this critical regulatory
region in SR-BI gene transcription upon activation of FXR and LXR
in a synergistic manner. Experiments in normolipidemic hamsters
showed that hepatic gene expression of SR-BI was not effectively
activated by individual treatment with OCA nor the LXR agonist
GW3965, whereas the co-activation of FXR and LXR by the combined
treatment resulted in significant increases in hepatic mRNA and
protein levels of SR-BI. Taken together, the findings demonstrated
the unprecedented concerted activation of SR-BI gene transcription
by the two nuclear receptors, FXR and LXR, which are master
regulators of bile acid and cholesterol metabolism.
Materials and methods
Cells and reagents
Human hepatoma HepG2 cells were obtained from
American Type Culture Collection (Manassas, VA, USA). Human primary
hepatocytes (HPH) were obtained from Invitrogen; Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). 6E-CDCA was purchased from
Abcam (Cambridge, MA, USA). GW4064 and GW3965 were purchased from
Tocris Bioscience (Bristol, UK). OCA was provided by Intercept
Pharmaceuticals, Inc. (New York, NY, USA). The expression plasmid
for human FXRα was provided by Dr Timothy F. Osborne from Sanford
Burnham Prebys Medical Discovery Institute (La Jolla, CA, USA).
Expression plasmids for human RXRα and LXRα were obtained from
GenScript Corporation (Piscataway, NJ, USA).
Cloning of hamster FXR responsive
intronic sequences and construction of luciferase reporters
Hamster genomic sequences were first analyzed
(NW_004801705. 1:3779075-3853729 Mesocricetus auratus
isolate Golden Hamster female 1 unplaced genomic scaffold,
MesAur1.0 scaffold00102, whole genome shotgun sequence) and three
segments were identified, termed A, B and C, in the first intron of
the SR-BI gene that are homologous to the IR-1-containing regions
of the mouse SR-BI gene (4).
Fragments A, B and C are located from +9,659 to +10,483, from
+19,849 to +20,687, and from +33,252 to +34,052 relative to the
translation start codon, respectively. All fragments were amplified
from hamster genomic DNA (50 ng) by polymerase chain reaction
(PCR). The thermocycling conditions were as follows: Dual-lock DNA
polymerase, 95°C for 2 min (1 cycle); denaturation at 95°C (30
cycles); annealing at 60°C for 1 min (30 cycles) followed by
extension at 72°C for 10 min (1 cycle). The PCR fragment was
initially cloned into the Topo 2.1 vector, and then subcloned into
the pGL4.23 mini luciferase reporter vector to generate reporter
plasmids, denoted as pGL4-ham-SRBI-site A, pGL4-ham-SRBI-site B and
pGL4-ham-SRBI-site C, respectively. Following transformation and
propagation in Escherichia coli, four independent clones of
each reporter plasmid were sequenced to verify the sequence and
orientation of the IR-1-containing fragment.
Using the QuikChange II XL Site-Directed Mutageneis
kit (Agilent Technologies, Inc., Santa Clara, CA, USA), the site B
FXRE mutant plasmid was produced by inducing mutations in four
bases within the IR-1 site, and the site B LXRE mutant plasmid was
produced by mutating three bases in the DR4 site. The cloning
primers and mutated sequences are listed in Table SI.
Genomic DNA sequence alignment
MatInspector version 8.0 (24-26) was used to analyse the sequence of
interest in the site B intronic region of different species.
Constructions of human SR-BI promoter
luciferase reporters
For generation of the SR-BI promoter reporter, a DNA
fragment of 1,106 bp covering the human SR-BI proximal promoter
region between −869 and +237 relative to the transcription start
site (TSS) was amplified from HepG2 genomic DNA and was cloned into
the Topo 2.1 vector, followed by subcloning into pGL3-basic at the
SacI and XhoI sites to yield the promoter reporter
pGL3-hSRBI-1106. Similarly, the plasmid pGL3-hSRBI-1831 was made,
which covered the genomic region between −1,899 and −68. Following
transformation and propagation in E. coli, four independent
clones per reporter construct were sequenced to verify the sequence
and orientation of the promoter fragment.
Luciferase reporter assay
The luciferase reporter assay was performed using
the Firefly Luciferase Reporter® assay system (Promega
Corporation, Madison, WI, USA) according to the manufacturer's
protocol. The HepG2 cells were trans-fected with the designated
reporter plasmids using FuGene 6 transfection reagent (Promega
Corporation). As a control for differences in transfection
efficiency, the pRL-TK plasmid encoding the Renilla
luciferase gene was cotransfected with SR-BI reporter vectors and
was used to normalize the firefly luciferase signal across all
samples. Cells were cultured in MEM containing 0.5% fetal bovine
serum overnight at 37°C and treated with GW4064 (1 µM), OCA
(10 µM), or LXR agonist GW3965 (5 µM) for 24 h. At
~48 h post-transfection or 24 h post-treatment with FXR and LXR
agonists, the cells were lysed with 50 µl of lysis buffer,
followed by measurements of firefly luciferase and Renilla
luciferase activities. The firefly luciferase activity was
normalized to Renilla activity. Four wells were assayed for
each condition. In certain transfection assays, plasmid pCMV-β-gal
was co-transfected with the luciferase reporter (27). The cells were lysed in 50
µl of reporter lysis buffer per well, of which 20 µl
of cell lysate was used to measure β-galactosidase activity
according to the manufacturer's protocol using the β-Galactosidase
Enzyme assay system (cat. no. E2000; Promega Corporation). The
remaining 30 µl of lysate was used to measure the firefly
lucif-erase activity using the luciferase assay system. The
absolute luciferase activity was normalized against β-galactosidase
activity to correct for transfection efficiency.
Electrophoretic mobility shift assay
(EMSA)
Binding of FXRα/RXRα to the hamster SR-BI site
B-FXRE region was assessed using the LightShift Chemiluminescent
EMSA kit (Pierce; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. Human recombinant proteins (FXRα, LXRα and
RXRα) were purchased from Active Motif (Carlsbad CA, USA). Briefly,
a 5′ biotin end-labeled probe identical to the 49-nucleotide region
surrounding the putative FXRE element in the SR-BI intron 1 site B
region was synthesized. The single stranded biotin-labeled probe,
denoted SRBI-FXRE, was hybridized to an unlabeled complement, and
20 fmole of the double-stranded oligonucleotide was incubated for
20 min with purified human recombinant FXRα and RXRα proteins
(100:100 ng; FXRα:RXRα ratio) mixture in vitro. The
protein-DNA complex was resolved on 5% TBE gels and transferred
onto a nylon membrane. Following UV cross-linking and subsequent
washing with 1X wash buffer (included in the kit), the biotin
signal was visualized using chemiluminescence. For the competition
experiments, a 100-fold higher concentration of unlabeled wild-type
(WT) or FXRE-mutant oligos were incubated with the reaction mixture
containing FXRα/RXRα and the biotin-SRBI-FXRE probe. The binding of
LXRα/RXRα to the SRBI-LXRE site was assessed using purified human
recombinant LXRα plus RXRα proteins utilizing the same experimental
procedure as for the FXRE site. The sequences of the EMSA probes
are listed in Table SI.
Animals, diet and drug treatment
All animal experiments were performed according to
procedures approved by the VA Palo Alto Health Care System
Institutional Animal Care and Use Committee (Palo Alto, CA, USA).
Eight-week old male golden Syrian hamsters were purchased from
Harlan Sprague Dawley (Indianapolis, IN, USA). The hamsters were
housed (two animals/cage) under controlled temperature (22°C) and
lighting (12-h light/dark cycle). The animals had free access to
autoclaved water and food (normal chow). OCA or GW3965 was
suspended in 0.5% carboxyl-methyl cellulose (vehicle) and sonicated
at 4°C in a Bioruptor 300 instrument (Diagenode, Inc., Denville,
NJ, USA) for 4-6 cycles of 30 sec ON: 30 sec OFF on a 'medium'
setting with intermittent vortexing.
A total of 20 hamsters, fed a normal chow diet, were
divided into four groups (n=5 per group) and were gavaged once per
day at 9:00 a.m. for 10 days with vehicle, or 10 mg/kg/day OCA, or
GW3965 twice a day (30 mg/kg BID, 9:00 a.m. and 5:00 p.m.), or both
in combination. Following the final treatment, all animals were
sacrificed for collection of 16-h fasting serum and liver tissues
at 9:00 a.m. of day 11. The livers were immediately removed,
weighed, cut into small pieces, and stored at −80°C for RNA and
protein isolations and lipid extraction. Fecal samples were
collected over a 24 h period after 9 days of treatment. Health
parameters, including body weight and food intake, were monitored
and recorded throughout the experimental duration. Fig. S1 shows the experimental
protocol.
Measurement of serum lipids
Standard enzymatic methods were used to determine
the TC and HDL-C with kits purchased from Stanbio Laboratory
(Boerne, TX, USA).
Measurement of hepatic lipids
The frozen liver tissue (50 mg) were homogenized in
1 ml chloroform/methanol (2:1). Following homogenization, lipids
were extracted by rocking the samples overnight at room
temperature, followed by centrifugation at 2,300 × g for 10 min at
room temperature. The supernatant was transferred to a new tube and
mixed with 0.2 ml 0.9% saline. The mixture was then centrifuged at
300 × g for 5 min at room temperature and the lower phase
containing the lipids was transferred into a new tube. The lipid
phase was dried overnight and dissolved in 0.25 ml isopropanol
containing 10% Triton X-100. TC and triglycerides were measured
using kits from Stanbio Laboratory.
Measurement of fecal total bile
acids
The dried feces (20 mg) samples were homogenized and
extracted in 1 ml of 75% ethanol at 50°C for 2 h (18). The extract was centrifuged
(10,000, 3 min at room temperature) and the supernatant was used to
measure total bile acids using a kit from Diazyme, Poway, CA.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total RNA was extracted from liver tissue using the
Quick RNA mini prep kit (Zymo Research Corp., Irvine, CA, USA) and
was reverse-transcribed into cDNA. RT-qPCR analysis was performed
with 50 ng of cDNA template and specific primers using a SYBR-Green
PCR kit (5 µl; Power SYBR®-Green PCR Master mix)
and an ABI Prism 7700 system (Applied Biosystems®;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocols. The thermocycling conditions were as follows: UDG
activation, 50°C for 2 min (1 cycle); Dual-lock DNA polymerase,
95°C for 2 min (1 cycle); denaturation, 95°C for 15 sec (40
cycles); annealing, 60°C for 1 min (40 cycles) and extension, 72°C
for 10 min (1 cycle). The RT-qPCR primers (400 nM) for each gene
are listed in Table SI. Target
mRNA expression in each sample was normalized to the housekeeping
gene GAPDH. The 2−ΔΔCq method was used to calculate
relative mRNA expression levels (28).
Western blot analysis
Approximately 50 mg of frozen liver tissue was
homogenized in 0.3 ml RIPA buffer containing 1 mM PMSF and protease
inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). Protein
concentration was determined via a BCA assay kit (Pierce; Therm
Fisher Scientific, Inc.). The homogenate proteins (75 µg)
from individual liver samples were resolved by SDS-PAGE (10%
Criterion™ Tris-HCl Protein Gel (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) and transferred onto nitrocellulose membranes;
membranes were blocked with 5% non-fat milk (cat. no. 170-6404;
Bio-Rad Laboratories, Inc.). Anti-SR-BI antibody was purchased from
Abcam (cat. no. Ab52629). Anti-β-actin antibody was purchased from
Sigma-Aldrich; Merck KGaA (cat. no. A1978). Primary antibody was
used at 1:1,000 dilution and secondary antibodies (cat. nos. 7074P2
and 7076P2) were used at 1:10,000 dilution. Immunoreactive bands of
predicted molecular mass were visualized using SuperSignal West
substrate (Thermo Fisher Scientific) and quantified using Alpha
View software (version 3.3; Cell Biosciences, Inc., Santa Clara,
CA, USA) with normalization by signals of β-actin.
Statistical analysis
GraphPad Prism 5 (GraphPad Software, Inc., La Jolla,
CA, USA) was used to calculate averages and errors, generate graphs
and perform statistical tests. Values are presented as the mean ±
standard error of the mean. Unless otherwise indicated, on passing
Bartlett's test for equal variances, one-way analysis of variance
with Tukey's multiple comparison test was used to compare groups of
three or more. Student's unpaired two-tailed t-test was used for
two-group comparisons. P<0.05 was considered to indicate a
statistically significant difference.
Results
Identification of a functional FXR
response element (IR-1) in conjunction with an LXRE motif in the
first intron of the hamster SR-BI gene
Our previous study conducted in
hyper-cholesterolemic hamsters suggested that OCA increases the
hepatic expression of SR-BI at the transcriptional level (18). However, how FXR activates SR-BI
gene transcription in the hamster species remains to be
elucidated.
To identify FXR response sequences in the hamster
SR-BI gene, the present study first analyzed hamster genomic
sequences and identified three segments, termed A, B and C, in
intron 1 of the SR-BI gene, which are homologous to the IR-1
containing regions of the mouse SR-BI gene (4). The hamster site A and site B regions
contained a putative IR-1 motif that are identical to the mouse
IR-1 sequence, whereas the IR-1 site in segment C of the mouse
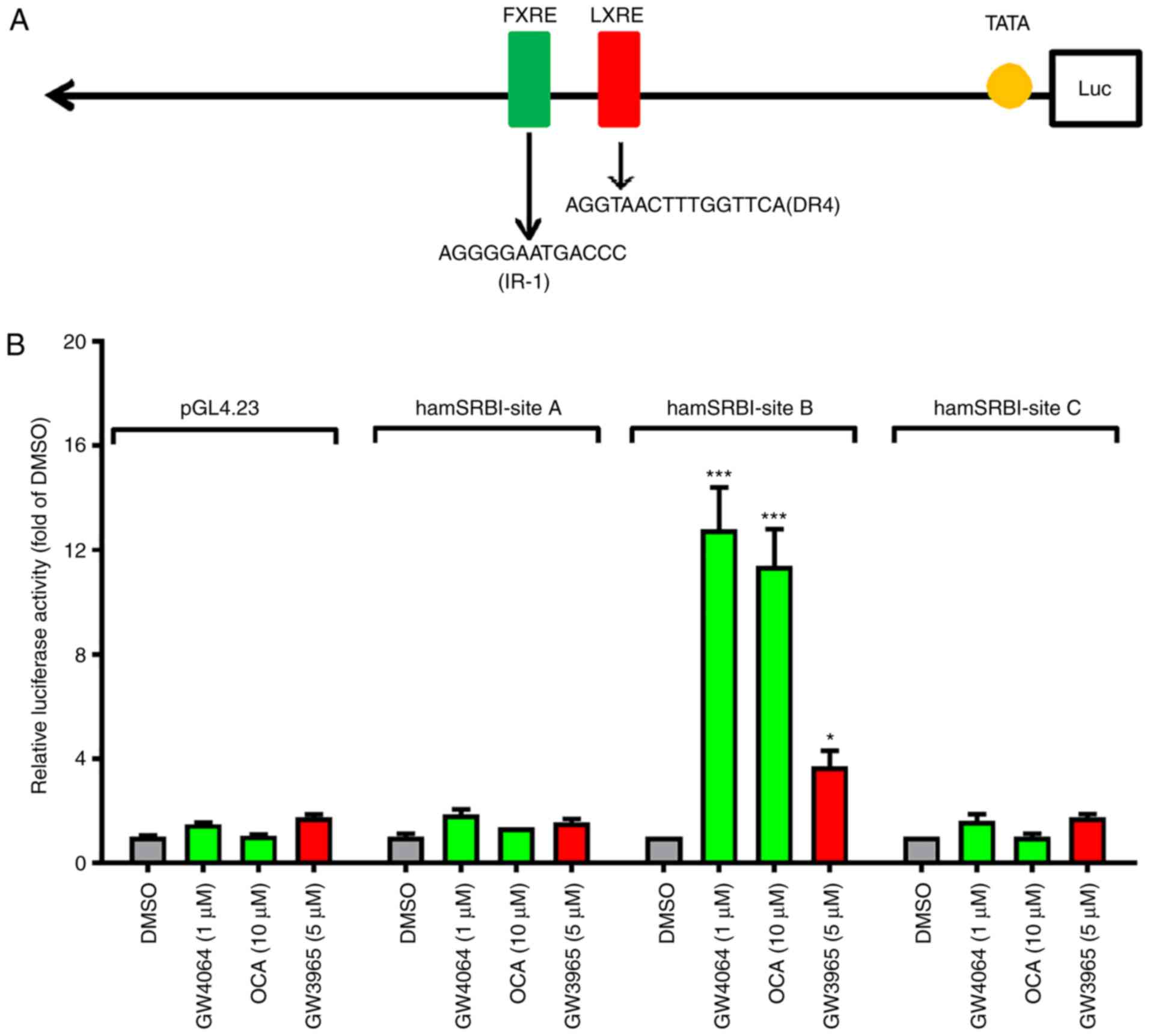
SR-BI gene was not found in the hamster sequence (Fig. 1A). The results of one
representative experiment of luciferase reporter assays conducted
in HepG2 cells are shown in Fig. 1B,
and C shows the summarized results from four separate
transfections. Together, these data demonstrate that the site B
segment was responsive to FXR agonists, and reporter activities
were induced ~15-fold by treating transfected cells with OCA or
GW4064. By contrast, the luciferase activities of site A and site C
were not induced by the activation of FXR over the control vector
pGL4.23 with a minimal promoter containing only a TATA element.
Therefore, these results identified the intron 1 site B region of
the hamster SR-BI gene as the functional regulatory segment that
responds to the ligand induced activation of FXR.
 | Figure 1FXR activates the transcription of
SR-BI via the FXRE motif located in the first intron of the hamster
SR-BI gene. (A) Schematic presentation of luciferase reporter
constructs with intronic sequences of the hamster SR-BI gene. (B)
Reporter constructs were transiently cotransfected with pCMV-β-gal
vector into HepG2 cells with four wells per condition. At 1 day
post-transfection, cells were incubated in 0.5% fetal bovine serum
medium overnight, followed by treatment with GW4064 (1 µM)
or OCA (10 µM) for 24 h. Cells were harvested, and the
luciferase and β-gal activities were measured. Following
normalization, relative luciferase activity of transfected cells
treated with vehicle DMSO is expressed as 1. Statistical
significance among all groups was assessed by one-way ANOVA with
Tukey's multiple comparison test. **P<0.01 and
***P<0.001 between control and ligand-treated group.
(C) Data are summarized results (mean ± standard error of the mean)
of four independent transfection assays. Following normalization,
relative luciferase activity of transfected cells treated with
vehicle DMSO is expressed as 1. Statistical significance among all
groups was assessed by one-way ANOVA with Tukey's multiple
comparison test. **P<0.01 and
***P<0.001 compared with the inducing effects of OCA
or GW4064 on control vector pGL4.23. FXR, farnesoid X receptor;
FXRE, FXR response element; SR-BI, scavenger receptor class B type
I; β-gal, β-galactosidase; OCA, obeticholic acid; ANOVA, analysis
of variance; n.s., not significant. |
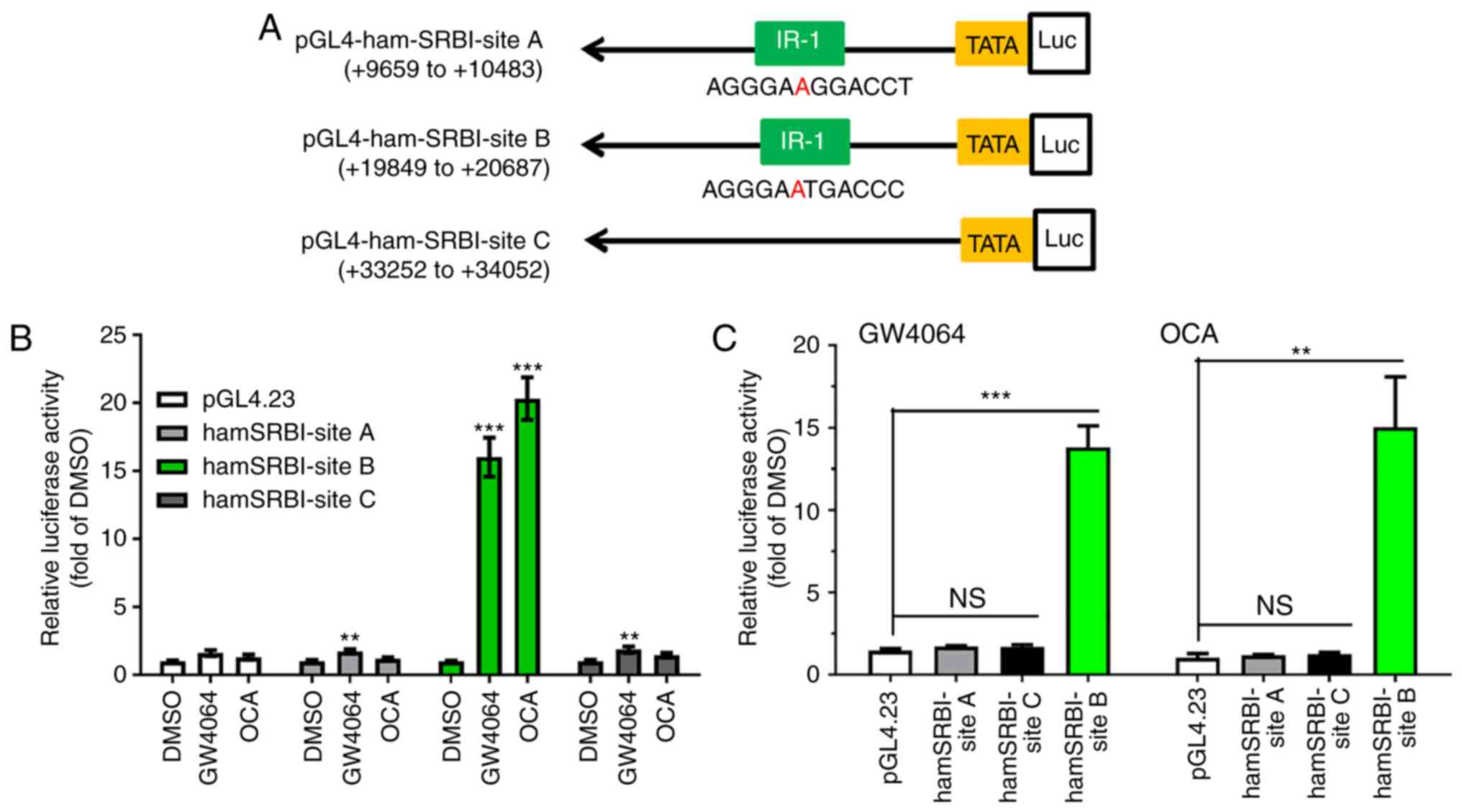
Analysis of the nucleotide sequence within this
824-bp segment of the site B intronic region using MatInspector
software revealed the presence of an LXR binding site located 57 bp
downstream from FXRE (Fig. 2A).
To assess the functionality of LXRE, the responses of hamster SR-BI
reporter genes to the LXR agonist GW3965 and FXR agonists were
examined. The results showed that treating the transfected cells
with LXR agonist GW3965 led to an increase of ~4-fold in hamster
site B reporter activity, without affecting site A and site C
reporters (Fig. 2B). Although the
4-fold increase in site B reporter activity induced by GW3965 was
relatively modest when compared with the effects of FXR agonists
OCA and GW4064, the stimulating effect of GW3965 on site B reporter
activity was reproducible in multiple transfection assays.
Concerted activation of SR-BI gene
transcription by FXR and LXR via intron bindings
Sequence alignments of site B regions of the SR-BI
gene of hamster, mouse, rat and human not only demonstrated that
the FXRE and LXRE sequences are identical, but spacings between the
two motifs are also strictly conserved among rodents (Fig. 3A). In contrast to rodents, this
intronic region is not conserved in the human SR-BI gene; in
particular, the FXRE sequence is absent in the human sequence.
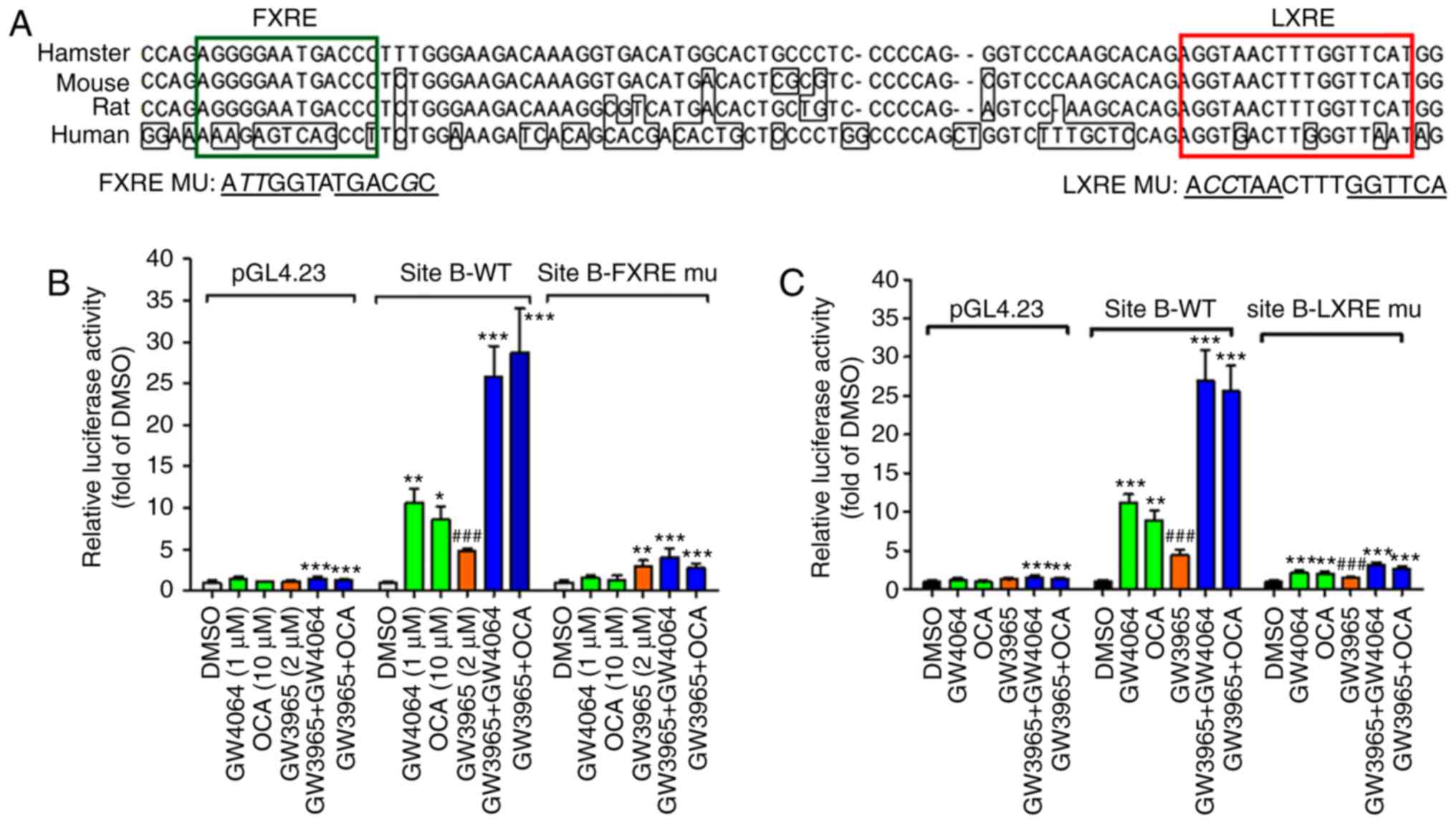 | Figure 3Novel FXR response element IR-1 and
LXR response element DR4 mediate the transcription of SR-BI upon
FXR and LXR coactivation. (A) Alignment of site B regions of intron
one of the hamster, mouse, rat and human SR-BI gene. (B) WT site B
reporter, FXRE-mu site B reporter and the control reporter pGL4.23
were transfected into HepG2 cells. The following day, cells were
cultured in MEM containing 0.5% fetal bovine serum overnight and
treated with GW4064 (1 µM), OCA (10 µM), GW3965 (5
µM), GW3965 + GW4064, or GW3965 + OCA for 24 h. Data are
presented as the mean ± standard error of the mean of four
replicates per treatment and are expressed as ratio of
firefly/Renilla activity from each sample where the relative
luminescence from DMSO-treated cells is set to 1. Statistical
significance among all groups was assessed by one-way analysis of
variance with Tukey's multiple comparison test. The data shown are
representative of three separate transfection experiments.
*P<0.05, **P<0.01 and
***P<0.001 compared with DMSO;
###P<0.001 compared with cotreated samples. (C)
Luciferase activities in HepG2 cells transfected with the WT or
LXRE-mu hamster site B plasmid and treated with compounds for 24 h.
Following normalization with Renilla activity, the relative
luciferase activity (fold of DMSO) is indicated for each reporter
plasmid. The data shown are representative of three separate
transfection experiments. **P<0.01 and
***P<0.001 compared with DMSO;
###P<0.001 compared with cotreated samples. FXR,
farnesoid X receptor; FXRE, FXR response element; LXR, liver X
receptor; LXRE, LXR response element; SR-BI, scavenger receptor
class B type I; WT, wild-type; mu, mutant; OCA, obeticholic
acid. |
The core nucleotide sequence of the FXRE site was
mutated and the mutated plasmid was transfected along with the WT
hamster site B reporter and the pGL4.23 basic vector individually
into HepG2 cells. The transfected cells were treated separately
with FXR agonists, LXR agonist or the combination of FXR and LXR
agonists. The site B WT reporter activity was increased ~10-fold by
GW4064 or OCA and 5-fold by GW3965; however, the coactivation of
both nuclear receptors increased the reporter activity almost
30-fold, indicating a synergistic effect (Fig. 3B). Mutation in FXRE not only
eliminated the induction by FXR agonists, but also almost
eradicated the effect of GW3965 alone, as well as the combined
inducing effects. Subsequently, the LXRE site was mutated and
similar inhibitory effects were observed of LXRE mutation on the
site B reporter activity in cells treated with GW3965, the FXR
agonists or their combination (Fig.
3C). Taken together, these results suggest that the binding of
FXR to its cis-acting element IR-1 in the site B region
depends on the interaction between activated LXR and the adjacent
LXRE motif.
In line with the hamster reporter activity data, it
was observed that the site B segment in the mouse SR-BI reporter
gene (4) was most responsive to
the FXR and LXR individual treatments and the concerted activations
(Fig. S2A and B).
It is well-demonstrated that FXR forms heterodimers
with RXRa and binds the IR-1 motif to activate its target gene
transcription (1). To determine
whether the observed increase in site B reporter activity by FXR
agonist treatment may be directly attributed to FXR binding to the
putative FXRE sequence, the present study first examined the
interaction of FXR with the SRBI-FXRE sequence by performing EMSA.
Using biotin-labeled probes, the binding of purified FXR-RXR
heterodimers to the 49-base pair region encompassing the hamster
SR-BI site B IR-1 sequence was demonstrated (Fig. 4, lane 2). Competition with the WT
unlabeled probe resulted in almost complete quenching of the
binding with the biotin-labeled probe, whereas competition with an
unlabeled probe harboring the mutant FXRE was less effective in
repressing the binding of the FXR-RXR complex to the SRBI-FXRE
probe (Fig. 4A, lanes 3 and
4).
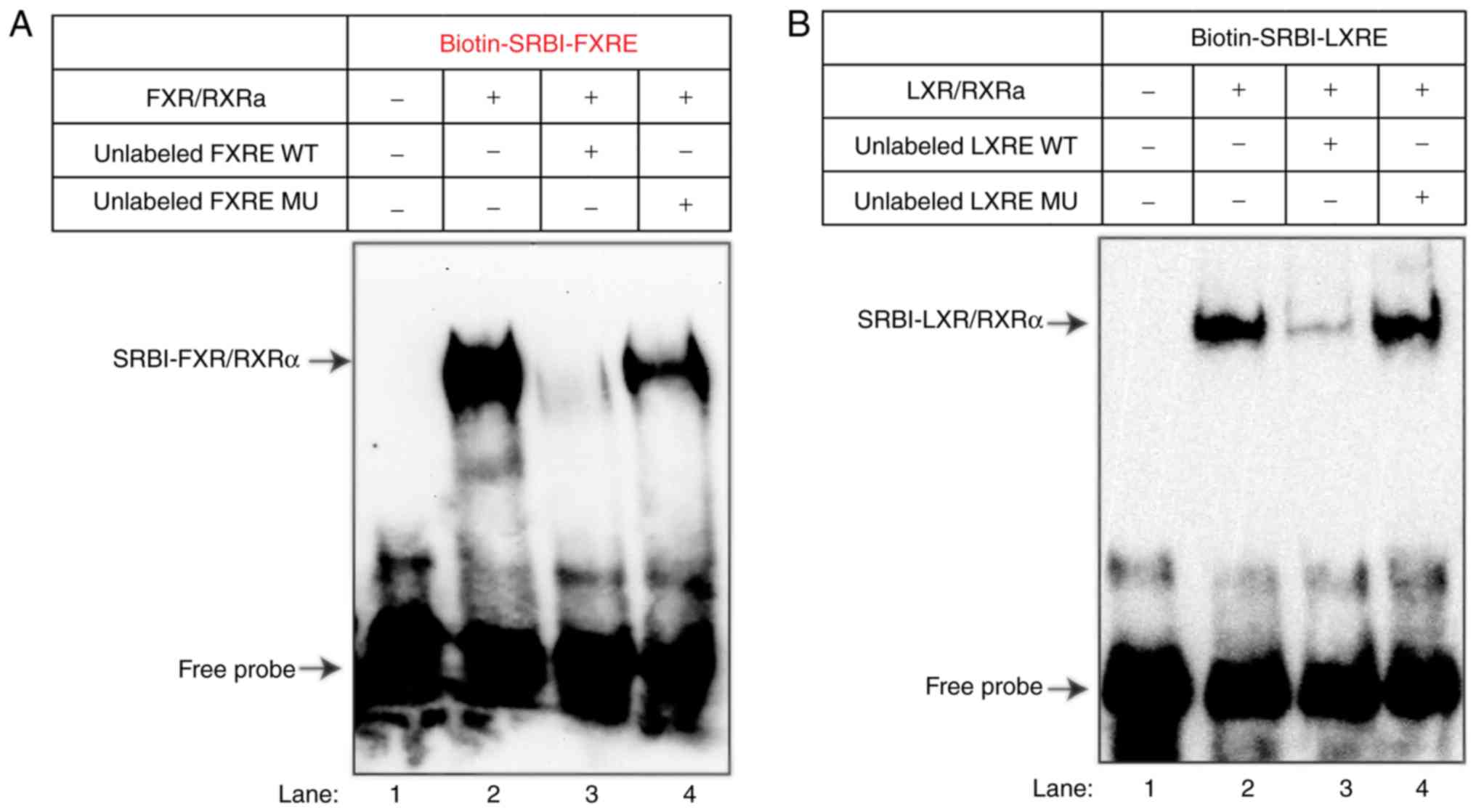 | Figure 4EMSA and reporter analyses of the
association of FXR and LXR with the regulatory intronic region of
site B segment of the hamster SR-BI gene. (A) EMSA to evaluate
DNA-binding by FXRα/RXRα. A biotin-5′ end-labeled SRBI-FXRE probe
was incubated without (lane 1) or with 100 ng of FXRα and 100 ng of
RXRα recombinant proteins in the absence (lane 2) or presence of
100-fold molar excess of unlabeled WT probe (lane 3) or MU probe
(lane 4). The protein-DNA complex was resolved on 5% TBE gels,
transferred onto a nylon membrane and UV-crosslinked to the
membrane prior to DNA visualization by avidin-chemiluminescent
probe. The data shown are representative of three separate EMSA
assays with comparable results. (B) EMSA to evaluate DNA-binding by
LXRα/RXRα. Biotin-5′ end labeled SRBI-LXRE probe was incubated
without (lane 1) or with 100 ng of LXRα and 100 ng of RXRα
recombinant proteins in the absence (lane 2) or presence of
100-fold molar excess of unlabeled WT probe (lane 3) or MU probe
(lane 4). The protein-DNA complex was resolved on 5% TBE gels,
transferred to a nylon membrane and UV-crosslinked prior to DNA
visualization by avidin-chemiluminescent probe. The data shown are
representative of two separate EMSA assays with comparable results.
(C) HepG2 cells were cotransfected with pGL3-basic,
pGL3-hSRBI-1831, pGL4.23, or hamster site B WT plasmid and
indicated nuclear receptor expression plasmids or the control
vector (pCMV-empty). At 2 days post-transfection, cell lysates were
prepared. Following normalization with Renilla activity, the
relative luciferase activity (fold of mock) is indicated for each
reporter plasmid. (D) Human and hamster SRBI reporter vectors and
their respective control vectors were transfected into HepG2 cells
along with pRL-TK. The following day, cells were cultured in MEM
containing 0.5% fetal bovine serum overnight and treated with OCA
(10 µM), GW3965 (2 µM), or GW3965 + OCA for 24 h
prior to cell lysis. Data are presented as the mean ± standard
error of the mean of four replicates per treatment and are
expressed as ratio of firefly/Renilla activity from each
sample where the relative luminescence from DMSO-treated cells is
set to 1. Statistical significance among all groups was assessed by
one-way analysis of variance with Tukey's multiple comparison test.
**P<0.01 and ***P<0.001 compared with
DMSO-treated samples. The data shown are representative of three
separate transfection experiments. FXR, farnesoid X receptor; FXRE,
FXR response element; LXR, liver X receptor; LXRE, LXR response
element; SR-BI, scavenger receptor class B type I; WT, wild-type;
mu, mutant; OCA, obeticholic acid. |
Subsequently, EMSA was performed to assess the
specific binding of LXR/RXR to the identified LXRE site within this
intronic region. As shown in Fig.
4B, LXR/RXR was bound to the LXRE site with a high specificity.
These results demonstrated the direct interactions of the SRBI-FXRE
sequence with FXR and the SRBI-LXRE sequence with LXR in
vitro.
In addition to the in vitro assay of direct
DNA-protein interactions, the effects of the exogenous
overexpression of nuclear receptors FXRα/RXRα and LXRα/RXRα on
SR-BI reporter gene transcription were examined. A previous study
reported that FXR binds to a putative FXRE site (DR8) that is
located from −816 to −796 of the human SR-BI promoter region
(17). In addition to the
putative DR8 site, it was reported that the human SR-BI promoter
contains a LXRE site located from −1,001 to −985 of the TSS
(29). Therefore, the human SR-BI
reporter construct pGL3-hSRBI-1109, containing the DR8 sequence,
and pGL3-hSRBI-1898, containing both DR8 and LXRE sites, were
constructed (Fig. S3A).
The hamster site B reporter and human SRBI-1893
reporter plasmids, and their respective control vectors, were
separately cotransfected with the mock vector (pCMV-empty), or
expression vectors for FXRα/RXRα, LXRα/RXRα, or FXRα/LXRα/RXRα into
HepG2 cells, and the luciferase activity was measured 48 h post
transfection (Fig. 4C). The
overexpression of FXR and LXR had marginal effects (~2-4-fold of
the mock control) in elevating luciferase activities in cells
transfected with the reporter control vector pGL4.23, pGL3-basic,
and in cells transfected with the human SR-BI promoter. By sharp
contrast, the luciferase activities in the hamster SRBI-site
B-transfected cells were increased to 60-fold by FXR, 46-fold by
LXR, and 210-fold by FXR and LXR co-expression compared with that
in cells transfected with the mock vector. These data are in line
with the findings of EMSA showing prominent bindings of FXR and LXR
to the intronic regulatory region of the hamster SR-BI gene.
The responses of the human SR-BI promoter and the
hamster reporter to ligand treatments were also examined (Fig. 4D and Fig. S3B and C). In contrast to the
hamster site B reporter, human SR-BI promoter reporter activity was
not significantly induced by OCA, GW3965 or the combination,
suggesting that this proximal promoter region of the human SR-BI
gene may not contain a high-affinity FXR binding sequence.
Subsequently, HPH were treated with OCA, GW3965 and
their combination for 24 h. RT-qPCR analysis showed that GW3965
treatment alone did not increase the mRNA levels of SR-BI, however,
it potentiated the OCA-induced gene expression of SR-BI (Fig. 5A). No such synergistic effects
were detected in the classical FXR target gene ABCB4 or the LXR
target gene ABCA1. In addition to HPH, HepG2 cells were treated
with FXR and LXR agonists and similar synergistic effects of OCA
and GW3965 were observed on the gene expression of SR-BI (Fig. 5B). These results suggest that the
gene expression of SR-BI in human liver cells can also be
co-activated by FXR and LXR via unknown genomic sequences that are
different from the regulatory elements found in the hamster and
mouse SR-BI genes.
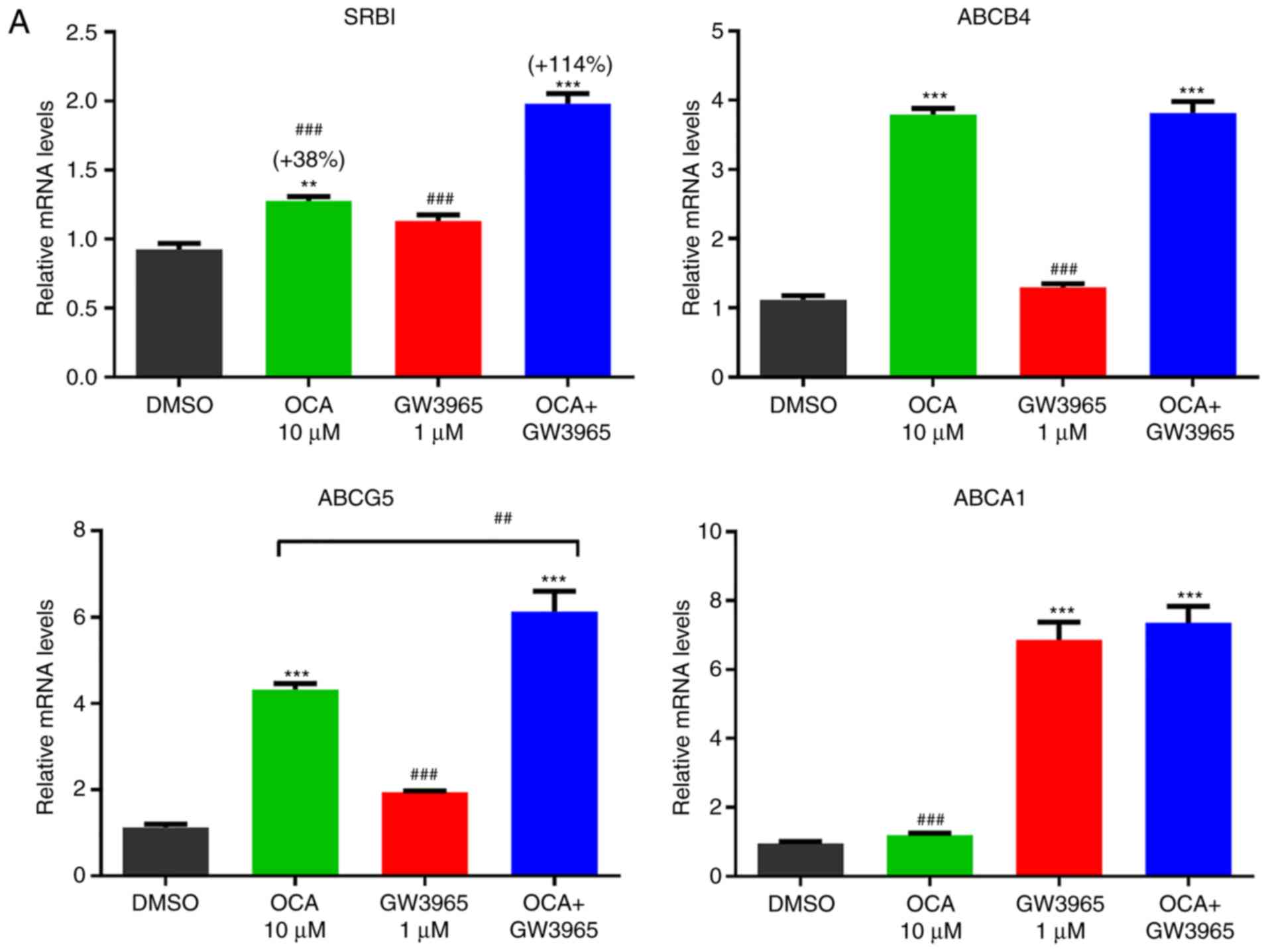 | Figure 5Synergistic activation of the gene
expression of SR-BI by farnesoid X receptor and liver X receptor in
HPH and HepG2 cells. (A) HPHs were treated with OCA (10 µM)
or GW3965 (1 µM) or OCA + GW3965 for 24 h prior to isolation
of total RNA. Triplicate wells were used in each treatment
condition. RT-qPCR analysis was performed to measure relative mRNA
levels of indicated genes with duplicate measurement of each cDNA
sample. Statistical signifi-cance among all groups was assessed by
one-way ANOVA with Tukey's multiple comparison test.
**P<0.01 and ***P<0.001 compared with
DMSO-treated samples; ##P<0.01 and
###P<0.001 compared with cotreated samples. (B) HepG2
cells in triplicate wells were cultured overnight in culture medium
containing 0.5% fetal bovine serum, followed by treatment of OCA
(10 µM), GW3965 (5 µM), or GW3965 + OCA for 24 h.
RT-qPCR analysis was performed to measure relative mRNA levels of
indicated genes with duplicate measurement of each cDNA sample.
One-way ANOVA with Tukey's multiple comparison test was used. HPH,
human primary hepatocytes; OCA, obeticholic acid; GW, GW3965;
SR-BI, scavenger receptor class B type I; ABC, ATP binding
cassette; ANOVA, analysis of variance; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction. |
Co-activation of SR-BI gene expression by
OCA and GW3965 in hamsters fed a normal chow diet
To examine whether this newly identified
transcriptional synergy between FXR and LXR on the gene expression
of SR-BI operates in vivo in the liver, chow-fed hamsters
(n=5 per group) were treated with vehicle, 10 mg/kg/day OCA, 30
mg/kg/day GW3965 or OCA + GW3965 for 10 days. The body weight, food
intake, liver weight and liver index were not significantly
affected by the treatment (Fig.
S4A-D). Examination of the protein expression of SR-BI in the
liver by western blotting showed that, consistent with our previous
study (18), OCA or GW3965 alone
were not able to upregulate the liver expression of SR-BI in
chow-fed hamsters. By contrast, the combined treatment
significantly increased the protein levels of SR-BI to 1.75-fold of
the vehicle control (P<0.001; Fig.
6A and B). Hepatic gene expression analysis showed that the
mRNA levels of SR-BI were only increased by the combined treatment
to 1.8-fold of the vehicle (P<0.001; Fig. 6C). It was observed that the mRNA
level of endothelial lipase was also elevated by the combination,
whereas typical FXR target genes SHP and bile salt export pump
(BSEP) were only responsive to OCA treatment. The mRNA levels of
CYP7A1 in the hamster liver were repressed by OCA and GW3965
individual treatments, and the combined treatment produced the most
marked inhibition. These in vivo results of the protein and
mRNA expression measurements of SR-BI support the in vitro
findings of a novel transcriptional mechanism for the upregulation
of SR-BI by the integrated activation of FXR and LXR via this
functional intronic region of the hamster SR-BI gene.
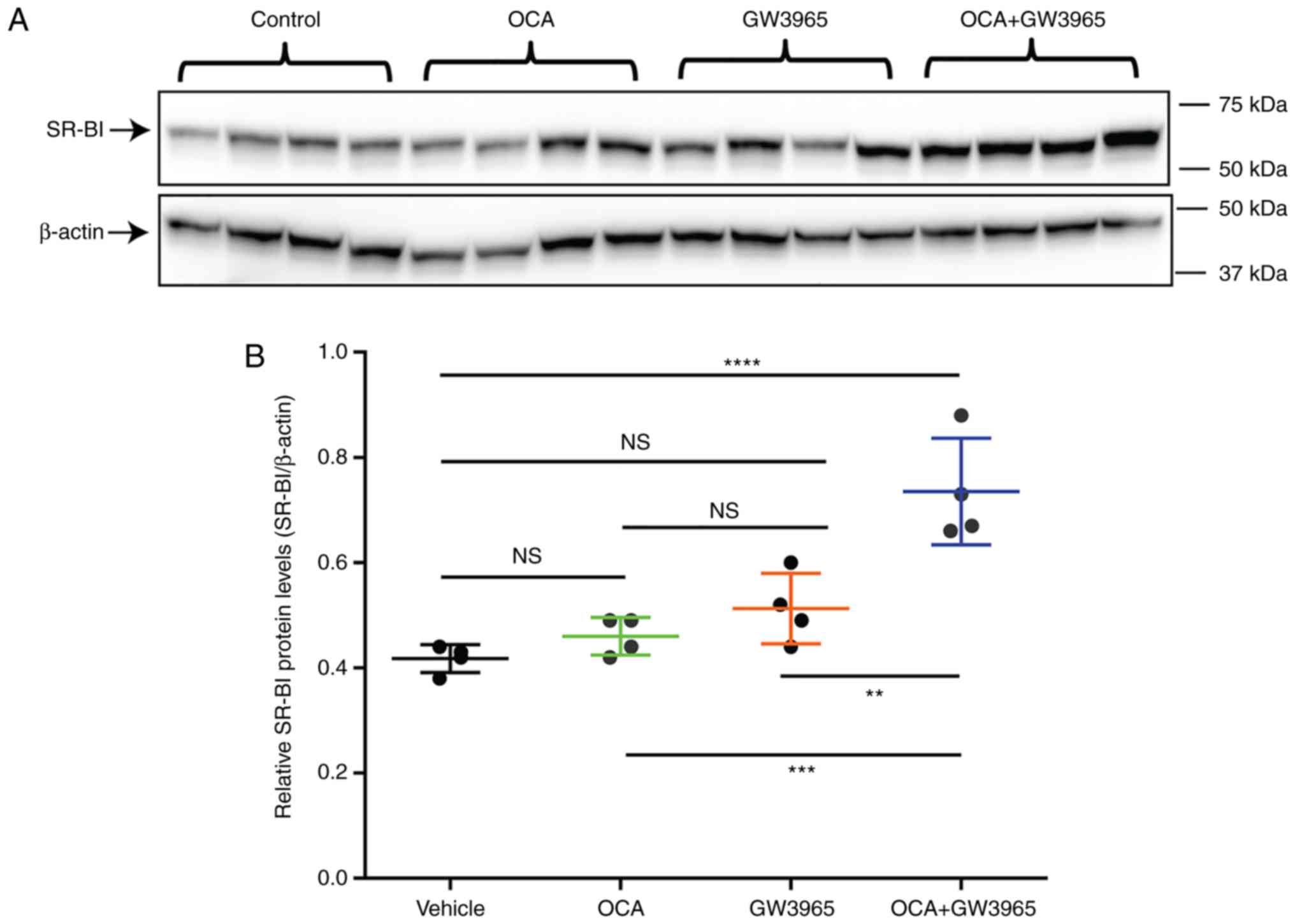 | Figure 6Synergistic activation of the gene
expression of SR-BI by FXR and liver X receptor in hamster liver
tissue. Hamsters fed a normal chow diet were treated with OCA (10
mg/kg/day, n=5), GW3965 (30 mg/kg/day, n=5), OCA + GW3965 (n=5), or
vehicle control (n=5) for 10 days. Overnight fasting serum samples
and liver tissues were collected. (A) Western blot analysis of the
protein expression of SR-BI in liver samples from hamsters treated
with vehicle, OCA, GW3965, and OCA + GW3965. Values are presented
as the mean ± standard error of the mean of four randomly chosen
liver samples per group. (B) Protein quantification of western
blots; protein levels of SR-BI were normalized to levels of
β-actin. Statistical significance among all groups was assessed by
one-way ANOVA with Tukey's multiple comparison test.
****P<0.0001 compared with the vehicle control group;
**P<0.01 and ***P<0.001 compared with
the combination group. N=4 hamsters per group. (C) Reverse
transcription-quantitative polymerase chain reaction analysis of
hepatic gene expression of SR-BI and other FXR-modulated genes.
Statistical significance among all groups was assessed by one-way
ANOVA with Tukey's multiple comparison test. *P<0.05,
**P<0.01 and ***P<0.001 compared with
the vehicle control group; #P<0.05 and
##P<0.01 compared with the combination group. N=5
hamsters per group. FXR, farnesoid X receptor; OCA, obeticholic
acid; SR-BI, scavenger receptor class B type I; ANOVA, analysis of
variance; n.s., not significant. |
In addition to affecting gene expression in the
liver, ligand treatments moderately altered cholesterol levels in
the serum and liver tissue of the chow-fed hamsters. Serum TC and
HDL-C levels were increased in the GW3965 group, which was not
observed in the combination group (Fig. S5A and B). Hepatic TC contents
were modestly increased in the livers of OCA-treated or
GW3965-treated hamsters, as observed in previous studies (18,30); however, hepatic cholesterol was
not increased in the combination group (Fig. S5C).
Consistent with the suppression of the mRNA
expression of CYP7A1, the fecal BA contents were reduced by 26%
(P<0.05) in the OCA-treated hamsters and were further reduced to
almost half of that in the vehicle control (P<0.01) by the
combinational treatment, despite no changes by GW3965 single
treatment (Fig. S5D). Overall,
these changes are consistent with the increased hepatic expression
of SR-BI by FXR and LXR co-activation.
Discussion
FXR regulates several aspects of bile acid
metabolism (3,31), whereas LXR regulates cholesterol
metabolism (19,32). Together, FXR and LXR maintain bile
acid and cholesterol homeostasis by translating hormonal, metabolic
and nutritional signals into changes in target gene expression
(6,33-35). It is well recognized that both FXR
and LXR partner with RXR, but bind to distinct DNA recognition
motifs and regulate different sets of metabolic genes. In certain
cases, they can modulate the expression of the same gene with
opposite effects. For example, in mice, the activation of FXR leads
to marked suppression of the gene expression of Cyp7A1, inhibiting
bile acid synthesis, whereas the gene expression of Cyp7A1 is
induced in mice by LXR agonists to promote the synthesis of bile
acids (36).
SR-BI is known as an FXR-modulated gene and the
major physiological receptor for HDL-C. However, unlike other
typical FXR target genes, including BSEP and SHP that are
unfailingly induced by FXR agonists in liver cells across species,
reports on the activation of FXR by various agonists on the gene
expression of SR-BI have been inconsistent and conflicting in
studies performed in cultured liver cells (16,17) and in animals (14). Our previous study demonstrated
that OCA upregulated the hepatic mRNA and protein expression of
SR-BI in hamsters fed a cholesterol-enriched diet, but not in
hamsters fed a normal chow diet (18). It is now clear that FXR activates
SR-BI gene transcription effectively in the presence of activated
LXR via their adjacent recognition sequences located in the site B
segment of the first intron of the hamster SR-BI gene. Notably,
this intronic region containing FXRE and LXRE motifs is conserved
in the hamster, mouse and rat SR-BI gene, but is not conserved in
the human SR-BI gene (Fig.
3A).
The mutagenesis performed in the present study
further showed that the disruption of LXRE attenuated FXR-mediated
gene transcription, and, similarly, the ablation of FXRE IR-1
almost eliminated LXR-induced reporter activity. These data suggest
a co-dependency of FXR and LXR binding to this regulatory region of
the hamster SR-BI gene. By performing in vitro DNA binding
assays, it was showed that FXR binds to the SRBI-FXRE sequence with
high specificity, and the strong binding of LXR to the newly
identified SRBI-LXRE motif in this intronic region was confirmed.
Due to the limitation of EMSA and the length of the biotin-labeled
probe, it was not possible to examine the possibility of a direct
interaction of FXR and LXR on this genomic region using the in
vitro assay. However, the in vivo transactivation
reporter assays clearly showed that the exogenous overexpression of
FXR or LXR substantially increased the activities of the reporter
containing the site B sequence without notable effects on the
control reporter without the intronic sequence. In addition, the
coexpression of FXR and LXR synergistically increased reporter
activity. It was also shown that treating normolipidemic hamsters
with OCA and the LXR agonist GW3965 individually did not
effectively increase the expression of SR-BI, whereas the combined
treatment significantly increased the mRNA and protein levels of
SR-BI in liver tissues, which provided direct evidence that this
synergistic activation of SR-BI gene transcription by FXR and LXR
operates in liver tissue. Future investigations are required to
elucidate the precise mechanism underlying this observed
cooperativity between FXR and LXR in the context of the SR-BI
gene.
In conclusion, the present study identified a
critical regulatory region in the hamster SR-BI gene that mediates
the gene transcription of SR-BI upon FXR activation by OCA
treatment under LXR-activated states. This occurs either by
endogenous sterols in the cholesterol-enriched liver of HCHFD-fed
hamsters or by synthetic agonist GW3965 cotreatment, an effect that
increases cholesterol output and consequently overrides hepatic
cholesterol accumulation caused by individual treatments. These
novel findings provide a molecular explanation for the potent
inducing effect of OCA on the expression of SR-BI in
hypercholesterolemic hamsters. The present study identified a novel
transcriptional mechanism for the expression of SR-BI through an
integrated activation by FXR and LXR via intron bindings in
hamsters and mice.
Regarding the human SR-BI gene, although the
intronic regulatory region of SR-BI was not found, and the proximal
promoter region up to −1,899 bp upstream of the TSS did not
significantly respond to the activation of FXR or LXR, higher mRNA
levels of SR-BI were detected in HPH and HepG2 cells upon OCA and
GW3965 cotreatment, thereby suggesting that FXR and LXR may
coactivate the human SR-BI gene through their interactions with
currently unidentified regulatory sequences in the human SR-BI
genome. Furthermore, it has been reported that the expression of
SR-BI is regulated transcriptionally by other nuclear receptors,
including peroxisome proliferator-activated receptor (PPAR)γ and
liver receptor homologue 1 (LRH-1) (37,38). Therefore, it is possible that the
activation of FXR may indirectly upregulate the expression of SR-BI
in human liver cells via interacting with the PPAR pathway or
LRH-1, and this interaction may be potentiated by LXR.
Supplementary Materials
Abbreviations:
|
EMSA
|
electrophoresis mobility shift
assay
|
|
FXR
|
farnesoid X receptor
|
|
FXRE
|
FXR response element
|
|
HPH
|
human primary hepatocytes
|
|
LXR
|
liver X receptor
|
|
LXRE
|
LXR response element
|
|
OCA
|
obeticholic acid
|
|
SR-BI
|
scavenger receptor class B type I
|
|
SRE
|
sterol regulatory element
|
|
SREBP
|
SRE-binding protein
|
Funding
This study was supported by the Department of
Veterans Affairs (Office of Research and Development, Medical
Research Service; grant no. I01 BX001419, J.L.) and by a grant
(grant no. 1R01AT006336-01A1, J.L) from the National Center for
Complementary and Integrative Health.
Availability of data and materials
The authors declare that all supporting data are
available within the article and its online supplementary
files.
Authors' contributions
BD and ABS performed the experiments. GLG, MY and JL
analyzed and interpreted the data. JL made substantial
contributions to the design supervision of the present study, and
wrote the manuscript. All authors reviewed the results and approved
the final version of the manuscript.
Ethics approval and consent to
participate
All animal experiments were performed according to
procedures approved by the VA Palo Alto Health Care System
Institutional Animal Care and Use Committee (Palo Alto, CA,
USA).
Patient consent for publication
Not applicable.
Competing interests
MY was an employee and stockholder in Intercept
Pharmaceuticals, Inc. at the time of contribution. The other
authors have no competing interests.
Acknowledgments
The authors would like to thank Dr Timothy F.
Osborne from Sanford Burnham Prebys Medical Discovery Institute for
providing the FXRα expression plasmid, and Dr Fredric B. Kraemer of
Stanford University of Medicine, and Dr Progga Sen of the VA Palo
Alto Health Care System for their critical review of the
manuscript.
References
|
1
|
Forman BM, Goode E, Chen J, Oro AE,
Bradley DJ, Perlmann T, Noonan DJ, Burka LT, McMorris T, Lamph WW,
et al: Identification of a nuclear receptor that is activated by
farnesol metabolites. Cell. 81:687–693. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lefebvre P, Cariou B, Lien F, Kuipers F
and Staels B: Role of bile acids and bile acid receptors in
metabolic regulation. Physiol Rev. 89:147–191. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sinal CJ, Tohkin M, Miyata M, Ward JM,
Lambert G and Gonzalez FJ: Targeted disruption of the nuclear
receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell.
102:731–744. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li G, Thomas AM, Williams JA, Kong B, Liu
J, Inaba Y, Xie W and Guo GL: Farnesoid X receptor induces murine
scavenger receptor Class B type I via intron binding. PLoS One.
7:e358952012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Laffitte BA, Kast HR, Nguyen CM, Zavacki
AM, Moore DD and Edwards PA: Identification of the DNA binding
specificity and potential target genes for the farnesoid
X-activated receptor. J Biol Chem. 275:10638–10647. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Calkin AC and Tontonoz P: Transcriptional
integration of metabolism by the nuclear sterol-activated receptors
LXR and FXR. Nat Rev Mol Cell Biol. 13:213–224. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thomas AM, Hart SN, Kong B, Fang J, Zhong
XB and Guo GL: Genome-wide tissue-specific farnesoid X receptor
binding in mouse liver and intestine. Hepatology. 51:1410–1419.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhan L, Liu HX, Fang Y, Kong B, He Y,
Zhong XB, Fang J, Wan YJ and Guo GL: Genome-wide binding and
transcriptome analysis of human farnesoid X receptor in primary
human hepatocytes. PLoS One. 9:e1059302014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Acton S, Rigotti A, Landschulz KT, Xu S,
Hobbs HH and Krieger M: Identification of scavenger receptor SR-BI
as a high density lipoprotein receptor. Science. 271:518–520. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rigotti A, Trigatti BL, Penman M, Rayburn
H, Herz J and Krieger M: A targeted mutation in the murine gene
encoding the high density lipoprotein (HDL) receptor scavenger
receptor class B type I reveals its key role in HDL metabolism.
Proc Natl Acad Sci USA. 94:12610–12615. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brodeur MR, Luangrath V, Bourret G,
Falstrault L and Brissette L: Physiological importance of SR-BI in
the in vivo metabolism of human HDL and LDL in male and female
mice. J Lipid Res. 46:687–696. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zanoni P, Khetarpal SA, Larach DB,
Hancock-Cerutti WF, Millar JS, Cuchel M, DerOhannessian S, Kontush
A, Surendran A, Saleheen D, et al: Rare variant in scavenger
receptor BI raises HDL cholesterol and increases risk of coronary
heart disease. Science. 351:1166–1171. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rizzo G, Passeri D, De Franco F, Ciaccioli
G, Donadio L, Orlandi S, Sadeghpour B, Wang XX, Jiang T, Levi M, et
al: Functional characterization of the semisynthetic bile acid
derivative INT-767, a dual farnesoid X receptor and TGR5 agonist.
Mol Pharmacol. 78:617–630. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hambruch E, Miyazaki-Anzai S, Hahn U,
Matysik S, Boettcher A, Perović-Ottstadt S, Schlüter T, Kinzel O,
Krol HD, Deuschle U, et al: Synthetic farnesoid X receptor agonists
induce high-density lipoprotein-mediated transhepatic cholesterol
efflux in mice and monkeys and prevent atherosclerosis in
cholesteryl ester transfer protein transgenic low-density
lipoprotein receptor (-/-) mice. J Pharmacol Exp Ther. 343:556–567.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Yin L, Anderson J, Ma H, Gonzalez
FJ, Willson TM and Edwards PA: Identification of novel pathways
that control farnesoid X receptor-mediated hypocholesterolemia. J
Biol Chem. 285:3035–3043. 2010. View Article : Google Scholar :
|
|
16
|
Chong HK, Infante AM, Seo YK, Jeon TI,
Zhang Y, Edwards PA, Xie X and Osborne TF: Genome-wide
interrogation of hepatic FXR reveals an asymmetric IR-1 motif and
synergy with LRH-1. Nucleic Acids Res. 38:6007–6017. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chao F, Gong W, Zheng Y, Li Y, Huang G,
Gao M, Li J, Kuruba R, Gao X, Li S and He F: Upregulation of
scavenger receptor class B type I expression by activation of FXR
in hepatocyte. Atherosclerosis. 213:443–448. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dong B, Young M, Liu X, Singh AB and Liu
J: Regulation of lipid metabolism by obeticholic acid in
hyperlipidemic hamsters. J Lipid Res. 58:350–363. 2017. View Article : Google Scholar :
|
|
19
|
Repa JJ and Mangelsdorf DJ: The role of
orphan nuclear receptors in the regulation of cholesterol
homeostasis. Annu Rev Cell Dev Biol. 16:459–481. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Apfel R, Benbrook D, Lernhardt E, Ortiz
MA, Salbert G and Pfahl M: A novel orphan receptor specific for a
subset of thyroid hormone-responsive elements and its interaction
with the retinoid/thyroid hormone receptor subfamily. Mol Cell
Biol. 14:7025–7035. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jakobsson T, Treuter E, Gustafsson JA and
Steffensen KR: Liver X receptor biology and pharmacology: New
pathways, challenges and opportunities. Trends Pharmacol Sci.
33:394–404. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maxwell KN, Soccio RE, Duncan EM, Sehayek
E and Reslow JL: Novel putative SREBP and LXR target genes
identified by micro-array analysis in liver of cholesterol-fed
mice. J Lipid Res. 44:2109–2119. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Grefhorst A, Oosterveer MH, Brufau G,
Boesjes M, Kuipers F and Groen AK: Pharmacological LXR activation
reduces presence of SR-B1 in liver membranes contributing to
LXR-mediated induction of HDL-cholesterol. Atherosclerosis.
222:382–389. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cartharius K, Frech K, Grote K, Klocke B,
Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M and Werner T:
MatInspector and beyond: Promoter analysis based on transcription
factor binding sites. Bioinformatics. 21:2933–2942. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cartharius K: MatInspector: Analysing
Promoters for Transcription Factor Binding Sites. The Nuts &
Bolts Series. DNA Press. 2005.
|
|
26
|
Quandt K, Frech K, Karas H, Wingender E
and Werner T: MatInd and MatInspector: New fast and versatile tools
for detection of consensus matches in nucleotide sequence data.
Nucleic Acids Res. 23:4878–4884. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li H, Dong B, Park SW, Lee HS, Chen W and
Liu J: Hepatocyte nuclear factor 1alpha plays a critical role in
PCSK9 gene transcription and regulation by the natural
hypocholesterolemic compound berberine. J Biol Chem.
284:28885–28895. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
29
|
Malerød L, Juvet LK, Hanssen-Bauer A,
Eskild W and Berg T: Oxysterol-activated LXRalpha/RXR induces
hSR-BI-promoter activity in hepatoma cells and preadipocytes.
Biochem Biophys Res Commun. 299:916–923. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dong B, Kan CF, Singh AB and Liu J:
High-fructose diet down-regulates long-chain acyl-CoA synthetase 3
expression in liver of hamsters via impairing LXR/RXR signaling
pathway. J Lipid Res. 54:1241–1254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schuetz EG, Strom S, Yasuda K, Lecureur V,
Assem M, Brimer C, Lamba J, Kim RB, Ramachandran V, Komoroski BJ,
et al: Disrupted bile acid homeostasis reveals an unexpected
interaction among nuclear hormone receptors, transporters, and
cytochrome P450. J Biol Chem. 276:39411–39418. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schultz JR, Tu H, Luk A, Repa JJ, Medina
JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, et al:
Role of LXRs in control of lipogenesis. Genes Dev. 14:2831–2838.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ding L, Pang S, Sun Y, Tian Y, Yu L and
Dang N: Coordinated actions of FXR and LXR in metabolism: From
pathogenesis to pharmacological targets for type 2 diabetes. Int J
Endocrinol. 2014:7518592014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu TT, Makishima M, Repa JJ, Schoonjans K,
Kerr TA, Auwerx J and Mangelsdorf DJ: Molecular basis for feedback
regulation of bile acid synthesis by nuclear receptors. Mol Cell.
6:507–515. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Matsukuma KE, Bennett MK, Huang J, Wang L,
Gil G and Osborne TF: Coordinated control of bile acids and
lipogenesis through FXR-dependent regulation of fatty acid
synthase. J Lipid Res. 47:2754–2761. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Peet DJ, Turley SD, Ma W, Janowski BA,
Lobaccaro JM, Hammer RE and Mangelsdorf DJ: Cholesterol and bile
acid metabolism are impaired in mice lacking the nuclear oxysterol
receptor LXR alpha. Cell. 93:693–704. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Malerød L, Sporstøl M, Juvet LK, Mousavi
A, Gjøen T and Berg T: Hepatic scavenger receptor class B, type I
is stimulated by peroxisome proliferator-activated receptor gamma
and hepatocyte nuclear factor 4alpha. Biochem Biophys Res Commun.
305:557–565. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schoonjans K, Annicotte JS, Huby T,
Botrugno OA, Fayard E, Ueda Y, Chapman J and Auwerx J: Liver
receptor homolog 1 controls the expression of the scavenger
receptor class B type I. EMBO Rep. 3:1181–1187. 2002. View Article : Google Scholar : PubMed/NCBI
|