Introduction
Atherosclerosis, resulting in lipid accumulation,
extracellular matrix protein deposition and calcification of the
arterial intima and media, causes arterial stiffness and reduces
elasticity (1). Atherosclerosis
is a progressive pathological process that involves vascular
endothelial injury, lipid infiltration, vascular smooth muscle cell
proliferation, inflammatory immune response and oxidative stress,
eventually leading to a series of coronary artery diseases
including ischemic stroke, heart attack, and peripheral vascular
disease which remain a predominant cause of morbidity and morbidity
in aged people worldwide (1).
Although atherosclerosis is a multifactorial disease, vascular
endothelial cell injury is a driving force in its initiation and
pathogenesis (2). Oxidized
low-density lipoprotein (Ox-LDL), a particularly important risk
factor in the development of atherosclerosis, has been reported to
induce vascular endothelial dysfunction via triggering accumulation
of abundant lipids in arterial walls, endothelial cell apoptosis,
oxidative stress, mitochondrial dysfunction and permeability
(3). Up to date, control of the
development of atherosclerosis remains a great challenge due to the
incomplete understanding of the involved molecular signaling
pathways (4). Therefore,
mitigating ox-LDL-induced endothelial cell injury and further
elucidating the underlying mechanism may be potential therapeutic
strategies for atherosclerosis prevention and therapy.
Oxidative stress has been recognized as one of the
causative factors in atherosclerosis and reactive oxygen species
(ROS) are considered to be signaling molecules that lead to
endothelial dysfunction in clinical and experimental
atherosclerosis (5). The
mitochondrion is one of the main sources of chronic ROS production
under physiological conditions (6). Mitochondrial dysfunction is
conducive to the process of atherosclerosis as evidenced by human
and animal models of oxidative stress (7). Oxidative stress and mitochondrial
dysfunction are closely linked, giving rise to a vicious cycle of
endothelial cell damage leading to the development of
atherosclerosis (8-10). Therefore, therapeutic approaches
involving inhibiting oxidation and improving mitochondrial function
may prevent atherosclerosis progression.
Salidroside is an active component isolated from
Rhodiola rosea, a well-known herb in traditional Chinese
medicine, exerts various pharmacological properties including
anti-inflammation (11),
anti-apoptosis (12),
antioxidative stress (13) and
anti-endothelial dysfunction (14). In recent years, the beneficent
effects of salidroside on atherosclerosis have attracted attention
(15,16). The study from Xing et al
(17) revealed that salidroside
has been demonstrated to attenuate endothelial cellular senescence
through decreasing the inflammatory response in an atherosclerosis
model. In addition, salidroside can protect against ox-LDL-induced
foam cell formation and apoptosis in THP1 human acute monocytic
leukemia cells (12). Although a
number of studies have reported on the anti-atherosclerotic effect
of salidroside, the molecular mechanisms underlying this effect are
not well understood.
SIRT1, a member of the conserved sirtuin family is a
key regulator in the progression of atherosclerosis, regulates a
well-known survival mechanism in atherosclerosis (18). Studies have demonstrated that
endothelial SIRT1 serves a protective role in the development of
atherosclerosis through a variety of mechanisms including reducing
apoptosis, improving endothelial function and defending against
oxidative stress (19,20). SIRT1 deficiency in endothelial
cells contributes to promoting the process of atherosclerosis
(21,22). Adenosine monophosphate-activated
protein kinase (AMPK), a fuel-sensing enzyme, takes part in the
anti-atherosclerotic process through improving endothelial function
in cardiovascular disease (23,24). Notably, it is reported that AMPK
can transcriptionally activate nicotinamide phosphoribosyl
transferase and then induce the activation of SIRT1 (25). Growing evidence has demonstrated
that the AMPK/SIRT1 pathway serves pivotal roles in
anti-atherosclerosis pathogenesis. However, the role of the
AMPK/SIRT1 pathway in the cardioprotective action of salidroside
against atherosclerosis has not been determined and remains to be
investigated.
Therefore, the present study investigated the
protective effect of salidroside on ox-LDL-induced human umbilical
vein endothelial cells (HUVECs) injury and focused on the roles of
the AMPK/SIRT1 pathway in these processes. A successful study may
provide a potential strategy for atherosclerosis therapy.
Materials and methods
Reagents and antibodies
Salidroside (cat. no. 43866-25, Purity >98%),
ox-LDL (cat. no. BP368) and dichloro-dihydro-fluorescein diacetate
(DCFH-DA; cat. no. D6883) were obtained from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany). Salidroside and ox-LDL were diluted with
DMSO and PBS, respectively, and stored at −20°C. Dulbecco's
modified Eagle's medium (DMEM; cat. no. 10569010), fetal bovine
serum (FBS; no. 10099-141), and streptomycin/penicillin (cat. no.
15140122) were purchased from Gibco (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Small interfering (si)RNA targeting SIRT1
(siSIRT1) and AMPK (siAMPK), and control siRNA (siControl) were
obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). The
MTT assay (cat. no. C0009) and bicinchoninic acid (BCA) protein
assay (cat. no. P0010) were purchased from the Beyotime Institute
of Biotechnology (Beijing, China). The kits for the measurement of
lactate dehydrogenase (LDH) release (cat. no. A020-2), methane
dicarboxylic aldehyde (MDA) content (cat. no. A003-1), superoxide
dismutase (SOD) activity (cat. no. A001-3) and glutathione
peroxidase (GSH-Px) level (cat. no. A005) were purchased from
Nanjing Jiancheng Bioengineering Institute (Nanjing, China). 5, 58,
6, 68-Tetraethylbenzimidazolcarbocyanine iodide (JC-1) was supplied
by BioVision (Inc., Milpitas, CA, USA; cat. no. 4999-100). The
caspase-3 colorimetric assay kit was supplied by Enzo Life Sciences
Inc. (Farmingdale, NY, USA; cat. no. BML-AK703-0001). The Annexin
V-Fluorescein Isothiocyanate (FITC)/Propidium Iodide (PI) Apoptosis
Detection kit was obtained from BD Biosciences (Franklin, Lakes,
NJ, USA; cat. no. 556547). Anti-SIRT1 (cat. no. ab104833) and
anti-NADPH oxidase 2 (NOX2; cat. no. ab133303) antibodies were
obtained from Abcam (Cambridge, UK). Anti-phosphorylated (p)-AMPK
(cat. no. 2537), AMPK (cat. no. 2793), cytochrome c (cat.
no. 11940), Bax (cat. no. 774), Bcl-2 (cat. no. 15071) and GAPDH
(cat. no. 2118) antibodies were obtained from Cell Signaling
Technology, Inc., (Danvers, MA, USA).
Cell culture and treatment
HUVECs were from the American Type Culture
Collection (Manassas, VA, USA; cat. no. CRL-1730) and cultured in
DMEM with 10% (v/v) FBS and 1% streptomycin/penicillin (v/v) in a
humidified 95% air-5% CO2 incubator at 37°C. For ox-LDL
treatment experiments, HUVECs were grown to 70-80% confluence and
then switched to different concentrations of ox-LDL (10, 25, 50,
100 and 200 µg/ml) for 12, 24, or 72 h. For protection
experiments, HUVECs were pretreated with salidroside (0.1, 1 and 10
µg/ml) for 1 h and incubated continuously for 24 h with
ox-LDL (100 µg/ml). For mechanism research studies, the
cells were pre-transfected with siSIRT1, siAMPK and siControl, and
then treated with salidroside (1 µg/ml) for 1 h followed by
co-incubation with ox-LDL (100 µg/ml) for 24 h.
Cell transfection
HUVECs at the density of 4×105 cells/well
were seeded in a six-well plate and then transfected with siSIRT1
(50 nM), siAMPK (100 nM) or siControl (100 nM; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) in accordance with the manufacturer's protocol.
The siSIRT1 sequences were 5′-CCCUCAAAGUAAGACCAGUTT-3′ (sense) and
5′-ACUGGUCUUACUUUGAGGGAA-3′ (antisense). The siAMPK sequences were
5′-GGACAGGGAAGCCUUAAAUTT-3′ (sense) and 5′-AUUUAAGGCUUCCCUGUCCTT-3′
(antisense). The negative control siRNA sequences were
5′-UUCUCCGAACGUGUCACGUTT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAATT-3′
(antisense). Briefly, cells were grown to 70-90% confluence at the
time of transfection. Lipofectamine® 2000 (10 µl)
was gently agitated and diluted in Opti-MEM® I Medium
(cat. no. 31985-070; Invitrogen; Thermo Fisher Scientific, Inc.)
without serum (150 µl) in a separate vessel and the mixture
was incubated for 5 min at room temperature. siRNA or control
siControl (150 pmol) was diluted into in Opti-MEM® I
Medium without serum (150 µl). Subsequently, diluted DNA was
added to diluted Lipofectamine® 2000 Reagent (1:1
ratio). Following incubation for 5 min at room temperature, the
siRNA-lipid complex (250 µl) was added to each well
containing cells and medium. Following transfection for 6 h, the
transfection mixture was replaced with fresh growth medium.
Transfection efficiency was measured using western blot analysis.
Subsequent experiments with transfected cells were performed
following transfection for 24 h.
HUVECs viability assay
The viability of HUVECs was determined by 3-(4,
5-dimethylthiazol-2-yl)-2, 5-diphenyltetra-zolium bromide (MTT)
assay according to the manufacturer′s protocol. Cells at a density
of 1×104 cells/well were plated onto a 96-well culture
plate and treated as discussed above. Following treatment for 24 h,
the MTT regent (0.5 mg/ml) was supplemented added and co-incubated
for 3 h at 37°C. Subsequently, DMSO (150 µl) was added to
each well to fully dissolve the crystal formazan. The optical
density (OD) value at 450 nm was then measured using a microplate
reader (Bio-Tek Instruments, Inc., Winooski, VT, USA). Each
experiment was performed three times.
LDH release assay
The release of LDH was determined using the LDH
assay kit following the manufacturer's protocol. Briefly, cells
were seeded in 96-well plates at a density of 1×104
cells/well and treated as discussion above for 24 h at 37°C. Then
the supernatant was collected. After centrifuging at 1,000 × g for
10 min at room temperature, the supernatant (20 µl) was
mixed with 2,4-dinitrophenylhydrazine (20 µl), transferred
at 37°C for 15 min and then NaOH (0.4 M, 250 µl) was added
into the mixture and co-incubated for a further 15 min at 37°C. The
absorbance was detected at 450 nm using a microplate reader
(Bio-Tek Instruments, Inc.).
Flow cytometric assay for cell
apoptosis
HUVECs apoptosis was performed using an Annexin
V-FITC/PI kit according to the manufacturer's protocol. Briefly,
cells were cultured in 6-well plates at the density of
1×105 cells/well and treated as discussion above. After
incubation for 24 h, cells were harvested using 0.05% trypsin and
washed three times with ice-cold PBS, and then re-suspended in 1X
binding buffer (500 µl) included in the kit followed by
mixed and incubated with 5 µl of Annexin V-FITC reagent and
5 µl of PI at room temperature for 15 min. Cell apoptosis
was analyzed on a FACScan flow cytometer (BD Biosciences). The
percentage of cells in quadrant 2 (Q2) and 4 (Q4) were
quantitatively analyzed using FCS Express 3.0 software (De Novo
Software, Glendale, CA, USA), generating the total percentage of
apoptotic cells at both early and late apoptotic stage or apoptotic
index. All experiments were performed in triplicate.
Measurement of intracellular ROS
generation
The intracellular ROS production was determined by a
redox-sensitive fluorescent probe dichloro-dihydro-fluorescein
diacetate (DCFH-DA) according to the protocol. Briefly, following
treatment for 24 h, HUVECs were washed twice with ice-cold PBS and
then incubated with DCFH-DA (10 µM) for 20 min at 37°C.
After washing twice with PBS, the fluorescence values were detected
by a F97 PRO fluorescence spectrophotometer (Shanghai Lengguang
Technology Co., Ltd., Shanghai, China). In addition, following
incubation with DCFH-DA (10 µM) for 20 min at 37°C, the
sample was collected and centrifuged at 800 × g for 5 min at room
temperature and washed twice with PBS, and then the fluorescence
intensity was measured using FACScan flow cytometer at excitation
and emission wavelengths of 485 and 528 nm, respectively.
Measurements of intracellular MDA
content, SOD and GSH-Px activities
The MDA content, SOD and GSH-Px activities in HUVECs
were measured using colorimetric assay kits according to the
manufacturer's protocols. The MDA level was measured using the
thiobarbituric acid method with a maximum absorbance at 532 nm. The
SOD activity was based on the combination of xanthine and xanthine
oxidase and the absorbance was recorded at a wavelength of 550 nm.
GSH-Px activity was determined using the enzyme-catalyzed reaction
product (reduced glutathione) at a wavelength of 412 nm.
Assessment of mitochondrial membrane
potential (MMP)
MMP was assessed using a cationic fluorescent
indicator JC-1 which can selectively enter mitochondria and change
color from red to green when MMP decreases. Briefly, HUVECs were
washed twice with PBS and then incubated with JC-1 (200 µM)
for 20 min at 37°C followed by washing with PBS to remove excess
JC-1. The fluorescence was observed by a F97 PRO fluorescence
spectrophotometer (Shanghai Lengguang Technology Co., Ltd.) at an
excitation wavelength of 529 nm and an emission wavelength of 590
nm.
Assessment of caspase-3 activity
The caspase-3 activity was determined using
caspase-3 colorimetric assay kit carried out as described by the
protocol provided by the manufacturer. The principle of the assay
is that acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-рNA) could
be catalyzed by caspase-3 to produce a yellow substance
p-nitroaniline, which reflects caspase-3 activity. In brief, HUVECs
were collected and lysed with lysis buffer (provided by the kit)
for 30 min on ice followed by centrifuging at 16,000 × g for 15 min
at 4°C. The supernatant was then incubated with Ac-DEVD-рNA at 37°C
for 2 h. Finally, the absorbance was detected at 405 nm and the
relative caspase-3 activities were calculated as follows: Caspase-3
activity (%) = (ODtreatment − ODtreatment
blank)/(ODcontrol − ODcontrol blank) ×
100%.
Western blot analysis
HUVECs were harvested and lysed in pre-cooled RIPA
buffer (cat. no. P0013B; Beyotime Institute of Biotechnology)
containing pheylmethanesulfonyl fluoride (0.2 mM). The cell lysate
was then centrifuged at 13,400 × g for 10 min at 4°C and the total
protein concentration was determined by the BCA protein assay kit
according to the protocol provided by the manufacturer. Equal
amounts of protein (30 µg) were separated by a 12% SDS-PAGE
and then transferred to a polyvinylidene difluoride membrane. The
membrane was blocked with TBS Tween-20 (TBST) buffer [20 mM
Tris-HCl (pH 8.0)/150 mM NaCl/0.05% Tween-20] containing 5% non-fat
dairy milk for 2 h at room temperature and then incubated overnight
at 4°C with the following antibodies: Rabbit anti-SIRT1 (1:1,000),
rabbit anti-phosphorylated AMPK (1:2,000), rabbit anti-AMPK
(1:2,000), rabbit anti-Bax (1:1,000), rabbit anti-Bcl-2 (1:1,000),
rabbit anti-NOX2 (1:1,000), rabbit anti-cytochrome c
(1:1,000) or rabbit anti-GAPDH (1:1,000). After washing five times
with TBST buffer, the membrane was incubated with horseradish
peroxidase-conjugated goat anti-rabbit secondary antibody (1:5,000;
cat. no. 7074; Cell Signaling Technology, Inc.) at for 2 h at room
temperature. The membrane was finally visualized with the BeyoECL
Plus detection kit (cat. no. P0018M; Beyotime Institute of
Biotechnology). Scanned images were visualized with the LI-COR
Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE,
USA).
Statistical analysis
Results are expressed as the mean ± standard
deviation from at least three separate experiments. Statistical
analyses were performed using SPSS 13.0 software (SPSS, Inc.,
Chicago, IL, USA). The significance of differences was calculated
with one-way analysis of variance followed by Tukey's test as
appropriate. P<0.05 was considered to indicate a statistically
significant difference.
Results
Salidroside prevents ox-LDL-induced
HUVECs injuries
To determine the cytotoxic potential of salidroside
on HUVECs, the effects of salidroside on cell viability was
evaluated. The MTT assay result demonstrated that incubation with
low concentrations of salidroside (0.1, 1, 5, or 10 µM) for
24 h did not affect cell viability, whereas high concentrations of
salidroside (20, 40, or 80 µM) significantly reduced cell
viability (P<0.05; Fig. 1A).
Therefore, three concentrations of salidroside (0.1, 1 and 10
µM) were selected for subsequent experiments. Next, in order
to investigate the damaging effects of ox-LDL on HUVECs, cells were
treated with ox-LDL (25, 50, 100 and 200 µg/ml) for 12, 24
or 72 h. The MTT assay results revealed that cell viability of
HUVECs in response to ox-LDL (100 and 200 µg/ml) for 12, 24
and 72 h was significantly reduced (P<0.05; Fig. 1B). Notably, incubation with ox-LDL
(100 µg/ml) for 24 h could reduce cell viability to
51.37±7.63%, which was selected as the next experimental condition.
Subsequently, it was demonstrated that pretreatment with
salidroside (0.1, 1, or 10 µM) significantly reversed
ox-LDL-induced the downregulation of cell viability (P<0.05;
Fig. 1C). Furthermore, the LDH
release assay result revealed that ox-LDL treatment significantly
increased the LDH release in HUVECs (P<0.01), while this effect
was also significantly inhibited by pretreatment with salidroside
(P<0.05; Fig. 1D). Flow
cytometry with Annexin V-FITC/PI staining demonstrated that
salidroside pretreatment significantly reversed the ox-LDL-induced
upregulation of the apoptosis percentage (P<0.05; Fig. 1E and F). These results suggested
that salidroside protects HUVECs from ox-LDL injury.
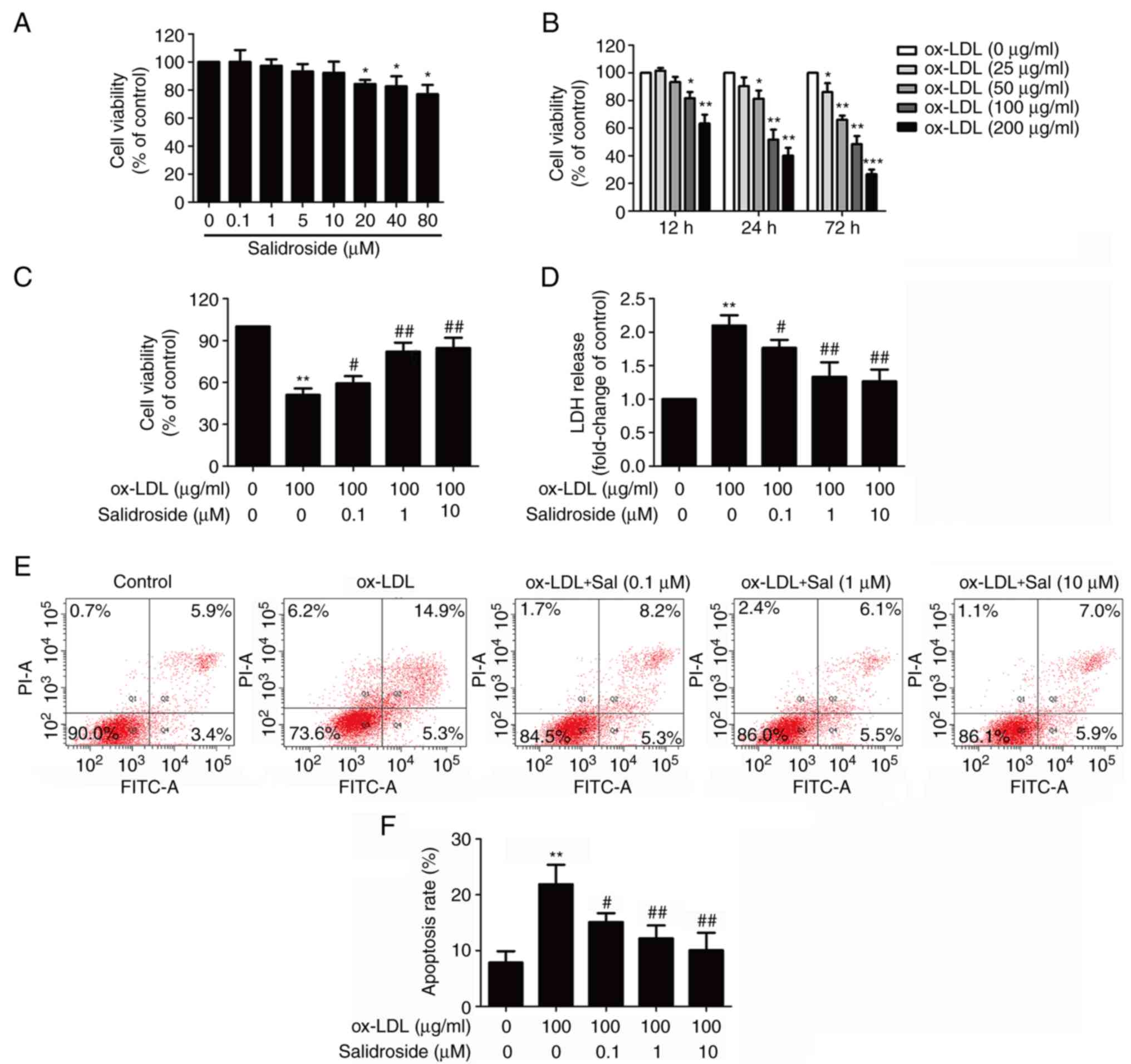 | Figure 1Effects of Sal on cytotoxicity in the
presence or absence of ox-LDL in HUVECs. (A) HUVECs were incubated
with Sal (0.1, 1, 10, 20, 40, or 80 µM) for 24 h and the
cell viability was assessed by performing an MTT assay. (B) HUVECs
were treated with different concentrations of ox-LDL (50, 100 and
200 µg/ml) for 24 h, and the cell viability was measured by
MTT assay. HUVECs were pretreated with salidroside (0.1, 1, or 10
µM) for 1 h and then exposed to ox-LDL (100 µg/ml)
for 24 h. (C) Cell viability was detected using an MTT assay. (D)
LDH release was determined using an LDH assay kit. (E) The
apoptosis ratio was measured using flow cytometry with an Annexin
V-FITC/PI staining kit. (F) Quantitative analysis of Annexin
V-FITC(+)/PI(-) and Annexin V-FITC(+)/PI(+). Data are expressed as
the mean ± standard deviation; n=3, *P<0.05,
**P<0.01 and ***P=0.001 vs. the control
group; #P<0.05 and ##P<0.01 vs. the ox-LDL group.
HUVECs, human umbilical vascular endothelial cells; ox-LDL,
oxidized-low density lipoprotein; FITC, fluorescein isothiocyanate;
PI, propidium iodide; LDH, lactate dehydrogenase; Sal,
salidroside. |
SIRT1 mediates the protection of
salidroside from cytotoxicity and apoptosis induced by ox-LDL in
HUVECs
SIRT1 serves a vital role in promoting cell survival
in the development of atherosclerosis (26). In the present study, it was
demonstrated that ox-LDL (100 µg/ml) treatment significantly
reduced the expression of SIRT1, while this function was
significantly abolished by pretreated with salidroside (0.1, 1, or
10 µM; P<0.05; Fig.
2A). According to the Fig. 1
and this result, it was demonstrated that salidroside (10
µM) has a protective effect on ox-LDL injury and
ox-LDL-induced SIRT expression downregulation. Therefore,
salidroside (10 µM) was selected for subsequent experiments.
To further confirm the role of SIRT1 in the cardioprotection of
salidroside, HUVECs were transfected with siSIRT1 or siControl. The
transfection efficiency was detected using western blotting
analysis and the result demonstrated that the expression of SIRT1
in siSIRT1 transfected cells was decreased compared with in the
siControl transfected cells (Fig.
2B), indicating that SIRT1 knockdown was achieved by siSIRT1
transfection. In addition, it was demonstrated that ox-LDL-induced
downregulation of cell viability was exacerbated by SIRT1 knockdown
and the salidroside-induced increase in the cell viability was also
significantly inhibited by siSIRT1 transfection in ox-LDL-treated
HUEVCs (Fig. 2C). siSIRT1
transfection also further induced the increase in LDH release and
reversed salidroside-induced the decrease in the LDH release in
ox-LDL-treated HUEVCs (Fig. 2D).
In addition, salidroside eliminated, while siSIRT1 exacerbated,
ox-LDL-induced the upregulation of apoptosis ratio (Fig. 2E and F) and caspase-3 activity
(Fig. 2G). Notably, the
inhibition of salidroside on apoptosis was inhibition by
transfection with siSIRT1. Together, the results suggested that
SIRT1 contributes to the protection of salidroside against ox-LDL
injury in HUVECs.
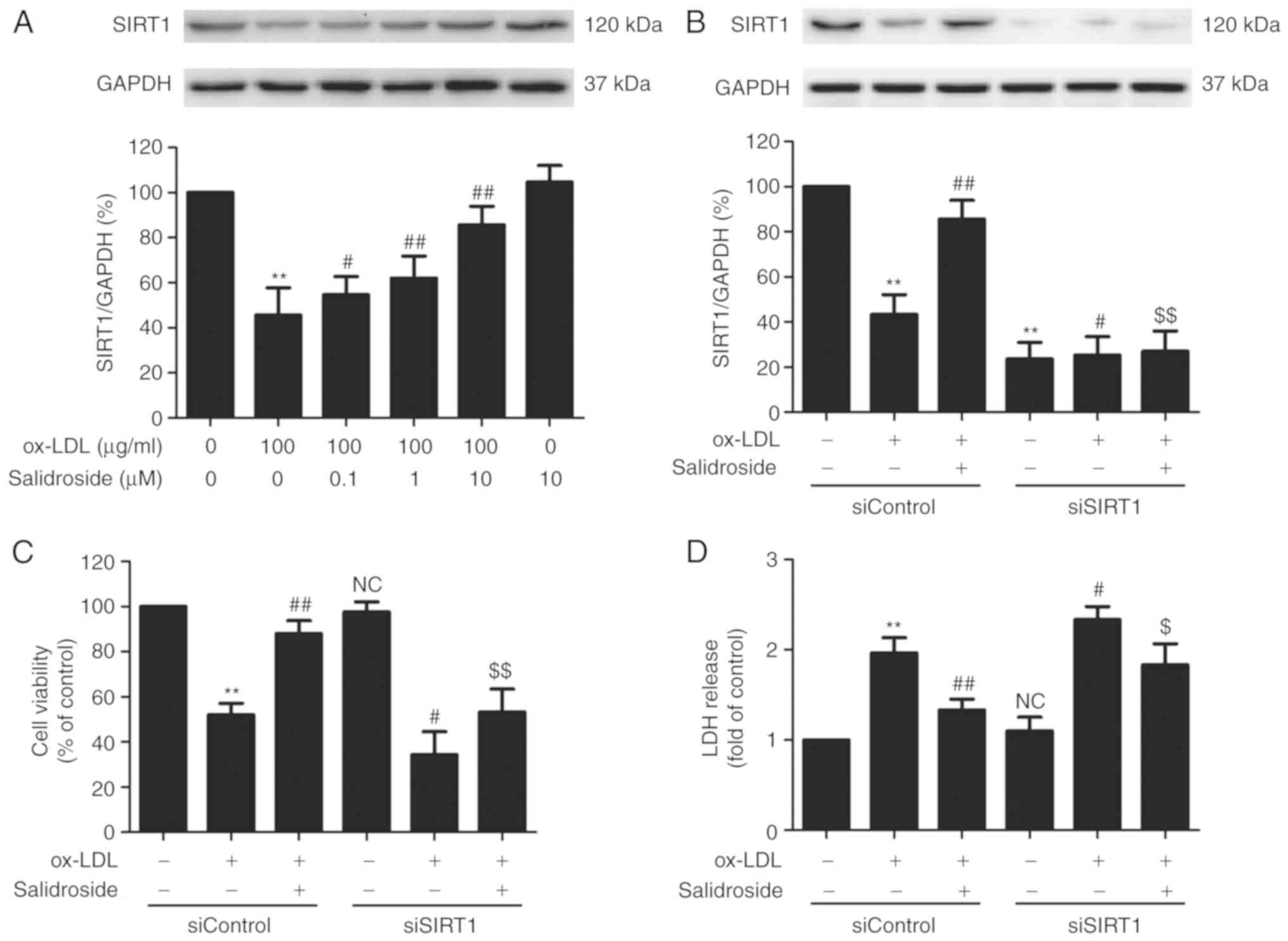 | Figure 2Effects of SIRT1 knockdown on the
protection of Sal on ox-LDL-induced HUVECs injury. (A) Cells were
pretreated with Sal (0.1, 1, or 10 µM) for 1 h and then
stimulated with ox-LDL (100 µg/ml) for 24 h, and the
expression of SIRT1 was measured by western blot analysis. HUVECs
were pre-transfected with siSRT1 or siControl and incubated with
Sal (10 µM) for 1 h followed by treatment with ox-LDL (100
µg/ml) for 24 h. (B) The transfection efficiency was
determined by western blot analysis. (C) Cell viability was
detected using MTT assay. (D) The LDH release was determined using
an LDH assay kit. (E) The apoptosis was detected using the Annexin
V-FITC/PI staining kit. (F) Quantitative analysis of Annexin
V-FITC(+)/PI(-) and Annexin V-FITC(+)/PI(+). (G) The caspase-3
activity was measured by the caspase-3 colorimetric assay kit. Data
are expressed as the mean ± standard deviation; n=3,
**P<0.01 vs. the control group; #P<0.05
and ##P<0.01 vs. the ox-LDL group;
$P<0.05 and $$P<0.01 vs. the ox-LDL and
Sal co-treatment group. si, small interfering; HUVECs, human
umbilical vascular endothelial cells; ox-LDL, oxidized low density
lipoprotein; FITC, fluorescein isothiocyanate; PI, propidium
iodide; LDH, lactate dehydrogenase; Sal, salidroside; NC, not
significant; SIRT1, sirtuin 1. |
SIRT1 participates in the inhibitory
action of salidroside against oxidative stress induced by ox-LDL in
HUVECs
Oxidative stress is involved in the progression of
atherosclerosis, which leads to ox-LDL-induced endothelial
dysfunction (8). The effects of
salidroside on the oxidative stress index including ROS generation,
MDA content and NOX2 expression were further investigated with
ox-LDL treatment. The result demonstrated that ox-LDL treatment
results in an increase in ROS green fluorescence (Fig. 3A), DCF fluorescence percentage
(Fig. 3B), MDA content (Fig. 3C) and NOX2 expression (Fig. 3D and E), which were reversed by
salidroside. Notably, the effects of salidroside were reversed by
SIRT1 knockdown induced by siSIRT1 transfection. Furthermore,
siSIRT1 transfection further aggravated ox-LDL-induced oxidative
stress. The antioxidants produced endogenously including SOD and
GSH-Px serve important roles in preventing oxidative damage in
atherosclerosis (27). It was
demonstrated that salidroside diminished the ox-LDL-induced
downregulation of SOD (Fig. 3F)
and GSH-Px activities (Fig. 3G).
However, the functions of salidroside were inhibited by siSIRT1
transfection. Taken together, these results indicated that SIRT1
mediates the protective effect of salidroside against oxidative
stress and inhibition of antioxidant defense system induced by
ox-LDL.
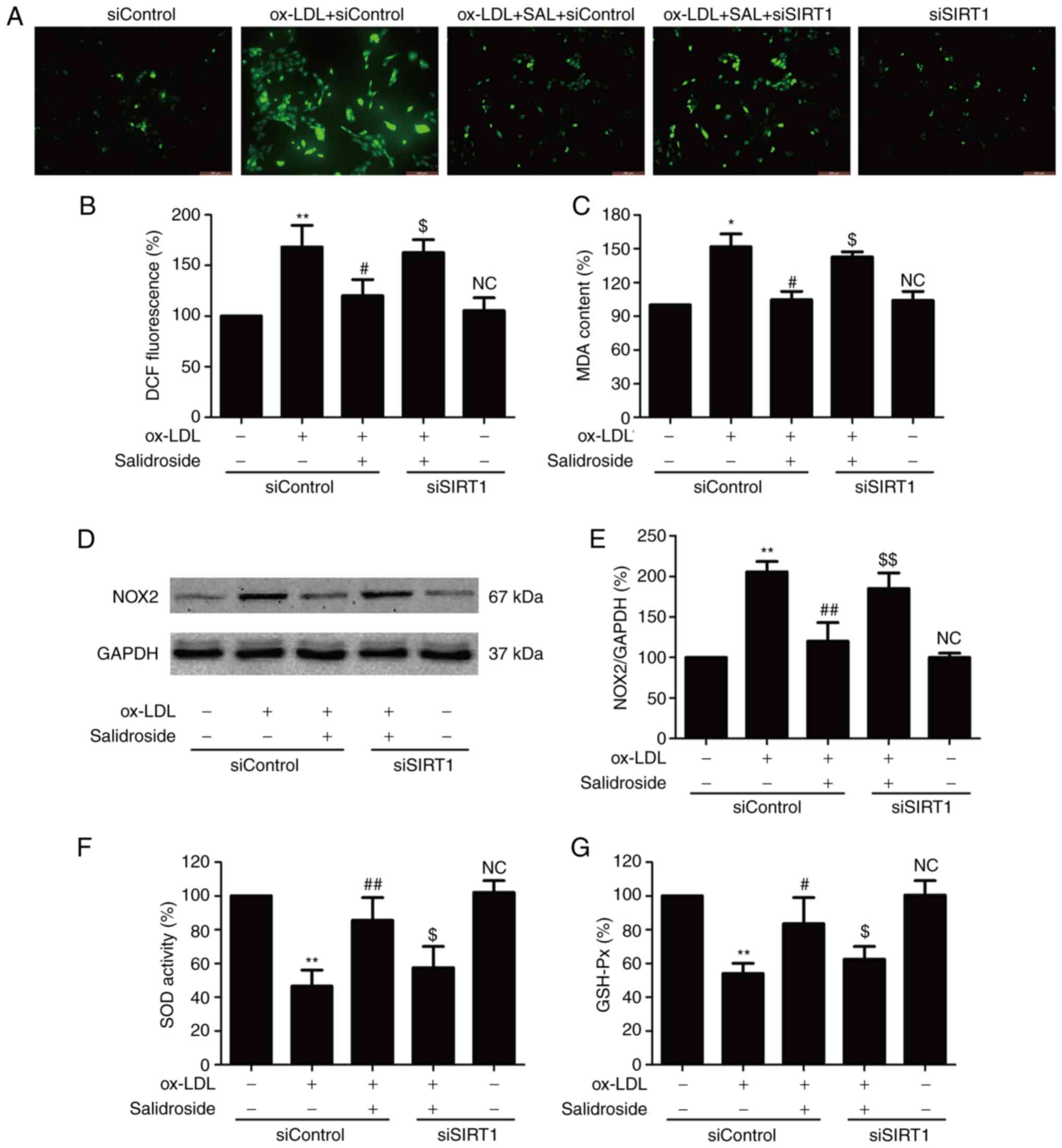 | Figure 3Effects of SIRT1 knockdown on the
inhibition of salidroside on ox-LDL-induced oxidative stress in
HUVECs. HUVECs were pre-transfected with siSRT1 or siControl and
incubated with SAL (10 µM) for 1 h followed by treatment
with ox-LDL (100 µg/ml) for 24 h. (A) The intracellular ROS
production was determined by a redox-sensitive fluorescent probe
DCFH-DA staining. Scale bar, 200 µM. (B) The fluorescence
intensity of DCFDA-stained sample was quantified by flow cytometry.
(C) The intracellular MDA content was measured using the
thiobarbituric acid method with a maximum absorbance at 532 nm. (D)
The expression of NOX2 was determined using western blot analysis.
(E) Bar charts demonstrate the quantification of NOX2 expression.
(F) The SOD activity was detected using a colorimetric assay kit
based on the combination of xanthine and xanthine oxidase. (G) The
GSH-Px level was determined using a colorimetric assay kit
depending on the enzyme-catalyzed reaction product (reduced
glutathione). Data are expressed as the mean ± standard deviation;
n=3, *P<0.05 and **P<0.01 vs. the
control group; #P<0.05 and ##P<0.01 vs.
the ox-LDL treatment group; $P<0.05 and
$$P<0.01 vs. the ox-LDL and SAL co-treatment group.
HUVECs, human umbilical vascular endothelial cells; ox-LDL,
oxidized-low density lipoprotein; GSH-Px, glutathione peroxidase;
SOD, superoxide dismutase; DCFH-DA, dichloro-dihydro-fluorescein
diacetate; NOX2, NADPH oxidase 2; MDA, methane dicarboxylic
aldehyde; ROS, reactive oxygen species; SAL, salidroside; NC, not
significant; si, small interfering. |
Salidroside attenuates ox-LDL-induced
mitochondrial dysfunction via upregulation of SIRT1 in HUVECs
ROS produced by mitochondrial dysfunction affecting
cell survival and resulting in apoptosis and oxidative stress have
been implicated in the induction of atherosclerosis (28,29). Therefore, whether salidroside
effects mitochondrial function in ox-LDL-treated HUVECs and whether
SIRT1 pathway is involved in this effect was investigated. JC-1
staining revealed that ox-LDL treatment resulted in the decrease in
red fluorescence and the increase in green fluorescence indicating
a lowering of MMP, while salidroside pretreatment resulted in the
increase red fluorescence and the decrease green fluorescence,
leading to increased MMP (Fig.
4A). However, the effect of salidroside was inhibited by siSIRT
transfection. As an increase in cytochrome c expression and
Bax/Bcl-2 ratio are associated with MMP disruption and
mitochondrial dysfunction (29),
the effects of salidroside on these markers of mitochondrial
function were analyzed. The results of western blot analyses
(Fig. 4B) demonstrated that
salidroside significantly reduced the expression of cytochrome
c (P<0.05; Fig. 4C) and
the ratio of Bax/Bcl-2 compared with the ox-LDL treatment group,
while these roles were abolished by transfection with siSIRT1
(Fig. 4D). These results
suggested that salidroside improves mitochondrial dysfunction under
ox-LDL injury conditions through promoting SIRT1 in HUVECs.
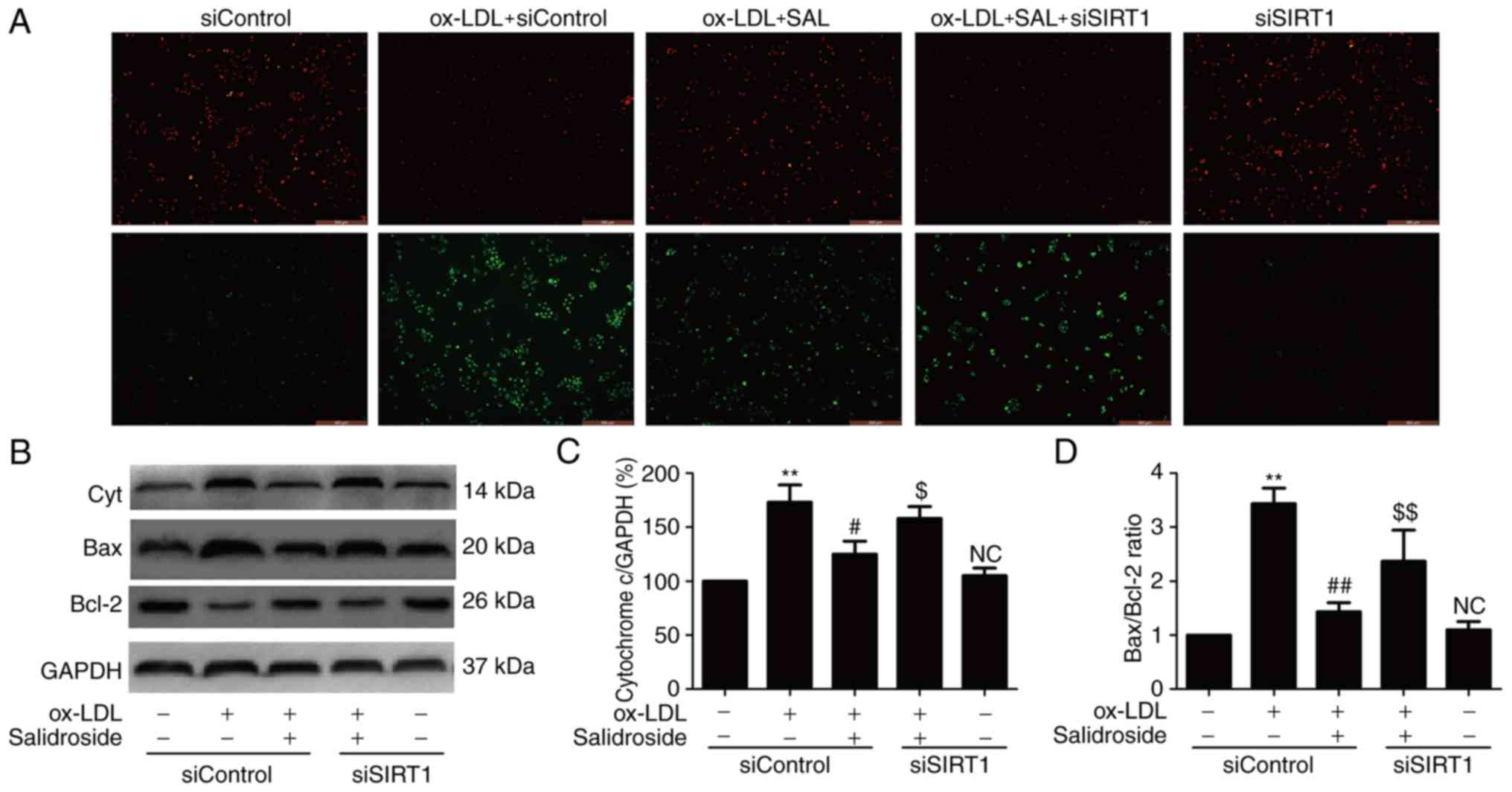 | Figure 4Effects of SIRT1 knockdown on the
protection of salidroside-induced the improvement of mitochondrial
dysfunction induced by ox-LDL in HUVECs. HUVECs were
pre-transfected with siSIRT1 or siControl and incubated with
salidroside (10 µM) for 1 h followed by treatment with
ox-LDL (100 µg/ml) for 24 h. (A) MMP was determined by the
specific dye 5, 58, 6, 68-tetraethylbenzimidazolcarbocyanine iodide
staining. Scale bar, 200 µM. (B) The expression of Cyt, Bax
and Bcl-2 were measured by western blot analyses. Bar charts
demonstrate the quantification of (C) Cyt and (D) the ratio of
Bax/Bcl-2. Data are expressed as the mean ± standard; n=3.
**P<0.01 vs. the control group;
##P<0.01 vs. the ox-LDL treatment group;
$P<0.05 and $$P<0.01 vs. the ox-LDL and
salidroside co-treatment group. NC, not significant; Cyt,
cytochrome c; MMP, mitochondrial membrane potential; HUVECs, human
umbilical vascular endothelial cells; ox-LDL, oxidized low density
lipoprotein; si, small interfering; SIRT1, sirtuin 1. |
AMPK/SIRT1 pathway activation mediates
the protective effect of salidroside against ox-LDL-induced HUVEC
injury
AMPK is a critical regulator of mitochondrial
biogenesis and is reported to enhance SIRT1 (30). To further certify the role of the
AMPK pathway in cardioprotection by salidroside the effects of
salidroside on the AMPK pathway following ox-LDL treatment were
measured. It was demonstrated that ox-LDL decreased the p-AMPK
protein expression in HUVECs, however salidroside significantly
upregulated the p-AMPK expression in ox-LDL-treated HUVECs
(P<0.05; Fig. 5A and B),
indicating that AMPK was activated by salidroside. To further
investigate the roles of the AMPK pathway in the protective action
of salidroside on ox-LDL injury, HUVECs were transfected with
siAMPK. Western blotting analyses validation demonstrated that
siAMPK transfection reduced the expression of AMPK compared with
siControl transfection (Fig. 5C)
following ox-LDL treatment or ox-LDL + salidroside co-treatment. In
addition, it was demonstrated that transfection with siAMPK leads
to the elimination of the salidroside-induced increase in cell
viability (Fig. 5D) and the
decrease in LDH release (Fig. 5E)
in ox-LDL-treated HUVECs. Notably, it was demonstrated that siAMPK
transfection further decreased the expression of SIRT1 compared
with the siControl transfection and also reversed
salidroside-induced the upregulation of SIRT1 expression (Fig. 5F). In conclusion, the results of
the present study suggested that salidroside protects HUVECs
against ox-LDL-induced injury through activating the AMPK/SIRT1
pathway.
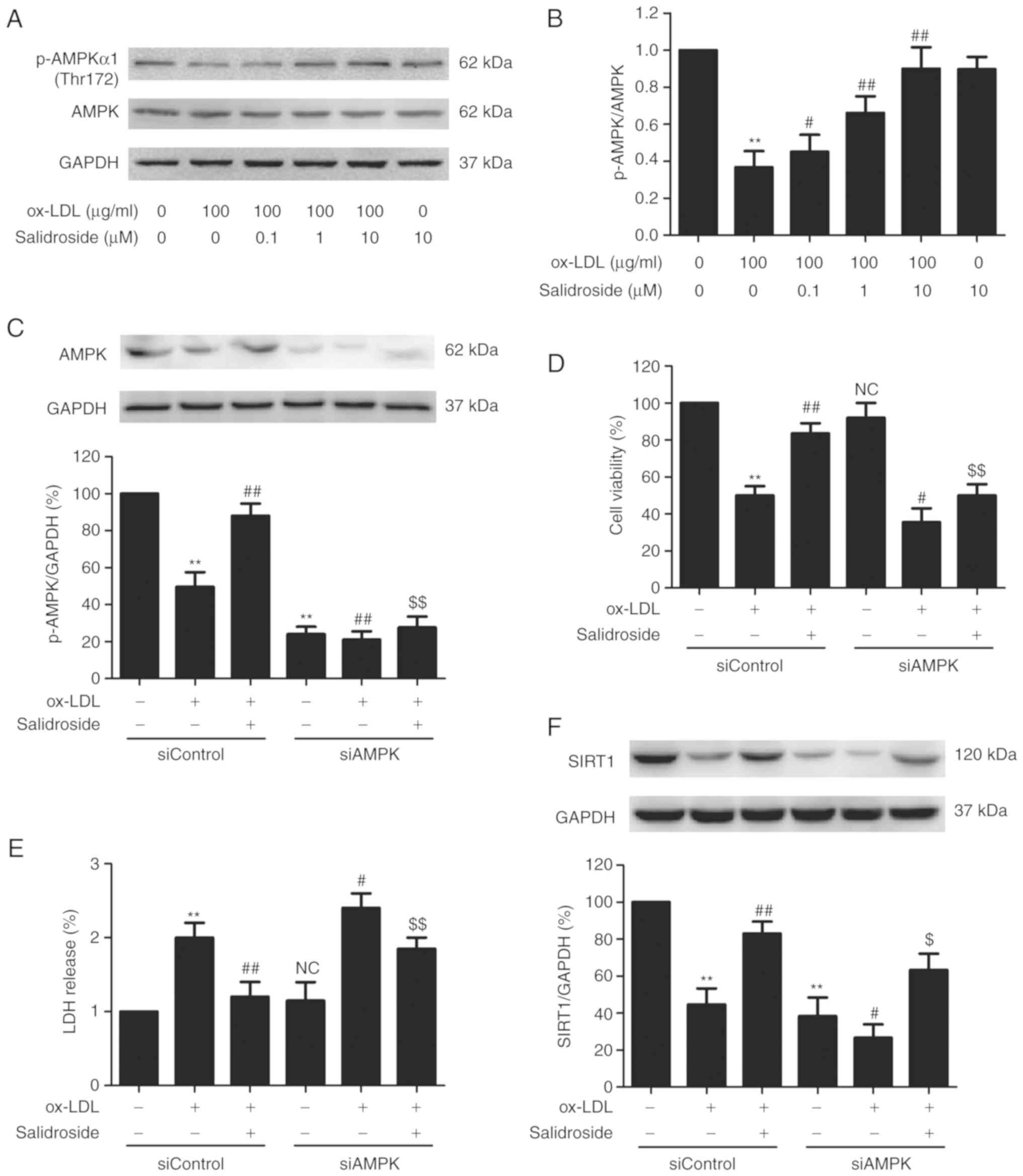 | Figure 5Effects of AMPK knockdown on the
protection of salidroside on ox-LDL-induced HUVECs injury and SIRT1
expression in HUVECs. (A) Cells were pretreated with salidroside
(0.1, 1, or 10 µM) for 1 h and then stimulated with ox-LDL
(100 µg/ml) for 24 h, and the expression of p-AMPK and AMPK
were measured by western blot analysis. (B) Bar charts illustrate
the quantification of p-AMPK and AMPK proteins. HUVECs were
pre-transfected with siAMPK or siControl and incubated with
salidroside (10 µM) for 1 h followed by treatment with
ox-LDL (100 µg/ml) for 24 h. (C) The transfection efficiency
was determined by western blot analysis. (D) The viability was
detected using MTT assay. (E) The LDH release was determined using
LDH Cytotoxicity Assay kit. (F) The expression of SIRT1 was
determined by western blot analysis. Data are expressed as the mean
± standard; n=3. **P<0.01 vs. the control group;
#P<0.05 and ##P<0.01 vs. the ox-LDL
treatment group; $P<0.05 and $$P<0.01
vs. the ox-LDL and salidroside co-treatment group. p,
phosphorylated; AMPK, adenosine monophosphate-activated protein
kinase; HUVECs, human umbilical vascular endothelial cells; ox-LDL,
oxidized low density lipoprotein; si, small interfering; SIRT1,
sirtuin 1; LDH, lactate dehydrogenase; NC, not significant. |
Discussion
Mounting evidence points to endothelial cell damage
as one of the major pathophysiological links between cardiovascular
risk factors and the development of atherosclerosis (31). Salidroside, a major active
ingredient from the medicinal plant Rhodiola rosea L.,
possesses anti-inflammatory, anti-apoptotic antioxidative and
cardioprotective effects (32-35). Although salidroside has been
proven to be useful in treating various types of diseases including
atherosclerosis, its effects on endothelial cell damage in
atherosclerosis and the possible underlying molecular mechanisms
remain unclear. In the present study, it was demonstrated that
salidroside was capable of protecting endothelial cells from damage
from oxidative stress and mitochondrial dysfunction induced by
exogenous ox-LDL. Furthermore, the investigation focused on the
underlying cardioprotective molecular mechanisms of salidroside,
which were involved in promoting the AMPK/SIRT1 pathway.
SIRT1, a member of the conserved sirtuin family,
serves a pivotal role in the pathogenesis and therapy of heart
disease through a variety of mechanisms, including reducing
apoptosis, improving endothelial function, and defending against
oxidative stress (19,20). During the recent years, a number
of investigators have proved that salidroside exhibits a
cardio-protective effect against atherosclerosis (12,17,36). Notably, evidence confirms that
SIRT1 participates in the beneficial effect of salidroside in lung
injury and nervous system disease through inhibiting apoptosis,
inflammation, and oxidative stress (37-39). However, the role of SIRT1 in the
cardioprotective effects of salidroside has not yet been reported.
In the present study, it was demonstrated that salidroside
mitigates ox-LDL-induced HUVEC injury as demonstrated by the
increase of cell viability and downregulation of LDH release.
Notably, salidroside reversed the ox-LDL-induced decrease in the
expression of the SIRT1 protein, which was in line with the
previous study by Donato et al (40), indicating the possible involvement
of SIRT1 in the cardioprotective effect of salidroside in
atherosclerosis. In addition, the present study confirmed that
knockdown of SIRT1 by siSIRT1 transfection inhibits the
salidroside-induced cardioprotective effect against ox-LDL-induced
HUVECs injury. These results suggested that SIRT1 mediates the
cardioprotective effect of salidroside against atherosclerosis.
A number of studies have revealed that oxidative
stress is a pivotal feature of atherosclerosis and is produced by
an imbalance between antioxidant capability and the presence of
species including ROS (41,42). These conditions cause cell injury
and serve a critical role in apoptosis via cell death signaling
pathways (43). Salidroside can
improve cardiac dysfunction by suppressing excessive oxidative
stress and cardiomyocyte apoptosis (33,44). However, there are few studies
about the effects of salidroside on oxidative stress and apoptosis
in atherosclerosis. In the current study, it was demonstrated that
salidroside reduces ox-LDL-induced oxidative stress as demonstrated
by the decrease of ROS generation, MDA content and NOX2 expression
and improves the antioxidant defense system as demonstrated by the
upregulation of SOD and GSH-Px activities. On the other hand, it is
confirmed in the literature that SIRT1 deficiency in endothelial
cells contributes to increased oxidative stress, inflammation, foam
cell formation, apoptosis and autophagy, thereby promoting
atherosclerosis (22,45), and multiple myocardial protective
drugs-induced beneficial processes were mediated by SIRT1 within
the molecular circuitry underlying endothelial dysfunction via
inhibiting oxidative stress in atherosclerosis (46-48). Importantly, a recent study
elucidated that SIRT1 mediates salidroside-elicited protection
against apoptosis and oxidative stress in Parkinson's disease
(37). In line with these
studies, the results of the present study also demonstrated that
SIRT1 knockdown mitigates salidroside-induced inhibition of
oxidative stress and promotion of the antioxidant defense system in
ox-LDL-treated HUVECs, suggesting that SIRT1 mediates the
protective effect of salidroside against oxidative stress in
atherosclerosis.
The mitochondrion is the main source of chronic ROS
production under physiological conditions (6). In addition, enhanced mitochondrial
ROS generation and functional disorder are conducive to the
development of atherosclerosis (7,49).
Previous studies revealed that salidroside can rescue mitochondrial
dysfunction caused by various stimuli in HUVECs (50,51). Cai et al (52) proved that salidroside protects the
liver against ischemia reperfusion injury by regulating the
antioxidant response and mitochondrial permeability transition. In
addition, salidroside can prevent
H2O2-induced HUVEC injury through promoting
mitochondrial function, thereby reducing the overactivation of
oxidative stress-associated downstream signaling pathways (50). Furthermore, salidroside can
improve endothelial function and alleviate atherosclerosis
depending on mitochondria-associated pathways (15). Additionally, research has
confirmed the regulatory effect of SIRT1 on mitochondrial dynamics,
which has gained much attention (53,54). Ma et al (55) confirmed that SIRT1 activation
alleviates cardiac dysfunction via the restoration of mitochondrial
biogenesis and function. In line with these studies, the present
study demonstrated that knockdown of SIRT1 attenuates the
salidroside-induced inhibition on mitochondrial dysfunction induced
by ox-LDL as illustrated by the increase in MMP and the decreases
in cytochrome c expression and the Bax/Bcl-2 ratio,
indicating the contribution of SIRT1 to the improvement of
salidroside on mitochondrial dysfunction in ox-LDL-treated
HUVECs.
AMPK is an important metabolic switch that is
involved in the modulation of cellular and whole-body metabolism.
Previous evidence indicates that AMPK is essential for the
maintenance of cardiovascular health and has a regulatory effect on
endothelial function and vascular structure (56,57). For instance, activation of AMPK
mitigates the generation of ROS induced by mitochondrial
dysfunction, endoplasmic reticulum stress and NADPH oxidase
(58). The mechanism of action of
a number of therapeutic agents commonly used in cardiovascular and
diabetic diseases, including metformin, thiazolidinediones and
statins, may involve AMPK (59),
although the precise downstream pathways remain poorly defined.
Activation of AMPK also has a number of potentially beneficial
anti-atherosclerotic effects. Notably, the study from Xing et
al (15) revealed that the
activation of AMPK-dependent pathway contributes to the
salidroside-mediated result of improving endothelial function and
alleviating atherosclerosis. Similarly, in the present study it was
demonstrated that salidroside reversed the ox-LDL-induced increase
in the expression of p-AMPK, indicating the that the activation of
AMPK was induced by salidroside. In addition, knockdown of AMPK
induced by transfection with siAMPK inhibited the cardioprotective
effect of salidroside on ox-LDL-induced HUVEC injury, which was
consistent with the study from Zheng et al (60), which demonstrated that the
inhibition of AMPK activity induced by the AMPK inhibitor Compound
C and siRNA suppressed the beneficial effects of salidroside in
hepatocytes. Notably, it was demonstrated that salidroside
increases the expression of p-AMPK but has little effect on the
level of AMPK in ox-LDL-treated HUVECs, indicating that salidroside
elicited a cardioprotective effect against atherosclerosis via
directly increasing p-AMPK expression and activating the AMPK
pathway. As SIRT1 exerts an NAD+-dependent deacetylase
action, its activity is regulated by the proteins modulating the
cellular NAD+ level. Among them, AMPK is an important
protein. It has been confirmed that AMPK phosphorylation regulates
SIRT1 activity (61).
Importantly, the results of the present study demonstrated that
knockdown of AMPK also attenuates the salidroside-induced
upregulation of SIRT1 in ox-LDL-treated HUVECs. These results
indicated that salidroside improves AMPK activation, which causes
SIRT1 expression, leading to cardioprotection in
atherosclerosis.
However, there are still certain limitations in the
present study, which require further explanation. First, the
effects of a wide range of salidroside concentrations (0.1, 1 and
10 µM) on cell viability under ox-LDL condition were
investigated, but a more accurate concentration of salidroside on
the viability of ox-LDL-treated HUVECs is required. In addition,
only the effect of siAMPK on SIRT1 expression was demonstrated,
whether AMPK affects SIRT1 expression directly or indirectly was
not very clear. This will be investigated by co-immunoprecipitation
in the authors' future studies. In addition, in the present study,
only the effect of salidroside on ox-LDL induced endothelial
injures were investigated not the impact of salidroside on
endothelial function under ox-LDL conditions. In addition, the
results of the present study were based on HUVECs in vitro,
whether the AMPK/SIRT1 pathway contributes to the cardioprotective
effect of salidroside on atherosclerosis in vivo is needed
to be validated further, which is the main focus of future studies.
In conclusion, all the findings from the current study indicated
that salidroside protects HUVECs from injury caused by ox-LDL by
inhibiting oxidation and improving mitochondrial function. The
cardio-protection elicited by salidroside is focused on promoting
the AMPK/SIRT1 pathway. The results of the present study will
provide experimental evidence for the future clinical applications
of salidroside to prevent atherosclerosis, giving new insights into
the cardiovascular benefits of the AMPK/SIRT1 pathway.
Funding
No funding received.
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
DZ performed the experiment and was the major
contributor in writing the manuscript. XS, SL and MS analyzed and
interpreted the data. HG, YZ and ZW contributed to the design the
work and to article revision. PD, LZ and MY made substantial
contributions to the conception and design of the study. XW
designed the entire experiment, revised the article for publication
and gave final approval of the version to be published. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nature. 473:317–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gao S and Liu J: Association between
circulating oxidized low-density lipoprotein and atherosclerotic
cardiovascular disease. Chronic Dis Transl Med. 3:89–94. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pirillo A, Norata GD and Catapano AL:
LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm.
2013:1527862013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pahwa R and Jialal I: Atherosclerosis.
StatPearls Publishing Treasure; Island, FL: 2019
|
|
5
|
Taniyama Y and Griendling KK: Reactive
oxygen species in the vasculature: Molecular and cellular
mechanisms. Hypertension. 42:1075–1081. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sorescu D and Griendling KK: Reactive
oxygen species, mitochondria, and NAD(P)H oxidases in the
development and progression of heart failure. Congest Heart Fail.
8:132–140. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Madamanchi NR and Runge MS: Mitochondrial
dysfunction in atherosclerosis. Circ Res. 100:460–473. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Panth N, Paudel KR and Parajuli K:
Reactive oxygen species: A key hallmark of cardiovascular disease.
Adv Med. 2016:91527322016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Victor VM, Apostolova N, Herance R,
Hernandez-Mijares A and Rocha M: Oxidative stress and mitochondrial
dysfunction in atherosclerosis: Mitochondria-targeted antioxidants
as potential therapy. Curr Med Chem. 16:4654–4667. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lum H and Roebuck KA: Oxidant stress and
endothelial cell dysfunction. Am J Physiol Cell Physiol.
280:C719–C741. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun P, Song SZ, Jiang S, Li X, Yao YL, Wu
YL, Lian LH and Nan JX: Salidroside regulates inflammatory response
in raw 264.7 macrophages via TLR4/TAK1 and ameliorates inflammation
in alcohol binge drinking-induced liver injury. Molecules.
21:E14902016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ni J, Li Y, Li W and Guo R: Salidroside
protects against foam cell formation and apoptosis, possibly via
the MAPK and AKT signaling pathways. Lipids Health Dis. 16:1982017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ju L, Wen X, Wang C, Wei Y, Peng Y, Ding
Y, Feng L and Shu L: Salidroside, a natural antioxidant, improves
β-cell survival and function via activating AMPK pathway. Frontiers
Pharmacol. 8:7492017. View Article : Google Scholar
|
|
14
|
Zhang P, Li Y, Guo R and Zang W:
Salidroside protects against advanced glycation end
products-induced vascular endothelial dysfunction. Med Sci Monit.
24:2420–2428. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xing SS, Yang XY, Zheng T, Li WJ, Wu D,
Chi JY, Bian F, Bai XL, Wu GJ, Zhang YZ, et al: Salidroside
improves endo-thelial function and alleviates atherosclerosis by
activating a mitochondria-related AMPK/PI3K/Akt/eNOS pathway.
Vascul Pharmacol. 72:141–152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Panossian A, Hamm R, Wikman G and Efferth
T: Mechanism of action of Rhodiola, salidroside, tyrosol and
triandrin in isolated neuroglial cells: An interactive pathway
analysis of the downstream effects using RNA microarray data.
Phytomedicine. 21:1325–1348. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xing SS, Li J, Chen L, Yang YF, He PL, Li
J and Yang J: Salidroside attenuates endothelial cellular
senescence via decreasing the expression of inflammatory cytokines
and increasing the expression of SIRT3. Mech Ageing Dev. 175:1–6.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kitada M, Ogura Y and Koya D: The
protective role of Sirt1 in vascular tissue: Its relationship to
vascular aging and atherosclerosis. Aging. 8:2290–2307. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Winnik S, Auwerx J, Sinclair DA and Matter
CM: Protective effects of sirtuins in cardiovascular diseases: From
bench to bedside. Eur Heart J. 36:3404–3412. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma L and Li Y: SIRT1: Role in
cardiovascular biology. Clin Chim Acta. 440:8–15. 2015. View Article : Google Scholar
|
|
21
|
Chong ZZ, Shang YC, Wang S and Maiese K:
SIRT1: New avenues of discovery for disorders of oxidative stress.
Expert Opin Ther Targets. 16:167–178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ota H, Eto M, Ogawa S, Iijima K, Akishita
M and Ouchi Y: SIRT1/eNOS axis as a potential target against
vascular senescence, dysfunction and atherosclerosis. J Atheroscler
Thromb. 17:431–435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang L, Cong HL, Wang SF and Liu T:
AMP-activated protein kinase mediates the effects of
lipoprotein-associated phospholipase A2 on endothelial dysfunction
in atherosclerosis. Exp Ther Med. 13:1622–1629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang S, Song P and Zou MH: AMP-activated
protein kinase, stress responses and cardiovascular diseases. Clin
Sci. 122:555–573. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Canto C, Jiang LQ, Deshmukh AS, Mataki C,
Coste A, Lagouge M, Zierath JR and Auwerx J: Interdependence of
AMPK and SIRT1 for metabolic adaptation to fasting and exercise in
skeletal muscle. Cell Metab. 11:213–219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stein S and Matter CM: Protective roles of
SIRT1 in atherosclerosis. Cell Cycle. 10:640–647. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Martin-Ventura JL, Rodrigues-Diez R,
Martinez-Lopez D, Salaices M, Blanco-Colio LM and Briones AM:
Oxidative stress in human atherothrombosis: Sources, markers and
therapeutic targets. Int J Mol Sci. 18:E23152017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Georgieva E, Ivanova D, Zhelev Z, Bakalova
R, Gulubova M and Aoki I: Mitochondrial dysfunction and redox
imbalance as a diagnostic marker of 'Free Radical Diseases'.
Anticancer Res. 37:5373–5381. 2017.PubMed/NCBI
|
|
29
|
Vásquez-Trincado C, García-Carvajal I,
Pennanen C, Parra V, Hill JA, Rothermel BA and Lavandero S:
Mitochondrial dynamics, mitophagy and cardiovascular disease. J
Physiol. 594:509–525. 2016. View Article : Google Scholar
|
|
30
|
Salminen A, Kaarniranta K and Kauppinen A:
Age-related changes in AMPK activation: Role for AMPK phosphatases
and inhibitory phosphorylation by upstream signaling pathways.
Ageing Res Rev. 28:15–26. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mudau M, Genis A, Lochner A and Strijdom
H: Endothelial dysfunction: The early predictor of atherosclerosis.
Cardiovasc J Afr. 23:222–231. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zou H, Liu X, Han T, Hu D, Wang Y, Yuan Y,
Gu J, Bian J, Zhu J and Liu ZP: Salidroside protects against
cadmium-induced hepatotoxicity in rats via GJIC and MAPK pathways.
PLoS One. 10:e01297882015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu Y, Shi YP, Wu D, Ji YJ, Wang X, Chen
HL, Wu SS, Huang DJ and Jiang W: Salidroside protects against
hydrogen peroxide-induced injury in cardiac H9c2 cells via PI3K-Akt
dependent pathway. DNA Cell Biol. 30:809–819. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tan CB, Gao M, Xu WR, Yang XY, Zhu XM and
Du GH: Protective effects of salidroside on endothelial cell
apoptosis induced by cobalt chloride. Biol Pharm Bull.
32:1359–1363. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu YL, Piao DM, Han XH and Nan JX:
Protective effects of salidroside against acetaminophen-induced
toxicity in mice. Biol Pharm Bull. 31:1523–1529. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun L, Dou F, Chen J, Chi H, Xing S, Liu
T, Sun S and Chen C: Salidroside slows the progression of EA.hy926
cell senescence by regulating the cell cycle in an atherosclerosis
model. Mol Med Rep. 17:257–263. 2018.
|
|
37
|
Wang CY, Sun ZN, Wang MX and Zhang C:
SIRT1 mediates salidroside-elicited protective effects against
MPP+-induced apoptosis and oxidative stress in SH-SY5Y
cells: Involvement in suppressing MAPK pathways. Cell Biol Int.
42:84–94. 2018. View Article : Google Scholar
|
|
38
|
Wang Y, Xu CF, Liu YJ, Mao YF, Lv Z, Li
SY, Zhu XY and Jiang L: Salidroside attenuates ventilation induced
lung injury via SIRT1-dependent inhibition of NLRP3 inflammasome.
Cell Physiol Biochem. 42:34–43. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Si PP, Zhen JL, Cai YL, Wang WJ and Wang
WP: Salidroside protects against kainic acid-induced status
epilepticus via suppressing oxidative stress. Neurosci Lett.
618:19–24. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Donato AJ, Magerko KA, Lawson BR, Durrant
JR, Lesniewski LA and Seals DR: SIRT-1 and vascular endothelial
dysfunction with ageing in mice and humans. J Physiol.
589:4545–4554. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang X, Li Y, Li Y, Ren X, Zhang X, Hu D,
Gao Y, Xing Y and Shang H: Oxidative stress-mediated
atherosclerosis: Mechanisms and therapies. Front Physiol.
8:6002017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang M, Pan H, Xu Y, Wang X, Qiu Z and
Jiang L: Allicin decreases lipopolysaccharide-induced oxidative
stress and inflammation in human umbilical vein endothelial cells
through suppression of mitochondrial dysfunction and activation of
Nrf2. Cell Physiol Biochem. 41:2255–2267. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mukherjee N, Parida PK, Santra A, Ghosh T,
Dutta A, Jana K, Misra AK and Sinha Babu SP: Oxidative stress plays
major role in mediating apoptosis in filarial nematode Setaria
cervi in the presence of trans-stilbene derivatives. Free Radic
Biol Med. 93:130–144. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang XL, Wang X, Xiong LL, Zhu Y, Chen HL,
Chen JX, Wang XX, Li RL, Guo ZY, Li P, et al: Salidroside improves
doxorubicin-induced cardiac dysfunction by suppression of excessive
oxidative stress and cardiomyocyte apoptosis. J Cardiovasc
Pharmacol. 62:512–523. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sosnowska B, Mazidi M, Penson P,
Gluba-Brzózka A, Rysz J and Banach M: The sirtuin family members
SIRT1, SIRT3 and SIRT6: Their role in vascular biology and
atherogenesis. Atherosclerosis. 265:275–282. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tsai KL, Hung CH, Chan SH, Hsieh PL, Ou
HC, Cheng YH and Chu PM: Chlorogenic acid protects against
oxLDL-induced oxida-tive damage and mitochondrial dysfunction by
modulating SIRT1 in endothelial cells. Mol Nutr Food Res.
62:e17009282018. View Article : Google Scholar
|
|
47
|
Chan SH, Hung CH, Shih JY, Chu PM, Cheng
YH, Lin HC, Hsieh PL and Tsai KL: Exercise intervention attenuates
hyper-homocysteinemia-induced aortic endothelial oxidative injury
by regulating SIRT1 through mitigating NADPH oxidase/LOX-1
signaling. Redox Biol. 14:116–125. 2018. View Article : Google Scholar
|
|
48
|
Zhu X, Yue H, Guo X, Yang J, Liu J, Liu J,
Wang R and Zhu W: The preconditioning of berberine suppresses
hydrogen peroxide-induced premature senescence via regulation of
Sirtuin 1. Oxid Med Cell Longev. 2017:23918202017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ballinger SW, Patterson C, Knight-Lozano
CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R,
Hunter GC, et al: Mitochondrial integrity and function in
atherogenesis. Circulation. 106:544–549. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xing S, Yang X, Li W, Bian F, Wu D, Chi J,
Xu G, Zhang Y and Jin S: Salidroside stimulates mitochondrial
biogenesis and protects against H2O2-induced
endothelial dysfunction. Oxid Med Cell Longev. 2014:9048342014.
View Article : Google Scholar
|
|
51
|
Xu MC, Shi HM, Wang H and Gao XF:
Salidroside protects against hydrogen peroxide-induced injury in
HUVECs via the regulation of REDD1 and mTOR activation. Mol Med
Rep. 8:147–153. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cai L, Li Y, Zhang Q, Sun H, Yan X, Hua T,
Zhu Q, Xu H and Fu H: Salidroside protects rat liver against
ischemia/reperfusion injury by regulating the GSK-3β/Nrf2-dependent
antioxidant response and mitochondrial permeability transition. Eu
J Pharmacol. 806:32–42. 2017. View Article : Google Scholar
|
|
53
|
Dolinsky VW: The role of sirtuins in
mitochondrial function and doxorubicin-induced cardiac dysfunction.
Biol Chem. 398:955–974. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tang BL: Sirt1 and the Mitochondria. Mol
Cells. 39:87–95. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Feng Ma S, Zhang J, Chen R, Han J, Li D,
Yang X, Li B, Fan X, Li MC, et al: SIRT1 activation by resveratrol
alleviates cardiac dysfunction via mitochondrial regulation in
diabetic cardiomyopathy mice. Oxid Med Cell Longev.
2017:46027152017.PubMed/NCBI
|
|
56
|
Bairwa SC, Parajuli N and Dyck JR: The
role of AMPK in cardiomyocyte health and survival. Biochim Biophys
Acta. 1862:2199–2210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shirwany NA and Zou MH: AMPK in
cardiovascular health and disease. Acta Pharmacol Sin.
31:1075–1084. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gao F, Chen J and Zhu H: A potential
strategy for treating atherosclerosis: Improving endothelial
function via AMP-activated protein kinase. Sci China Life Sci.
61:1024–1029. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ewart MA and Kennedy S: AMPK and
vasculoprotection. Pharmacol Ther. 131:242–253. 2011. View Article : Google Scholar
|
|
60
|
Zheng T, Yang X, Li W, Wang Q, Chen L, Wu
D, Bian F, Xing S and Jin S: Salidroside attenuates high-fat
diet-induced nonalcoholic fatty liver disease via AMPK-dependent
TXNIP/NLRP3 pathway. Oxid Med Cell Longev. 2018:85978972018.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ruderman NB, Xu XJ, Nelson L, Cacicedo JM,
Saha AK, Lan F and Ido Y: AMPK and SIRT1: A long-standing
partnership. Am J Physiol Endocrinol Metab. 298:E751–E760. 2010.
View Article : Google Scholar : PubMed/NCBI
|














