Introduction
Chronic hepatitis B (CHB) is a major health problem
worldwide (1). It is estimated that at least one-third of the world
population have been infected with hepatitis B virus (HBV) (2) and
240 million individuals are chronic carriers; however, a curative
therapy remains unavailable (3). HBV, a hepadnaviridae, stabilizes
in hepatocytes by forming covalently closed circular DNA (cccDNA)
(4,5). At present, nucleos(t)ide analogues (NAs) are one of the two
major options for CHB treatment (6,7). The NAs approved for HBV
treatment include tenofovir alafenamide, entecavir (ETV) and
adefovir dipivoxil (ADV). Although ADV has not been recommended as
the first-line therapy, it is commonly used in numerous Asian
countries due to the relatively lower resistance rate and lower
cost compared with those of other therapies (6-10).
NAs markedly inhibit reverse transcriptase to reduce
the DNA levels of HBV (11,12). However, an increasing body of
evidence has indicated that cccDNA stably attaches to the host
hepatocyte genome in order to avoid elimination by NA (4,11,13,14).
Thus, the majority of patients require long-term NA therapy, even
if HBV DNA has decreased to undetectable levels for a short time.
Therefore, it is important to study the safety of NA therapy and
the re-treatment efficacy following a relapse (15). By using
specific assays, studies have determined that the nucleoside
analogue ETV has genotoxic (16) and carcinogenic effects (17).
Various types of DNA lesions, including single-strand DNA breaks,
double-strand DNA breaks (DSBs), alkylation of DNA bases and
covalent links between bases (intrastrand and inter-strand
crosslinks), may be caused by genotoxic chemicals (18). Unrepaired
or incorrectly repaired lesions result in mutations and/or genetic
instability, which may then be risk factors of carcinogenesis. In
addition, the US prescription information sheet (19) states that
ADV was indicated to be mutagenic in an in vitro mouse
lymphoma cell assay. However, the underlying mechanisms of the
genotoxicity of ADV remain elusive. Further studies are required to
gain a better understanding of the genetic toxicity mechanisms of
ADV. The DT40 cell line originates from a chicken B-lymphocyte line
(20) and TK6 lymphoblastoid cells are a human-derived cell line
(21-24). By using the wild-type (WT) or specific gene
knockout variants of these cell lines, the toxicity of ADV was
evaluated and the underlying mechanisms were investigated in the
present study.
Materials and methods
Chemicals
ADV (purity, ≥99%) and camptothecin (CPT; purity,
≥99%) were purchased from MedChemExpress (Monmouth Junction, NJ,
USA). These chemicals were dissolved in dimethyl sulfoxide (DMSO).
Stock solutions of ADV (10 mM) and CPT (100 μM) were stored
at -20°C in aliquots. In each experiment, the final concentration
of DMSO never exceeded 0.1%.
Cell lines and cell culture
All of the cell lines used in the present study were
provided by Professor Shunichi Takeda (Kyoto University, Kyoto,
Japan). The DT40 cell lines (25-30) with different phenotypes used
in the present study are summarized in Table I. The DT40 cells were incubated in
RPMI-1640 medium containing 10% newborn calf serum, 1% chicken
serum, 1% penicillin streptomycin (all Wisent, Inc., St. Bruno, QC,
Canada), 200 mM L-glutamine and 50 μM β-mercaptoethanol
(both Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) with
5% CO2 at 39.5°C. The TK6 cells were routinely
maintained in RPMI-1640 medium (Wisent, Inc.) including 10% horse
serum (Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin (Wisent Inc.) at 37°C in the presence of 5%
CO2. These sera (except chicken serum) were
heat-deactivated at 56°C for 30 min prior to use.
 | Table IDNA repair genes mutated in the
analyzed DT40 clones. |
Table I
DNA repair genes mutated in the
analyzed DT40 clones.
| Author, year | Gene | Function | (Refs.) |
|---|
| Sonoda et
al, 2003 | Rev3 | TLS, HR (catalytic
subunit of Polζ) | (25) |
| Okada et al,
2002 | Xpa | Initial step of
NER | (26) |
| Masson et
al, 1998 | Parp1 | Poly (adenosine
diphosphate) ribosylation, associated with single-strand break and
BER | (27) |
| Qing et al,
2011; Rosen, 2013 | Brca1 | HR, NHEJ | (28,30) |
| Takata et
al, 1998 | Ku70 | Initial step for
NHEJ-dependent DSB repair | (29) |
Cell viability assay
The anti-proliferative effects of ADV and CPT on
cells was determined with MTT assay (31-33). Briefly, the cells
(1×104 cells/ml) were seeded into 96-well plates in
complete medium, followed by incubation in the presence of various
concentrations of ADV or CPT for 72 h. The concentrations of ADV
were 0, 0.06, 0.12, 0.18, 0.24 and 0.3 μM for DT40 cells or
0, 1, 2, 3, 4 and 5 μM for TK6 cells. The CPT concentrations
were 0, 10, 20, 30, 40 and 50 nM for DT40 cells, or 0, 2, 4, 6 and
8 nM for TK6 cells. The concentrations of CPT were 0, 1, 2, 3, 4
and 5 nM, or 0, 10, 20, 30, 40 and 50 nM for DT40 cells
specifically for sensitivity experiments. DMSO (<0.1%) was
applied as a solvent and vehicle control with 3 replicates for each
concentration of the drugs. Following 69 h treatment at 39.5°C for
DT40 and at 37°C for TK6 cells, 20 μl of 5 mg/ml MTT
(Amresco, LLC, Solon, OH, USA) was added to the cells for 3 h at
39.5°C for DT40 and at 37°C for TK6 cells, and subsequently, the
formazan dye crystals were dissolved by 50 μl of 20% sodium
dodecyl sulfate overnight at 39.5°C for DT40 and at 37°C for TK6
cells. The absorbance was detected at a wavelength of 570 nm by a
microplate reader. The wells without cells served as a blank
control. All the experiments were repeated at least 3 times. The
50% inhibitory concentration (IC50) of the drugs on the
cells was calculated with SPSS 25.0 software (IBM Corp, Armonk, NY,
USA).
Chromosome aberration (CA) analysis
Preparation of chromosome samples was performed as
described previously (34-37), with certain modifications. Briefly,
cells (2×105 cells/ml) were seeded in 6-well plates and
were treated with ADV or CPT in complete medium. DT40 cells were
treated with 0.2 or 0.4 μM ADV for 9-30 h and 5 or 25 nM CPT
for 6-24 h at 39.5°C. TK6 cells were performed with 5 μM ADV
for 12-36 h and 2.5 μM ADV for 12 h at 37°C. Colcemid (0.2
μg/ml; Gibco; Thermo Fisher Scientific, Inc.) was added 3 h
prior to harvesting in order to enrich mitotic cells. The cells
were then incubated in 1 ml hypotonic KCl solution (75 mM) for 25
min at room temperature. Subsequently, the cells were fixed with 3
ml Carnoy's solution [methanol/acetic acid, 3:1, (v/v)] for 35 min.
A drop of this suspension was placed onto ethanol-washed glass
slides and immediately dried by a flame. Finally, the dried slides
were dyed with 5% Giemsa solution for 5 min at room temperature and
carefully rinsed with water prior to air-drying. In the present
study, 50 metaphase cells per each experiment were analyzed under a
light microscope (magnification, ×1,000). All assays were performed
3 times. A break or gap in a chromosome was evaluated and defined
according to The International System for Human Cytogenetic
Nomenclature (ISCN) (37).
Immunofluorescence analysis
γ-H2A histone family member X (H2AX) formation is a
rapid and sensitive cellular response to DSBs (38).
Immunofluorescent staining was performed as reported previously
(39). Briefly, cells (2×105 cells/ml) cultured on
12-well-plates. DT40 cells were treated with 0.1 μM ADV or
0.1 μM CPT for 3, 6 and 9 h at 39.5°C, and TK6 cells were
treated with 1 μM ADV or 20 nM CPT for 3, 6 and 9 h at 37°C
and then harvested on glass slides. The cells were fixed with 3%
paraformaldehyde for 10 min at room temperature followed by washing
with PBS 3 times. The fixed cells were permeabilized with 0.1%
Nonidet P-40 for 20 min at room temperature and washed with PBS
again. Following blocking with 3% bovine serum albumin (cat. no.
A8020; Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China) for 30 min at room temperature, the cells were
probed with anti-γ-H2AX mouse monoclonal antibody (cat. no. 05-636;
1:1,000 dilution; EMD Millipore, Billerica, MA, USA) in a
humidified environment at 4°C overnight. Following washing with
PBS, the cells were incubated for 1 h at 37°C with Alexa Fluor
488-labeled goat anti-mouse IgG (H+L) (cat. no. A0428; 1:1,000
dilution; Beyotime Institute of Biotechnology, Wuhan, China). The
nuclei were stained with 4′,6-diamidino-2-phenylindole for 10 min
at room temperature. Finally, a fluorescence microscope (IX81;
Olympus Corporation, Tokyo, Japan) was used to visualize the γ-H2AX
foci (magnification, ×1,000). The foci in 100 nuclei were counted.
The visible foci as described in previous reports were counted by
eye using Photoshop (version 12.0.3; Adobe Systems, Inc., San Jose,
CA, USA) (40). The experiments were performed 3 times.
Statistical analysis
Statistical analysis was performed with SPSS 25.0
software (IBM Corp.). The statistically significant differences
were determined with a Student's t-test or two-way analysis of
variance with Tukey's post hoc test. Values are expressed as the
mean ± standard deviation. P<0.05 was considered to indicate a
statistically significant difference.
Results
ADV induces DSBs in WT DT40 and TK6
cells
Adefovir was previously demonstrated to induce CAs
by a human peripheral blood lymphocyte assay in vitro
without metabolic activation (19). As a diester prodrug of adefovir
(19), it cannot be excluded that ADV may also has mutagenic
effects. DSBs, one type of DNA damage, may be reliably identified
by γ-H2AX foci detection (41). In the present study, DT40 and TK6
cells were continuously exposed to various concentrations of ADV
for 72 h and CPT was used as a positive control. The results of the
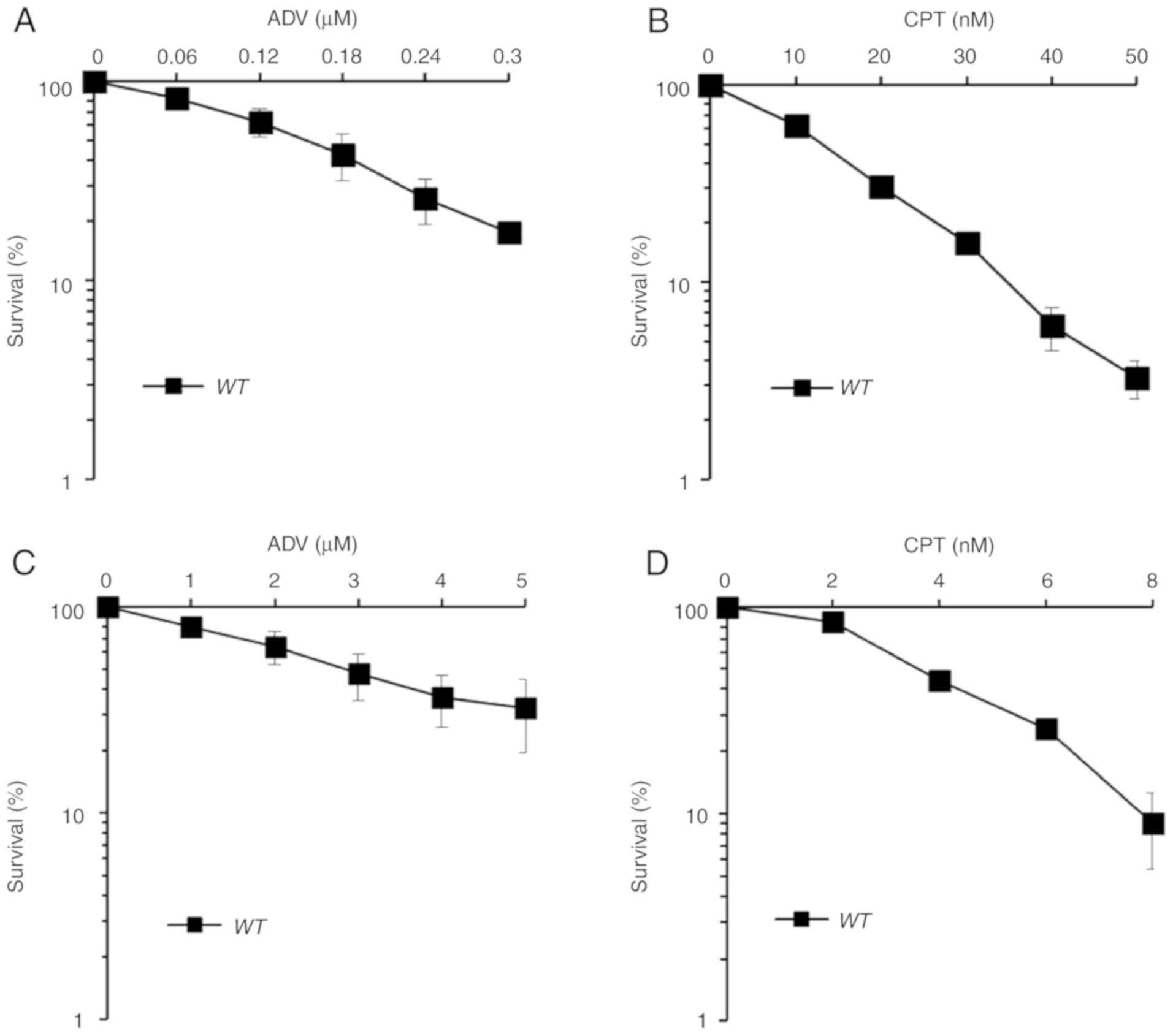
MTT assay indicated that ADV exerts a notable cytotoxic effect in
WT DT40 and TK6 cells (Fig.
1). Furthermore, DT40 cells were treated with 0.1 μM ADV
for 3, 6 or 9 h to dynamically investigate the changes in the
number of γ-H2AX foci. In addition, TK6 cells were also exposed to
1 μM ADV for the same durations. As presented in Figs. 2 and S1, the number of γ-H2AX foci was
significantly increased following treatment with ADV and exhibited
a peak at 6 h. According to the quantitative distribution of γ-H2AX
foci in WT DT40 and TK6 cells (Fig. S1), ADV induced a greater
percentage of γ-H2AX-positive cells.
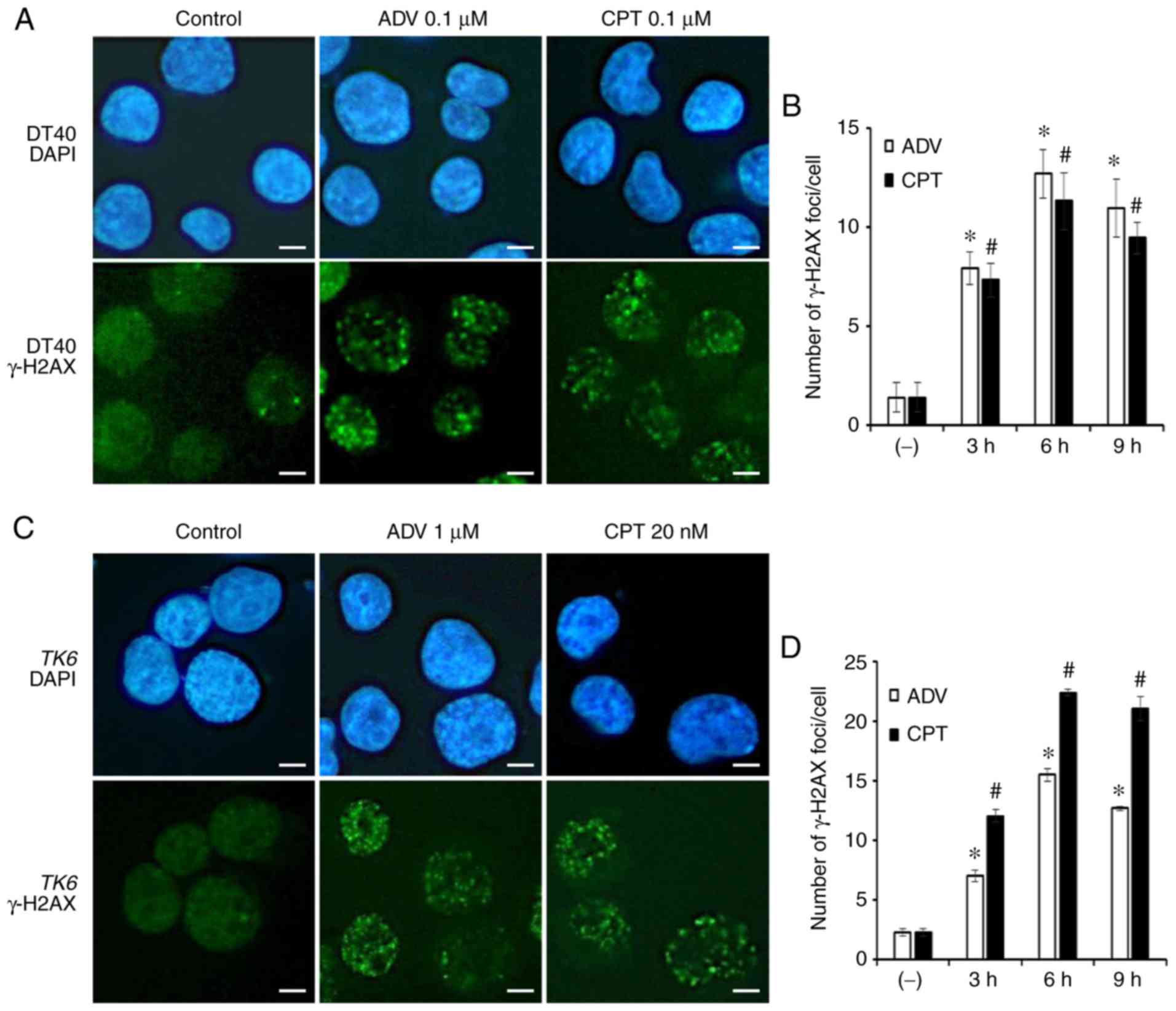 | Figure 2ADV induces the accumulation of
γ-H2AX in the nuclei of WT DT40 and TK6 cells. (A)
Immunostaining of WT DT40 cells using an anti-γ-H2AX
antibody and DAPI. Cells were treated with 0.1 μM ADV or 0.1
μM CPT for 6 h (scale bar, 10 μM; magnification,
×1,000). (B) Quantification of γ-H2AX foci in the nuclei of
WT DT40 cells treated with 0.1 μM ADV or 0.1
μM CPT for different durations. (C) Immunostaining of
WT TK6 cells using an anti-γ-H2AX antibody and DAPI. Cells
were exposed to 1 μM ADV or 20 nM CPT for 6 h (scale bar, 10
μM; magnification, x1,000). (D) Quantification of γ-H2AX
foci in the nuclei of WT TK6 cells treated with 1 μM
ADV or 20 nM CPT for different durations. Values are expressed as
the mean ± standard deviation. *P<0.05 vs. ADV
control; #P<0.05 vs. CPT control. ADV, adefovir
dipivoxil; γ-H2AX, γ-H2A histone family member X; WT,
wild-type; CPT, camptothecin; DAPI,
4′,6-diamidino-2-phenylindole. |
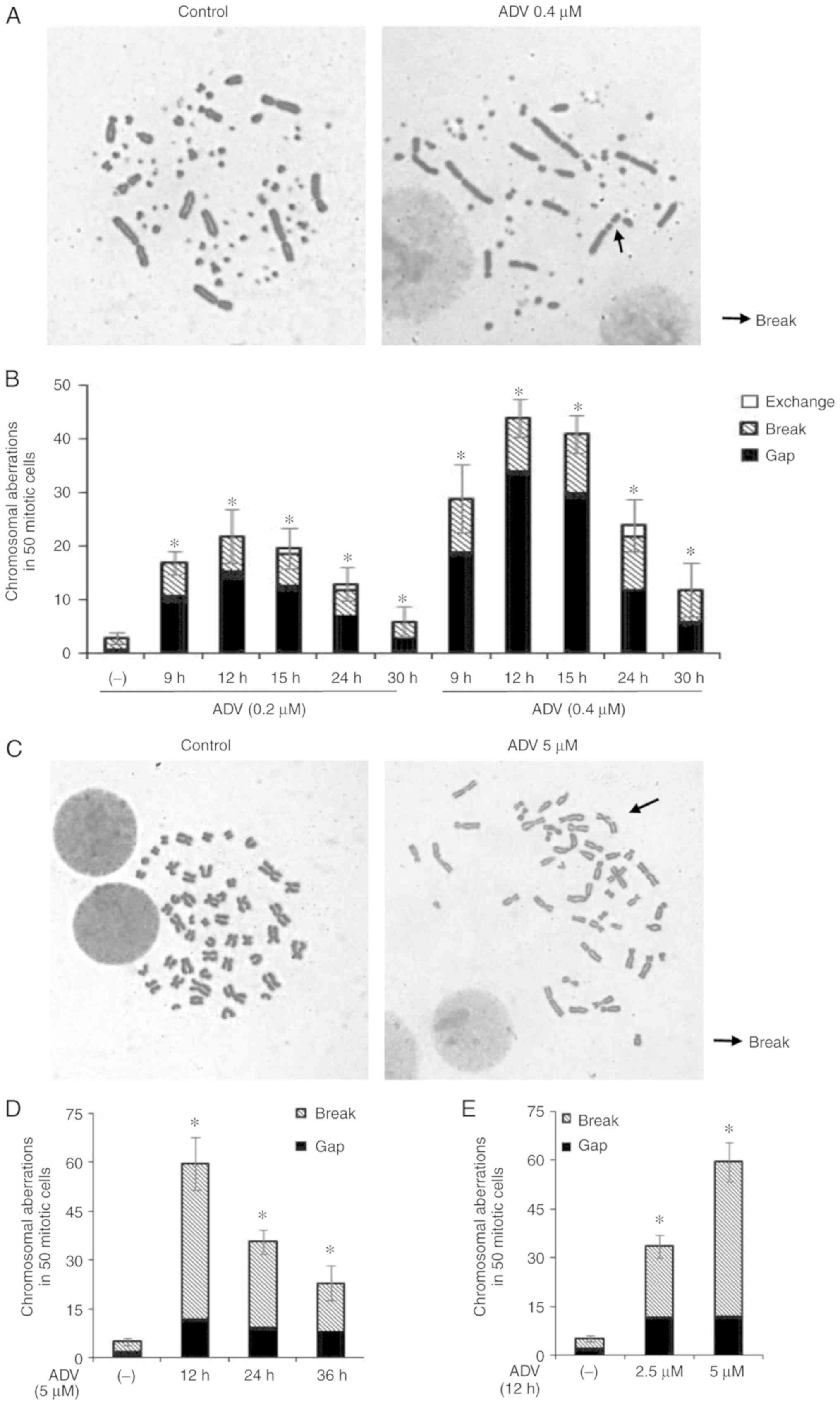
In addition, DNA damage was analyzed by measuring
cytologically detectable CAs in mitotic cells. WT DT40 cells
were exposed to 0.2 or 0.4 μM ADV, and CAs were determined
at 9, 12, 15, 24 and 30 h. The maximum number of CAs was observed
at 12 h of ADV treatment (Fig. 3A and
B). Similarly, TK6 cells were exposed to 5 μM ADV for
12-36 h and with 2.5 μM ADV for 12 h. CAs were determined at
12, 24 and 36 h. The maximum number of chromosomal breaks was
observed at 12 h (Fig. 3C-E).
These results confirmed that ADV was able to generate DNA DSBs in
DT40 and TK6 cells.
Brca1−/− cells defective in
DNA repair pathways are sensitive to ADV
To study the genotoxic mechanisms of ADV, an MTT
assay using WT and mutant DT40 cells was performed. The
sensitivity of ADV was assessed in a panel of DT40 clones, each of
which was deficient in a different DNA repair pathway (Table I) (25-30). DT40 cells were treated
with ADV at various concentrations for 72 h. It was revealed that
ADV inhibited cell growth in a dose-dependent manner (Fig. 4). As presented in Fig. 4A-C, only
Brca1−/− (28,30) cells exhibited significant
sensitivity to ADV. The present study also exposed WT and
Brca1−/− DT40 cells to ADV and evaluated their
cellular response using a colony survival assay (Data S1). Compared
with WT DT40 cells, the colony survival of
Brca1−/− cells was significantly reduced at 14
days (Fig. S2). The results of
the clonogenic assays also confirmed this sensitivity (Fig. S2). The cell variants deficient in
other DNA repair genes, including poly (adenosine
diphosphate-ribose) polymerase 1 (Parp1−/−) (27),
REV3-like DNA directed polymerase ζ catalytic subunit
(Rev3−/−) (25), Ku autoantigen 70 kDa
(Ku70−/−) (29) and xeroderma pigmentosum group
A-complementing protein (Xpa−/−) (26), were not
more sensitive to ADV than WT cells (Fig. 4). In the present experiments, CPT
was selected as a positive control (Fig. 4D-F).
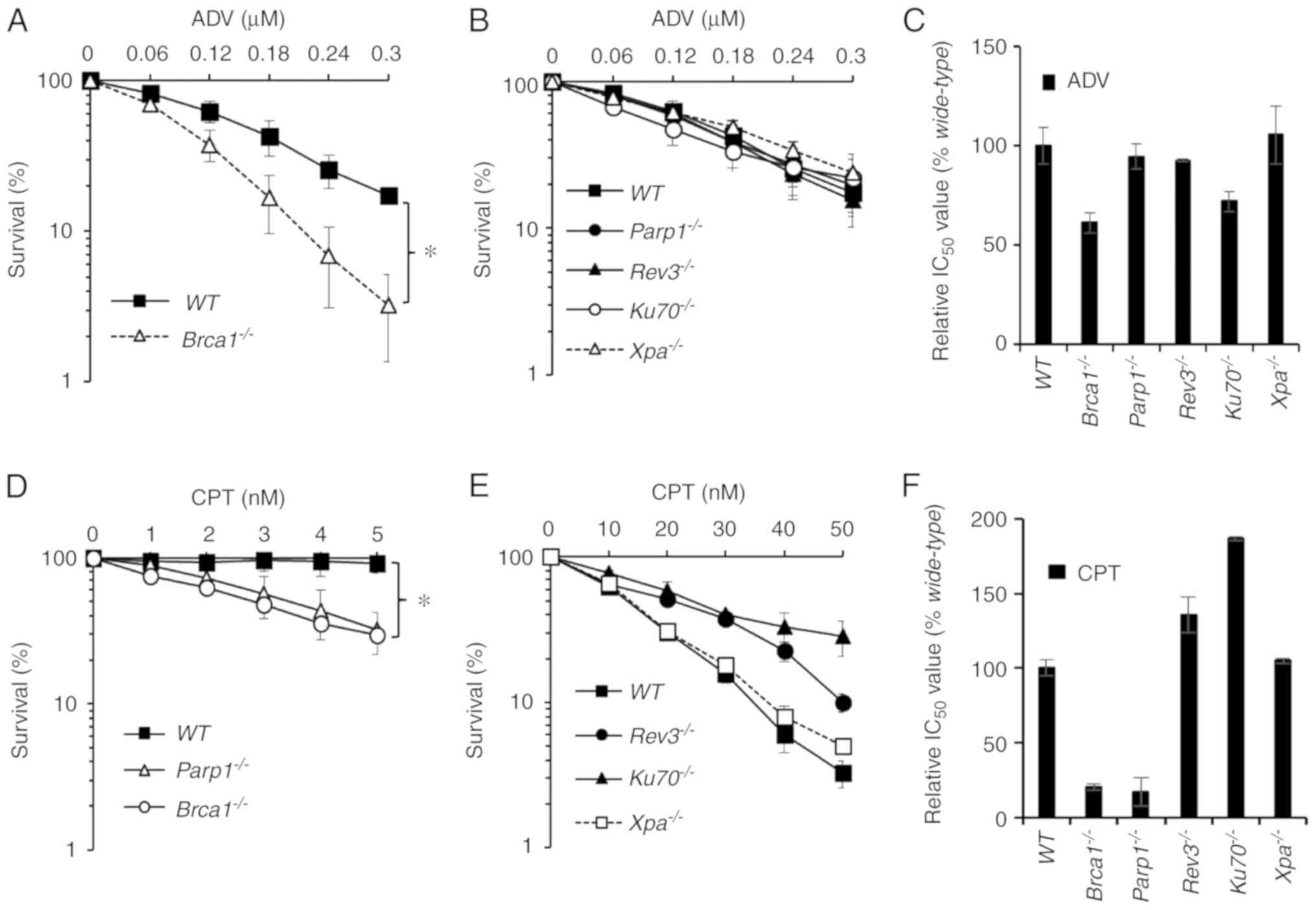 | Figure 4Brca1−/− cells were
sensitive to ADV. (A and B) Sensitivity of
Parp1−/−, Xpa−/−,
Rev3−/−, Brca1−/− and
Ku70−/− cells to ADV. The x-axis represents the
concentration of ADV and the y-axis represents the relative
percentage of surviving cells at 72 h. (C) Relative IC50
values of ADV in DT40 cells. (D and E) Sensitivity of
Parp1−/−, Xpa−/−,
Rev3−/−, Brca1−/− and
Ku70−/− cells to CPT. The x-axis represents the
concentration of CPT and the y-axis represents the relative
percentage of surviving cells at 72 h. (F) Relative IC50
values of CPT in DT40 cells. Survival data were log-transformed to
approximate normality. Two-way analysis of variance was used to
test for differences in the linear dose-response curves between
WT cells and mutant cells. *P<0.05, as
indicated. ADV, adefovir dipivoxil; CPT, camptothecin; WT,
wild-type; IC50, 50% inhibitory concentration;
Parp1−/−, poly (adenosine diphosphate-ribose)
polymerase deficient cells; Rev3−/−, REV3-like
DNA directed polymerase ζ catalytic subunit deficient cells;
Ku70−/−, Ku autoantigen 70 kDa deficient cells;
Xpa−/−, xeroderma pigmentosum group
A-complementing protein deficient cells. |
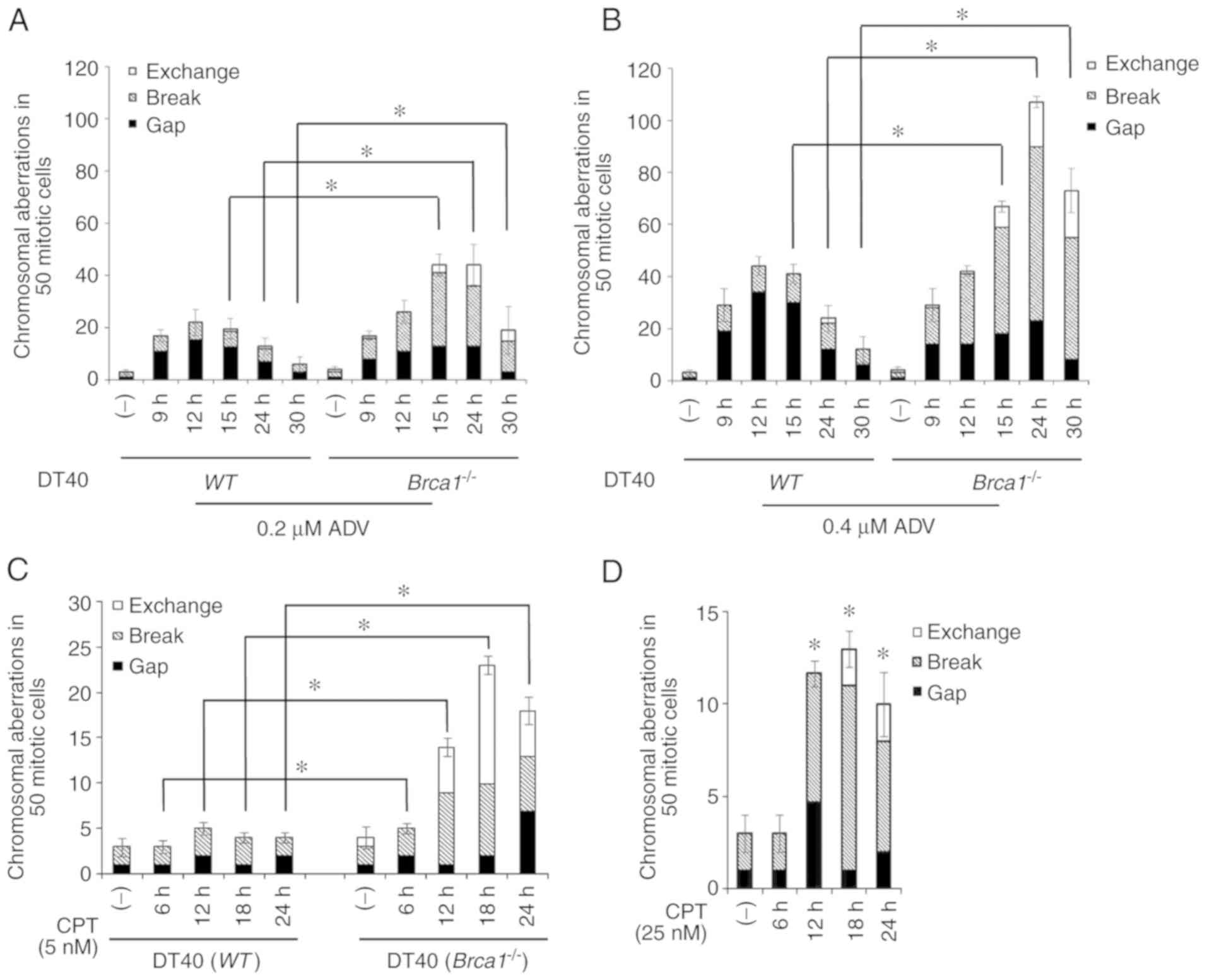
Brca1 deficiency sensitizes DT40 cells to
ADV-induced CAs
In the present study, CAs were classified as
chromosome or chromatid gaps, breaks and exchanges according to The
ISCN (37). WT or Brca1−/− DT40 cells were
exposed to 0.2 or 0.4 μM ADV for 9, 12, 15, 24 or 30 h.
Compared with WT DT40 cells, Brca1−/−
cells exhibited significantly increased CAs at 15, 24 and 30 h, and
the higher the dose the greater the increase in CAs observed
(Fig. 5A and B). Notably, a
monophasic pattern of CAs was induced in WT and
Brca1−/− cells. In these 2 cell lines, a peak was
detectable at 12 h for WT and 24 h for
Brca1−/− cells, respectively, and it was much
higher in Brca1−/− cells than in WT cells
(Fig. 5A and B). As a positive
control, WT and Brca1−/− DT40 cells were
treated with 5 nM CPT for 6, 12, 18 and 24 h (Fig. 5C). At the same time, WT
DT40 cells were treated with 25 nM CPT (Fig. 5D). The results indicated that CPT
induced more DNA damage in Brca1−/− cells when
compared with WT DT40 cells. These results further confirmed
that Brca1 participates in repairing ADV-induced DNA
damage.
Discussion
NAs are effective inhibitors against reverse
transcriptase to inhibit HBV DNA replication; however, NAs,
including ETV, adefovir and ADV, have been reported to have
genotoxic effects by various studies (16,19). In the present study,
the possible mechanisms underlying the genotoxic effects of ADV
were assessed, and it was indicated that DSBs were generated
following ADV treatment in WT DT40 and TK6 cells.
Furthermore, the increase in γ-H2AX foci and CAs confirmed that DNA
damage was induced by ADV. By determining the quantitative
distribution of γ-H2AX foci in WT DT40 and TK6 cells, the
DNA damage induced by ADV was described in detail.
To the best of our knowledge, the present study was
the first to screen the sensitivity of a series of isogenic DNA
repair-deficient cell lines to ADV in a quantitative manner. These
cell lines include a base excision repair-mutant
(Parp1−/−), a homologous recombination (HR)
repair mutant (Brca1−/−), a non-homologous
end-joining (NHEJ) repair mutant (Ku70−/−), a
translesion DNA synthesis repair-mutant (Rev3−/−)
and a nucleotide excision repair-mutant (Xpa−/−).
Notably, the sensitivity profile (IC50) evaluation
indicated that only Brca1−/− cells displayed a
higher sensitivity to ADV compared with any other DNA
repair-deficient cell lines. The increased CAs in
Brca1−/− cells compared with the WT cell
line reflected that Brca1 may have a critical role in preventing
ADV-induced CAs. These results suggested that ADV may be genotoxic
with Brca1 dependence. The results of the clonogenic assays also
confirmed that Brca1−/− cells were significantly
more sensitive to ADV when compared with WT DT40 cells.
Brca1 protein is generally considered as not only a
tumor suppressor but a DNA repair factor involved in multiple DNA
repair and genome stability processes (42-44). Much of the research
on DNA repair associated with Brca1 has focused on HR repair as
well as NHEJ (30,45,46). Firstly, Brca1 has an important role in
the HR of DSBs, which is comprised of the dynamic removal of NHEJ
proteins from DSBs to prevent inappropriate end-ligation and
promote reliable sister chromatid repair (47-50). Secondly, Brca1
is also associated with NHEJ (51). DNA breaks in
Brca1−/− cells are abnormally connected to
complex chromosome rearrangements via NHEJ factor tumor protein p53
binding protein 1 (53BP1). 53BP1 is able to block the resection of
DNA breaks and inhibit HR in Brca1−/− cells. The
capacity of Brca1−/− cells to accurately repair
DSBs is limited by the presence of 53BP1. HR and NHEJ compete to
deal with DNA breaks (51). Brca1 and 53BP1 adjust the balance
between HR and NHEJ, which may be used to selectively protect or
kill Brca1−/− cells (51).
The present results illustrated that
Ku70−/− cells did not have an increased
sensitivity to ADV compared with WT DT40 cells. Furthermore,
the results also demonstrated that 53BP1-deficient cells had a
similar viability following treatment with ADV compared with
WT TK6 cells (data not shown). Based on all of these
results, it was concluded that Brca1 took part in the repair of
ADV-induced DNA damage, and the molecular mechanisms may mainly be
associated with HR.
In conclusion, ADV-induced cellular genotoxicity in
DT40 and TK6 cells, and Brca1 are involved in tolerance to DNA
damage induced by ADV. The present results suggest that it is
necessary to monitor the genotoxicity of ADV and to restrict the
usage or limit the treatment period. A better understanding of the
mechanisms underlying ADV-induced genotoxicity may contribute to
the development of novel drugs against CHB with higher therapeutic
efficacy and less genotoxicity. To date, at least 6 NAs have been
approved for HBV treatment, including telbivudine, lamivudine, ETV,
ADV, tenofovir alafenamide and tenofovir disoproxil fumarate (6-8).
However, further research on their molecular mechanisms is
worthwhile.
Supplementary Materials
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81373431) and the
Science and Technology Support Program of Sichuan Province, China
(grant no. 2012SZ0147).
Availability of data and materials
All data generated or analyzed during this study are
included in this article and in the supplementary materials.
Authors' contributions
YQ, XW and HL conceived and designed the
experiments. HL and ZC performed the experiments. HL, YQ, XW, YW
and FH analyzed the data. ZZ, CX, XF and XB performed part of
chromosome aberration analysis work for TK6 cells and analyzed the
data. HL, XW, YQ, YW and FH wrote the paper. ST generated the gene
knockout cells. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
HBV
|
hepatitis B virus
|
|
cccDNA
|
covalently closed circular DNA
|
|
NAs
|
nucleos(t)ide analogues
|
|
CHB
|
chronic hepatitis B
|
|
ETV
|
entecavir
|
|
ADV
|
adefovir dipivoxil
|
|
DSBs
|
double-strand DNA breaks
|
|
CPT
|
camptothecin
|
|
DMSO
|
dimethyl sulfoxide
|
|
IC50
|
50% inhibiting concentration
|
|
ISCN
|
International System for Human
Cytogenetic Nomenclature
|
|
WT
|
wild-type
|
|
CAs
|
chromosomal aberrations
|
|
BER
|
base excision repair
|
|
HR
|
homologous recombination
|
|
NHEJ
|
non-homologous end joining
|
|
TLS
|
translesion DNA synthesis
|
|
NER
|
nucleotide excision repair
|
Acknowledgments
Not applicable.
References
|
1
|
Zhou TC, Li X, Chen LJ, Fan JH, Lai X,
Tang Y, Zhang L and Wei J: Differential expression profile of
hepatic circular RNAs in chronic hepatitis B. J Viral Hepat.
25:1341–1351. 2018.PubMed/NCBI
|
|
2
|
Sarin SK, Kumar M, Lau GK, Abbas Z, Chan
HL, Chen CJ, Chen DS, Chen HL, Chen PJ, Chien RN, et al:
Asian-Pacific clinical practice guidelines on the management of
hepatitis B: A 2015 update. Hepatol Int. 10:1–98. 2016.
|
|
3
|
Hu J, Cheng J, Tang L, Hu Z, Luo Y, Li Y,
Zhou T, Chang J and Guo JT: Virological basis for the cure of
chronic hepatitis B. ACS Infect Dis. 2018.
|
|
4
|
Yang HC and Kao JH: Persistence of
hepatitis B virus covalently closed circular DNA in hepatocytes:
Molecular mechanisms and clinical significance. Emerg Microbes
Infect. 3:e642014.
|
|
5
|
Zoulim F: New insight on hepatitis B virus
persistence from the study of intrahepatic viral cccDNA. J Hepatol.
42:302–308. 2005.PubMed/NCBI
|
|
6
|
European Association for the Study of the
Liver: EASL clinical practice guidelines: Management of chronic
hepatitis B virus infection. J Hepatol. 57:167–185. 2012.PubMed/NCBI
|
|
7
|
Terrault NA, Bzowej NH, Chang KM, Hwang
JP, Jonas MM and Murad MH; American Association for the Study of
Liver Diseases: AASLD guidelines for treatment of chronic hepatitis
B. Hepatology. 63:261–283. 2016.
|
|
8
|
Lok AS, McMahon BJ, Brown RS Jr, Wong JB,
Ahmed AT, Farah W, Almasri J, Alahdab F, Benkhadra K, Mouchli MA,
et al: Antiviral therapy for chronic hepatitis B viral infection in
adults: A systematic review and meta-analysis. Hepatology.
63:284–306. 2016.
|
|
9
|
European Association for the Study of the
Liver: Electronic address easloffice@easloffice.eu; European
Association for the Study of the Liver: EASL 2017 clinical practice
guidelines on the management of hepatitis B virus infection. J
Hepatol. 67:370–398. 2017.
|
|
10
|
Terrault NA, Lok ASF, McMahon BJ, Chang
KM, Hwang JP, Jonas MM, Brown RS Jr, Bzowej NH and Wong JB: Update
on prevention, diagnosis, and treatment of chronic hepatitis B:
AASLD 2018 hepatitis B guidance. Hepatology. 67:1560–1599.
2018.PubMed/NCBI
|
|
11
|
Tong S and Revill P: Overview of hepatitis
B viral replication and genetic variability. J Hepatol. 64(Suppl
1): S4–S16. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ghany MG: Current treatment guidelines of
chronic hepatitis B: The role of nucleos(t)ide analogues and
peginterferon. Best Pract Res Clin Gastroenterol. 31:299–309. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lai CL, Wong D, Ip P, Kopaniszen M, Seto
WK, Fung J, Huang FY, Lee B, Cullaro G, Chong CK, et al: Reduction
of covalently closed circular DNA with long-term nucleos(t)ide
analogue treatment in chronic hepatitis B. J Hepatol. 66:275–281.
2017. View Article : Google Scholar
|
|
14
|
Nassal M: HBV cccDNA: Viral persistence
reservoir and key obstacle for a cure of chronic hepatitis B. Gut.
64:1972–1984. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tacke F and Kroy DC: Treatment for
hepatitis B in patients with drug resistance. Ann Transl Med.
4:3342016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang L, Wu X, He F, Liu Y, Hu X, Takeda S
and Qing Y: Genetic evidence for genotoxic effect of entecavir, an
anti-Hepatitis B virus nucleotide analog. PLoS One.
11:e01474402016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Choi J, Kim HJ, Lee J, Cho S, Ko MJ and
Lim YS: Risk of hepatocellular carcinoma in patients treated with
entecavir vs tenofovir for chronic hepatitis B: A Korean nationwide
cohort study. JAMA Oncol. 5:30–36. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cartus A and Schrenk D: Current methods in
risk assessment of genotoxic chemicals. Food Chem Toxicol.
106:574–582. 2017. View Article : Google Scholar
|
|
19
|
US Food Drug Administration: Draft
guidance on Adefovir Dipivoxil. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM136541.pdf.
|
|
20
|
Baba TW, Giroir BP and Humphries EH: Cell
lines derived from avian lymphomas exhibit two distinct phenotypes.
Virology. 144:139–151. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bryce SM, Bemis JC, Avlasevich SL and
Dertinger SD: In vitro micronucleus assay scored by flow cytometry
provides a comprehensive evaluation of cytogenetic damage and
cytotoxicity. Mutat Res. 630:78–91. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ellinger-Ziegelbauer H, Fostel JM, Aruga
C, Bauer D, Boitier E, Deng S, Dickinson D, Le Fevre AC, Fornace AJ
Jr, Grenet O, et al: Characterization and interlaboratory
comparison of a gene expression signature for differentiating
genotoxic mechanisms. Toxicol Sci. 110:341–352. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fellows MD and O'Donovan MR: Etoposide,
cadmium chloride, benzo[a]pyrene, cyclophosphamide and colchicine
tested in the in vitro mammalian cell micronucleus test (MNvit) in
the presence and absence of cytokinesis block using L5178Y mouse
lymphoma cells and 2-aminoanthracene tested in MNvit in the absence
of cytokinesis block using TK6 cells at AstraZeneca UK, in support
of OECD draft Test Guideline 487. Mutat Res. 702:163–170. 2010.
View Article : Google Scholar
|
|
24
|
Platel A, Nesslany F, Gervais V and Marzin
D: Study of oxidative DNA damage in TK6 human lymphoblastoid cells
by use of the in vitro micronucleus test: Determination of
no-observed-effect levels. Mutat Res. 678:30–37. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sonoda E, Okada T, Zhao GY, Tateishi S,
Araki K, Yamaizumi M, Yagi T, Verkaik NS, van Gent DC, Takata M and
Takeda S: Multiple roles of Rev3, the catalytic subunit of polzeta
in maintaining genome stability in vertebrates. EMBO J.
22:3188–3197. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Okada T, Sonoda E, Yamashita YM, Koyoshi
S, Tateishi S, Yamaizumi M, Takata M, Ogawa O and Takeda S:
Involvement of vertebrate polkappa in Rad18-independent
postreplication repair of UV damage. J Biol Chem. 277:48690–48695.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Masson M, Niedergang C, Schreiber V,
Muller S, Menissier- de Murcia J and de Murcia G: XRCC1 is
specifically associated with poly(ADP-ribose) polymerase and
negatively regulates its activity following DNA damage. Mol Cell
Biol. 18:3563–3571. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qing Y, Yamazoe M, Hirota K, Dejsuphong D,
Sakai W, Yamamoto KN, Bishop DK, Wu X and Takeda S: The epistatic
relationship between BRCA2 and the other RAD51 mediators in
homologous recombination. PLoS Genet. 7:e10021482011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takata M, Sasaki MS, Sonoda E, Morrison C,
Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A and Takeda S:
Homologous recombination and non-homologous end-joining pathways of
DNA double-strand break repair have overlapping roles in the
maintenance of chromosomal integrity in vertebrate cells. EMBO J.
17:5497–5508. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rosen EM: BRCA1 in the DNA damage response
and at telomeres. Front Genet. 4:852013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Carmichael J, DeGraff WG, Gazdar AF, Minna
JD and Mitchell JB: Evaluation of a tetrazolium-based semiautomated
colorimetric assay: Assessment of chemosensitivity testing. Cancer
Res. 47:936–942. 1987.PubMed/NCBI
|
|
32
|
Hu X, Wu X, Liu H, Cheng Z, Zhao Z, Xiang
C, Feng X, Takeda S and Qing Y: Genistein-induced DNA damage is
repaired by nonhomologous end joining and homologous recombination
in TK6 cells. J Cell Physiol. 234:2683–2692. 2019. View Article : Google Scholar
|
|
33
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: Application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sonoda E, Sasaki MS, Buerstedde JM,
Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y and
Takeda S: Rad51-deficient vertebrate cells accumulate chromosomal
breaks prior to cell death. EMBO J. 17:598–608. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
An international system for human
cytogenetic nomenclature 1978 ISCN 1978 Report of the standing
commitee on human cytogenetic nomenclature. Cytogenet Cell Genet.
21:309–409. 1978.
|
|
36
|
Yamamoto KN, Hirota K, Kono K, Takeda S,
Sakamuru S, Xia M, Huang R, Austin CP, Witt KL and Tice RR:
Characterization of environmental chemicals with potential for DNA
damage using isogenic DNA repair-deficient chicken DT40 cell lines.
Environ Mol Mutagen. 52:547–561. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ji K, Kogame T, Choi K, Wang X, Lee J,
Taniguchi Y and Takeda S: A novel approach using
DNA-repair-deficient chicken DT40 cell lines for screening and
characterizing the genotoxicity of environmental contaminants.
Environ Health Perspect. 117:1737–1744. 2009. View Article : Google Scholar
|
|
38
|
Dickey JS, Redon CE, Nakamura AJ, Baird
BJ, Sedelnikova OA and Bonner WM: H2AX: Functional roles and
potential applications. Chromosoma. 118:683–692. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu Y, Wu X, Hu X, Chen Z, Liu H, Takeda S
and Qing Y: Multiple repair pathways mediate cellular tolerance to
resveratrol-induced DNA damage. Toxicol In Vitro. 42:130–138. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ibuki Y and Toyooka T: Evaluation of
chemical phototoxicity, focusing on phosphorylated histone H2AX. J
Radiat Res. 56:220–228. 2015. View Article : Google Scholar :
|
|
41
|
Solovjeva L, Firsanov D, Pleskach N and
Svetlova M: Immunofluorescence analysis of gamma-H2AX Foci in
mammalian fibroblasts at different phases of the cell cycle.
Methods Mol Biol. 1644:187–194. 2017. View Article : Google Scholar
|
|
42
|
Scully R and Livingston DM: In search of
the tumour-suppressor functions of BRCA1 and BRCA2. Nature.
408:429–432. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tarapore P, Hanashiro K and Fukasawa K:
Analysis of centrosome localization of BRCA1 and its activity in
suppressing centrosomal aster formation. Cell Cycle. 11:2931–2946.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pathania S, Bade S, Le Guillou M, Burke K,
Reed R, Bowman-Colin C, Su Y, Ting DT, Polyak K, Richardson AL, et
al: BRCA1 haploinsufficiency for replication stress suppression in
primary cells. Nat Commun. 5:54962014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Moynahan ME, Chiu JW, Koller BH and Jasin
M: Brca1 controls homology-directed DNA repair. Mol Cell.
4:511–518. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Turner N, Tutt A and Ashworth A: Hallmarks
of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 4:814–819. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Prakash R, Zhang Y, Feng W and Jasin M:
Homologous recombination and human health: The roles of BRCA1,
BRCA2, and associated proteins. Cold Spring Harb Perspect Biol.
7:a0166002015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen L, Nievera CJ, Lee AY and Wu X: Cell
cycle-dependent complex formation of BRCA1.CtIP.MRN is important
for DNA double-strand break repair. J Biol Chem. 283:7713–7720.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sartori AA, Lukas C, Coates J, Mistrik M,
Fu S, Bartek J, Baer R, Lukas J and Jackson SP: Human CtIP promotes
DNA end resection. Nature. 450:509–514. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yu X, Wu LC, Bowcock AM, Aronheim A and
Baer R: The C-terminal (BRCT) domains of BRCA1 interact in vivo
with CtIP, a protein implicated in the CtBP pathway of
transcriptional repression. J Biol Chem. 273:25388–25392. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bunting SF, Callén E, Wong N, Chen HT,
Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao
L, et al: 53BP1 inhibits homologous recombination in
Brca1-deficient cells by blocking resection of DNA breaks. Cell.
141:243–254. 2010. View Article : Google Scholar : PubMed/NCBI
|