Introduction
A number of studies have indicated that the brains
of patients with neurodegenerative disease (such as brain stroke,
Parkinson’s and Alzheimer’s diseases) is subjected to increased
oxidative stress, induced by an imbalanced redox states (1,2). The
excessive generation of reactive oxygen species (ROS) and the
dysfunction of the antioxidant system are widely condisered to be
responsible for neuronal degeneration (3). Neurodegenerative
processes are mostly mediated by ROS, as well as hydrogen peroxide
(H2O2), superoxide anion and hydroxyl
radicals, which are generated as products of normal or altered
metabolic processes that utilize molecular oxygen. ROS, produced by
oxidative stress-caused mitochondrial damage, can damage the
proteins, nucleic acids and polyunsaturated fatty acids of cell
membranes. The consequent alterations of cellular functions and
membrane integrity lead to cellular apoptosis or necrosis (4).
Antioxidant therapy has been suggested for the
prevention and treatment of neurodegenerative diseases; in
particular, an increased dietary intake of polyunsaturated fatty
acids (PUFAs) is considered an efficient strategy for the
prevention of neurological disorders (5). In this regard, the use
of docosahexaenoic acid (DHA), the main ω-3-PUFA in the mammalian
brain, plays a crucial role in the development, function and
protection of brain neurons (6). In fact, several studies have
highlighted that the consumption of DHA, but not that of other n-3
fatty acids, is associated with a reduced risk of stroke and
cognitive decline or dementia (7,8). Although the molecular
mechanisms regarding its efficacy in the prevention of
neurodegenerative disease are not yet entirely clear, it is
considered that DHA protects the cells from oxida-tive stress by
the synthesis of factors responsible for cellular defense
mechanisms (9). Among these elements, nuclear factor
(erythroid-derived 2)-like 2 (NFE2L2) represents one of the most
important protective agents against oxidative stress (10-12). The
activation of NFE2L2 induces the transcriptional regulation of
various antioxidant enzymes, such as heme-oxygenase-1 (HO-1) and
NAD(P)H:quinone oxidoreductase 1 (NQO1), as well as the modulation
of apoptosis-related genes (13).
Within this framework, the present study was
designed with an aim to obtain a better understanding of the
mechanisms underlying the cellular protective effects of DHA.
Therefore, using an in vitro
H2O2-induced PC12 cell oxidative damage
model, we examined the associations among the antioxidant effects
of DHA pre-treatment and ROS production, mitochondrial membrane
potential (ΔΨm), apoptosis and the activation of the
NFE2L2/antioxidant response element (ARE) signaling pathway, in
order to provide a possible explanation for the protective effects
of DHA.
Materials and methods
Cell line, culture and treatments
PC12 cells (a cellular line of rat pheochromocytoma
from Sigma-Aldrich Co., St. Louis, MO, USA. Cat. no. 88022401) were
cultured in a 5% CO2 atmosphere at 37°C in RPMI with 10
mM HEPES, 1.0 g/l glucose, 3.7 g/l NaHCO3, 100 U/ml
penicillin, 100 µg/ml streptomycin, 10% fetal calf serum and
15% horse serum. Once grown to 85% confluence, the cells were
subcultured at an appropriate density according to each
experimental procedure.
The cells were treated with a physiological
concentration (60 µM) of DHA (purchased from Sigma-Aldrich
Co.) 24 h prior to and during H2O2 exposure
(for a further 24 h). For the control groups, medium or
H2O2 alone were used where appropriate. DHA
was dissolved in 99.5% ethanol to prepare a 500 mM stock solution;
prior to use, DHA was complexed (1:2) to bovine serum albumin (BSA)
and diluted in RPMI to the required concentration. The
concentration of H2O2 used was 300 µM;
since, in a previous series of dose-response experiments, described
in detail in (14), we demonstrated that after 24 h,
H2O2 resulted in a reduction of the viability
of PC12 cells of 50% at this concentration. At the end of all the
experiments, the cells were collected and stored at 37°C (unless
otherwise stated) for the analytical experiments.
Measurement of cell viability and
intracellular ROS production
For the determination of cell viability and ROS
production, the PC12 cells were plated in 96-well plates at a
density of 10,000 cells/well and incubated with DHA 24 h prior to
exposure to H2O2. Cell survival was evaluated
by the 3-[(4,5-dimethylthiazol-2-yl)- 5,3-carboxymethoxyphenyl]- 2-
(4- sulfophenyl)-2H tetrazolium, inner salt reduction assay (MTS).
The MTS assay (Promega srl, Padova, Italy) is a sensitive
measurement of the normal metabolic status of cells, which reflects
early cellular redox changes. The intracellular soluble formazan
produced by the cellular reduction of the MTS was determined by
recording the absorbance of each 96-well plate using the automatic
microplate photometer (BioTek™ Elx800 -Box 998; BioTek Instruments,
Winooski, VT, USA) at a wavelength of 490 nm. The morphological
features of the PC12 cells subjected to the different treatments
were analyzed and photographed by phase-contrast microscopy (Nikon
eclipse ts100 microscope; Nikon, Tokyo, Japan), at a magnification
of x20.
The detection of ROS production was performed using
the 2′,7′-dichlorofluorescein diacetate (DCFDA)-Cellular ROS
Detection Assay kit (Abcam, Cambridge, UK). DCFDA is initially
non-fluorescent and is converted by oxidation to DCF, a highly
fluorescent compound. DCF was quantified using a CytoFluor
Multiwell Plate Reader (Victor 3 Wallac 1420; Perkin Elmer Waltham,
MA, USA), with 485 nm excitation and 538 nm emission filters. ROS
production was expressed as the fluorescence intensity and
presented as a percentage relative to the control.
Measurement of enzymatic and
non-enzymatic antioxidant levels
The cell cultures (1×106) were washed
twice in phosphate buffered saline (PBS, pH 7.4) and subsequently
centrifuged at 1,890 × g for 10 min at 4°C, producing a cell pellet
and the supernatant. Cell pellets were processed for the
determination of the enzymatic activity of total glutathione
peroxidase (GSH-Px), catalase and total superoxide dismutase (SOD),
by adding 500 µl/106 cells of a hypotonic buffer
containing 15 mM KCl + 1 mM KH2PO4, pH 7.4.
After vigorous vortexing for 60 sec, the cell suspensions were
sonicated at 4°C for 3 cycles of 20 sec each with 20 sec intervals
between each pulse of sonication. Subsequently, the samples were
centrifuged at 20,690 × g for 15 min at 4°C to remove any cell
debris. Enzymatic activities were performed spectrophotometrically
(using an Agilent 89090A spectrophotometer; Agilent Technologies,
Santa Clara, CA, USA) on cell lysates. Total GSH-Px activity was
assayed as previously described (15). Briefly, 1 ml of reaction
mixture had the following composition: 0.1 M Tris-HCl, pH 8.0, 4 mM
EDTA, 2 mM reduced glutathione (GSH), 1 IU glutathione reductase,
0.3 mM NADPH and 10 µl of cell extract. Following 10 min of
incubation at 37°C, the reaction began by the addition of 70
µM tert-butyl hydroperoxide (TBH) and monitored
spectro-photometrically (Agilent 89090A) at 340 nm by following for
10 min NADPH oxidation; blank without TBH and additional blank
without cell lysate were used. The millimolar extinction
coefficient of 6.3×10-3 of NADPH was used for the
calculation. Catalase activity was examined as previously described
(15); the blank reaction mixture (1 ml final volume) was composed
by 10 mM KH2PO4 pH 7.4 and
H2O2 10 mM; the reaction began by the
addition of 5 µl of cell extract and monitored
spectrophotometrically (Agilent 89090A) at 240 nm by following
H2O2 dismutation. The millimolar extinction
coefficient of 3.94×10-3 of H2O2
was used for the calculation. Total SOD activity was analyzed as
previously described (16). The reaction mixture (1 ml final volume)
was formed by 50 mM KH2PO4 pH 7.8, 10
µM oxidized cytochrome c, 1.25 mM xanthine, 0.006
µM xanthine oxidase and 4 mM sodium azide (to inactivate
catalase). The reaction began by the addition of 20 µl of
cell extract and monitored spectrophotometrically (Agilent 89090A)
at 550 nm following the cytochrome c reduction. The
millimolar extinction coefficient of 8.4x10-3 of
oxidized cytochrome c was used for the calculation. All
enzymatic activities were normalized for the protein content
determined by Bradford assay (17) and expressed as IU/mg of protein
(1 IU=1 µmol substrated consumed/min). In a different set of
experiments, cell pellets, obtained as described above, were
treated for the determination of non-enzymatic antioxidant levels
with a precipitating solution composed of CH3CN 75% and
KH2PO4 25% (10 mM) at pH 7.4, and then
centrifuged at 20,690 × g for 15 min at 4°C. The supernatants were
collected and subjected to 2 chloroform washings in order to obtain
an upper aqueous phase that was directly injected onto the HPLC.
Each sample was analyzed to determine the concentration of GSH,
ascorbate, malondialdehyde (MDA), nitrites and nitrates
(NO2 and NO3) according to an ion-pairing
HPLC method previously set up in our laboratories (18,19). All
concentration values were normalized with respect to the protein
amount and expressed as nmol/mg protein (17).
Detection of apoptotic cells by Annexin
V
The cells were seeded at a density of
1×106 cells/well on a 24-well plate and incubated with
DHA 24 h prior to the exposure to H2O2 (300
µM for 24 h). The medium was then aspirated, and the cells
were washed with PBS. After the cells were incubated at room
temperature, for 15 min in a dark room with Annexin V-FITC (TACS
Annexin V kit; Trevigen, Gaithersburg, MD, USA). The cells were
then examined under a fluorescence microscope (Nikon Eclipse TE300
Inverted Fluorescence Microscope; Nikon) with a ×20 objective
lens.
Detection of ΔΨm
ΔΨm, an early marker of the induction of apoptosis,
was assessed using the Mitolight™ Apoptosis Detection kit (APT142;
Chemicon International, Temecula, CA, USA). The cells
(4×104 cells/well in a PLL-coated black-bottom 96-well
plate) (treated as described above) were incubated with 100
µl of prediluted Mitolight™ dye solution for 15 min (37°C,
5% CO2, 95% air). In healthy cells, the dye accumulates
and aggregates in the mitochondria, giving off a bright red
fluorescence (λem=585 nm). In apoptotic cells with an altered ΔΨm,
the dye in monomeric form remains in the cytoplasm, fluorescing
green (λem=530 nm) and thus providing a ready discrimination
between apoptotic and non-apoptotic cells. The fluorescence
intensities were measured with a Victor multilabel plate reader
(PerkinElmer, Waltham, MA, USA) at an excitation wavelength 485 nm
and an emission wavelength 530 nm to monitor apoptotic cells and
585 nm for healthy cells, respectively. The data were expressed as
a fluorescence ratio (585/530) representative of the arbitrary ΔΨm.
The cells were also observed using a Nikon Eclipse TE300 Inverted
Fluorescence Microscope (Nikon) (data not shown).
RNA isolation and semi-quantitative
PCR
Total RNA was isolated using the SV Total RNA
Isolation System (Promega srl). The RNA concentration was evaluated
by spectrophotometric reading at 280 and 260 nm. Total RNA was used
for first strand cDNA synthesis with the HyperScript, First strand
Synthesis kit and Oligo-dT, as random primer (GeneAll Biotechnology
Co. Ltd., Seoul, Korea). PCR was performed with about 150 ng of
cDNA using PCRBIO Classic Taq (PCR Biosystems Ltd., London, UK).
The following primer sequences were used for amplification: Actin β
(β-actin) forward, 5′-TGCTATGTTGCCCTAGACTTCG-3′ and reverse,
5′-GTTGGCATAGAGGTCTTTACGG-3′ (240 bp); Bax forward,
5′-GCAGGGAGGATGGCTGGGGAG-3′ and reverse,
5′-TCCAGACAAGCAGCCGCTCACG-3′ (352 bp); Bcl2 forward,
5′-CACCCCTGGCATCTTCTCCT-3′ and reverse, 5′-GTTGACGCTCCCCACACACA-3′
(349 bp); NFE2L2 forward, 5′-GCCAGCTGAACTCCTTAGAC-3′ and reverse,
5′-GATTCGTGCACAGCAGCA-3′ (466 bp); and HO-1 forward,
5′-GAAACAAGCAGAACCCAGTC-3′ and reverse, 5′-AGAGGTCACCCAGGTAGCG-3′
(225 bp).
The experimental protocols, in accordance with the
conditions reported in the literature (14,20) and slightly
modified, were as follows: An initial denaturation for 3 min at
95°C; amplification for 21 (β-actin), 20 (NFE2L2), 30 (HO-1) and 40
cycles (Bax and Bcl2) of denaturation, 15 sec, 95°C, annealing: 15
sec, at 61°C (β-Actin), 58°C (NFE2L2), 55°C (HO-1), 60°C (Bax),
59°C (Bcl2) and elongation at 72°C for 1 min; and a final
elongation for 10 min at 72°C. The PCR products were then analyzed
by 1.5% agarose gel electrophoresis in TBE1X buffer. Image
acquisition and product analysis were made using Bio-Rad imaging
systems with Quantity One® 1-D analysis software.
Bax, Bcl2, NFE2L2 and HO-1 intracellular
detection
The detection of intracellular Bax, Bcl2 and NFE2L2
proteins was performed using colorimetric cell-based ELISA kits
from Assay Biotechnology (Sunnyvale, CA, USA) for Bax (cat. no.
CB5066) and for Bcl2 (cat. no. CB5068) and from LSBio/LifeSpan
Biosciences, Inc. (Seattle, WA, USA) for NFE2L2 (cat. no.
LS-F2192). In brief, the cells were seeded at 3×104
cells/well in a 96-well dish and treated according to the
experimental protocol; at the end of the experiment, the cells were
fixed with 4% formaldehyde. Successively, quenching buffer,
blocking buffer and finally primary antibodies (rabbit anti-Bax,
rabbit anti-Bcl2, rabbit anti-NFE2L2 according to the experimental
design, or mouse anti-GAPDH; the latter used as a positive internal
control) were added followed by incubation overnight at 4°C. All
primary antibodies were diluted 1:100 according to the Technical
Manual (the antibodies were included with the kit). Following this
overnight incubation, the secondary antibodies (included with the
kit; anti-rabbit IgG for Bax, Bcl2 and NFE2L2; Anti-mouse IgG for
GADPH) conjugated to peroxidase were added and subsequently the
samples were read with a microplate reader (BioTek™ Elx800 -Box
998; BioTek Instruments) [optical density (OD) at 450 nm]. The
positive controls were performed in the same plates with the target
experiments. All values obtained, normalized to GAPDH OD450, were
expressed as a percentage relative to the control (untreated
cells).
For the intracellular detection of HO-1, the PC12
cells were examined using a kit sandwich ELISA (Abcam, Cambridge,
UK, cat. no. ab213968). The ELISA was performed according to the
manufacturer’s protocol. Briefly, 1x106 cells, after
treating, were lysed (using 1 ml extraction reagent, provided with
the kit, supplemented with protease inhibitors) and centrifuged at
10,000 × g, at 4°C for 10 min. The collected supernatants,
following protein measurement and normalization, were plated into
plates precoated (with monoclonal anti-rat HO-1 antibody, ready to
use). HO-1 immobilization was detected with a specific polyclonal
antibody and successively with a secondary antibody (Rat HO-1,
ready to use) conjugated to the peroxidase. The intensity of the
color was measured on a microplate reader (BioTek™ Elx800 -Box 998;
BioTek Instruments) at 450 nm. To generate the standard curve,
provided rat HO-1 standard (5 µg/ml) was stepwise diluted
(25, 12, 5, 6, 5, 3, 13, 1, 56, 0, 78, 0, 39 and 0 ng/ml) and the
readers were plotted (using a linear scale) and thus:
Concentrations (ng/ml) on the x-axis and absorbance measurements on
the y-axis. HO-1 concentrations from the samples were quantified by
interpolating the absorbance readings from a standard curve. The
data were expressed as a percentage relative to the control
(untreated cells).
Statistical analysis
Each experiment was repeated at least 3 times,
separately. All the results are presented as the means ± SEM of (n)
replicates per experimental group. Data were subsequently analyzed
by one-way ANOVA, followed by the post hoc Newman-Keuls test for
comparisons between group means, using a Prism™ computer program
(GraphPad, San Diego, CA, USA). Differences were considered
statistically significant at P<0.05.
Results
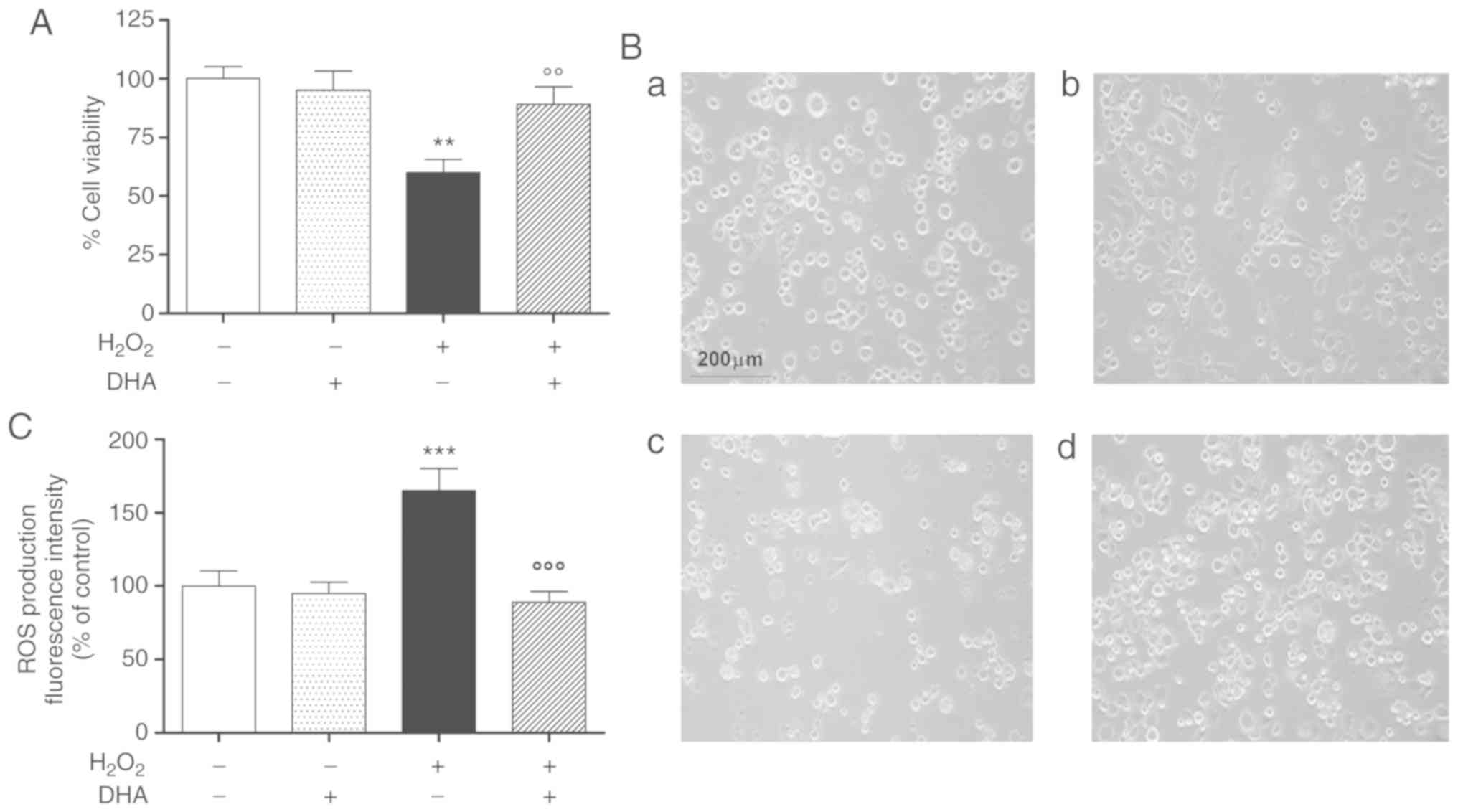
DHA pre-treatment attenuates
H2O2-induced cell death and oxidative
damage
In preliminary experiments, the protective effects
of DHA against H2O2-induced oxidative stress
were investigated in the pheochromocytoma cell line, PC12, by MTS
assay. The cells were pre-treated with physiological concentrations
of DHA (60 µM) for 24 h and then treated with
H2O2 for a further 24 h, with the exception
of the control groups. A significant and consistent decrease
(approximately 40%) in cell viability was observed following 24 h
of exposure to H2O2. Under these conditions,
pre-treatment with DHA was able to antagonize the effects of
H2O2. DHA alone did not affect cellular
viability in the same experimental paradigm (Fig. 1A).
The cell viability data were also confirmed by
subsequent morphological experiments conducted in the same
experimental paradigm (Fig. 1B).
The number of cells was markedly reduced and many of these had a
rounded appearance following exposure to 300 µM
H2O2 (Fig.
1B, panel c), compared to the controls and to cells treated
with DHA alone (Fig. 1B, panels a
and b); pre-treatment with DHA exerted potent protective effects
against H2O2-induced oxidative damage
(Fig. 1B, panel D).
A second series of experiments were performed to
ascertain whether the protective effects observed post-DHA
treatment were associated with the reduction of ROS production and
oxidative damage. Under these conditions, pre-treatment (24 h) with
DHA was able to reduce ROS production induced by
H2O2, while it was not able to alter the
intracellular ROS levels under baseline conditions (Fig. 1C).
Additional experiments were performed to investigate
the biochemical mechanisms underlying the antioxidant effects of
DHA: Enzymatic (SOD, catalase and GSH-Px) and non-enzymatic (GSH
and ascorbic acid) antioxidants, an indicator of lipid peroxidation
(MDA) and nitrates and nitrites were measured in the presence or
absence of DHA under basal conditions and in response to oxidative
stress. The mean concentration levels of enzymatic antioxidants
were markedly lower in the cells exposed to
H2O2 than in the controls (Table I). DHA pre-treatment increased the
intracellular levels of SOD and GSH‑Px in a significant manner
under basal conditions and following
H2O2-induced oxidative stress (Table I). The catalase levels were
increased by DHA pre‑treatment under oxidative stress conditions
compared to H2O2 exposure alone, although the
difference was not statistically significant. The catalase basal
intracellular concentrations remained unaltered (P>0.05) in the
presence of DHA (Table I).
 | Table IIntracellular levels of enzymatic
antioxidants in PC12 cells in both the presence and absence of DHA
under different experimental conditions. |
Table I
Intracellular levels of enzymatic
antioxidants in PC12 cells in both the presence and absence of DHA
under different experimental conditions.
| Compound | Control mean ± SEM
(n) | DHA mean ± SEM
(n) |
H2O2 mean ± SEM
(n) | DHA +
H2O2 mean ± SEM (n) |
|---|
| SOD | 42.20±2.291
(5) | 58.72±2.348
(5)b | 25.20±2.652
(5)a | 40.00±6.611
(5)c |
| CAT | 23.42±3.149
(5) | 19.278±1.332
(5) | 10.890±0.599
(5)b | 12.036±0.579
(5) |
| GSH-Px | 18.36±1.592
(5) | 36.55±4.759
(5)b | 9.15±0.913
(5)a | 25.69±2.671
(5)d |
In addition, Table
II shows the effects of DHA pretreatment on GSH, ascorbic acid,
MDA, nitrates and nitrites under basal and oxidative conditions. It
was observed that: i) DHA significantly increased the GSH levels
both under baseline conditions and in the presence of
H2O2; ii) DHA increased the intracellular
concentrations of ascorbic acid and decreased those of MDA in a
statistically significant manner only in the presence of
H2O2; iii) the intracellular levels of
nitrates and nitrites remained unaltered following pre‑treatment
with DHA under both experimental conditions. The results obtained
above support the hypothesis that DHA exerts antioxidant effects on
H2O2-exposed PC-12 cells by modulating the
levels of antioxidant enzymes and non‑enzymatic scavengers, which
regulate the levels of ROS.
| Table IIIntracellular levels of GSH, ascorbic
acid, MDA, nitrate and nitrite in PC12 cells in both the presence
and absence of DHA under different experimental conditions. |
Table II
Intracellular levels of GSH, ascorbic
acid, MDA, nitrate and nitrite in PC12 cells in both the presence
and absence of DHA under different experimental conditions.
| Compound | Control mean ± SEM
(n) | DHA mean ± SEM
(n) |
H2O2 mean ± SEM
(n) | DHA +
H2O2 mean ± SEM (n) |
|---|
| GSH | 55.25±5.2 (5) | 86.65±6.8
(5)b | 52.47±5.3 (5) | 93.11±7.1
(5)b,d |
| Ascorbic acid | 2.25±0.52 (5) | 2.29±0.7 (5) | 0.45±0.09
(5)a | 0.65±0.1
(5)a |
| MDA | 0.01±0.001 (5) | 0.02±0.001 (5) | 0.03±0.009
(5)a | 0.01±0.002
(5)c |
| Nitrate | 7.45±1.0 (5) | 9.81±1.3 (5) | 8.89±0.9 (5) | 9.48±1.2 (5) |
| Nitrite | 1.28±0.8 (5) | 2.00±0.6 (5) | 2.91±0.8 (5) | 2.15±0.8 (5) |
DHA pre-treatment attenuates
oxidative-stress-induced apoptosis
PC12 cell morphology was observed by fluorescence
microscopy using an Annexin V staining assay to detect apoptotic
cells following pre-treatment with DHA in the presence or absence
of oxidative damage. As shown in Fig.
2A, the apoptosis of the cells pre-treated with DHA was visibly
decreased compared with that of the cells exposed to
H2O2 alone. The process of apoptosis is
strongly associated with the loss of ΔΨm in different cells.
Indeed, the opening of the mitochondrial permeability transition
pore induces the depolarization of the transmembrane potential, and
the release of apoptotic factors and the loss of oxidative
phosphorylation. Consequently, in this study, in order to determine
whether DHA inhibits H2O2-induced apoptosis
related to the mitochondrial pathway, PC12 cells were pre-treated
with DHA and exposed to H2O2 to induce
mitochondrial dysfunction. The ΔΨm of the cells was reduced with
H2O2 alone (60% less than the control), but
was enhanced follwoing pre-treatment (24 h) with DHA in a
statistically significant manner (Fig. 2B). These results confirmed our
hypothesis that DHA is able to inhibit
H2O2-induced mitochondrial dysfunction in
PC12 cells.
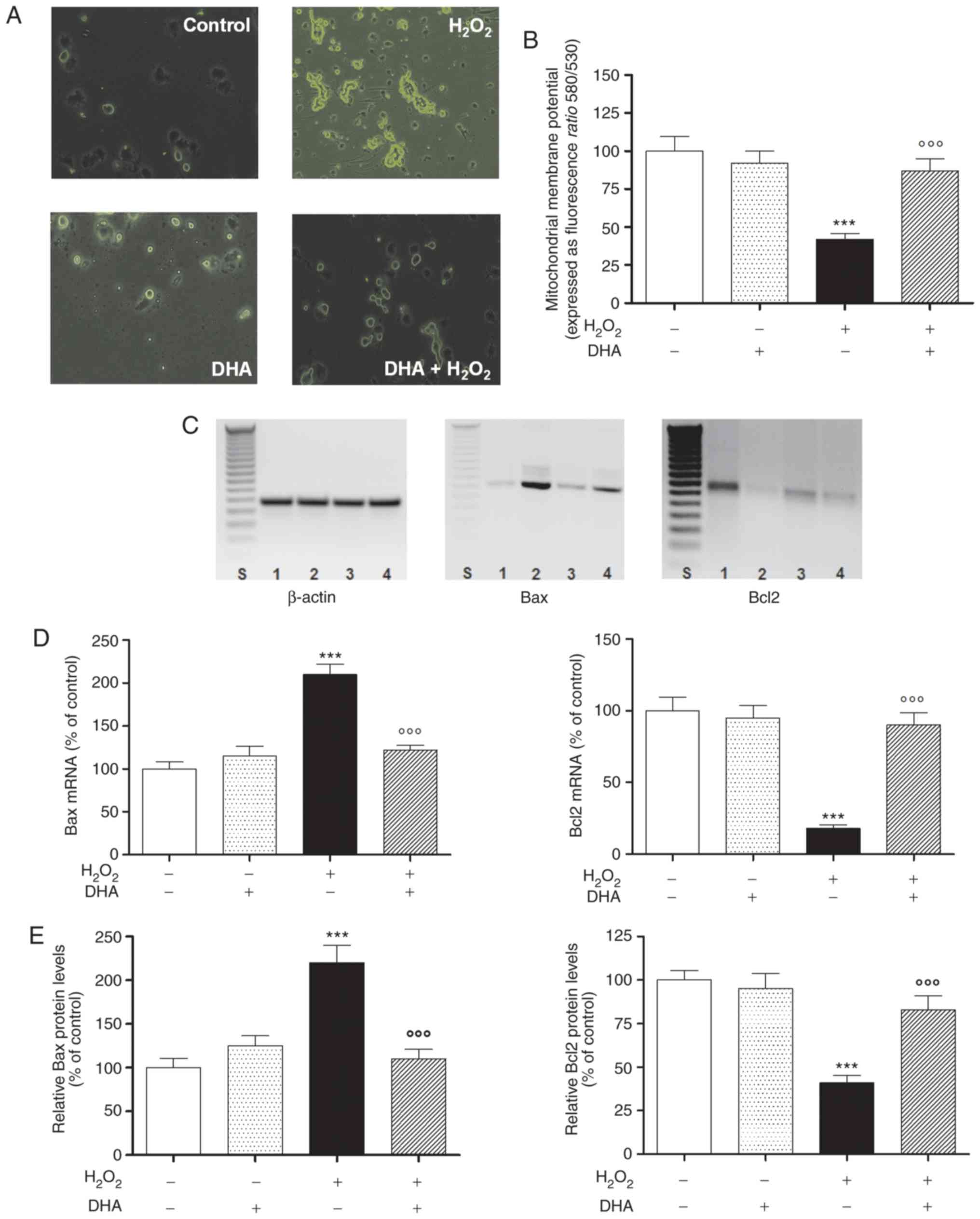 | Figure 2DHA and apoptosis. (A) Fluorescent
staining for the detection of the apoptosis of PC12 cells under
basal conditions (control) and following treatment with: i)
H2O2 (300 µM; 24 h of treatment); ii) DHA (60
µM; 24 h of treatment); iii) DHA plus H2O2
(cells pre-treated for 24 h with DHA and subsequently exposed to
H2O2 for a further 24 h). Apoptotic cells
were detected by Annexin staining assay. Representative images from
3 independent experiments are shown. (B) The mitochondrial membrane
potential (ΔΨm) was measured in PC12 cells by the Mitolight
Apoptosis detection kit (see the Materials and methods section for
details). The fluorescence intensities were measured at an emission
wavelength 580 nm to monitor aggregated molecules (apoptotic cells)
and 530 nm for monomer (healthy cells), respectively. Data are
expressed as fluorescence intensities ratio (580/530) and reported
as a percentage of the control. The means ± SEM of 3 determinations
in double are shown. (C) Agarose gels representing the mRNA
expression levels of β-actin (housekeeping gene, 240 bp), Bax (352
bp) and Bcl2 (349 bp). Lane S, DNA ladder (50 bp); lane 1,
untreated cells; lane 2, cells exposed to
H2O2 alone; lane 3, cells treated with DHA
alone; lane 4, cells pre-conditioned for 24 h with DHA prior to
exposure to H2O2 for a further 24 h. (D) Bax
(left panel) and Bcl2 (right panel) densitometric analysis of the
gels in (C) The density of the gel bands was divided by β-actin and
the results were expressed as a percentage of the control band
density. Results are from 3 independent experiments performed in
duplicate. (E) Effects of pre-treatment with DHA (24 h) on Bax
(left panel) and Bcl2 (right panel) protein levels in PC12 cells
under baseline conditions and under H2O2
stimulation. Bax and Bcl2 protein levels were detected by ELISA
assay and normalized against GADPH (details in Materials and
methods section). Results are from 2 independent experiments, each
including 3 replicates per experimental group. The data in the
figure are presented as the means ± SEM. ***P<0.001
vs. controls; °°°P<0.001 vs.
H2O2 alone. DHA, docosahexaenoic acid. |
To confirm this hypothesis further, the subsequent
experiments were aimed to investigate the effects of DHA
pre-treatment on Bax and Bcl2 gene and protein expression levels.
As shown (in Fig. 2C and D, left
panel), the expression of the pro-apoptotic gene, Bax, was
increased in the H2O2-exposed cells, while
DHA pre-treatment markedly reduced its expression. On the contrary,
the expression of anti-apoptotic gene, Bcl2, was decreased
following H2O2 exposure, while the gene
expression was reversed in the cells pre-treated with DHA (Fig. 2C and D, right panel). The same
trend was also observed for the protein levels of Bax and Bcl2 in
the same experimental paradigm (Fig.
2E).
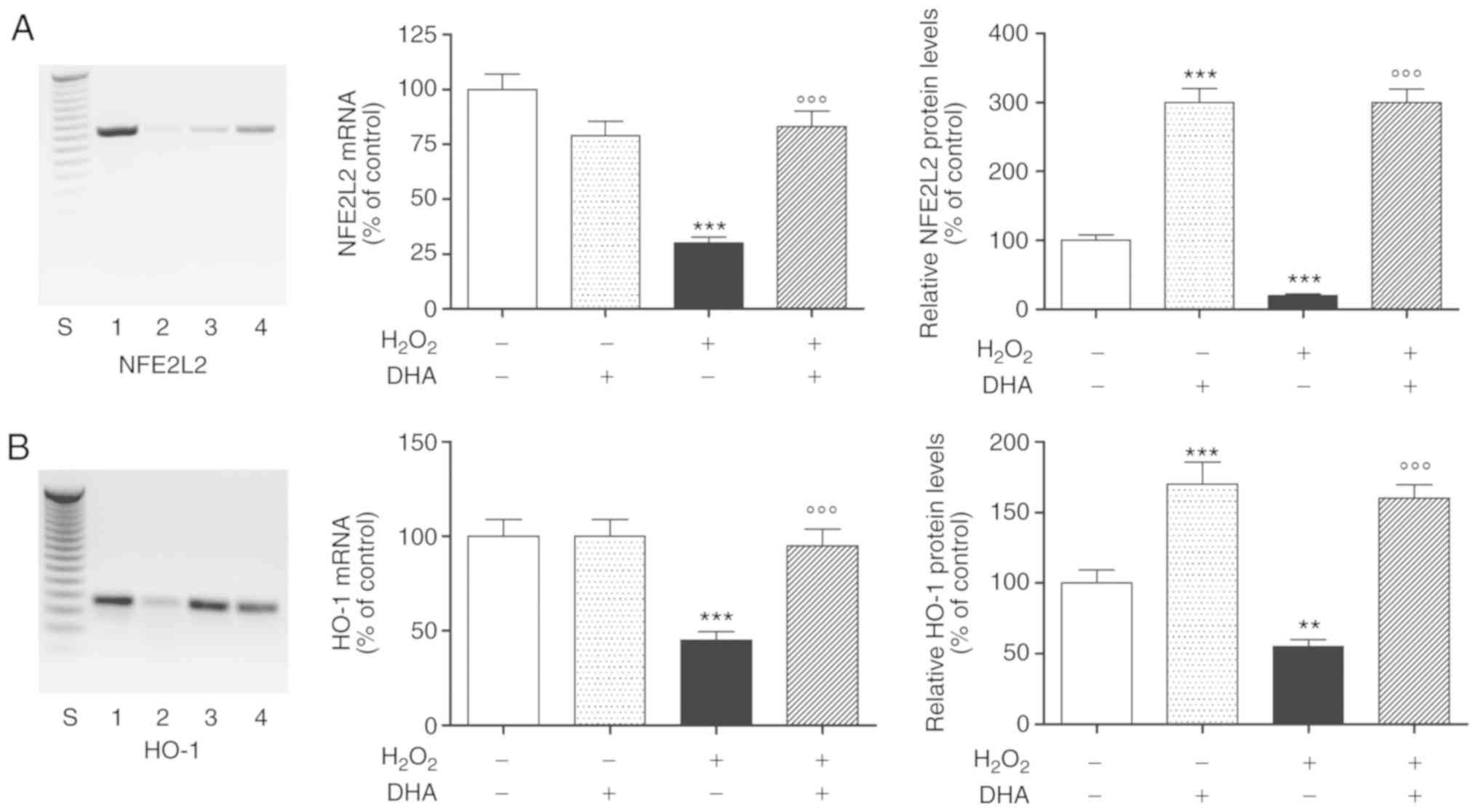
DHA protective effect involves the NFE2L2
antioxidant pathway
To elucidate the plausible signal transduction
pathways involved in the DHA-mediated protective effects, we
examined the involvement of NFE2L2, the principal transcription
factor that regulates the basal and inducible expression of a
number of antioxidant genes. In order to provide direct evidence of
the involvement of NFE2L2 activation, the PC12 cells were
pre-treated with DHA for 24 h, followed by exposure to
H2O2 for an additional 24 h. We observed in
our experimental paradigm that H2O2 markedly
downregulated NFE2L2 mRNA expression, which was antagonized by
pre-treatment with DHA (Fig. 3A,
left and middle panels). Notably, DHA was effective only in the
presence of oxidative damage; in fact, pre-treatment with DHA alone
did not stimulate NFE2L2 gene expression, but rather decreased it
(Fig. 3A, left and middle
panels). The results obtained were also confirmed by subsequent
experiments conducted to determine the NFE2L2 protein intracellular
levels. In particular, the NFE2L2 protein levels increased
>3-fold following DHA pre-treatment both under basal conditions
and following H2O2-induced oxidative damage
(Fig. 3A, right panel).
The transcriptional activation of NFE2L2 evokes the
activation of a series of phase II detoxifying enzymes, including
HO-1, which is able to protect cells against oxidative damage.
Therefore, to further investigate whether DHA-induced NFE2L2
activation modulate in turn HO-1, we examined the effects of DHA
pre-treatment on the activation of the NFE2L2/HO-1 pathway in PC12
cells in same experimental paradigm. The intracellular mRNA
expression and protein levels of HO-1 were markedly decreased to an
estimated 50% following exposure to H2O2 in
comparison to the untreated cells. The observed effect was markedly
inhibited by DHA pre-treatment (24 h), which increased the mRNA
expression and protein levels of HO-1 to values equal or higher
than those observed in the untreated cells, respectively (Fig. 3; pan B). Under baseline
conditions, the DHA alone significantly increased the HO-1 protein
levels, while it had no effect on the mRNA expression of the enzyme
(Fig. 3B).
Discussion
The wide variety of physiopathological relapses,
generated in the organism by redox imbalance, can explain why
oxidative stress has been implicated in the pathogenesis and
complications of a number of neurodegenerative diseases. Therefore,
evaluating oxidative stress and ROS (which are considered to be
mediators of oxidative damage) levels and attempting to counteract
the progression of damage, can be limited. A number of edible
natural compounds have been shown to modulate ROS production and
consequently, oxidative damage. DHA, a dietary fat, is a
representative n-3 PUFA, which has a wide array of health
beneficial effects; of note are its effects at the central nervous
system level (21-23), as it has been shown to decrease the risk of
developing Alzheimer's disease (24). Although the protective
effects of DHA have long been associated with the modification of
cell membrane fluidity, there is new evidence to suggest that the
action of this fatty acid may be attributed to the modulation of
anti-inflammatory gene expression and anti-oxidative pathways (25,
26). However, the biochemical mechanisms underlying the protective
effects of DHA remain unclear. Thus, in the present study, we
focused on investigating the potential protective effects of DHA at
physiological concentrations against
H2O2-induced oxidative damage in rat
pheochromocytoma cells (PC12), exploring cell viability, the
oxidation potential, and the anti-apoptotic and antioxidant
effects.
H2O2 is currently the most
widely used experimental model for simulating oxidative stress. In
fact, H2O2 belongs to ROS and is potentially
harmful to cell viability as it damages different cellular targets.
As expected, H2O2 was found to be toxic to
cells in our experimental model, leading to a marked decrease in
the overall cellular viability, an increase in ROS levels and to
the activation of the apoptotic cascade. These effects were
antagonized by treatment with DHA for 24 h prior to exposure to
H2O2. Pre-treatment with DHA increased the
antioxidant activity by an increase in cell viability and a
reduction in ROS levels, thus protecting the cells against
oxidative stress-induced cell damage. The cells have integrated
antioxidant systems, which include enzymatic and non-enzymatic
antioxidants, that are usually effective in blocking the noxious
effects of ROS (27). However, under pathological conditions, these
systems may be insufficient (28). In this study, we demonstrated
that DHA was able to reverse the H2O2-induced
intracellular levels of a few of these antioxidants (SOD,
cata-lase, GSH-Px, GSH, ascorbic acid and MDA). Furthermore, we
observed a clear reduction in the apoptotic signals by DHA, which
exerted both a protective effect at the mitochondrial level and
played a regulatory role in the modulation of Bax- and
Bcl2-mediated apoptosis, both negative and positive. Overall, these
findings suggest that DHA may be able to protect the cells against
stress-induced apoptosis, mainly via the protection of the
mitochondrial pathway.
Notably, it has been recently identified that NFE2L2
is able to activate antioxidant cellular defences and to inhibit
apoptotic pathways (29). NFE2L2 is a regulator of cellular
resistance against oxidants. It controls the gene expression of a
large variety of antioxidant/cytoprotective enzymes and
detoxification phase II enzymes, regulating the physiological and
pathophysiological outcomes of oxidant exposure. In fact, oxidative
stress dissociates NFE2L2 from its cytoplasmic inhibitor,
Kelch-like ECH-associated protein 1 (KEAP1), promoting its
translocation into the nucleus, where it binds to AREs leading to
the transcriptional activation of antioxidant/cytoprotective
enzymes, such as HO-1, SOD and subunits which are necessary for GSH
biosynthesis (30). In particular, HO-1 antagonizes oxidative stress
by activating the expression of SOD (31) and through the
degradation of the pro-oxidant, heme, leading to the production of
bilirubin and CO, that are important antioxidants (32-34).
Moreover, under stress conditions, NFE2L2 directly prevents
apoptosis by activating Bcl-2, inhibiting Bax and through the
activation of p62, and reduces mitochondrial dysfunctions. The
findings obtained in this study coincide with the aforementioned:
We observed a significant increase in the cellular levels of NFE2L2
following pre-incubation with DHA in our experimental model, and we
thus hypothesized that this PUFA can increase the levels of key
antioxidant enzymes, such as SOD, GSH-Px and HO-1, as well as
modulate apoptotic pathways.
In this study, we measured the level of NFE2L2 in
the whole cell, based on previously published data (35,36)
demonstrating that when PC12 cells are subjected to oxidative
stress, NFE2L2 is exclusively present at nuclear level. However the
observed mechanism may be associated mainly with a direct action of
DHA on availability of NFE2L2 in monomeric form, rather than on
gene expression, as evidenced by the data on gene expression and
protein levels of NEF2L2. In fact, the mRNA expression levels of
both NFE2L2 and HO-1 remained almost similar to the controls
following pre-treatment with DHA, in the presence of
H2O2 but, above all, under basal conditions.
This direct action at the post-transcriptional level could also
explain the directness with which DHA, after 24 h of pre-treatment,
counteracted the oxidizing and apoptotic effects exerted by
H2O2. Notably, while the exposure of cells to
H2O2 is a well-established experimental model
of oxidative damage, the effects of H2O2 on
NEF2L2 activation and the expression of its downstream gene
products remains controversial and unclear. In fact, on the one
hand, it has been shown that H2O2 stabilizes
and activates NEF2L2 by the oxidation and de-activation of KEAP1
(35), while on the other hand, it has been observed that exposure
to H2O2 reduces the mRNA and protein levels
of NEF2L2 (35-41), hypothesizing an involvement of NF-κB (a
negative regulator of the expression of NEF2L2) (38), or rather an
upregulation of KEAP1 during oxidative stress, thus leading to the
inhibition of NEF2L2 (40,41). Consistent with literature data on
PC12 cells (35,36), we observed in our experimental paradigm, that
H2O2 markedly downregulated NFE2L2
expression, which was antagonized by pre-treatment with DHA.
In conclusion, in this study, we demonstrate that
DHA is able to protect PC12 cells from
H2O2-induced oxidative damage through a
mechanism involving the activation of the NFE2L2/HO-1 signaling
pathway. These findings suggest that a strong link could exist
between an adequate supply in the diet of this essential PUFA and
the maintenance of the antioxidant potential of the neuronal cells.
Thus, in light of these findings, it can be hypothesized that DHA
may prove to be a potential therapeutic agent for the prevention of
diseases characterized by inflammation and oxidative stress at the
central nervous system level.
Funding
The present study was financed by the Italian
National Research Council (CNR) funding to MEC and by internal
funding (Fondi di Ateneo D1:2013-2018) to GT.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors′ contributions
MEC, GL and GT conceived and designed the study,
performed the experiments, contributed to data analysis and wrote
the manuscript. GL reviewed and supported MEC and GT in revisions
of the manuscript. BS and AB performed the experiments,
conceptualized the study design, and contributed to data analysis
and experimental materials. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
DHA
|
docosahexaenoic acid
|
|
NFE2L2
|
nuclear factor (erythroid-derived
2)-like 2
|
|
HO-1
|
heme-oxygenase-1
|
|
ARE
|
antioxidant response element
|
|
GSH
|
reduced glutathione
|
|
Keap1
|
Kelch-like ECH-associated protein
1
|
|
NQO1
|
NAD(P)H:quinone oxidoreductase 1
|
|
CAT
|
catalase
|
|
GSH-Px
|
glutathione peroxidase
|
|
SOD
|
superoxide dismutase
|
Acknowledgments
Not applicable.
References
|
1
|
Yan MH, Wang X and Zhu X: Mitochondrial
defects and oxidative stress in Alzheimer disease and Parkinson
disease. Free Radic Biol Med. 62:90–101. 2013. View Article : Google Scholar :
|
|
2
|
Cheng YC, Sheen JM, Hu WL and Hun YC:
Polyphenols and oxidative stress in atherosclerosis-related
ischemic heart disease and stroke. Oxid Med Cell Longev.
2017:85264382017. View Article : Google Scholar
|
|
3
|
Li J, O W, Li W, Jiang ZG and Ghanbari HA:
Oxidative stress and neurodegenerative disorders. Int J Mol Sci.
14:24438–24475. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shi H, Hudson LG and Liu KJ: Oxidative
stress and apoptosis in metal ion-induced carcinogenesis. Free
Radic Biol Med. 37:582–593. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wysoczański T, Sokoła-Wysoczańska E,
Pękala J, Lochyński S, Czyż K, Bodkowski R, Herbinger G,
Patkowska-Sokoła B and Librowski T: Omega-3 fatty acids and their
role in central nervous system-a review. Curr Med Chem. 23:816–831.
2016. View Article : Google Scholar
|
|
6
|
Miller E, Kaur G, Larsen A, Loh SP,
Linderborg K, Weisinger HS, Turchini GM, Cameron-Smith D and
Sinclair AJ: A short-term n-3 DPA supplementation study in humans.
Eur J Nutr. 52:895–904. 2013. View Article : Google Scholar
|
|
7
|
Siegel G and Ermilov E: Omega-3 fatty
acids: Benefits for cardio-cerebro-vascular diseases.
Atherosclerosis. 225:291–295. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cardoso C, Afonso C and Bandarra NM:
Dietary DHA and health: Cognitive function ageing. Nutr Res Rev.
29:281–294. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang W, Yang H, Johnson D, Gensler C,
Decker E and Zhang G: Chemistry and biology of ω-3 PUFA
peroxidation-derived compounds. Prostaglandins Other Lipid Mediat.
132:84–91. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Smith RE, Tran K, Smith CC, McDonald M,
Shejwalkar P and Hara K: The Role of the Nrf2/ARE antioxidant
system in preventing cardiovascular diseases. Diseases. 4:E342016.
View Article : Google Scholar
|
|
11
|
Vomund S, Schäfer A, Parnham MJ, Brüne B
and von Knethen A: Nrf2, the master regulator of anti-oxidative
responses. Int J Mol Sci. 18:E27722017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dinkova-Kostova AT, Kostov RV and
Kazantsev AG: The role of Nrf2 signaling in counteracting
neurodegenerative diseases. FEBS J. 285:3576–3590. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cong P, Liu Y, Liu N, Zhang Y, Tong C, Shi
L, Liu X, Shi X, Liu Y, Tong Z and Hou M: Cold exposure induced
oxidative stress and apoptosis in the myocardium by inhibiting the
Nrf2-Keap1 signaling pathway. BMC Cardiovasc Disord. 18:362018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Clementi ME, Pani G, Sampaolese B and
Tringali G: Punicalagin reduces H2O2-induced cytotoxicity and
apoptosis in PC12 cells by modulating the levels of reactive oxygen
species. Nutr Neurosci. 21:447–454. 2018. View Article : Google Scholar
|
|
15
|
Lazzarino G, Viola AR, Mulieri L, Rotilio
G and Mavelli I: Prevention by fructose-1,6-bisphosphate of cardiac
oxidative damage induced in mice by subchronic doxorubicin
treatment. Cancer Res. 47:6511–6516. 1987.PubMed/NCBI
|
|
16
|
McCord JM and Fridovich I: Superoxide
dismutase. An enzymic function for erythrocuprein (hemocuprein). J
Biol Chem. 244:6049–6055. 1969.PubMed/NCBI
|
|
17
|
Noble JE: Quantification of protein
concentration using UV absor-bance and coomassie dyes. Methods
Enzymol. 536:17–26. 2014. View Article : Google Scholar
|
|
18
|
Lazzarino G, Amorini AM, Fazzina G,
Vagnozzi R, Signoretti S, Donzelli S, Di Stasio E, Giardina B and
Tavazzi B: Single-sample preparation for simultaneous cellular
redox and energy state determination. Anal Biochem. 322:51–59.
2003. View Article : Google Scholar
|
|
19
|
Amorini AM, Giorlandino C, Longo S, D'Urso
S, Mesoraca A, Santoro ML, Picardi M, Gullotta S, Cignini P,
Lazzarino D, et al: Metabolic profile of amniotic fluid as a
biochemical tool to screen for inborn errors of metabolism and
fetal anomalies. Mol Cell Biochem. 359:205–216. 2012. View Article : Google Scholar
|
|
20
|
Innamorato NG, Rojo AI, García-Yagüe AJ,
Yamamoto M, de Ceballos ML and Cuadrado A: The transcription factor
Nrf2 is a therapeutic target against brain inflammation. J Immunol.
181:680–689. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Simopoulos AP: Omega-3 fatty acids in
health and disease and in growth and development. Am J Clin Nutr.
54:438–463. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Uauy R and Dangour AD: Nutrition in brain
development and aging: Role of essential fatty acids. Nutr Rev.
64:S24–S33. 2006. View Article : Google Scholar
|
|
23
|
Moore SA: Polyunsaturated fatty acid
synthesis and release by brain-derived cells in vitro. J Mol
Neurosci. 16:195–200. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Heras-Sandoval D, Pedraza-Chaverri J and
Pérez-Rojas JM: Role of docosahexaenoic acid in the modulation of
glial cells in Alzheimer's disease. J Neuroinflammation. 13:612016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Molz P and Schröder N: Potential
therapeutic effects of lipoic acid on memory deficits related to
aging and neurodegeneration. Front Pharmacol. 8:8492017. View Article : Google Scholar
|
|
26
|
Yadav RK, Singh M, Roy S, Ansari MN,
Saeedan AS and Kaithwas G: Modulation of oxidative stress response
by flaxseed oil: Role of lipid peroxidation and underlying
mechanisms. Prostaglandins Other Lipid Mediat. 135:21–26. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sakai C, Ishida M, Ohba H, Yamashita H,
Uchida H, Yoshizumi M and Ishida T: Fish oil omega-3
polyunsaturated fatty acids attenuate oxidative stress-induced DNA
damage in vascular endothelial cells. PLoS One. 12:e01879342017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matschke V, Theiss C and Matschke J:
Oxidative stress: The lowest common denominator of multiple
diseases. Neural Regen Res. 14:238–241. 2019. View Article : Google Scholar :
|
|
29
|
Jiang S, Deng C, Lv J, Fan C, Hu W, Di S,
Yan X, Ma Z, Liang Z and Yang Y: Nrf2 weaves an elaborate network
of neuroprotection against stroke. Mol Neurobiol. 54:1440–1455.
2017. View Article : Google Scholar
|
|
30
|
Vomhof-Dekrey EE and Picklo MJ Sr: The
Nrf2-antioxidant response element pathway: A target for regulating
energy metabolism. J Nutr Biochem. 23:1201–1206. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Frankel D, Mehindate K and Schipper HM:
Role of heme oxygenase-1 in the regulation of manganese superoxide
dismutase gene expression in oxidatively-challenged astroglia. J
Cell Physiol. 185:80–86. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mo L, Yang C, Gu M, Zheng D, Lin L, Wang
X, Lan A, Hu F and Feng J: PI3K/Akt signaling pathway-induced heme
oxygenase-1 upregulation mediates the adaptive cytoprotection of
hydrogen peroxide preconditioning against oxidative injury in PC12
cells. Int J Mol Med. 30:314–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Giudice A, Arra C and Turco MC: Review of
molecular mechanisms involved in the activation of the Nrf2-ARE
signaling pathway by chemopreventive agents. Methods Mol Biol.
647:37–74. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bellezza I, Giambanco I, Minelli A and
Donato R: Nrf2-Keap1 signaling in oxidative and reductive stress.
Biochim Biophys Acta Mol Cell Res. 1865:721–733. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xu J, Gan S, Li J, Wand DB, Chen Y, Hu X
and Yang GZ: Garcinia xanthochymus extract protects PC12 cells from
H2O2-induced apoptosis through modulation of PI3K/AKT and NRF2/HO-1
pathways. Chin J Nat Med. 15:825–833. 2017.
|
|
36
|
Li M, Xu T, Zhou F, Wang M, Song H, Xiao X
and Lu B: Neuroprotective effects of four phenylethanoid glycosides
on H2O2-induced apoptosis on PC12 Cells via the Nrf2/ARE pathway.
Int J Mol Sci. 19:E11352018. View Article : Google Scholar
|
|
37
|
Fourquet S, Guerois R, Biard D and
Toledano MB: Activation of NRF2 by nitrosative agents and H2O2
involves KEAP1 disulfide formation. J Biol Chem. 285:8463–8471.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cheng J, Wang H, Zhang Z and Liang K:
Stilbene glycoside protects osteoblasts against oxidative damage
via Nrf2/HO-1 and NF-κB signaling pathways. Arch Med Sci.
15:196–203. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Maralani MN, Movahedian A and Javanmard
ShH: Antioxidant and cytoprotective effects of L-Serine on human
endothelial cells. Res Pharm Sci. 7:209–215. 2012.PubMed/NCBI
|
|
40
|
Kim HJ and Vaziri ND: Contribution of
impaired NRF2-KEAP1 pathway to oxidative stress and inflammation in
chronic renal failure. Am J Physiol Renal Physiol. 298:F662–F671.
2010. View Article : Google Scholar
|
|
41
|
Liu X, Liu H, Zhai Y, Li Y, Zhu X and
Zhang W: Laminarin protects against hydrogen peroxide-induced
oxidative damage in MRC-5 cells possibly via regulating NRF2. Peer
J. 5:e36422017. View Article : Google Scholar : PubMed/NCBI
|