Introduction
Alzheimer's disease (AD) is a progressive,
irreversible neurodegenerative disease and is characterized by
neuronal loss, neurofibrillary tangles (NFTs) of
hyperphosphorylated Tau and amyloid β-peptide (Aβ) protein
accumulation (1,2). Progressive synapse loss and cell
death, particularly in the frontal cortex and the hippocampus
region, is characteristic of AD and eventually leads to severe
memory loss. The progressive accumulation of Aβ peptides and Tau
hyper-phosphorylation can enhance the assembly of NFTs and the
formation of senile plaques (SPs); the latter exert additional
toxic effects on neuronal function and cognitive function (3,4).
In recent years, a number of studies have found that
glycogen synthase kinase3β (GSK3β) is a key modulator of the
pathogenesis of AD. It has been demonstrated that GSK 3β
contributes to Aβ-induced neuronal toxicity, and Aβ production and
accumulation is promoted by GSK3β activators and is reduced by
GSK3β inhibitors (5-8). Additionally, the overexpression of
GSK3β increases Tau hyperphosphorylation (9,10),
and the inhibition of GSK3β activity reduces Tau
hyperphosphorylation, restores spatial memory deficits, and
decreases neuronal cell death (11). GSK3β is also a pivotal regulator
of the Wnt/β-catenin signalling pathway, which is closely related
to the pathogenesis of AD (12,13).
Increasing evidence has indicated that the
activation of mammalian target of rapamycin (mTOR) signalling
contributes to the progression of AD and eventually exacerbates AD
pathology and clinical manifestation (14,15). Therefore, mTOR inhibitors may be
effective drugs for the treatment of AD (16). GSK3β is also involved in the mTOR
signalling pathway. However, it remains unclear as to whether the
inhibition of the activation of mTOR via the regulation of the
function of GSK3β can prevent the progression of AD. In this study,
we found that GSK3β activity was inhibited following treatment with
rapamycin, which decreased Aβ deposition and increased Tau
clearance. The data of this study suggest that rapamycin can
attenuate the development of AD by activating the Wnt pathway and
inhibiting GSK3β, in addition to inducing autophagy, thus providing
a novel target for AD therapy.
Materials and methods
Mice and drug treatment
A total of 20 4-month-old amyloid precursor protein
(APP)/presenilin-1 (PS1) double-transgenic
mice overexpressing mouse/human APP (Mo/Hu Aβ
PP695swe) and mutant human PS1-Δ9, weighing 22-25 g,
were purchased from Beijing HFK Bioscience Co. Ltd. Mice were
housed in individually ventilated cage (IVC) systems in a 12-h
light/dark cycle, with free access to water and food. All
procedures involving animals were performed under institutional
guidelines. This study was approved by The Ethics Committee of
Chongqing Medical University (SCXK 2014-0004). Ten male mice were
randomly divided into the rapamycin group and the control group, 5
mice per group. The mice in the rapamycin group were treated with
rapamycin at 2 mg/kg per day for 4 weeks, while the mice in the
control group were administered the same volume of the solvent as
the rapamycin group. Rapamycin (Gene Operation, Ann Arbor, MI, USA)
was first dissolved in dimethyl sulfoxide (DMSO) and then diluted
with double deionized water containing 5% Tween-80 and 5% PEG 400
immediately prior to intragastric administration. The body weight
of the mice was recorded.
Routine blood analysis and chemical
detection
Drug safety was evaluated by aroutine blood
examination and chemical detection. Blood was obtained from the
lateral caudal vein of the mice (approximately 200 μl per
time). The supernatants of the blood samples obtained by
centrifugation (1,500 × g for 15 min) were kept at room temperature
for 1 h and were then collected for chemical testing, including the
quantification of alanine aminotransferase (ALT), aspartate
aminotransferase (AST), blood urea nitrogen (BUN), triglyceride
(TG), serum creatinine (Scr) and total bilirubin (TBIL). Routine
blood tests consisted of a red blood cell (RBC) count, mean
corpuscular volume (MCV), white blood cell (WBC) count, haemoglobin
(HGB), mean platelet volume (MPV) and a platelet (PLT) count.
Brain tissue preparation
Mice were intraperitoneally injected with 10%
chloral hydrate (400 mg/kg) and sacrificed by carbon dioxide
(CO2) euthanasia (CO2 percentage volume
displacement is 20%/min). For each mouse, half of the brain was
used for protein extraction, and the other half was post-fixed in
cold 4% paraformaldehyde (PFA) in 0.1 M phosphate-buffered saline
(PBS) for 24 h. Following dehydration and embedding with optimal
cutting temperature (OCT) compound (Sakura Finetek USA, Inc.,
Torrance, CA, USA), the fixed brain tissues were cut into
10-μm-thick coronal sections using a microtome (Leica
Microsystems, Wetzlar Germany).
4G8 staining and thioflavin S
staining
The slides were developed with methanoic acid for 10
min at room temperature before being blocked in 0.1% Triton and 5%
BSA. The sections were then incubated at 4°C with mouse anti-Aβ,
17-24 (4G8) antibody (800704; 1:250; BioLegend, San Diego, CA, USA)
overnight. After washing, the slides were incubated at 37°C for 40
min with cy3-conjugated goat anti-mouse antibody (A0521; 1:200;
Beyotime Biotechnology, Haimen, China). The mounted slides were
observed using a TCS-TIV confocal laser scanning microscope (Leica
Microsystems).
The slides were deparaffinised with clearing acetone
for 15 min and hydrated by a series of graded ethanol (EtOH)
solutions as follows: 100-80-70%. The sections were then washed
with double distilled water followed by 0.1% potassium permanganate
solution. The slides were subsequently incubated in filtered 1%
aqueous thioflavin-S (1326-12-1; Sigma-Aldrich, Darmstadt, Germany)
for 15 min at room temperature in the dark. The slides were then
washed in a series of graded EtOH solutions of 80-70% and then
washed 3 times with distilled water. The coverslip was mounted with
aqueous mounting media, and the samples were observed under a
TCS-TIV confocal laser scanning microscope (Leica
Microsystems).
We counted senile plaques in half of the brains of
the mice, including the whole cortex and hippocampus on each slice,
and 3 mice in each group. All counts are made by the same
individual who was blinded to the experimental conditions.
Cells, cell culture and treatment
Differentiated SH-SY5Y cells stably transfected with
the APPsw gene (gift from the Laboratory of Translational Medical
Research in Cognitive Development and Learning and Memory
Disorders, Children's Hospital of Chongqing Medical University,
Chongqing, China) were confirmed to be human through STR profiling.
Cells were cultured in DMEM (Gibco/Thermo Fisher Scientific,
Waltham, MA, USA) at 37°C with 5% CO2 and 95% air (v/v)
supplemented with 10% foetal bovine serum (FBS, Biological
Industries, USA), 100 U/ml penicillin and 100 μg/ml
streptomycin. Rapamycin (Gene Operation) was dissolved in DMSO and
then diluted in the culture medium. The final concentration of DMSO
was <0.1%. The control group was treated with the same
concentration of DMSO. The cells were incubated in 96-well
flat-bottom plates (1.0×104 cells/well) with 50 or 100
nM rapamycin for 24 h before cell proliferation was assessed.
Determination of cell proliferation
The cell proliferative ability was determined by a
Cell-Light™ EdU Apollo® 488 In Vitro Imaging kit
(RiboBio Co., Ltd., Guangzhou, China). Following treatment with 50
and 100 nM rapamycin, the cells were treated with 50 μM EdU
for 2 h at 37°C. The cells were then fixed, permeabilized and
exposed to 1X Apollo® reaction solution for 30 min. The
cell nuclei were then counterstained with DAPI. After washing with
phosphate-buffered saline (PBS), the samples were examined under a
fluorescence microscope (Leica Microsystems). Each experimental
condition was repeated in at 6 wells, and data were calculated from
3 independent experiments.
Detection of autophagy by mRFP-GFP-LC3
adenoviral vector
Differentiated SH-SY5Y cells stably transfected with
the APPswe gene were plated in 24-well plates and trans-fected with
mRFP-GFP-LC3 adenoviral vectors (Hanbio Biotechnology Co., Ltd.
Shanghai, China) at 70-80% confluence. On the 2nd day following
transfection, 50 or 100 nM rapamycin was added to the medium for 24
h. After fixing the cells with PFA, autophagy was observed under a
TCS-TIV confocal laser scanning microscope (Leica
Microsystems).
Western blot analysis
Tissues from the half brains of 3 mice from each
group were lysed in RIPA lyses buffer (Pierce/Thermo Fisher
Scientific). The protein concentration was determinated by BCA.
Subsequently, 10 or 12% SDS-PAGE gels were used to separate
proteins. The proteins were then transferred to polyvinylidene
fluoride (PVDF) membranes (Immobilon-P, Millipore, Bedford, MA,
USA). Subsequently, the membranes were blocked in 5% BSA (dissolved
in double deionized water). The membranes were incubated with
rabbit anti-LC3B antibody (L7543; 1:1,000; Sigma-Aldrich, St.
Louis, MO, USA), rabbit anti-NeuN antibody (D4G40), rabbit
anti-β-catenin antibody (D10A8), rabbit anti-p62 antibody (5114)
(all 1:1,000; all from Cell Signaling Technology, Danvers, MA,
USA), rabbit anti-Beclin 1 antibody (ab62557), mouse anti-Tau5
antibody (ab80579), rabbit anti-insulin degrading enzyme (IDE)
antibody (ab133561), rabbit anti-PHF-1 antibody (ab184951), mouse
anti-GSK3β antibody (ab93926), rabbit anti-BACE1 antibody (ab2077),
rabbit anti-PS1 antibody (ab71181), mouse anti-APP mouse antibody
(ab32136) (1:1,000; all purchased from Abcam, Cambridge, MA, USA),
anti-phosphorylated GSK3β (1:1,000; sc81495; Santa Cruz
Biotechnology, Santa Cruz, CA, USA), mouse biotin anti-Aβ, 17-24
antibody (1:250; 800704; BioLegend) and mouse anti-β-actin antibody
(1:1,000; ABM0001-50; Zoonbio Biotechnology, Nanjing, China) at 4°C
overnight. After washing, the membranes were incubated at 37°C for
40 min with horseradish peroxidase-conjugated secondary antibody
(goat anti-mouse IgG, S0002 or goat anti-rabbit IgG, S0001,
Affinity Biosciences, USA) at a 1:5,000 dilution. The membranes
were washed again and treated with enhanced chemiluminescence (ECL)
substrate (K12045; Advansta, Menlo Park, CA, Cincinnati, OH, USA).
The membranes were then visualized with Gel Imaging Systems
software and quantified by Quantity One image software (both from
Bio-Rad, Hercules, CA, USA).
Statistical analysis
Data were analysed with GraphPad Prism 5.0 software.
All results are expressed as the means ± standard error of the mean
(SEM). Statistical comparisons between the control and rapamycin
treatment groups were determined using a two-sample t-test or
one-way ANOVA, followed by a Bonferroni test. The level of
significance was P<0.05.
Results
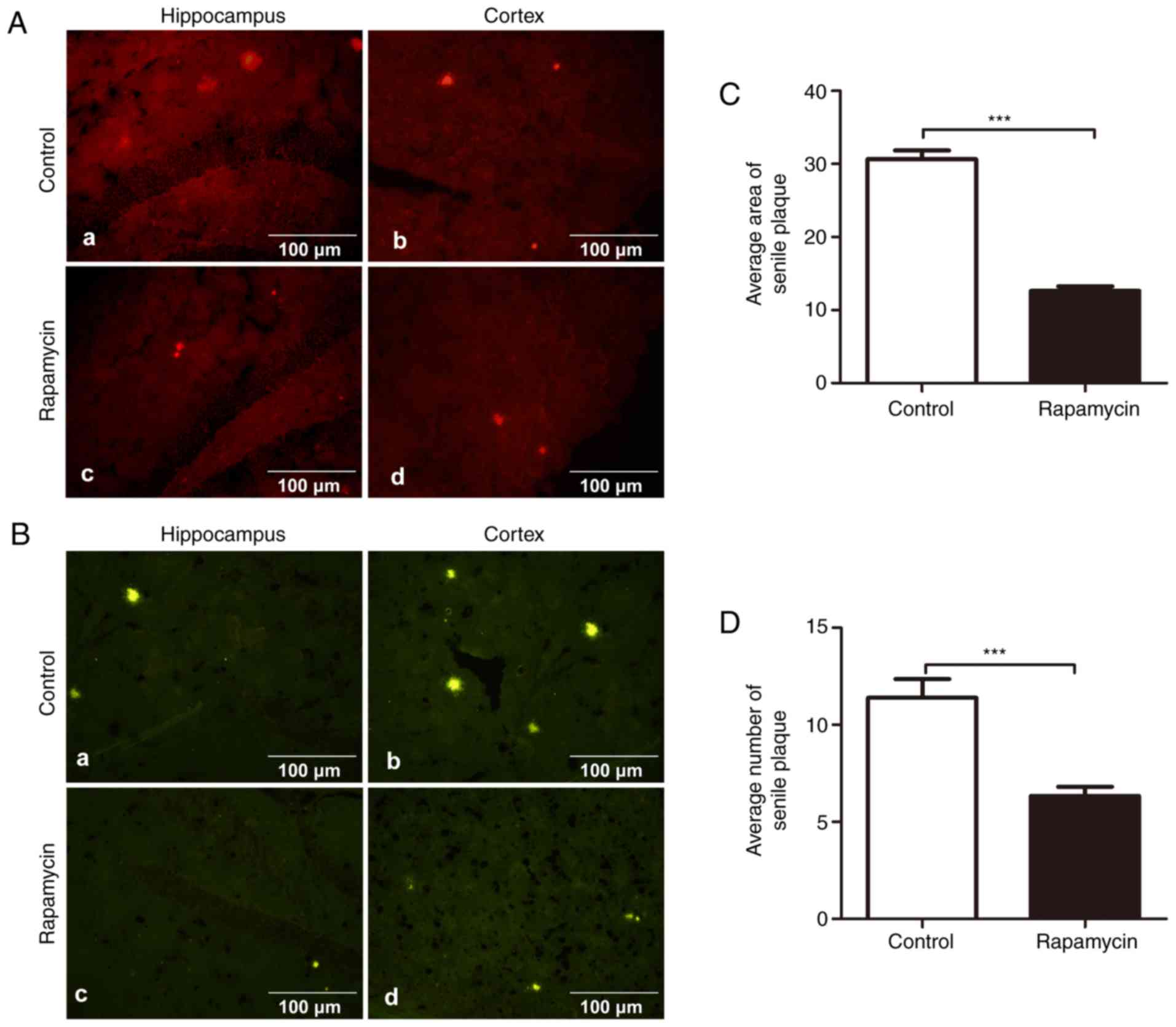
Treatment with rapamycin decreases the
deposition of senile plaques in APP/PS1 transgenic mice
To assess whether rapamycin treatment results in Aβ
aggregation and deposition in the brain, coronal sections of
rapamycin- and vehicle-treated mice were immunohistochemically and
fluorescently stained for Aβ plaques using 4G8 antibody and
thioflavin S staining, respectively. The results revealed that
rapamycin treatment significantly reduced the number (6.333±0.471
vs. 11.390±0.964, t=4.712, P<0.001) and the area of SPs
(12.610±0.622 vs. 30.670±1.193, t=13.420, P<0.001) in both the
hippocampus and cortex of the mouse brains compared with the
control treatment. Thioflavin S staining revealed that the
rapamycin group had fewer fibrillary Aβ deposits than the control
group (Fig. 1).
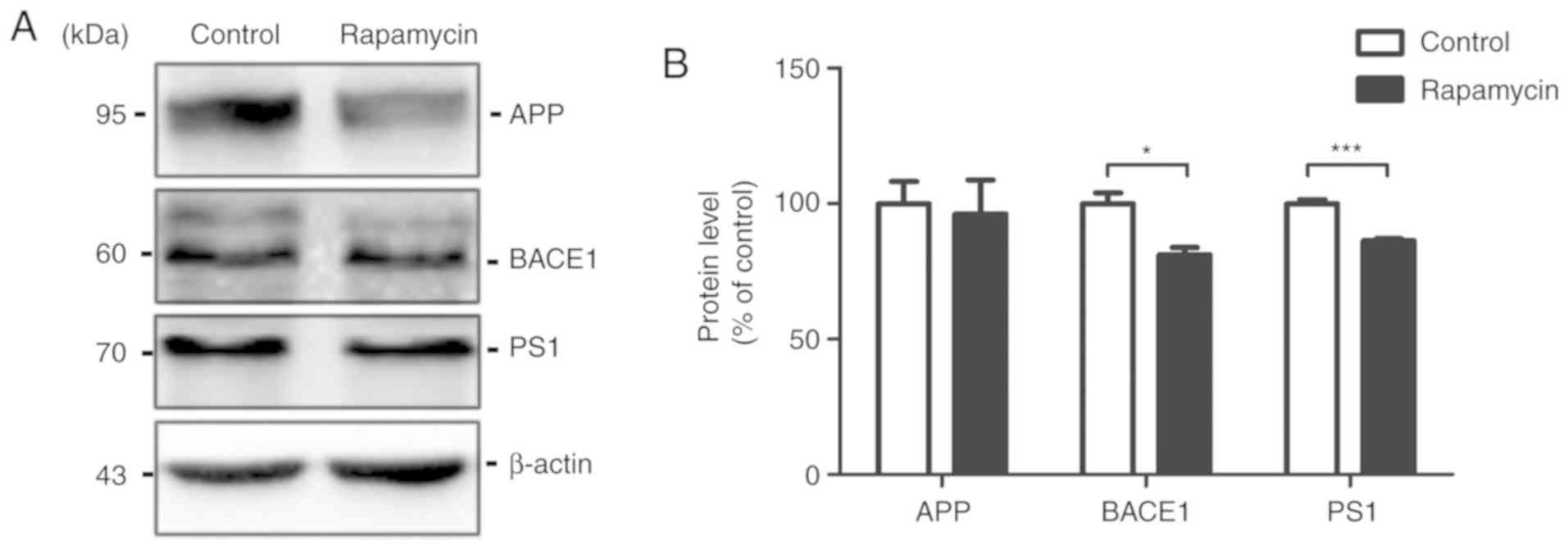
Rapamycin decreases Aβ generation
The expression levels of APP, BACE1 and PS1 in the
mouse brains were measured by western blot analysis. Rapamycin
treatment had no effect on the level of APP (100.000±8.173 vs.
96.220±12.590, t=0.252, P=0.806). However, the APP cleaving
enzymes, BACE1 and PS1, were both decreased following treatment
with rapamycin (100.000±3.900 vs. 81.150±2.637, t=4.005, P=0.016;
and 100.000±0.520 vs. 86.320±0.766, t=14.790, P<0.001,
respectively; Fig. 2).
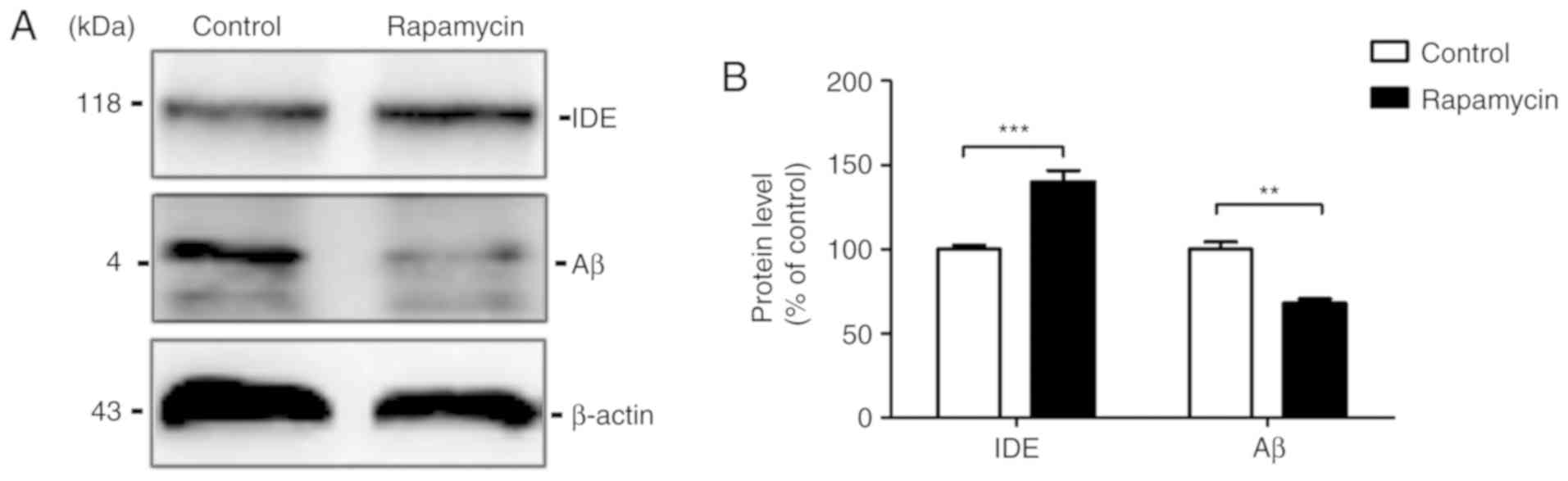
Rapamycin accelerates the degradation of
Aβ
The Aβ levels in the mouse brains were then
examined. The results of western blot analysis revealed that the Aβ
level was markedly decreased by rapamycin treatment (100.000±4.185
vs. 68.060±2.442, t=6.591, P=0.003) compared with the control
group. To examine the possibility that the Aβ reduction induced by
rapamycin treatment is associated with Aβ clearance, western blot
analysis was used to measure the levels of the Aβ degradation
enzyme, IDE, in the mouse brains. The results revealed that IDE
expression increased in the rapamycin group compared with the
control group (100.000±2.418 vs. 139.900±6.851, t=5.495,
P<0.001; Fig. 3).
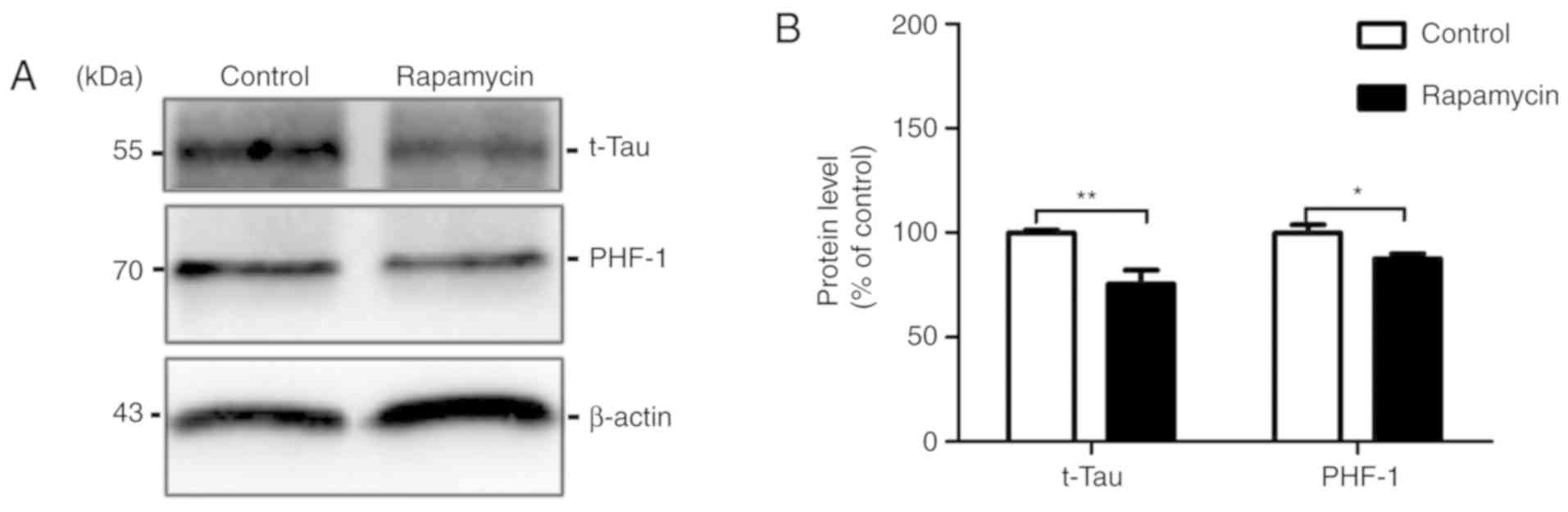
Rapamycin decreases the levels of total
Tau and abnormal hyperphosphorylated Tau
The anti-Tau antibody, Tau 5, was used to measure
the total Tau level. We found that the total Tau level in the
rapamycin treatment group was significantly decreased by 10.488%
compared with that in the control group (100.000±1.187 vs.
75.450±6.672, t=3.622, P=0.005). In AD-affected brains, the
aberrant hyperphosphorylation of Tauinduces the formation of paired
helical filaments (PHFs), which contributes to the formation of
NFTs (17). In the present study,
the PHF-1 levels were decreased by 12.430% in the rapamycin group
compared with the control group (100.000±3.776 vs. 87.570±2.320,
t=2.805, P=0.049; Fig. 4).
Rapamycin increases cell
proliferation
The effects of rapamycin on cell proliferation were
assessed by an EdU incorporation assay. Quantitative analysis
revealed that the number of EdU-incorporated cells was
significantly increased by treatment with 50 nM (43.020±0.303 vs.
47.280±0.831, t=4.943, P<0.01; F=42.72; P<0.001) and 100 nM
(43.020±0.303 vs. 50.980±0.578, t=9.236, P<0.001; F=42.72;
P<0.001) rapamycin (Fig. 5A and
B). The neuronal nuclei (NeuN) antigen is a neuronal nuclear
marker. The results of western blot analysis revealed that
rapamycin treatment elevated NeuN expression in the brain tissues
compared with the control group (100.000±4.618 vs. 189.600±4.156,
t=14.420, P<0.001) (Fig. 5C and
D).
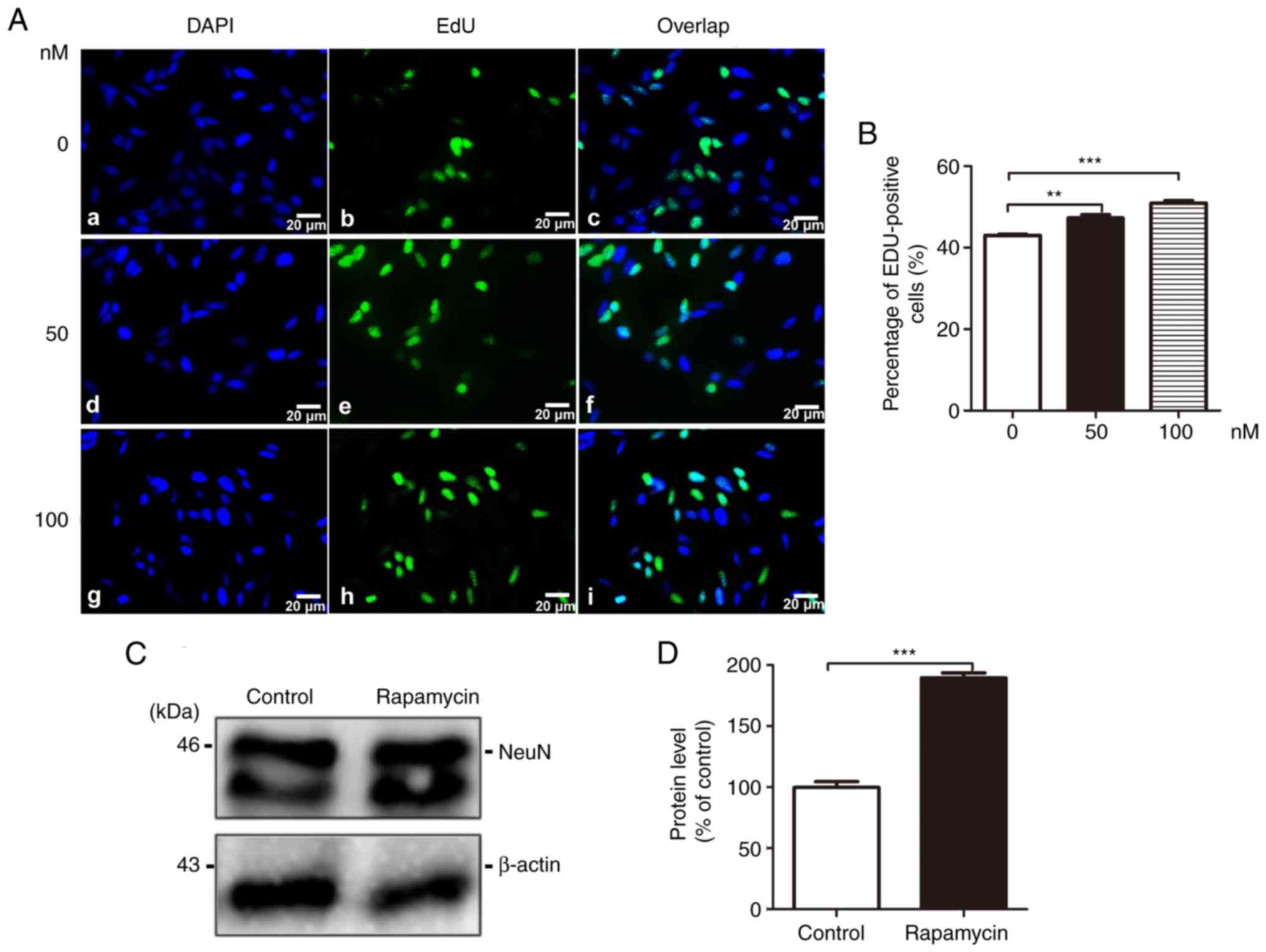 | Figure 5Rapamycin increases cell
proliferation. (A) Cell proliferation and viability were determined
by an EdU incorporation assay after SH-SY5Y cells stably
transfected with the APPsw gene were incubated with various
concentrations of rapamycin for 24 h. Cell nuclei were
counterstained with DAPI (panels a, d and g, DAPI; panels b, e and
h, EdU; panels c, f and i, overlap; scale bar, 20 μm). (B)
Quantitative analysis of the EdU-positive cells was performed by
counting in 5 randomly selected fields of each well. Data were
plotted as the means ± SEM of 3 independent experiments were
analysed by one-way ANOVA (followed by Bonferroni test)
(**P<0.01 and ***P<0.001, compared with
control group, no treatment). (C) A representative western blot of
NeuN levels in brain from the control group and rapamycin group.
(D) Relative grey density analysis of the protein expression levels
in 2 groups, and data are presented as the means ± SEM and were
analysed by Student's t-test (***P<0.001, compared
with control group). |
Rapamycin induces autophagic
activities
The treatment of differentiated SH-SY5Y cells stably
transfected with the APPswe gene with 50 and 100 nM rapamycin
markedly increased the number of mRFP-positive cells (1.333±0.882
vs. 42.670±3.712, t=5.369, P<0.01; 1.333±0.882 vs. 36.000±8.622,
t=4.503, P<0.05, respectively; F=16.62, P=0.004) and 100 nM
rapamycin treatment also increased the number of GFP-positive
puncta (1.000±0.577 vs. 20.330±5.783, t=4.028, P<0.05; F=8.270,
P=0.019; Fig. 6A and B).
Rapamycin treatment at 50 nM increased the number of autolysosomes
(0.333±0.333 vs. 34.330±2.963, t=9.016, P<0.001; F=40.770,
P<0.001), and 100 nM treatment increased the number of both
autophagosomes and autolysosomes (1.000±0.577 vs. 20.330±5.783,
t=4.028, P<0.05; F=8.272, P=0.019; 0.333±0.333 vs. 15.670±3.528,
t=4.066, P<0.05; F=40.77, P<0.001; Fig. 6C). Additionally, the levels of
autophagy-related markers were examined by western blot analysis.
Compared with the control group, rapamycin treatment increased the
ratio of LC3-II/I (100.000±0.672 vs. 145.900±12.850, t=3.565,
P=0.024) and the level of Beclin-1 in the brains of mice with AD
(100.00±7.571 vs. 188.4±16.590, t=4.844, P=0.008), both of which
are markers of autophagy initiation. Subsequently, we measured the
expression of the p62/SQSTM1 protein, which is a lysosomal
degradation substrate and dysregulated autophagy increases its
elimination (18). The p62 level
in the rapamycin group was decreased compared with that of the
control group (100.00±2.988 vs. 144.200±18.590, t=2.346, P=0.041;
Fig. 6D and E).
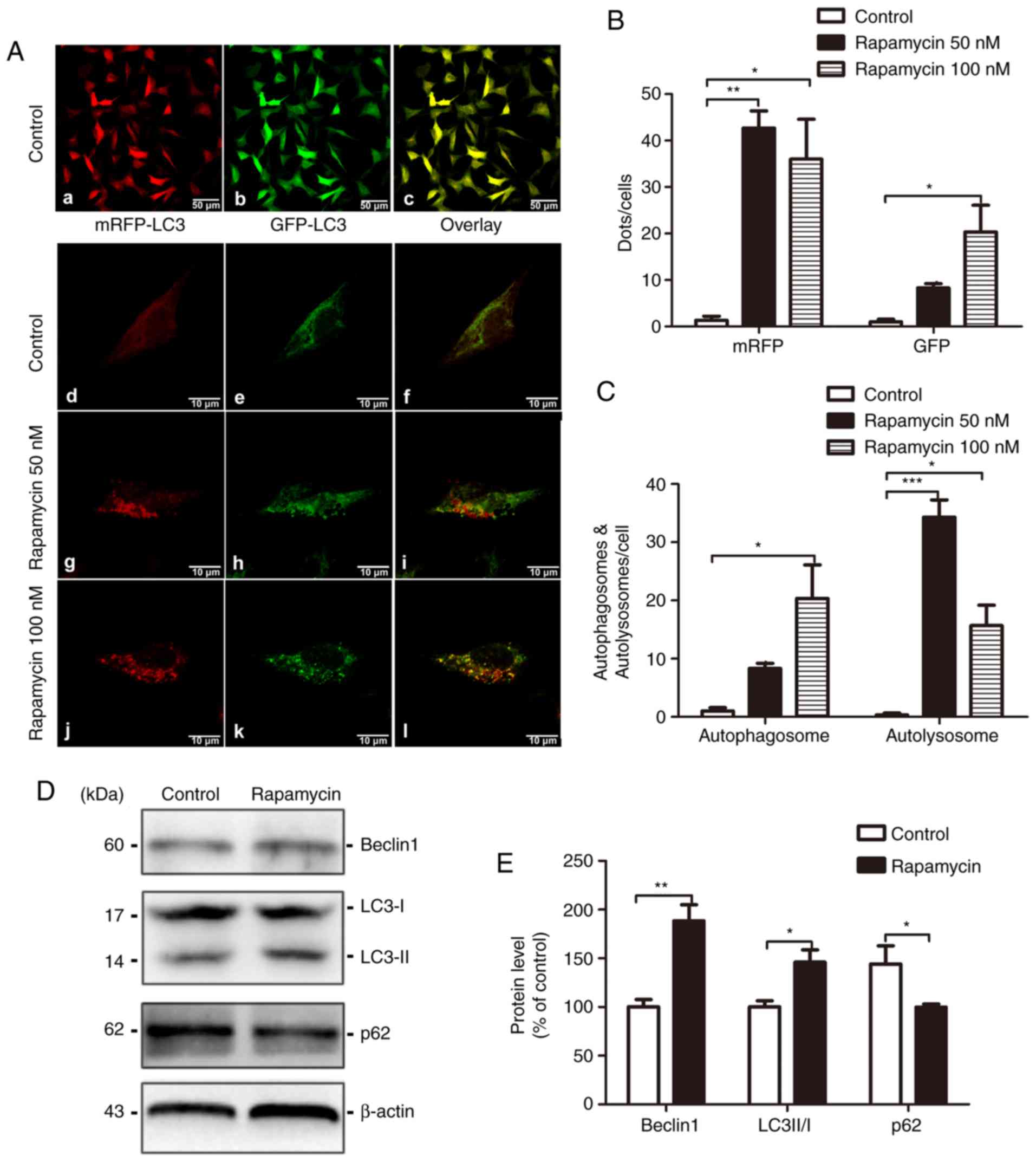 | Figure 6Rapamycin activates autophagy. (A)
Fluorescent microscopy images of differentiated SH-SY5Y cells
stably transfected with the APPsw gene expressing mRFP-GFP-LC3 and
treated with 50 or 100 nM rapamycin or vehicle for 24 h. The
efficiency of transfection were also shown (panels a, d, g and j,
mRFP; panels b, e, h and k, GFP; panels c, f, i and l, overlap;
scale bar, 10 μm). (B) Quantification of mRFP and GFP puncta
per cell. Five randomly selected fields were counted and the data
are presented as the means ± SEM and were analysed by one-way ANOVA
(followed by Bonferroni test) (*P<0.05,
**P<0.01 and ***P<0.001, compared with
the control group). (C) Quantification of autophagosomes and
autolysosomes per cell. Five randomly selected fields were counted.
The data were presented as the means ± SEM and were analysed by
one-way ANOVA (followed by Bonferroni test). (D) Representative
western blots of Beclin1, LC3 and p62 levels in the brain in the
control group and the rapamycin group. (E) Relative grey density
analysis of protein expression levels in the 2 groups is shown, and
the data are presented as the means ± SEM and were analysed by a
Student's t-test (*P<0.05 and **P<0.01,
compared with the control group). |
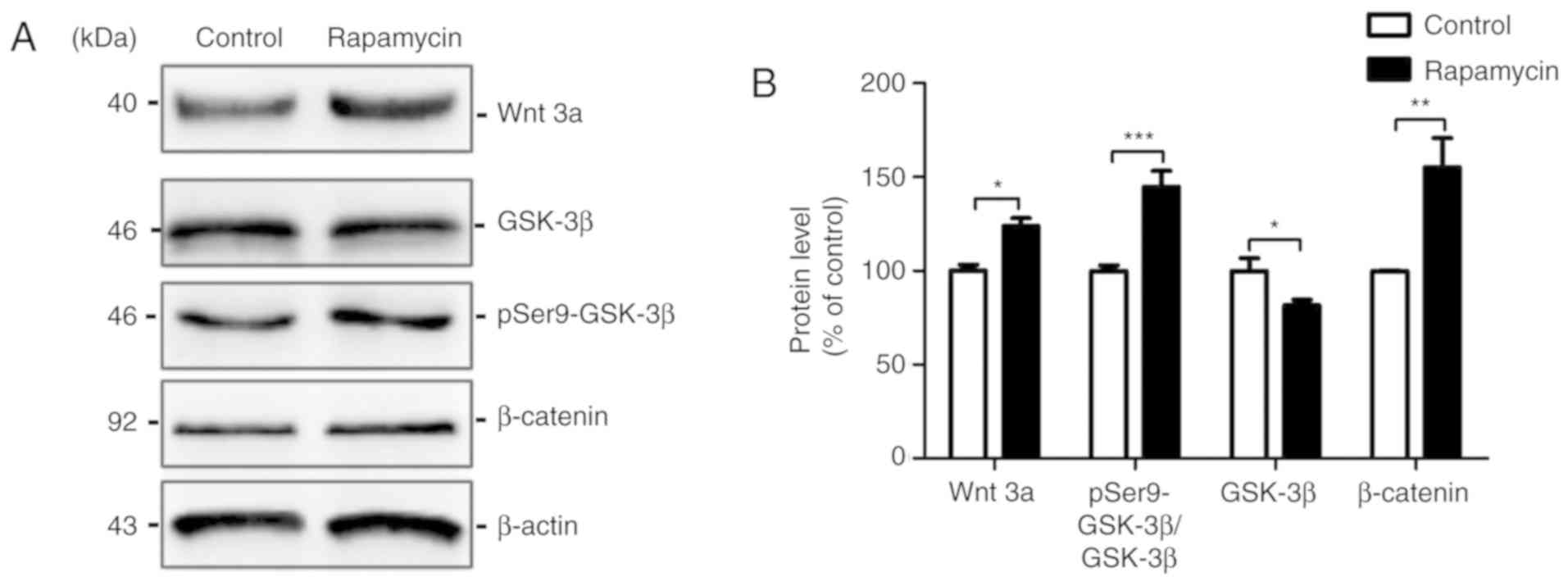
Rapamycin activates the
Wnt/GSK3β/β-Catenin signalling pathway
The Wnt and mTOR pathways have been confirmed to be
linked to GSK 3β in heart disease (19). Therefore, in this study, we
detected whether rapamycin, an mTOR inhibitor, alters the Wnt
pathway in AD. The results of western blot analysis revealed that
Wnt3a was activated by rapamycin treatment (100.000±3.202 vs.
124.000±4.119, t=4.593, P=0.010) and that its downstream target
protein, GSK3β, was inhibited by rapamycin treatment (100.00±6.805
vs. 81.600±3.051, t=2.467, P=0.033). Phosphorylation at the serine
9 residues of GSK3β downregulated its kinase activity, and
rapamycin treatment increased phospho-GSK3β (Ser9) levels
(100.00±4.607 vs. 144.700±8.587, t=4.590, P=0.001), compared with
GSK3β. β-catenin is a target protein of GSK3β, and the activation
of GSK3β inhibits its activity. The β-catenin expression level in
the rapamycin group was elevated compared to the control group
(100.000±0.211 vs. 155.000±15.680, t=3.509, P=0.006). All these
data suggest that rapamycin activates the Wnt pathway via the
suppression of GSK3β (Fig.
7).
Drug safety of rapamycin as reflected by
routine blood analysis and biochemical detection
Following treatment with rapamycin for 4 weeks, we
first measured the drug safety of rapamycin by routine blood
analysis (Table I) and chemical
detection (Table II). The
results of the routine blood tests and chemical detections did not
differ significantly between the control and rapamycin-treated
groups (all P>0.05), which indicates that this dosage of
rapamycin caused no toxic effects in vivo compared with the
control group.
 | Table IRoutine blood analysis following
rapamycin treatment in mice (compared with the control group
(P>0.05). |
Table I
Routine blood analysis following
rapamycin treatment in mice (compared with the control group
(P>0.05).
| Indexes | Control group | Rapamycin
group |
|---|
| RBC
(×1012/l) | 10.7±0.6 | 10.5±0.3 |
| HGB (g/l) | 147.7±16.2 | 145.7±2.3 |
| MCV (fl) | 43.4±1.4 | 44.9±0.2 |
| WBC
(×109/l) | 10.4±2.3 | 8.5±6.6 |
| PLT
(×109/l) | 989.3±278.6 | 1,140.0±50.8 |
| MPV (fl) | 6.5±0.1 | 6.7±0.1 |
| Table IIBiochemical detection of rapamycin
administration effects on mice (compared with control group,
P>0.05). |
Table II
Biochemical detection of rapamycin
administration effects on mice (compared with control group,
P>0.05).
| Indexes | Control group | Rapamycin
group |
|---|
| ALT (U/l) | 41.3±4.0 | 43.3±4.2 |
| AST (U/l) | 143.7±44.8 | 137.3±38.9 |
| BUN (mmol/l) | 11.2±0.6 | 10.8±2.0 |
| Scr
(μmol/l) | 11.1±0.6 | 11.8±1.0 |
| TBIL
(μmol/l) | 0.8±0.8 | 0.6±0.7 |
| TG (mmol/l) | 1.0±0.1 | 1.1±0.5 |
Discussion
The increased production and accumulation of Aβ are
the primary pathological features of AD and are closely associated
with neurotoxicity and the decline in cognitive function. APP/PS1
double transgenic AD model mice are an ideal animal model with
which to investigate the pathogenesis and drug targets of AD.
Mutations associated with the familial form of AD are present in
the amyloid precursor gene APP and in the presenilin genes 1 and 2,
which are involved in the cleavage of APP and thus in the
generation of Aβ. Aβ generated within cells is the suspected source
of fibrillar Aβ plaque deposits, and elevated intracellular Aβ in
the absence of SP formation has been reported to be accompanied by
cognitive deficits (20). Aβ is
produced from APP processing, in which APP can be sequentially
cleaved by β-secretase (BACE1) and γ-secretase (PS1) to produce
amyloidogenic peptides. In addition, the dynamic balance of Aβ is
maintained by its production and degradation. Several proteases can
regulate the degradation of Aβ, such as neprilysin (NEP), zinc
metal-lopeptidases, and the carefully studied IDE (21,22). IDE is a zinc-endopeptidase that is
well known for its activity against insulin and glucagon (23). However, recent studies have
confirmed that IDE is a major endogenous Aβ-degrading enzyme.
Additionally, IDE has been found to increase the secretion induced
by Aβ in the presence of autophagy from astrocytes (24). Rapamycin-activated autophagy in
conjunction with increased IDE secretion from astrocytes and IDE
located in autophagosomes was discovered in a previous study. These
findings imply that the IDE secretion induced by Aβ is regulated by
autophagy, which is in accordance with the conclusion of this
study. In this study, the results of immunofluorescence revealed
that rapamycin treatment reduced SP formation and deposition. The
results of western blot analysis demonstrated that rapamycin
decreased the expression levels of BACE1, PS1 and Aβ, and increase
the level of IDE, which illustrates that rapamycin can ameliorate
Aβ accumulation, by not only enhancing autophagy activity, but also
by reducing APP cleavage and increasing the proteolytic degradation
of Aβ.
Tau is the most abundant microtubule-associated
protein in the brain. Under physiological conditions, the function
of Tau is to combine with microtubules to promote microtubule
stabilization. However, by contrast, under pathological conditions,
Tau exhibits altered solubility properties, is abnormally
hyperphosphorylated at certain residues and forms PHF structures,
which are prone to form NFTs (17,25,26). It has been confirmed that Tau can
be eliminated by autophagy under pathological conditions (27,28). The inactivation of autophagy leads
to the accumulation of hyperphosphorylated Tau, and the activation
of autophagy results in a reduction in hyperphosphorylated Tau
levels (29). Notably, it has
been demonstrated that different lengths of Tau may be degraded
through different autophagic pathways; the full-length Tau is
preferentially degraded through macroautophagy, whereas truncated
Tau (for one, TauRDΔK280) is cleared through chaperone-mediated
autophagy (30). Although the
APP/PS1/Tau tri-transgenic AD mouse model is an ideal model with
which to study the pathology of Aβ and Tau, the tau-induced
pathology in the APP/PS1 double-transgenic AD mouse model was also
found in our previous study and in other reports (31-33). This study examined the effects of
rapamycin on Tau clearance. The results of western blot analysis
indicated that rapamycin treatment decreased the expression levels
of Tau-5 and PHF-1, suggesting that rapamycin contributed to the
degradation of not only full-length Tau, but also phosphorylated
Tau.
AD is characterized by neuronal cell death, which
eventually leads to memory loss. Neurons are post-mitotic cells
that are unable to degrade the toxic substances accumulated during
the ageing processes with mitosis; thus autophagy is an important
mechanism for clearing abnormally aggregated proteins in neuronal
cells (34). It is becoming
evident that autophagy is a protective reaction in neurons and that
dysfunctional autophagy aggravates neuronal death in AD (35-37). NeuN, a marker of neuronal
differentiation, is a nuclear protein expressed in most
post-mitotic neurons in the nervous system and plays an important
role in nervous system development and function (38). This study found that rapamycin
treatment elevated NeuN expression levels and the number of
EdU-positive cells, which indicates that rapamycin can increase
cell proliferation and may exert a neuroprotective effect in
AD.
Autophagy is a catabolic mechanism responsible for
the clearance of intracellular cytosolic components, including
misfolded proteins, aggregates and organelles, and it has now been
confirmed to play a crucial role in AD pathology. Properly
functioning autophagy is essential for neuronal metabolism, and the
lack of autophagy in the central nervous system (CNS) leads to
neurodegeneration; for example, inactivating autophagy impairs
neuronal homeostasis (29,39).
Autophagy is initiated when an 'isolation membrane' appears.
Subsequently, the membrane elongates, engulfing cellular components
to form autophagosomes (APs) and lysosomes, which can digest the
cargo in their acidic environment, fuse with APs to generate
autolysosomes (ALs). Recent studies have found that the expression
of autophagy-related genes is downregulated with ageing; a
deficiency in Beclin1, a key protein in autophagy induction,
results in increases in Aβ levels, whereas its overexpression
reduces the accumulation of Aβ (40-43). These studies have indicated that
autophagic activity is reduced with ageing, and the reduction of
autophagy contributes to the onset of AD. Several studies which
have focused on autophagy have discovered that the activation of
autophagy can decrease abnormal Aβ aggregation in neurons and can
alleviate neurotoxicity (44-48), which is consistent with the
results of this study.
The transfection of cells with the mRFP-GFP-LC3
adenoviral vector allows for the visualization of autophagic
activity. Upon rapamycin exposure, the cells accumulate dense red
and yellow puncta, which represents autolysosomes and
autophagosomes, respectively. The present study on rapamycin found
that treatment with rapamycin increased the number of autolysosomes
and autophagosomes. However, the results of western blot analysis
indicated that rapamycin treatment increased the ratio of LC3-II/I
and elevated the expression of Beclin1, both of which are crucial
mediators of autophagy induction. p62 protein is a lysosomal
degradation substrate that reduces when autophagy begins and the
autophagic flux proceeds smoothly (18). In this study, the p62 expression
level decreased following rapamycin treatment, which demonstrated
the increased degradation mediated by autophagy.
GSK3β is one of the phosphorylating glycogen
synthase enzymes (49). In the
brain, GSK3β is involved in a number of cellular processes and
mediates several signalling pathways (50-52). Researchers have reported that Aβ
leads to GSK3β activation, and the inhibition of GSK3β has been
verified to reduce Aβ production and decrease Aβ-induced
neurotoxicity (53,54). It is well known that GSK 3β has
the exact site for the hyperphosphorylation of Tau protein. The
excessive activation of GSK 3β increases hyperphosphorylated Tau
protein production, and GSK3β itself is involved in the initiation
and development of AD (55). GSK
3β is most commonly mediated by inhibitory phosphorylation at the
Ser9 site residue, and this serine can be phosphorylated by several
kinases; thus, multiple signalling pathways are involved in its
regulation, such as the PI3k/Akt/mTOR pathway and the Wnt pathway.
It has been reported that GSK3β functions in several pathways which
regulate the cellular homeostasis of energy and growth, including
those mediated by PI3K and AKT, mTOR and MAPK. The inhibition of
mTOR by rapamycin leads to the inactivation of its target kinases,
including AKT, p70S6K and p85S6K, which in turn promotes GSK3β
activation (56-59). The Wnt pathway is also involved in
the onset of AD. Wnt signalling is an autocrine signal transduction
pathway that participates in brain development (60,61). The ablation of Wnt-3a, a Wnt
ligand, results in the disappearance of the hippocampus (62,63). Wnt signalling can be mainly
divided into two types: β-catenin-dependent signalling
(Wnt/β-catenin) and β-catenin-independent signalling (64,65). GSK3β is a crucial regulator of the
Wnt/β-catenin pathway. The activation of GSK3β by Wnt signalling
results in the phosphorylation and degradation of its downstream
target, β-catenin (66). Without
Wnt stimulation, the phosphorylation of β-catenin prevents
transcriptional activity in the nucleus.
In this study, 4 weeks of rapamycin administration
significantly activated the Wnt pathway, which led to an increased
Wnt3a and a reduced GSK 3β expression. The phosphorylation of GSK3β
at Ser9 reduced the activation of GSK 3β, and treatment with
rapamycin seemed to reduce the activity of GSK3β. It has been
reported that the overexpression of GSK3β in neurons decreases
nuclear β-catenin levels and increases Tau hyperphosphorylation in
the hippocampus (10). In this
study, the results of western blot analysis indicated that
rapamycin treatment increased the expression level of
β-catenin.
It has been reported that rapamycin improves
learning after sepsis by enhancing autophagy and may be a
potentially effective therapeutic agent for the treatment of
sepsis-induced cognitive impairment (67,68). In this study, the data imply that
rapamycin can increase cell proliferation as well as reduce Aβ and
Tau pathology by inducing autophagy and activating the
Wnt/GSK3β/β-catenin signalling pathway. Thus, rapamycin may be a
potential therapy with which to prevent the progression of AD.
Rapamycin is a macrolide antibiotic produced by
Streptomyces hygroscopicus, and a previous study found that
rapamycin is an inhibitor of mTOR, which is a major signalling
molecule that suppresses the initiation step of autophagy (69). Notably, GSK3β is a key regulator
of many cellular signalling pathways, and the mTOR pathway is one
of these pathways. However, the effect of rapamycin on the GSK3β
signalling pathway has not yet been clearly elucidated. The purpose
of the present study was to analyse the effects of rapamycin on AD
progression, autophagy activity, and GSK3β function, as well as the
underlying mechanisms of action of rapamycin in APP/PS1 double
transgenic mice.
Funding
This study was supported by the National Natural
Science Foundation of China (nos. 31670899, 81671257, 81371221 and
31600825); the Basic and Frontier Research Project of Chongqing
(cstc2016jcyjA0244; cstc2016jcyjA0069); and the Education
Commission Science and Technology Research Project of Chongqing
(no. KJ1500231).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
GH and SL conceived and designed the study. JC and
ZL performed the experiments and drafted the manuscript, YL and ML
conducted the statistical analysis. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
All procedures involving animals were performed
under institutional guidelines. This study was approved by The
Ethics Committee of Chongqing Medical University (SCXK
2014-0004).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Professor Kejian
Wang (Department of Anatomy, Chongqing Medical University),
Professor Weihong Song (University of British Columbia) and
Professor Zhifang Dong (Children's Hospital Affiliated to Chongqing
Medical University) for their assistance with performing the
experiments.
References
|
1
|
Hardy J and Selkoe DJ: The amyloid
hypothesis of Alzheimer's disease: Progress and problems on the
road to therapeutics. Science. 297:353–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mucke L: Neuroscience: Alzheimer's
disease. Nature. 461:895–897. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Näslund J, Haroutunian V, Mohs R, Davis
KL, Davies P, Greengard P and Buxbaum JD: Correlation between
elevated levels of amyloid beta-peptide in the brain and cognitive
decline. JAMA. 283:1571–1577. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Karran E, Mercken M and De Strooper B: The
amyloid cascade hypothesis for Alzheimer's disease: An appraisal
for the development of therapeutics. Nat Rev Drug Discov.
10:698–712. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun X, Sato S, Murayama O, Murayama M,
Park JM, Yamaguchi H and Takashima A: Lithium inhibits amyloid
secretion in COS7 cells transfected with amyloid precursor protein
C100. Neurosci Lett. 321:61–64. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li B, Ryder J, Su Y, Zhou Y, Liu F and Ni
B: FRAT1 peptide decreases Abeta production in swAPP(751) cells.
FEBS Lett. 553:347–350. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Su Y, Ryder J, Li B, Wu X, Fox N,
Solenberg P, Brune K, Paul S, Zhou Y, Liu F and Ni B: Lithium, a
common drug for bipolar disorder treatment, regulates amyloid-beta
precursor protein processing. Biochemistry. 43:6899–6908. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ly PT, Wu Y, Zou H, Wang R, Zhou W,
Kinoshita A, Zhang M, Yang Y, Cai F, Woodgett J and Song W:
Inhibition of GSK3β-mediated BACE1 expression reduces
Alzheimer-associated phenotypes. J Clin Invest. 123:224–235. 2013.
View Article : Google Scholar
|
|
9
|
Brownlees J, Irving NG, Brion JP, Gibb BJ,
Wagner U, Woodgett J and Miller CC: Tau phosphorylation in
transgenic mice expressing glycogen synthase kinase-3beta
transgenes. Neuroreport. 8:3251–3255. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lucas JJ, Hernández F, Gómez-Ramos P,
Morán MA, Hen R and Avila J: Decreased nuclear beta-catenin, tau
hyperphosphorylation and neurodegeneration in GSK-3beta conditional
transgenic mice. EMBO J. 20:27–39. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Engel T, Hernández F, Avila J and Lucas
JJ: Full reversal of Alzheimer's disease-like phenotype in a mouse
model with conditional overexpression of glycogen synthase
kinase-3. J Neurosci. 26:5083–5090. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ghanevati M and Miller CA:
Phospho-beta-catenin accumulation in Alzheimer's disease and in
aggresomes attributable to proteasome dysfunction. J Mol Neurosci.
25:79–94. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ferrari De GV, Papassotiropoulos A,
Biechele T, Wavrant De-Vrieze F, Avila ME, Major MB, Myers A, Sáez
K, Henríquez JP, Zhao A, et al: Common genetic variation within the
low-density lipoprotein receptor-related protein 6 and late-onset
Alzheimer's disease. Proc Natl Acad Sci USA. 104:9434–9439. 2007.
View Article : Google Scholar
|
|
14
|
Paccalin M, Pain-Barc S, Pluchon C, Paul
C, Besson MN, Carret-Rebillat AS, Rioux-Bilan A, Gil R and Hugon J:
Activated mTOR and PKR kinases in lymphocytes correlate with memory
and cognitive decline in Alzheimer's disease. Dement Geriatr Cogn
Disord. 22:320–326. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang Z and Klionsky DJ: Mammalian
autophagy: Core molecular machinery and signaling regulation. Curr
Opin Cell Biol. 22:124–131. 2010. View Article : Google Scholar
|
|
16
|
Spilman P, Podlutskaya N, Hart MJ, Debnath
J, Gorostiza O, Bredesen D, Richardson A, Strong R and Galvan V:
Inhibition of mTOR by rapamycin abolishes cognitive deficits and
reduces amyloid-beta levels in a mouse model of Alzheimer's
disease. PLoS One. 5:e99792010. View Article : Google Scholar
|
|
17
|
Alonso A, Zaidi T, Novak M, Grundke-Iqbal
I and Iqbal K: Hyperphosphorylation induces self-assembly of tau
into tangles of paired helical filaments/straight filaments. Proc
Natl Acad Sci USA. 98:6923–6928. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mathew R, Karp CM, Beaudoin B, Vuong N,
Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al:
Autophagy suppresses tumorigenesis through elimination of p62.
Cell. 137:1062–1075. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vigneron F, Dos Santos P, Lemoine S,
Bonnet M, Tariosse L, Couffinhal T, Duplaà C and Jaspard-Vinassa B:
GSK-3β at the crossroads in the signalling of heart
preconditioning: Implication of mTOR and Wnt pathways. Cardiovasc
Res. 90:49–56. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
LaFerla FM, Green KN and Oddo S:
Intracellular amyloid-beta in Alzheimer's disease. Nat Rev
Neurosci. 8:499–509. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Iwata N, Tsubuki S, Takaki Y, Shirotani K,
Lu B, Gerard NP, Gerard C, Hama E, Lee HJ and Saido TC: Metabolic
regulation of brain Abeta by neprilysin. Science. 292:1550–1552.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Farris W, Mansourian S, Chang Y, Lindsley
L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ and
Guenette S: Insulin-degrading enzyme regulates the levels of
insulin, amyloid beta-protein, and the beta-amyloid precursor
protein intracellular domain in vivo. Proc Natl Acad Sci USA.
100:4162–4167. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang
J, Podlisny MB, Rosner MR, Safavi A, Hersh LB and Selkoe DJ:
Insulin-degrading enzyme regulates extracellular levels of amyloid
beta-protein by degradation. J Biol Chem. 273:32730–32738. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Son SM, Cha MY, Choi H, Kang S, Choi H,
Lee MS, Park SA and Mook-Jung I: Insulin-degrading enzyme secretion
from astrocytes is mediated by an autophagy-based unconventional
secretory pathway in Alzheimer disease. Autophagy. 12:784–800.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ballatore C, Lee VM and Trojanowski JQ:
Tau-mediated neuro-degeneration in Alzheimer's disease and related
disorders. Nat Rev Neurosci. 8:663–672. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan
M, Wisniewski HM and Binder LI: Abnormal phosphorylation of the
microtubule-associated protein tau (tau) in Alzheimer cytoskeletal
pathology. Proc Natl Acad Sci USA. 83:4913–4917. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rubinsztein DC: The roles of intracellular
protein-degradation pathways in neurodegeneration. Nature.
443:780–786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chesser AS, Pritchard SM and Johnson GV:
Tau clearance mechanisms and their possible role in the
pathogenesis of Alzheimer disease. Front Neurol. 4:1222013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hara T, Nakamura K, Matsui M, Yamamoto A,
Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I,
Okano H and Mizushima N: Suppression of basal autophagy in neural
cells causes neurodegenerative disease in mice. Nature.
441:885–889. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang Y, Martinez-Vicente M, Krüger U,
Kaushik S, Wong E, Mandelkow EM, Cuervo AM and Mandelkow E: Tau
fragmentation, aggregation and clearance: The dual role of
lysosomal processing. Hum Mol Genet. 18:4153–4170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Long ZM, Zhao L, Jiang R, Wang KJ, Luo SF,
Zheng M, Li XF and He GQ: Valproic acid modifies synaptic structure
and accelerates neurite outgrowth via the glycogen synthase
kinase-3β signaling pathway in an Alzheimer's disease model. CNS
Neurosci Ther. 21:887–897. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kempf SJ, Metaxas A, Ibáñez-Vea M, Darvesh
S, Finsen B and Larsen MR: An integrated proteomics approach shows
synaptic plasticity changes in an APP/PS1 Alzheimer's mouse model.
Oncotarget. 7:33627–33648. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhong L, Liu H, Zhang W, Liu X, Jiang B,
Fei H and Sun Z: Ellagic acid ameliorates learning and memory
impairment in APP/PS1 transgenic mice via inhibition of β-amyloid
production and tau hyperphosphorylation. Exp Ther Med.
16:4951–4958. 2018.PubMed/NCBI
|
|
34
|
Mariño G, Madeo F and Kroemer G: Autophagy
for tissue homeostasis and neuroprotection. Curr Opin Cell Biol.
23:198–206. 2011. View Article : Google Scholar
|
|
35
|
Pivtoraiko VN, Harrington AJ, Mader BJ,
Luker AM, Caldwell GA, Caldwell KA, Roth KA and Shacka JJ: Low-dose
bafilomycin attenuates neuronal cell death associated with
autophagy-lysosome pathway dysfunction. J Neurochem. 114:1193–1204.
2010.PubMed/NCBI
|
|
36
|
Chu CT: Autophagy in different flavors:
Dysregulated protein degradation in neurological diseases.
Neurobiol Dis. 43:1–3. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Galluzzi L, Maiuri MC, Vitale I, Zischka
H, Castedo M, Zitvogel L and Kroemer G: Cell death modalities:
Classification and pathophysiological implications. Cell Death
Differ. 14:1237–1243. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mullen RJ, Buck CR and Smith AM: NeuN, a
neuronal specific nuclear protein in vertebrates. Development.
116:201–211. 1992.PubMed/NCBI
|
|
39
|
Komatsu M, Waguri S, Chiba T, Murata S,
Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E and
Tanaka K: Loss of autophagy in the central nervous system causes
neurodegeneration in mice. Nature. 441:880–884. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Omata Y, Lim YM, Akao Y and Tsuda L:
Age-induced reduction of autophagy-related gene expression is
associated with onset of Alzheimer's disease. Am J Neurodegener
Dis. 3:134–142. 2014.
|
|
41
|
Salminen A, Kaarniranta K, Kauppinen A,
Ojala J, Haapasalo A, Soininen H and Hiltunen M: Impaired autophagy
and APP processing in Alzheimer's disease: The potential role of
Beclin 1 interactome. Prog Neurobiol. 106-107:33–54. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jaeger PA, Pickford F, Sun CH, Lucin KM,
Masliah E and Wyss-Coray T: Regulation of amyloid precursor protein
processing by the Beclin 1 complex. PLoS One. 5:e111022010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pickford F, Masliah E, Britschgi M, Lucin
K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E,
Levine B and Wyss-Coray T: The autophagy-related protein beclin 1
shows reduced expression in early Alzheimer disease and regulates
amyloid beta accumulation in mice. J Clin Invest. 118:2190–2199.
2008.PubMed/NCBI
|
|
44
|
Sarkar S, Perlstein EO, Imarisio S, Pineau
S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O'Kane CJ,
Schreiber SL and Rubinsztein DC: Small molecules enhance autophagy
and reduce toxicity in Huntington's disease models. Nat Chem Biol.
3:331–338. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Williams A, Sarkar S, Cuddon P, Ttofi EK,
Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, et
al: Novel targets for Huntington's disease in an mTOR-independent
autophagy pathway. Nat Chem Biol. 4:295–305. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Balgi AD, Fonseca BD, Donohue E, Tsang TC,
Lajoie P, Proud CG, Nabi IR and Roberge M: Screen for chemical
modulators of autophagy reveals novel therapeutic inhibitors of
mTORC1 signaling. PLoS One. 4:e71242009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jimenez-Sanchez M, Thomson F, Zavodszky E
and Rubinsztein DC: Autophagy and polyglutamine diseases. Prog
Neurobiol. 97:67–82. 2012. View Article : Google Scholar
|
|
48
|
Bové J, Martinez-Vicente M and Vila M:
Fighting neurodegeneration with rapamycin: Mechanistic insights.
Nat Rev Neurosci. 12:437–452. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Embi N, Rylatt DB and Cohen P: Glycogen
synthase kinase-3 from rabbit skeletal muscle. Separation from
cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J
Biochem. 107:519–527. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Doble BW and Woodgett JR: Role of glycogen
synthase kinase-3 in cell fate and epithelial-mesenchymal
transitions. Cells Tissues Organs. 185:73–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Forde JE and Dale TC: Glycogen synthase
kinase 3: A key regulator of cellular fate. Cell Mol Life Sci.
64:1930–1944. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Llorens-Martin M, Jurado J, Hernández F
and Avila J: GSK-3β, a pivotal kinase in Alzheimer disease. Front
Mol Neurosci. 7:462014.
|
|
53
|
Rockenstein E, Torrance M, Adame A, Mante
M, Bar-on P, Rose JB, Crews L and Masliah E: Neuroprotective
effects of regulators of the glycogen synthase kinase-3beta
signaling pathway in a transgenic model of Alzheimer's disease are
associated with reduced amyloid precursor protein phosphorylation.
J Neurosci. 27:1981–1991. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Koh SH, Noh MY and Kim SH:
Amyloid-beta-induced neuro-toxicity is reduced by inhibition of
glycogen synthase kinase-3. Brain Res. 1188:254–262. 2008.
View Article : Google Scholar
|
|
55
|
Terwel D, Muyllaert D, Dewachter I,
Borghgraef P, Croes S, Devijver H and Van Leuven F: Amyloid
activates GSK-3beta to aggravate neuronal tauopathy in bigenic
mice. Am J Pathol. 172:786–798. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen
Y, Zhang SM, Lian G, Liu Q, Ruan K, et al: GSK3-TIP60-ULK1
signaling pathway links growth factor deprivation to autophagy.
Science. 336:477–481. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cohen P and Frame S: The renaissance of
GSK3. Nat Rev Mol Cell Biol. 2:769–776. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Woodgett JR and Ohashi PS: GSK3: An
in-Toll-erant protein kinase? Nat Immunol. 6:751–752. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang HH, Lipovsky AI, Dibble CC, Sahin M
and Manning BD: S6K1 regulates GSK3 under conditions of
mTOR-dependent feedback inhibition of Akt. Mol Cell. 24:185–197.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Salinas PC and Zou Y: Wnt signaling in
neural circuit assembly. Annu Rev Neurosci. 31:339–358. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Inestrosa NC and Arenas E: Emerging roles
of Wnts in the adult nervous system. Nat Rev Neurosci. 11:77–86.
2010. View Article : Google Scholar
|
|
62
|
Moon RT, Kohn AD, De Ferrari GV and Kaykas
A: WNT and beta-catenin signalling: Diseases and therapies. Nat Rev
Genet. 5:691–701. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ciani L and Salinas PC: WNTs in the
vertebrate nervous system: From patterning to neuronal
connectivity. Nat Rev Neurosci. 6:351–362. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Logan CY and Nusse R: The Wnt signaling
pathway in development and disease. Annu Rev Cell Dev Biol.
20:781–810. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gordon MD and Nusse R: Wnt signaling:
Multiple pathways, multiple receptors, and multiple transcription
factors. J Biol Chem. 281:22429–22433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Inestrosa NC, Montecinos-Oliva C and
Fuenzalida M: Wnt signaling: Role in Alzheimer disease and
schizophrenia. J Neuroimmune Pharmacol. 7:788–807. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lin AL, Jahrling JB, Zhang W, DeRosa N,
Bakshi V, Romero P, Galvan V and Richardson A: Rapamycin rescues
vascular, metabolic and learning deficits in apolipoprotein E4
transgenic mice with pre-symptomatic Alzheimer's disease. J Cereb
Blood Flow Metab. 37:217–226. 2017. View Article : Google Scholar
|
|
68
|
Liu W, Guo J, Mu J, Tian L and Zhou D:
Rapamycin protects sepsis-induced cognitive impairment in mouse
hippocampus by enhancing autophagy. Cell Mol Neurobiol.
37:1195–1205. 2017. View Article : Google Scholar
|
|
69
|
Sarbassov DD, Ali SM and Sabatini DM:
Growing roles for the mTOR pathway. Curr Opin Cell Biol.
17:596–603. 2005. View Article : Google Scholar : PubMed/NCBI
|