Introduction
The incidence of macrovascular complications in
diabetic patients is >50% and constitutes a high rate of
mortality (1). Hyperglycemia
resulting from uncontrolled glucose regulation is mainly considered
to form the link between diabetes and diabetic vasculopathy
(2). As components of blood
barriers, vascular endothelial cells (ECs) are more vulnerable to
hyperglycemia damage (3). During
the development of diabetes, the impaired EC functioning results in
decreased levels of vasodilation, weakened barrier function, and
increased reactive oxygen species (ROS) generation and inflammatory
activation (4). Therefore,
elucidating the mechanisms of endothelial injury is critical to
alleviating diabetic vasculopathy.
Mitochondria are not only the focus of cellular
energy generation, they are also involved in the regulation of
cellular differentiation, proliferation, senility and apoptosis.
Several studies have suggested that mitochondrial fission serves an
important role in mitochondrial homeostasis (5,6).
Furthermore, mitochondrial fission factor (Mff)-mediated
mitochondrial fission is considered to be an important facet of
diabetic complications (7).
Excessive mitochondrial fission leads to cellular energy metabolism
disorders, ROS bursts, increased mitochondrial permeability
transition pore (mPTP) opening volumes and the activation of
mitochondrial-dependent cell apoptotic procedures in response to HG
treatment (8). Therefore,
maintaining mitochondrial homeostasis by inhibiting mitochondrial
fission is necessary to protect endothelial function and survival
in diabetic vasculopathy. However, the upstream regulators of
mitochondrial fission in HG-induced endothelial injury remain to be
elucidated.
Convincing experimental data indicates that Sirt1
serves a critical function in the pathological progression of
diabetic vasculopathy by modulating endothelial cellular
physiological processes, including metabolism, migration,
senescence and apoptosis (9). The
activation of Sirt1 by resveratrol ameliorates HG-induced
endothelial damage by stabilizing mitochondrial energy metabolism
(10). Of note, studies have
reported a potential association between Sirt1 and mitochondrial
fission (11,12). However, how Sirt1 regulates
mitochondrial fission, particularly in HG-induced endothelial
injury, remains to be elucidated.
In addition to apoptosis, decreased levels of
endothelial cell migration are another key feature of endothelial
injury in diabetic vasculopathy (13,14). It is well known that F-actin
homeostasis is crucial in the regulation of cellular migration
(15,16). However, whether Sirt1 protects
cellular migration by regulating F-actin homeostasis remains
unclear. Accordingly, the aim of the present study was to examine
the role of Sirt1 in repairing diabetic vasculopathy, with a focus
on mitochondrial fission and cellular migration.
Materials and methods
Animal procedure and treatment
All experimental procedures described here were
conducted in accordance with the National Institutes of Health
Guidelines on the Care and Use of Laboratory Animals. All animal
experimental protocols were approved by the Institutional Animal
Care and Use Committee of Beijing Vital River Laboratory Animal
Technology Co., Ltd. (Beijing, China). ApoE−/− mice
(male; n=120; supplied by Beijing Vital River Laboratory Animal
Technology Co., Ltd; body weight, 18-22 g) were fed a high-fat diet
(45% kcal fat, 35% kcal carbohydrates and 20% kcal protein) from 4
weeks of age to the end of the study and housed at 23°C with 40-70%
humidity and a 12-h light/dark cycle. 8-week-old apoE−/−
mice were intraperitoneally injected with streptozotocin (STZ;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany; 50 mg/kg) for five
consecutive days. Following this, venous blood was collected from
the tails of diabetic mice; blood glucose levels >16.7 mmol/l
following a 6-h period of daytime fasting was considered successful
model establishment. The diabetic mice (12 weeks old), defined as
the diabetic group, were treated with SRT1720 (Selleck Chemicals,
Houston, TX, USA; 15 mg/kg/day, defined as the SRT1720 group) for
12 weeks. The mice were anesthetized by intraperitoneal injection
of pentobarbital sodium (60 mg/kg body weight) prior and whole
blood was drawn by cardiac puncture or from the eye retroorbital
sinus (17). The mice were
sacrificed with CO2 using an established CO2
euthanasia method, for endothelial function assessment and
histological examination. Briefly, the mice were placed in a
chamber and the flow rate of CO2 displacement was 2
l/min (<30% of the chamber volume/min), which was stopped when
the mice were no longer breathing and sacrifice was confirmed by
cervical dislocation (18,19).
Measurement of biochemical
parameters
The blood samples were collected following a 6-h
period of fasting. Aortic tissues were homogenized for further
biochemical analysis. Glycated hemoglobin (HbA1c) was detected
using the in2it A1C system (Bio-Rad Laboratories, Inc., Hercules,
CA, USA) as described previously (20). The levels of insulin, glucagon,
C-peptide, glutathione (GSH), superoxide dismutase (SOD) and
malondialdehyde (MDA) were determined using ELISA kits (Beyotime
Institute of Biotechnology, Beijing, China).
Measurement of endothelial relaxation
function of the thoracic aorta
The measurement of endothelial relaxation
functioning was conducted as previously described (3). In brief, the thoracic aorta was
immediately dissected and immersed in chilled Krebs-Henseleit
solution (Sigma-Aldrich; Merck KGaA) at 37°C and aerated with 95%
O2 and 5% CO2 (pH 7.4). The aortic rings were
then pre-contracted with U46619 (Sigma-Aldrich; Merck KGaA; 30 nM).
Endothelium-dependent and -independent vasodilation were determined
using ACh (Selleck Chemicals; 10−9-10−5 M)
and SNP (Selleck Chemicals; 10−10-10−6 M),
respectively.
Cell culture
Human umbilical vein endothelial cells (HUVECs) were
obtained from the National Infrastructure of Cell Line Resource
(Beijing, China; catalog no. 3111C0002000000024). The HUVECs
(1×105) were seeded on 6-well plates and cultured in
DMEM (high glucose; Gibco; Thermo Fisher Scientific, Inc.) with 10%
PBS (HyClone; GE Healthcare Life Sciences, Logan, UT, USA) at 37°C.
To activate mitochondrial fission, FCCP (5 µM, Selleck
Chemicals) was used for 2 h at 37°C prior to treatment. To suppress
and activate the JNK pathway, SP600125 (SP, 10 µM, Selleck
Chemicals) and anisomycin (Ani, 10 µM, Selleck Chemicals)
were used 2 h at 37°C prior to treatment, respectively. To inhibit
the F-action degradation, Jasplakinolide (2 µM; Selleck
Chemicals) was used 2 h before treatment at 37°C.
Construction of an adenovirus for the
overexpression of Sirt1
The Sirt1 adenovirus plasmids (ad-Sirt1) and control
adenovirus plasmids (ad-ctrl) were purchased from Vigene
Biosciences, Inc. (Rockville, MD, USA). The HUVEC cells
(0.5×106) were transfected with 20 nM ad-Sirt1 and
ad-ctrl with Lipofectamine® 2000 (Thermo Fisher
Scientific, Inc.) as described in a previous study (21). The expression of proteins in the
transfected cells were determined by western blot analysis.
ROS measurements
The cellular ROS was measured via dihydroethidium
(DHE, Invitrogen; Thermo Fisher Scientific, Inc.) staining and was
observed through a confocal microscope (Olympus Corporation, Tokyo,
Japan). DHE was alternately excited at the wavelengths of 300 and
535 nm following the manufacturer's protocol (22).
Mitochondrial fission analysis,
mitochondrial membrane potential (ΔΨm) measurements and mPTP
opening
Mitochondrial fission was analyzed according to a
previous study (4). The
mitochondrion was labelled with Tom20 (1:1,000, Abcam, Cambridge,
MA, USA; cat. no. ab56783) at room temperature (25°C) for 1 h and
the cell was observed under a confocal microscope. The ΔΨm was
measured using the JC-1 kit (Beyotime Institute of Biotechnology)
and the mPTP opening was measured as described previously (23). The images were analyzed using
ImageJ 1.47 version software (National Institutes of Health).
Cellular viability assay, terminal
deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)
staining and determination of caspase-9 activity
For the in vivo cellular viability assay, ECs
were isolated from the thoracic aorta as described previously
(24). Cellular viability was
quantitatively measured using an FITC Annexin V Apoptosis Detection
kit (BD Biosciences) (25). In
brief, the cells were incubated (1×105) with 5 µl
of FITC Annexin V and PI for 15 min at room temperature (25°C) in
the dark. The samples were then analyzed with a BD FACS-Calibur
cytometer (BD Biosciences).
A one-step TUNEL kit (Beyotime Institute of
Biotechnology) was used for TUNEL staining, as previously
described. Following treatment, the HUVECs were incubated with
fluorescein-dUTP (Invitrogen; Thermo Fisher Scientific, Inc.) to
stain apoptotic cell nuclei and with DAPI (5 mg/ml) to stain all
cell nuclei at room temperature for 3 min. Images of the slides
were captured under a confocal microscope with at least five random
separate fields. Cellular viability was also measured using an MTT
assay, according to the manufacturer's protocol. A caspase 9
activity kit (Beyotime Institute of Biotechnology) was used to
measure the activity of caspase 9, according to the manufacturer's
protocol (26).
Cell migration and wound healing
assay
Following treatment, the cells were seeded in 6-well
plates at a density of 0.5×106 cells/well. A wound track
was scored in each dish with a pipette head. Debris was removed by
rinsing the plates with PBS. Following culture for 24 h, the
migration distances were visualized and images were captured
(Olympus IX71; Olympus Corporation, Tokyo, Japan). Cell migration
was also analyzed using a Transwell chamber assay (24 wells with
8-µm pores and polycarbonate membranes), as previously
described (27).
Western blot analysis
Following treatment, the cells were lysed with
radioimmunoprecipitation assay buffer (Thermo Fisher Scientific,
Inc.) supplemented with phenylmethylsulfonyl fluoride. A
bicinchoninic acid protein assay was used to measure the protein
concentrations (28). The
proteins (50 µg) were separated by 10% SDS-PAGE and then
transferred onto polyvinylidene difluoride membranes. The membranes
were blocked with 5% nonfat milk for 1 h at room temperature (25°C)
and then incubated with primary antibodies overnight at 4°C. Then,
the membranes were washed 3 times with PBS and incubated with
secondary antibodies at room temperature (25°C) for 1 h. The
following antibodies were used: Caspase 3 (1:2,000, CST Biological
Reagents Co., Ltd., Shanghai, China; cat. no. 9662), caspase 9
(1:2,000, CST Biological Reagents Co., Ltd., cat. no. 9508), Bcl-2
(1:2,000, CST Biological Reagents Co., Ltd., cat. no. 3498),
X-linked inhibitor of apoptosis (x-IAP; 1:1,000, CST Biological
Reagents Co., Ltd., cat. no. 2042), phosphorylated (p)-JNK
(1:1,000, CST Biological Reagents Co., Ltd., cat. no. 9255), JNK
(1:1,000, CST Biological Reagents Co., Ltd., cat. no. 9252), Sirt1
(1:1,000, Abcam, cat. no. ab19A7AB4), dynamin-1-like protein (Drp1;
1:1,000, Abcam, cat. no. ab56788), mitochondrial fission 1 protein
(Fis1; 1:1,000, Abcam, cat. no. ab71498), mitofusin-(Mfn)2
(1:1,000, Abcam, cat. no. ab56889), Mfn1 (1:1,000, Abcam, cat. no.
ab57602), Mff (1:1,000, CST Biological Reagents Co., Ltd., cat. no.
86668), F-actin (1:1,000, Abcam, cat. no. ab205), G-actin (1:1,000,
Abcam, cat. no. ab200046), p-endothelial nitric oxide synthase
(eNOS; Ser1117; 1:1,000, Abcam, cat. no. ab184154), eNOS (1:1,000,
Abcam, cat. no. ab76198), Bad (1:1,000, Abcam, cat. no. ab32445),
Bax (1:2,000, Abcam, cat. no. ab32503), poly (ADP-ribose)
polymerase (1:2,000, Abcam, cat. no. ab32064), horseradish
peroxidase (HRP)-conjugated anti-mouse immunoglobulin (Ig)-G
(1:1,000; CST Biological Reagents Co., Ltd.; cat. no. 7076) and
HRP-conjugated anti-rabbit IgG (1:1,000; CST Biological Reagents
Co., Ltd.; cat. no. 7074). The blots were detected with an enhanced
chemiluminescence substrate kit (Thermo Fisher Scientific, Inc.),
and band intensity levels were analyzed using Quantity One 4.6
software (Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
RT-qPCR was performed according to the
manufacturer's instructions. Briefly, total RNA was extracted from
the cells using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and reverse transcribed with a the TaqMan
MicroRNA Reverse Transcription kit (Takara Bio, Inc., Otsu, Japan)
at 37°C for 30 min according to the manufacturer's instructions.
qPCR was performed using the SYBR-Green RT-PCR kit (Takara Bio,
Inc.). The following primers were used for polymerase chain
reaction: Sirt1, forward, 5′-GAGAGACGTCTGGTAGATCG-3′ and reverse
5′-GTGCCAGCATGTGTCGTAGT-3′; intercellular adhesion molecule 1
(ICAM-1), forward 5′-GAGACGCAGAGGACCTTAACAG-3′ and reverse
5′-GACGCCGCTCAGAAGAACCA-3′; CRP, forward 5′-GGATGGATTGCACAGCCATT-3′
and reverse 5′-GCGCCGACTCAGAGGTGT-3′; tumor necrosis factor (TNF)α,
forward 5′-AGATGGAGCAACCTAAGGTC-3′ and reverse
5′-GCAGACCTCGCTGTTCTAGC-3′; interleukin (IL)6, forward
5′-CAGACTCGCGCCTCTAAGGAGT-3′ and reverse 5′-GATAGCCGATCCGTCGAA-3′;
miR-195, forward 5′-UAGCAGCACAGAAAUAUUGGC-3′ and reverse
5′-CAAUAUUUCUGUGCUGCUAUU-3′; U6, forward 5′-CTCGCTTCGGCAGCACA-3′
and reverse 5′-AACGCTTCACGAATTTGCGT-3′; and GAPDH, forward
5′-AATGGTGAAGGTCGGTGTG-3′ and reverse
5′-GTGGAGTCATACTGGAACATGTAG-3′. The thermocycling conditions were
as follows: 95°C for 5 min, followed by 40 cycles at 95°C for 40
sec, 60°C for 30 sec and 72°C for 30 sec. U6 and GAPDH were
selected as internal controls for micro (mi)RNA and mRNA,
respectively (23,29). Fold-changes for mRNA expressions
were calculated using the 2−ΔΔCq method (30,31).
Transfection
The miR-195 mimics (agmir-195, forward
5′-UAGCAGCACAGAAAUAUUGGC-3′ and reverse
5′-CAAUAUUUCUGUGCUGCUAUU-3′; miR-Ctrl, forward
5′-UUCUCCGAACGUGUCACGU-3′ and reverse 5′-ACGUGACACGUUCGGAGAA-3′)
and miR-195 inhibitor (antagomir; 5′-GCCAAUAUUUCUGUGCUGCUA-3′) were
purchased from GenePharma Co., Ltd. (Shanghai, China). The HUVEC
cells (0.5×106) were transfected with 20 nM miR-195
mimic, miR-195 inhibitor and miR negative control (NC) with
Lipofectamine™ RNAiMAX (Thermo Fisher Scientific, Inc.) according
to the manufacturer's instructions (32). Transfection was performed for 48 h
and the transfection efficiency was evaluated by RT-qPCR.
Immunohistochemistry and
immunofluorescence staining
The change in expression of Sirt1 in the thoracic
aorta was measured via immunohistochemistry according to a previous
study (33,34). Immunofluorescence staining was
used to measure cytochrome-c (cyt-c) localization,
F/G-actin, and mitochondrial fission. Following treatment, the
cells were fixed with 3.7% paraformaldehyde for 10 min at room
temperature. Following blocking with 5% bovine serum albumin
(Sigma-Aldrich; Merck KGaA) in PBS for 1 h at room temperature, the
cells were incubated with primary antibodies for 4 h at room
temperature. The secondary antibodies were incubated at room
temperature for 1 h in the dark. Images were captured using a laser
confocal microscope (TcS SP5; Leica Microsystems, Inc., Buffalo
Grove, IL, USA). The primary antibodies, Sirt1 (1:500, Abcam, cat.
no. ab19A7AB4), cyt-c (1:500, Abcam, cat. no. ab90529), translocase
of outer mitochondrial membrane 20 (1:500, Abcam, cat. no. ab56783)
and F-actin (1:500, Abcam, cat. no. ab205) were used. The Alexa
Fluor® secondary antibodies, anti-mouse IgG (1:500; cat.
no. 4408; green) and anti-rabbit IgG (1:500; cat. no. 4412; green),
were purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA). DAPI (5 mg/ml; Sigma-Aldrich; Merck KGaA) was used to stain
the nucleus at room temperature for 3 min.
Luciferase activity assay
The wild-type Sirt1 3′-UTR (WT) and mutant Sirt1
3′-UTR (MUT) containing the putative binding site of miR-195 were
chemically synthesized and cloned downstream from the firefly
luciferase gene in a pGL3-promoter vector (Promega Corporation,
Madison, WI, USA). The luciferase plasmids and miR-195 or control
miRNA were co-transfected in HUVECs (0.5×106) in DMEM
supplemented with 10% FBS using Lipofectamine® 2000 at
37°C according to the manufacturer's instructions. pRL
Renilla control reporter vectors (Promega Corporation) were
used as an internal control to normalize the values of the
experimental reporter gene. Following 48 h of transfection, the
intensities were measured with a Luciferase Reporter Assay system
(Promega Corporation).
Statistical analysis
All analyses were performed with SPSS 20.0 software
(IBM Corp., Armonk, NY, USA). All experiments were repeated three
times. Corresponding data are presented as the mean ± standard
deviation, and the statistical significance of each variable was
estimated by a one-way analysis of variance followed by Tukey's
test for post hoc analysis. P<0.05 was considered to indicate a
statistically significant difference.
Results
Sirt1 attenuates glucose metabolic
abnormalities in diabetic mice
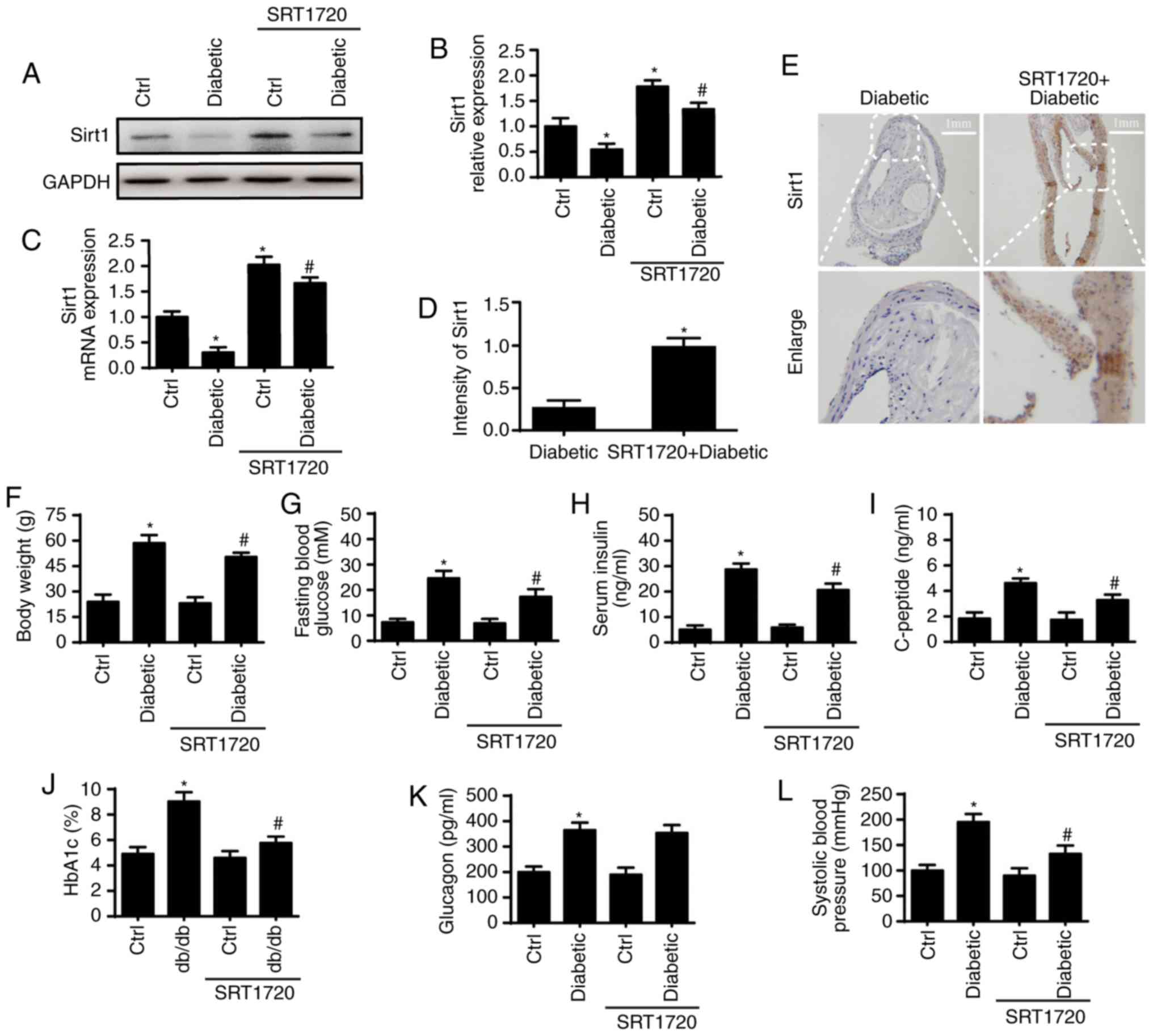
First, the expression of Sirt1 in the aorta of
diabetic and non-diabetic mice was analyzed via western blot and
qPCR analyses (Fig. 1A-C). The
results showed that the expression of Sirt1 was significantly
reduced at the protein and mRNA levels in the diabetic mice. A
previous study showed that the loss of Sirt1 contributed to glucose
metabolic abnormalities in diabetics (35). To examine the role of Sirt1 in the
glucose metabolic activities observed in diabetic mice, SRT1720, an
activator of Sirt1, was used to reactivate Sirt1 in the diabetic
mice. SRT1720 reversed the downregulation of Sirt1 in diabetic
mice, as indicated by the western blot (Fig. 1A and B), qPCR (Fig. 1C) and immunohistochemical
(Fig. 1D and E) analyses. The
effects of Sirt1 on the glucose metabolic activities observed in
diabetic mice were then evaluated. As is shown in Fig. 1F-K, diabetic mice exhibited higher
body weights, fasting blood glucose levels, serum insulin levels,
serum-peptide levels, glycosylated hemoglobin A1c (HbA1c) levels
and systolic blood pressure (SBP). As expected, the activation of
Sirt1 significantly reduced or abrogated the above changes.
However, the high levels of glucagon observed in diabetic mice were
not altered by SRT1720 (Fig. 1L).
These data suggest that the reduced expression of Sirt1 contributes
to the glucose metabolic abnormalities observed in diabetic
mice.
Sirt1 activation ameliorates aortic
endothelial dysfunction in diabetic mice
Endothelial dysfunction is an early marker of
chronic HG injury. Endothelial functioning was measured 4 weeks
following SRT1720 treatment in mice. As is shown in Fig. 2A, diabetes inhibited the
endothelial-dependent vasodilation in response to Ach, which was
reversed by SRT1720. By contrast, no significant difference in
endothelial-independent vasodilation levels were observed among the
four groups. To examine the underlying mechanism involved,
fundamental factors, including eNOS phosphorylation (Ser1177),
intercellular adhesion molecules, inflammation and oxidative
stress, were analyzed. Diabetes decreased the protein levels of
p-eNOS (Fig. 2B) and increased
the mRNA levels of ICAM1 and VCAM1 (Fig. 2C and D), and this effect was
nullified by SRT1720. SRT1720 also decreased the mRNA levels of
pro-inflammatory TNF-α, IL-6 and CRP in the diabetic thoracic
aortas (Fig. 2E-G). In addition,
diabetic mice generated more MDA and consumed more antioxidant
factors, including GSH/SOD, and this effect was reversed by SRT1720
(Fig. 2H-J). Together, these data
demonstrate that the activation of Sirt1 reduces aortic endothelial
dysfunction in diabetic mice.
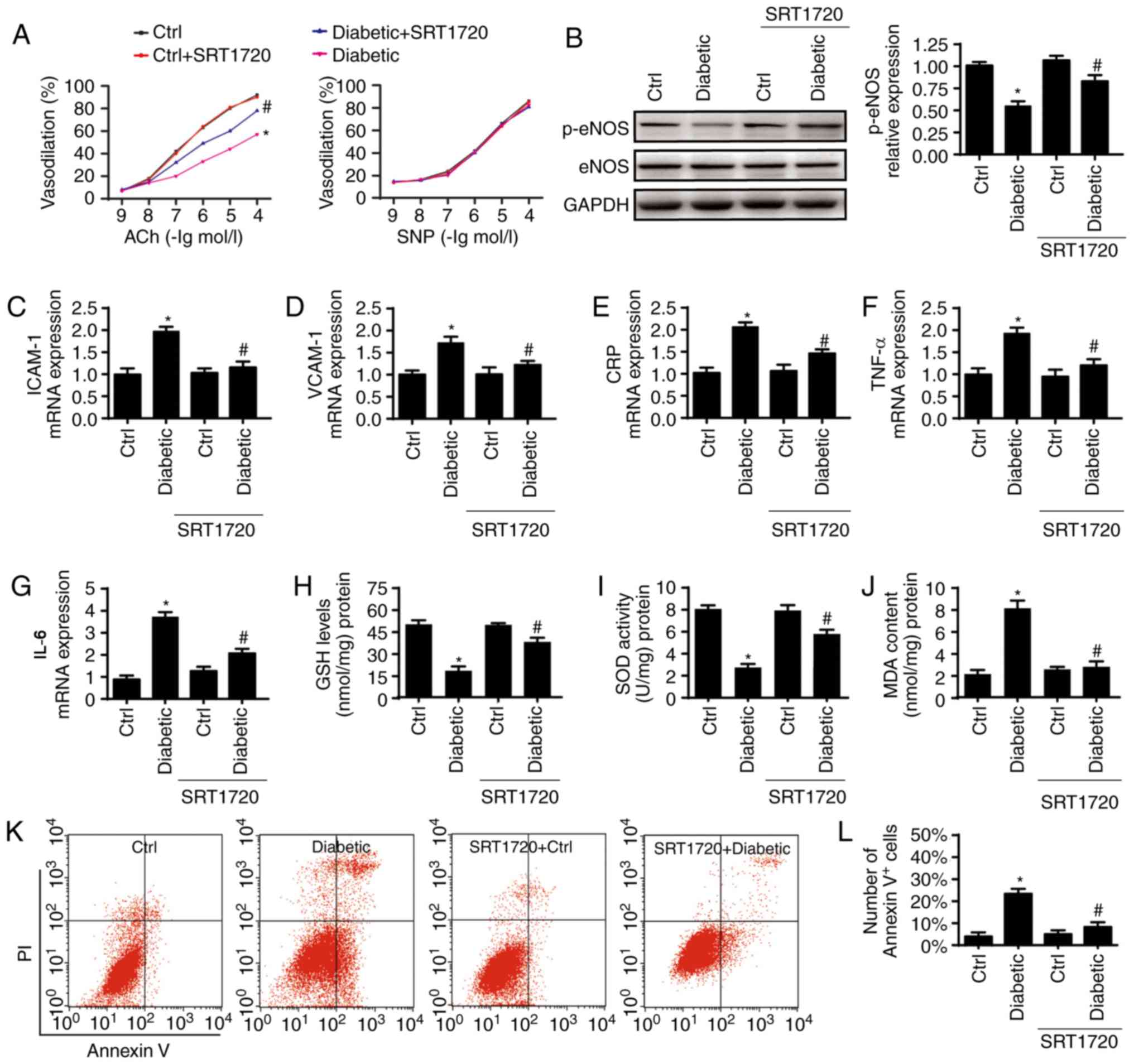 | Figure 2Sirt1 activation ameliorates aortic
endothelial dysfunction. (A) Endothelium-dependent and
endothelium-independent vasodilation to ACh or SNP. (B) Western
blot analysis was used to measure the expression of p-eNOS and
e-NOS. SRT1720 reduced the expression of (C) ICAM1 and (D) VCAM1,
which were enhanced in diabetics. Quantitative analysis of the mRNA
expression of pro-inflammatory factors (E) CRP, (F) TNF-α and (G)
IL-6 and in diabetic thoracic aortas. Diabetics consumed
anti-oxidant factors, including (H) GSH and (I) SOD and generated
more (J) MDA; these effects we reversed by SRT1720. (K) Effects of
Sirt1 on endothelial apoptosis were detected via flow cytometry
with Annexin V/PI staining and (L) quantified. (n=6/group)
*P<0.05, vs. Ctrl; #P<0.05, vs. SRT1720
+ Diabetic. Sirt1, sirtuin 1; Ctrl, control; Ach, acetylcholine;
SNP, sodium nitro-prusside; eNOS, endothelial nitric oxide
synthase; p-, phosphorylated; ICAM1, intercellular adhesion
molecule 1; VCAM1; TNF-α, tumor necrosis factor; IL-6, interleukin
6; CRP, C-reactive protein; GSH, glutathione; SOD, superoxide
dismutase; MDA, malondialdehyde. |
Downregulated Sirt1 promotes HG-mediated
endothelial cell death
The apoptosis of endothelial cells has been reported
to serve a critical role in HG-induced diabetic vasculopathy
(36,37). Therefore, the present study
measured endothelial cell apoptosis in vivo and in
vitro. Annexin V/PI flow cytometry was used to qualify
endothelial cell apoptosis in cells extracted from thoracic aortas.
Relative to the control group, the diabetic group exhibited clearly
elevated ratios of Annexin V+ cells, and this effect was
reduced by SRT1720 (Fig. 2K and
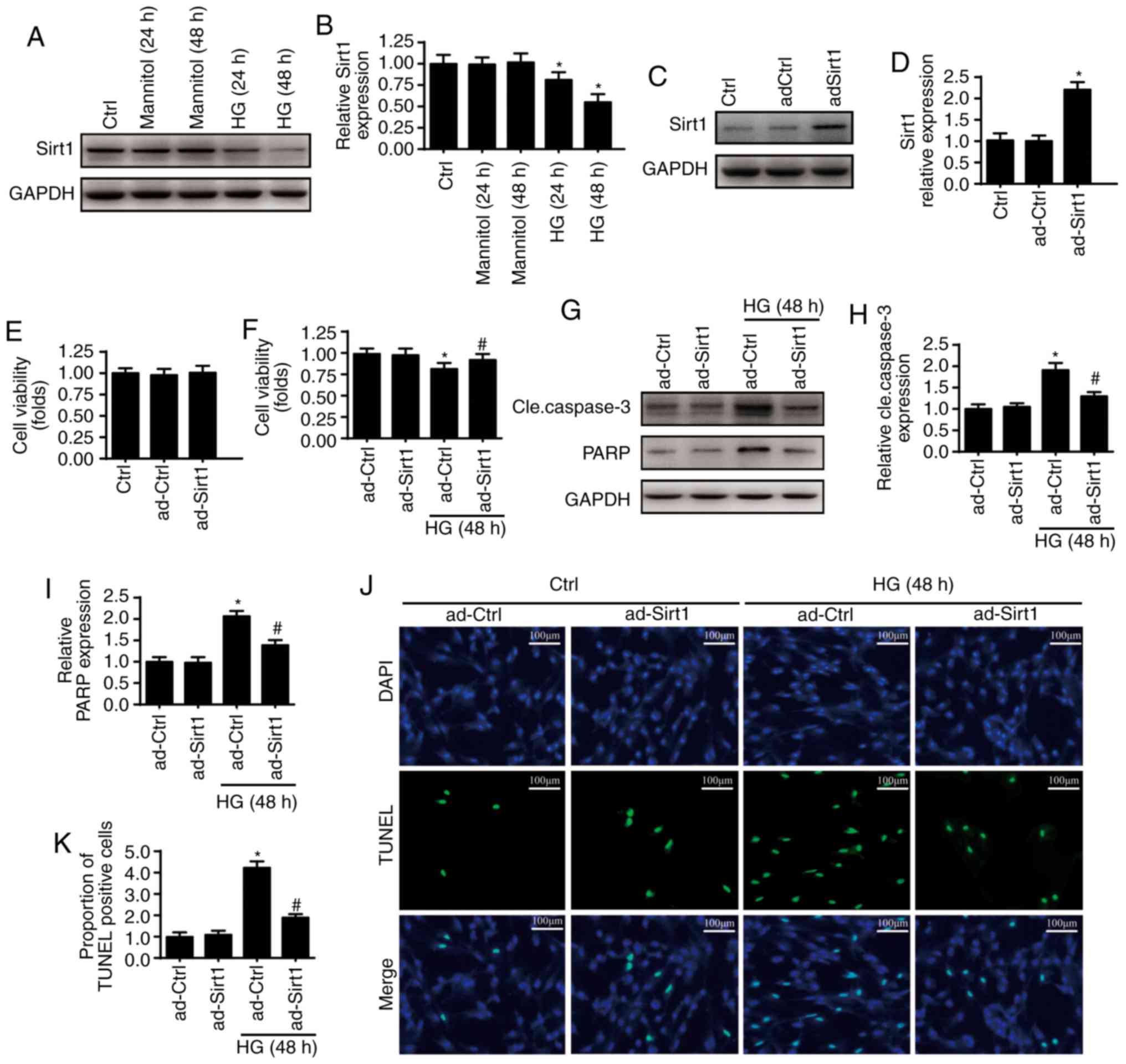
L). From the in vitro experiments, alterations of Sirt1
were observed in HUVECs prior to and following HG (30 mmol/l)
treatment. The expression of Sirt1 was downregulated following HG
treatment (Fig. 3A and B), with
the lowest expression levels of Sirt1 following 48 h of HG
treatment. Therefore, 48 h of HG treatment was used in the
following experiments. A gain-of function assay on Sirt1 was
performed via adenovirus vector transfection (ad-Sirt1) and the
transfection efficiency levels were confirmed via western blotting
(Fig. 3C and D). The
overexpression of Sirt1 had no influence on cellular viability
(Fig. 3E). However, the regaining
of Sirt1 reduced the occurrence of HG-induced cellular death
(Fig. 3F) as indicated by the
expression of cleaved caspase 3 and its substrate, which are
indicators of cellular apoptosis (Fig. 3G-I). To further determine whether
Sirt1 promotes cellular survival, TUNEL staining was performed
(Fig. 3K). The overexpression of
Sirt1 also reduced the percentage of TUNEL-positive cells present.
These data indicate that Sirt1 is an anti-apoptotic factor of
HUVECs under the treatment of HG.
Sirt1 deficiency triggers endothelial
death by activating mitochondria-dependent apoptotic pathways
To describe the protective role of Sirt1 in
endothelial apoptosis under HG, the present study focused on
mitochondria-dependent cellular apoptosis occurring through
cellular ROS release, mitochondrial potential collapse, mPTP
opening and mitochondrial pro-apoptotic factor leakage. ROS
alterations were analyzed through DHE staining. Compared with the
control group, HG increased the cellular ROS content levels, which
were reduced by the overexpression of Sirt1 (Fig. 4A). HG also induced higher
expression levels of Bax, Bad and Caspase 9 but lower expression
levels of Bcl-2 and x-IAP, suggesting the activation of
mitochondria-related apoptotic pathways (Fig. 4B-G). However, the overexpression
of Sirt1 inhibited the pro-apoptotic effects of HG. A JC-1 assay
was used to measure the mitochondrial electrochemical potential
(ΔΨm) (Fig. 4H and I). HG also
impaired ΔΨm, with evidence of decreased levels of red fluorescence
and increased levels of green fluorescence. However, the
overexpression of Sirt1 reversed the stability of ΔΨm. Furthermore,
the rate of mPTP opening was increased by HG but decreased by the
overexpression of Sirt1 (Fig.
4J). Following the dissipated ΔΨm and long-lasting mPTP
opening, HG increased the leakage of cyt-c from the mitochondria
into the cytoplasm, and cyt-c was even observed to migrate into the
nucleus (Fig. 4K), whereas an
increase in Sirt1 limited cyt-c leakage. These results showed that
Sirt1 prevents endothelial apoptosis by inhibiting the
mitochondria-dependent apoptotic pathway.
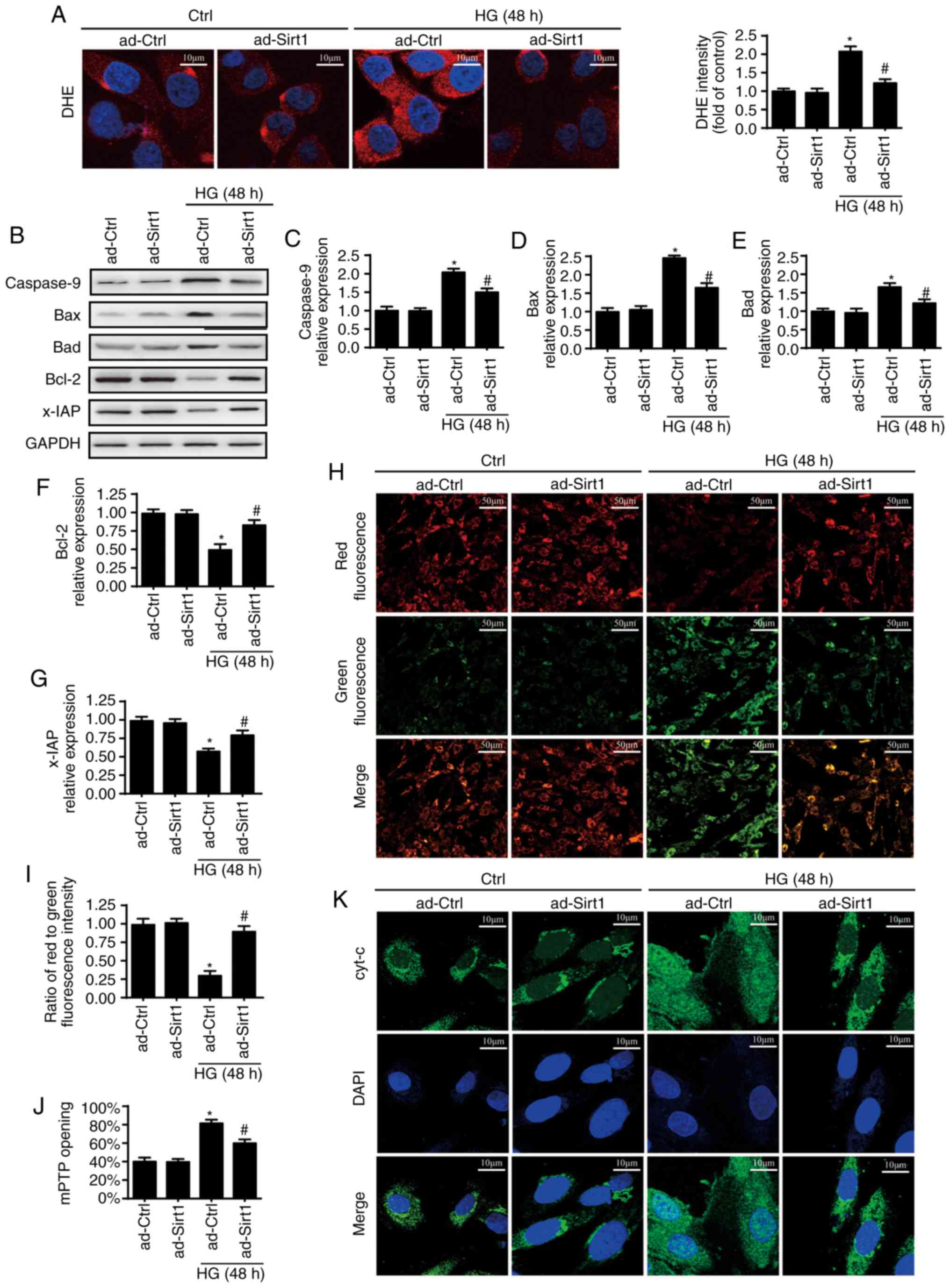 | Figure 4Sirt1 inhibits HG-induced apoptosis
associated via a mitochondria-dependent apoptotic pathway. (A)
Cellular ROS was measured by DHE staining. Scale bars, 10
µm. (B) Western blot analysis was used to measure expression
levels of (C) Caspase-9, (D) Bax, (E) Bad, (F) Bcl-2 and (G) x-IAP.
(H) Change in ΔΨm was determined by JC-1 (scale bars, 50 µm)
and (I) quantified. (J) mPTP opening. (K) Immunostaining of cyt-c
leakage from mitochondria into the cytoplasm; scale bars, 10
µm. *P<0.05, vs. ad-Ctrl;
#P<0.05, vs. HG + ad-Ctrl. Sirt1, sirtuin 1; HG, high
glucose; Ctrl, control; ROS, reactive oxygen species; x-IAP,
X-linked inhibitor of apoptosis; ΔΨm, mitochondrial electrochemical
potential; DHE, dihydroethidium; cyt-c, cytochrome-c; mPTP,
mitochondrial permeability transition pore. |
Mitochondrial fission is involved in
Sirt1 deficiency-mediated mitochondrial apoptosis under HG
treatment
Previous studies have reported that mitochondrial
fission serves a critical role in mitochondria-related apoptosis
(28,38,39). Therefore, the present study
examined whether Sirt1 prevents endothelial apoptosis through the
regulation of mitochondrial fission. As is shown in Fig. 5A, HG led to a markedly larger
volume of fragmented mitochondria in the Ad-ctrl group. However,
upregulating Sirt1 repressed the effects of HG on mitochondrial
fragments. Mitochondrial lengths were also recorded (Fig. 5B) to quantify the fission levels,
and the results were in accordance with the above findings. These
results indicate the role of Sirt1 in activating mitochondrial
fission. To examine whether mitochondria are responsible for
endothelial apoptosis, FCCP, a mitochondrial fission activator, was
used to reactivate mitochondrial fission in Ad-Sirt1 cells
(Fig. 5A and B). The protective
role of Sirt1 in mPTP opening and cellular apoptosis disappeared
following the re-activation of mitochondrial fission in ad-Sirt1
cells (Fig. 5C-E). These results
indicate that mitochondrial fission is responsible for Sirt1
deficiency-mediated mitochondrial apoptosis under HG treatment.
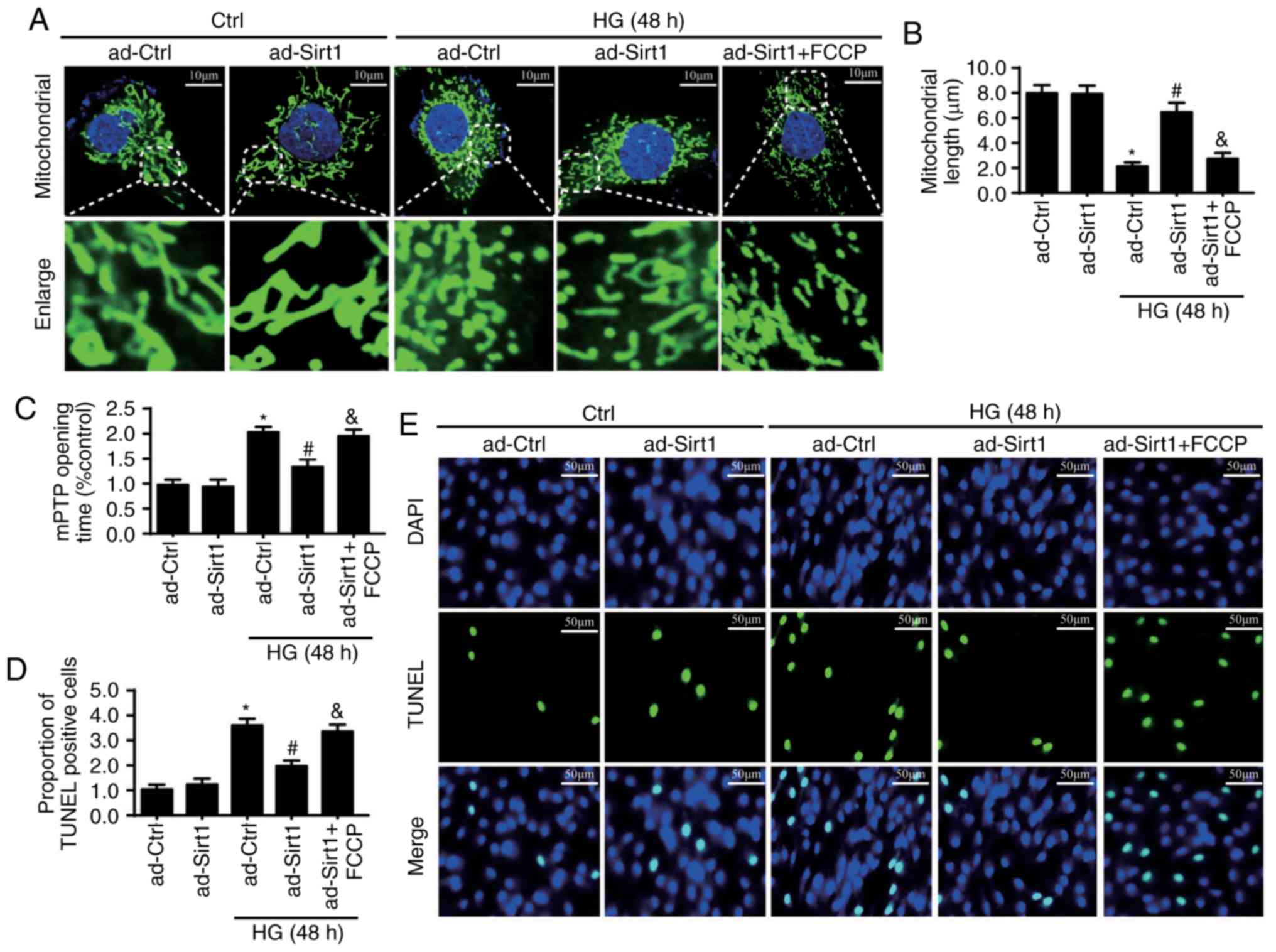 | Figure 5Sirt1 loss promotes cellular
apoptosis by activating mitochondrial fission. (A) Change in
mitochondrial morphology observed with Tom20 staining. Images shown
under each micrograph amplify mitochondrial fragments. FCCP, a
mitochondrial fission activator, was used to reactivate
mitochondrial fission in Ad-Sirt1 cells (scale bars, 10 µm).
(B) Mitochondrial lengths were also recorded for fission
quantification. (C) Protective role of the overexpression pf Sirt1
in mPTP opening time disappeared in ad-Sirt1 cells following the
application of FCCP. (D) Cellular apoptosis was measured by (E)
TUNEL staining (scale bars, 50 µm). *P<0.05
vs. ad-Ctrl; #P<0.05 vs. HG+ad-Ctrl;
&P<0.05 vs. HG+ad-Sirt1. Sirt1, sirtuin 1; HG,
high glucose; Ctrl, control; mPTP, mitochondrial permeability
transition pore; TUNEL, TUNEL, terminal deoxynucleotidyl
transferase dUTP nick end labeling. |
Sirt1 regulates mitochondrial fission by
activating the JNK/Mff pathway
To determine the means through which Sirt1 regulates
mitochondrial fission, changes in Drp1 and in its receptors (Mff,
Fis1, Mid49 and Mid51) required to regulate mitochondrial fission
were measured. It was first found that HG mainly enhanced the
expression of Mff in ad-ctrl cells (Fig. 6A and B) and that the
overexpression of Sirt1 reduced the expression of Mff rather than
other receptors. Furthermore, HG treatment enhanced the expression
of Drp1, which was reversed by the overexpression of Sirt1
(Fig. 6C).
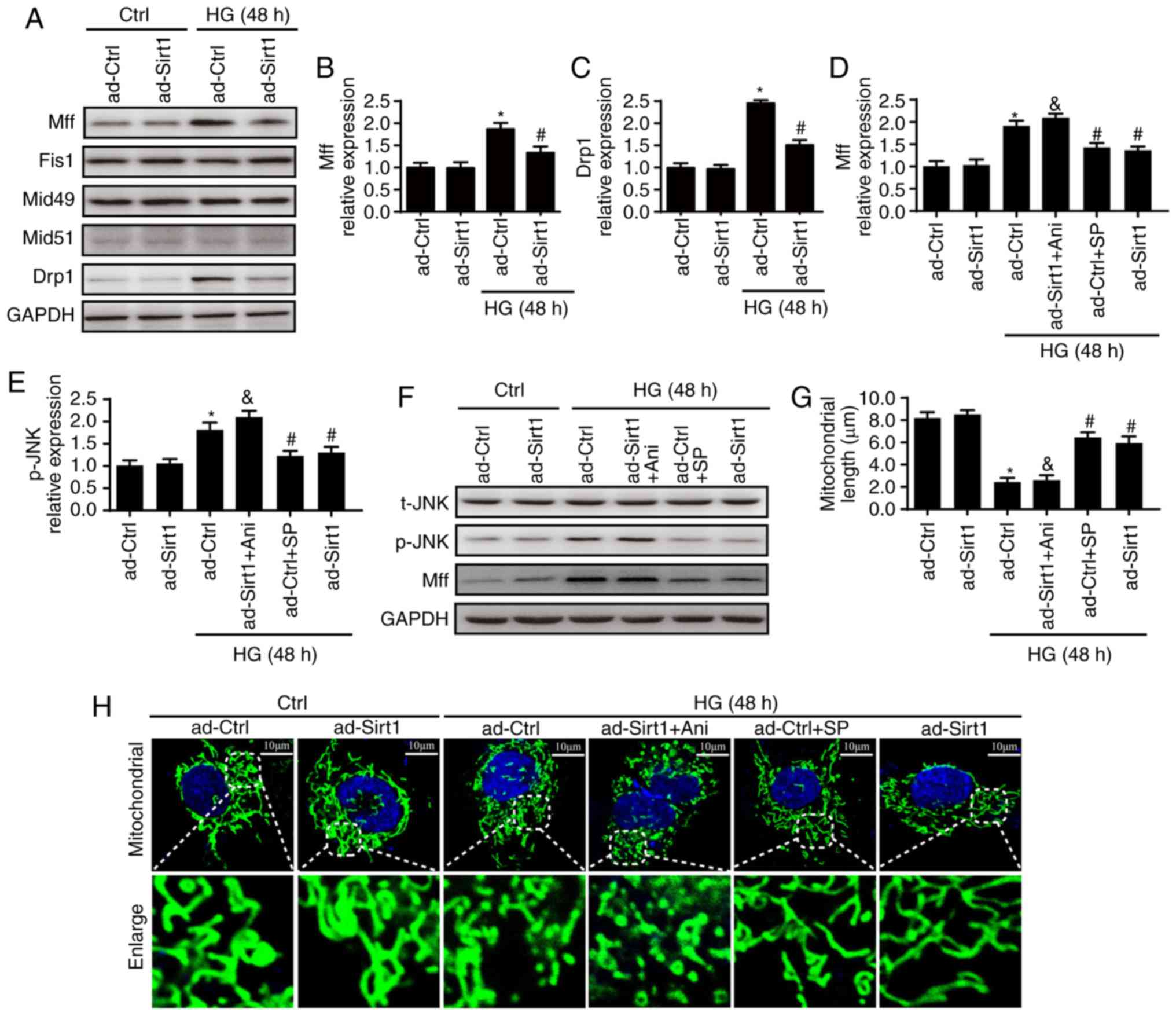 | Figure 6Sirt1 regulates mitochondrial fission
by activating the JNK/Mff signaling pathway. (A) Changes in
mitochondrial fission receptors and Drp1. (B) Changes in Mff; (C)
changes in Drp1. (D) Changes in Mff and (E) p-JNK following with
Ani or SP treatment. (F) Blots showing expression of JNK and Mff.
Sirt1 inactivated Mff via JNK. To examine the mechanism by which
Sirt1 inactivates Mff, SP, an inhibitor of JNK, was added to
ad-Ctrl cells to inhibit the JNK pathway. Ani, an activator of the
JNK pathway, was also added to ad-Sirt1 cells. (G) Changes in
mitochondrial length. (H) Changes in observed mitochondrial
morphologies. Sirt1 inactivated mitochondrial fission via the JNK
pathway (scale bars, 10 µm). *P<0.05 vs.
ad-Ctrl; #P<0.05 vs. HG + ad-Ctrl;
&P<0.05 vs. HG + ad-Sirt1. Mff, mitochondrial
fission factor; Drp1, dynamin-1-like protein; Fis1, mitochondrial
fission 1 protein; JNK, c-Jun N-terminal kinase; t-, total; p-,
phosphorylated; SP, SP600125; Ani, anisomycin; Sirt1, sirtuin 1;
HG, high glucose; Ctrl, control. |
Several studies have confirmed that the activity of
Mff is regulated by the JNK pathway (28,40,41). Therefore, the present study
examined the JNK pathway to examine the mechanism by which Sirt1
inactivates Mff. Relative to the control group, JNK was activated
by HG, as indicated by an increase observed in the expression of
p-JNK (Fig. 6D-F). This tendency
was reversed by the overexpression of Sirt1. To examine the role of
the activation of JNK in Sirt1-induced mitochondrial fission in the
present study, SP, an inhibitor of JNK, was added to ad-Ctrl cells
to inhibit the JNK pathway. Ani, an activator of the JNK pathway,
was also used in ad-Sirt1 cells. Through western botting, it was
found that the JNK pathway was triggered by HG treatment or Ani,
but was inhibited by the overexpression of Sirt1 or SP (Fig. 6D-F). The expression of Mff and
mitochondrial fission were enhanced following HG treatment, but
decreased to normal levels upon the overexpression of Sirt1 or SP
treatment (Fig. 6D-F). The
reactivation of JNK in ad-Sirt1 cells eliminated the inhibition of
Mff activation and the protective effects on mitochondrial fission
induced by the overexpression of Sirt1 (Fig. 6G and H). This experiment suggests
that Sirt1 regulates mitochondrial fission through the JNK
pathway.
Overexpression of Sirt1 maintains
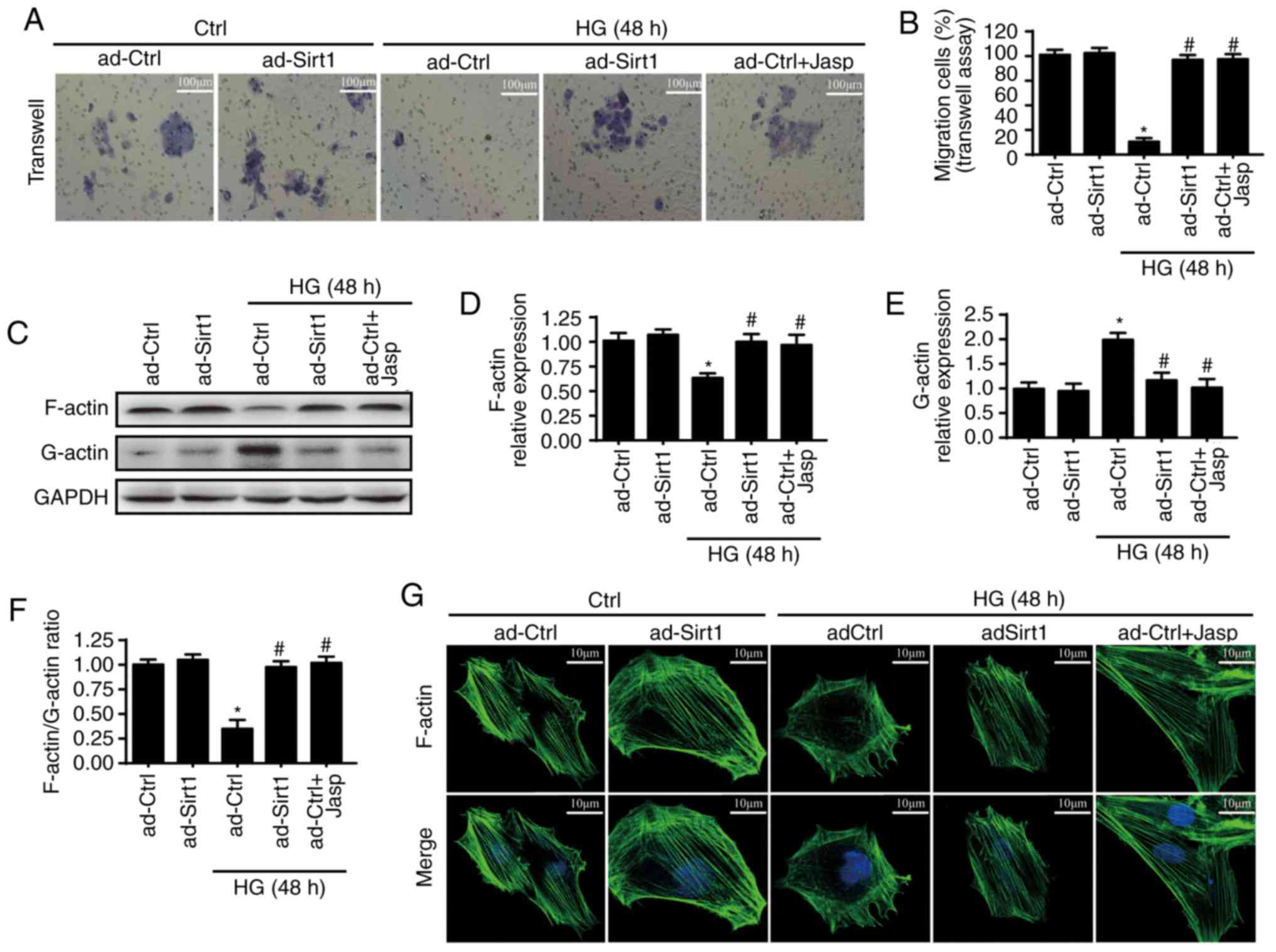
endothelial migration by sustaining F-actin homeostasis
Endothelial cell mobilization capacities are
critical for the repair of endothelial injury. Therefore, whether
Sirt1 is involved in cellular migration was measured. First, a
Transwell assay was used to investigate associations between HUVEC
migration and Sirt1. As is shown in Fig. 7A and B, HG treatment reduced the
migration of HUVECs, whereas the overexpression of Sirt1 prevented
this change from occurring. These results suggest that Sirt1 is
involved in cellular migration. F-actin has been reported to serve
a critical role in cellular mobilization. Therefore, the present
study examined whether Sir1 regulates cellular migration through
F-actin dyshomeostasis. Jasplakinolide, an F-actin depolymerization
inhibitor, was used to inhibit F-actin depolymerization in ad-Ctrl
cells. As F-actin is composed of G-actin, western blotting was
performed to measure the expression of F-actin and G-actin
following HG treatment. As is shown in Fig. 7C-F, HG treatment clearly reduced
the expression of F-actin and enhanced the expression of G-actin,
which was reversed by the overexpression of Sirt1 or jasplakinolide
application. Similar results for F-actin were observed by
fluorescence (Fig. 7G).
Jasplakinolide and the overexpression of Sirt1 significantly
enhanced the fluorescence intensity of F-actin, which was reduced
by HG. Jasplakinolide also promoted cellular migration under HG
treatment, similar to the overexpression of Sirt1 (Fig. 7A and B). These results suggested
that Sirt1 regulates cellular migration by inhibiting F-actin
depolymerization into G-actin under HG treatment.
Sirt1 is negatively regulated by miR-195
via hyperglycemic stimulus
To examine the mechanism by which Sirt1 is
downregulated through diabetic vasculopathy, miR-195 was examined.
It was found that the expression of miR-195 was significantly
enhanced in ad-ctrl cells under HG treatment (Fig. 8A). To elucidate the mechanism by
which HG down-regulates Sirt1, a mimic (agmir-195) and inhibitor
(antagomir) of miR-195 were used, and the transfection efficiency
was evaluated by RT-qPCR analysis (Fig. 8B). As is shown in Fig. 8C, agmir-195 reduced the expression
of Sirt1, echoing the results derived following HG treatment.
However, inhibitors of miR-195 reversed the downregulated level of
Sirt1 under treatment with HG. These results suggest that miR-195
negatively regulates Sirt1 under HG. Furthermore, as shown in
Fig. 8D, miR-195 was matched to
Sirt1. To examine the direct binding of miR-195 with the 3′UTR of
Sirt1, luciferase assays were used by co-transfecting miR-195 mimic
with luciferase reporter plasmids with cloned miR-195 binding sites
for WT Sirt1-3′UTR or MUT Sirt1-3′UTR (Fig. 8E). Compared with that of the MUT
type, luciferase activity was downregulated in cells co-transfected
with miR-195 mimics and in the WT Sirt1-3′UTR, suggesting that
Sirt1 is a target gene of miR-195. To provide more evidence, the
effects of miR-195 on mitochondrial fission, cellular apoptosis and
migration were analyzed. It was found that agmir-195 promoted
mitochondrial fission, enhanced the expression of caspase-9 and
inhibited cellular migration, consistent with the effects of HG
treatment (Fig. 8F-I). However,
the inhibition of miR-195 inhibited the HG-induced mitochondrial
fission, upregulation of caspase-9 and inhibited migration. These
results support the hypothesis that HG represses the expression of
Sirt1 by upregulating miR-195.
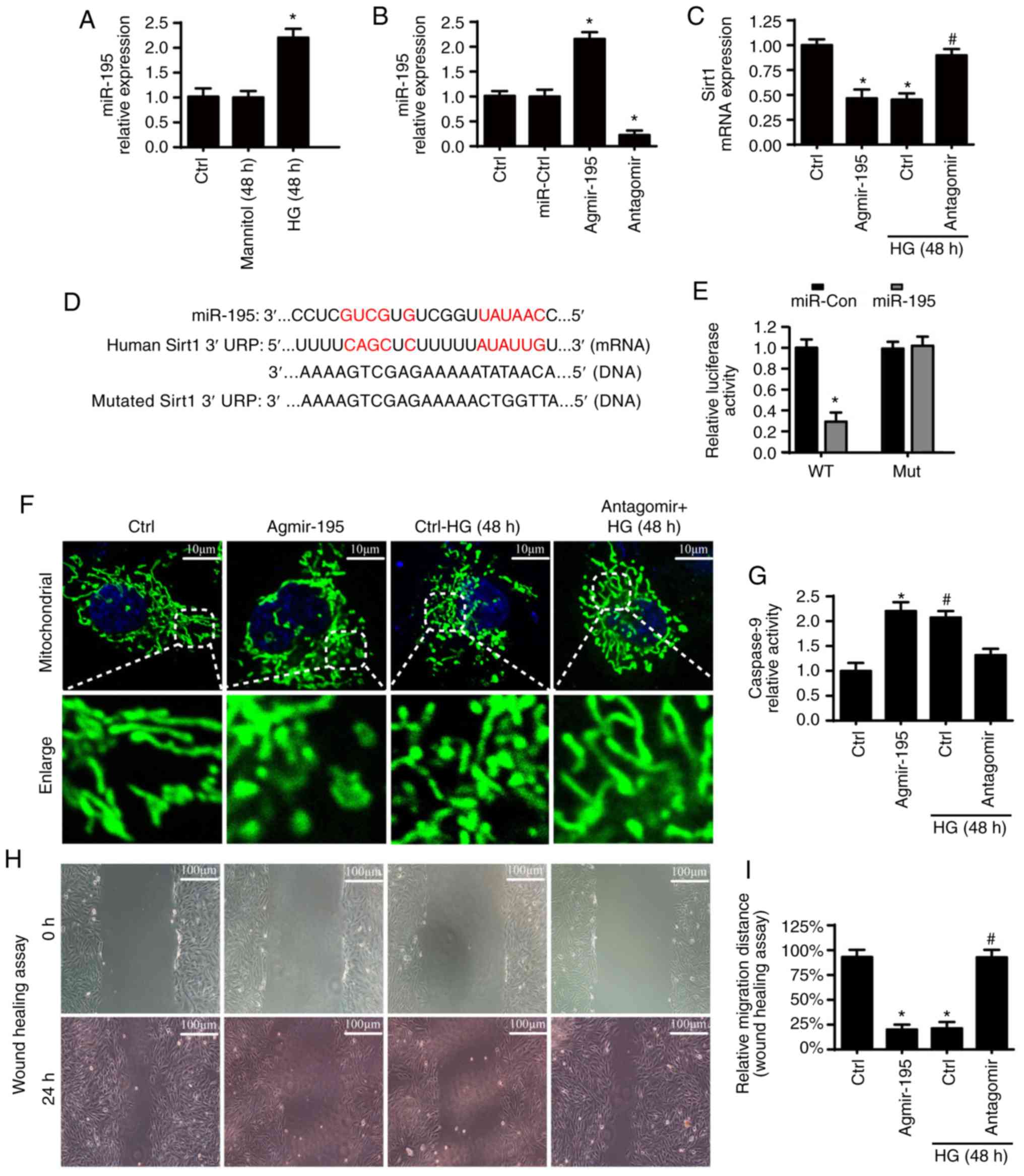 | Figure 8Sirt1 is negatively regulated by
miR-195. (A) Changes in miR-195 were detected by qPCR analysis.
miR-195 levels were significantly increased in ad-ctrl cells under
HG. (B) Transfection efficiency was evaluated by reverse
transcription-qPCR analysis. (C) mRNA expression of Sirt1. To
examine the mechanism by which HG downregulates Sirt1, a mimic
(agmir-195) and inhibitor (antagomir) of miR-195 were used. (D)
Alignment of the WT Sirt1-3′UTR (and MUT Sirt1-3′UTR) sequence with
mature miR-195 based on bioinformatics predictions. (E) Luciferase
assay for the post-transcriptional repression of Sirt1. (F) Change
of mitochondrial fragments or debris measured via Tom20
immunofluorescence (scale bars, 10 µm). (G) Caspase-9
relative activity. (H and I) Influence of miR-195 on the cellular
capacities of migration was measured by performing a wound healing
assay in vitro (scale bars, 100 µm).
*P<0.05, vs. ad-Ctrl; #P<0.05, vs. HG +
ad-Ctrl. miR, microRNA; qPCR, quantitative polymerase chain
reaction; Sirt1. Sirt1, sirtuin 1; HG, high glucose; Ctrl, control;
WT, wild-type; MUT, mutated. |
Discussion
Vascular endothelial injury caused by a
hyperglycemic environment serves an important role in the
occurrence and development of diabetic vasculopathy. Accumulating
evidence demonstrates the critical role of Sirt1 in HG-induced
endothelial injury. The present study confirmed that i) HG triggers
the downregulation of Sirt1 by activating miR-195; ii) reduced
expression of Sirt1 contributes to glucose metabolic abnormalities,
aortic endothelial dysfunction and EC apoptosis; iii) downregulated
Sirt1 triggers EC apoptosis by activating mitochondrial fission;
iv) mechanically, the loss of Sirt1 triggers mitochondrial fission
by evoking the JNK/Mff pathway; v) Sirt1 deficiency limits EC
migration through F-actin dyshomeostasis. These results further
enrich current understanding of the molecular pathways of
Sirt1-mediated endothelial protection in diabetic vasculopathy.
Sirt1 is related to cellular metabolism, migration,
senescence and survival, and is implicated in the pathophysiology
of diabetes, neurodegenerative disorders and cardiovascular disease
(9,42). In the present study, the
activation of Sirt1 with SRT1720 inhibited EC apoptosis and
endothelial dysfunction in diabetes. Previous reports have argued
that the protective endothelial effect of Sirt1 is closely
associated with the regulation of mitochondrial functions by
limiting mitochondrial DNA damage and preventing the activation of
mitochondrial damaging MMP-9 (11,43,44). Furthermore, mitochondrial
dysfunction caused by mitochondrial fission is reported to be
involved in the development of diabetic nephropathy (7,45)
and diabetic cardiomyopathy (46). These results indicate the
involvement of mitochondrial fission in the development of
HG-induced endothelial injury and diabetic vasculopathy.
An association between Sirt1 and mitochondrial
fission has been illustrated in several studies, which confirms the
regulation of Sirt1 on mitochondrial fission. However, how Sirt1
regulates mitochondrial fission in HG-induced endothelial injury
remains to be fully elucidated. It has been reported that the JNK
pathway is involved in the regulation of mitochondrial fission
(28,40,47). In addition, previous studies have
confirmed the association between Sirt1 and the JNK pathway
(48-50). SIRT1 has been reported to reduce
ROS-induced mouse embryonic stem cell apoptosis via the phosphatase
and tensin homolog/JNK/Forkhead box O1 pathway (51). Therefore, Sirt1 likely regulates
Mff-associated fission via the JNK pathway. The present study
confirmed that Sirt1 attenuates mitochondrial fission via the JNK
pathway in HG-induced endothelial injury.
Mitochondria are energy metabolism organelles
involved in the regulation of ATP generation, ROS production and
apoptosis (52-54). It is well known that mitochondrial
fission serves an important role in mitochondrial functional
regulation. Mechanistically, Drp1, the mitochondrial division
executive factor, binds to its corresponding receptor which is
located on the mitochondrial outer member, forming a ring on
mitochondrial surfaces that contracts and creates a large number of
mitochondrial fragments (55,56). In addition, accumulating evidence
shows that four proteins, Fis1, Mff, Mid49 and Mid51, are involved
in mitochondrial fission (4). As
one of the key findings of the present study, it was confirmed that
Sirt1 regulates mitochondrial fission mainly by regulating Mff in
HG-induced endothelial injury. HG induced the downregulation of
Sirt1, leading to the phosphorylated activation of the JNK pathway,
which promoted the expression of Mff. Additionally, Sirt1 loss
contributed to the upregulation of Drp1. Excessive mitochondrial
fission caused mitochondrial depolarization, followed by increased
mPTP opening periods and cyt-c leakage into the cytoplasm. As a
result of cyt-c release, mitochondria-associated apoptotic pathways
were activated, as evidenced by the imbalanced expression of pro-
and anti-apoptotic proteins.
The migration capacities of endothelial cells are
important in the repair of endothelial injury. Sirt1 is reported to
be involved in the regulation of cellular migration capacity,
however, the underlying mechanisms involved remain to be fully
elucidated. F-actin is a key fiber of cellular migration.
Therefore, it was hypothesized that Sirt1 may regulate the cellular
migration by regulating F-actin homeostasis. HG treatment reduced
the expression of F-actin, enhanced the expression of G-actin and
led to impaired migration capacities, which were reversed by the
overexpression of Sirt1 and jasplakinolide.
Previous studies have argued that miR-195 is
involved in the regulation of Sirt1 (57-59). In human dermal microvascular ECs,
Sirt1 is inactivated through the upregulation of miR-195 under HG
treatment. The results of the present study also show that miR-195
contributes to the downregulation of Sirt1 under HG treatment in
vitro. miR-195 inhibition enhanced the expression of Sirt1,
limited mitochondrial fission and cellular apoptosis, and improved
migration capacities. However, whether miR-195 contributes to
diabetic vasculopathy requires further examination in
vivo.
According to previous studies, SRT1720 was used in
the present study to reactivate Sirt1 and upregulate the expression
of Sirt1 in diabetic mice to examine the role of Sirt1 in diabetic
vasculopathy in vivo (30,60,61). However, SRT1720 has been reported
not to be a direct activator of SIRT1 (62). Therefore, to provide more accurate
evidence, Sirt1 transgenic mice should be used in future
studies.
Overall, the results of the present study illustrate
the protective mechanism of Sirt1 in diabetic vasculopathy, which
limits endothelial apoptosis by inhibiting JNK/Mff/mitochondrial
fission and enhances endothelial migration by stabilizing F-actin
homeostasis. The mechanisms underlying the downregulation of Sirt1
induced by HG were also examined. HG repressed the expression of
Sirt1 by upregulating miR-195. Further investigations are required
to examine the clinical value of Sirt1 in diabetic
vasculopathy.
Funding
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
BJ-2014-066).
Availability of data and materials
The data and materials used and/or analyzed during
the present study are available from the corresponding author on
reasonable request.
Authors' contributions
RQ, LZ and DL conceived and designed the
experiments, performed the experiments, and contributed
reagents/materials/analysis tools. FX and LG analyzed the data. RQ
and LG wrote the paper. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Experimental
Animal Ethics Committee of Beijing Vital River Laboratory Animal
Technology (Beijing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Booth GL, Kapral MK, Fung K and Tu JV:
Recent trends in cardiovascular complications among men and women
with and without diabetes. Diabetes Care. 29:32–37. 2006.
View Article : Google Scholar
|
|
2
|
Brownlee M: The pathobiology of diabetic
complications: A unifying mechanism. Diabetes. 54:1615–1625. 2005.
View Article : Google Scholar
|
|
3
|
Lu J, Xiang G, Liu M, Mei W, Xiang L and
Dong J: Irisin protects against endothelial injury and ameliorates
atherosclerosis in apolipoprotein E-Null diabetic mice.
Atherosclerosis. 243:438–448. 2015. View Article : Google Scholar
|
|
4
|
Zhou H, Wang S, Zhu P, Hu S, Chen Y and
Ren J: Empagliflozin rescues diabetic myocardial microvascular
injury via AMPK-mediated inhibition of mitochondrial fission. Redox
Biol. 15:335–346. 2018. View Article : Google Scholar :
|
|
5
|
Bohuszewicz O and Low HH: Structure of a
mitochondrial fission dynamin in the closed conformation. Nat
Struct Mol Biol. 25:722–731. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mansouri A, Gattolliat CH and Asselah T:
Mitochondrial dysfunction and signaling in chronic liver diseases.
Gastroenterology. 155:629–647. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sheng J, Li H, Dai Q, Lu C, Xu M, Zhang J
and Feng J: NR4A1 promotes diabetic nephropathy by activating
mff-mediated mitochondrial fission and suppressing parkin-mediated
mitophagy. Cell Physiol Biochem. 48:1675–1693. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu WJ, Jiang HF, Rehman FU, Zhang JW,
Chang Y, Jing L and Zhang JZ: Lycium barbarum polysaccharides
decrease hyperglycemia-aggravated ischemic brain injury through
maintaining mitochondrial fission and fusion balance. Int J Biol
Sci. 13:901–910. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wils J, Favre J and Bellien J: Modulating
putative endothelial progenitor cells for the treatment of
endothelial dysfunction and cardiovascular complications in
diabetes. Pharmacol Ther. 170:98–115. 2017. View Article : Google Scholar
|
|
10
|
Fang WJ, Wang CJ, He Y, Zhou YL, Peng XD
and Liu SK: Resveratrol alleviates diabetic cardiomyopathy in rats
by improving mitochondrial function through PGC-1alpha
deacetylation. Acta Pharmacol Sin. 39:59–73. 2018. View Article : Google Scholar
|
|
11
|
Ding M, Feng N, Tang D, Feng J, Li Z, Jia
M, Liu Z, Gu X, Wang Y, Fu F and Pei J: Melatonin prevents
Drp1-mediated mitochondrial fission in diabetic hearts through
SIRT1-PGC1alpha pathway. J Pineal Res. 65:e124912018. View Article : Google Scholar
|
|
12
|
Ou X, Lee MR, Huang X, Messina-Graham S
and Broxmeyer HE: Sirt1 positively regulates autophagy and
mitochondria function in embryonic stem cells under oxidative
stress. Stem Cells. 32:1183–1194. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shi Y, Lv X, Liu Y, et al: Elevating
ATP-binding cassette transporter G1 improves re-endothelialization
function of endothelial progenitor cells via Lyn/Akt/eNOS in
diabetic mice. FASEB J. Jun 12–2018.Epub ahead of print. View Article : Google Scholar
|
|
14
|
Wang W, Yang C, Wang XY, Zhou LY, Lao GJ,
Liu D, Wang C, Hu MD, Zeng TT, Yan L and Ren M: MicroRNA-129 and -
335 promote diabetic wound healing by inhibiting sp1-mediated mmp-9
expression. Diabetes. 67:1627–1638. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma TJ, Zhang ZW, Lu YL, Zhang YY, Tao DC,
Liu YQ and Ma YX: CLOCK and BMAL1 stabilize and activate RHOA to
promote F-actin formation in cancer cells. Exp Mol Med. 50:1302018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kilian P, Campbell S, Bilodeau L, Guimond
MO, Roberge C, Gallo-Payet N and Payet MD: Angiotensin II type 2
receptor stimulation increases the rate of NG10815 cell migration
via actin depolymerization. Endocrinology. 149:2923–2933. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chang CH, Hsu CC, Lee AS, Wang SW, Lin KT,
Chang WL, Peng HC, Huang WC and Chung CH: 4-Acetylantroquinonol B
inhibits lipopolysaccharide-induced cytokine release and alleviates
sepsis through of MAPK and NFkappaB suppression. BMC Complement
Altern Med. 18:1082018. View Article : Google Scholar
|
|
18
|
Li Y, Wang H, Zhang R, Zhang G, Yang Y and
Liu Z: Leukemia growth is inhibited by benzoxime without causing
any harmful effect in rats bearing RBL-1 xenotransplants. Oncol
Lett. 17:1934–1938. 2019.PubMed/NCBI
|
|
19
|
Cavariani MM, de Mello Santos T, Pereira
DN, de Almeida Chuffa LG, Felipe Pinheiro PF, Scarano WR and
Domeniconi RF: Maternal protein restriction differentially alters
the expression of AQP1, AQP9 and VEGFr-2 in the epididymis of rat
offspring. Int J Mol Sci. 20:E4692019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu Y, Bai L, Chen X, Li Y, Qin Y, Meng X
and Zhang Q: 6-Shogaol ameliorates diabetic nephropathy through
anti-inflammatory, hyperlipidemic, anti-oxidative activity in db/db
mice. Biomed Pharmacother. 97:633–641. 2018. View Article : Google Scholar
|
|
21
|
Liu Y and Zheng Y: Bach1 siRNA attenuates
bleomycin-induced pulmonary fibrosis by modulating oxidative stress
in mice. Int J Mol Med. 39:91–100. 2017. View Article : Google Scholar
|
|
22
|
Ekim Kocabey A, Kost L, Gehlhar M, Rodel G
and Gey U: Mitochondrial sco proteins are involved in oxidative
stress defense. Redox Biol. 21:1010792018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu P, Hu S, Jin Q, Li D, Tian F, Toan S,
Li Y, Zhou H and Chen Y: Ripk3 promotes ER stress-induced
necroptosis in cardiac IR injury: A mechanism involving calcium
overload/XO/ROS/mPTP pathway. Redox Biol. 16:157–168. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liang W, Buluc M, van Breemen C and Wang
X: Vectorial Ca2+ release via ryanodine receptors contributes to
Ca2+ extrusion from freshly isolated rabbit aortic endothelial
cells. Cell Calcium. 36:431–443. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pickart L and Thaler MM: Growth-modulating
human plasma tripeptide: Relationship between molecular structure
and DNA synthesis in hepatoma cells. FEBS Lett. 104:119–122. 1979.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi C, Cai Y, Li Y, Li Y, Hu N, Ma S, Hu
S, Zhu P, Wang W and Zhou H: Yap promotes hepatocellular carcinoma
metastasis and mobilization via governing
cofilin/F-actin/lamellipodium axis by regulation of
JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 14:59–71. 2018.
View Article : Google Scholar
|
|
27
|
Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D,
Hu S, Ren J, Cao F and Chen Y: Ripk3 induces mitochondrial
apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury.
Redox Biol. 13:498–507. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S, Ma
S, Zhu H, Ren J and Zhou H: DUSP1 alleviates cardiac
ischemia/reperfusion injury by suppressing the Mff-required
mitochondrial fission and Bnip3-related mitophagy via the JNK
pathways. Redox Biol. 14:576–587. 2018. View Article : Google Scholar
|
|
29
|
Shuang Y, Li C, Zhou X, Huang Y and Zhang
L: MicroRNA-195 inhibits growth and invasion of laryngeal carcinoma
cells by directly targeting DCUN1D1. Oncol Rep. 38:2155–2165. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ren Y, Du C, Shi Y, Wei J, Wu H and Cui H:
The Sirt1 activator, SRT1720, attenuates renal fibrosis by
inhibiting CTGF and oxidative stress. Int J Mol Med. 39:1317–1324.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
32
|
Zhao WJ, Zhang HF and Su JY:
Downregulation of microRNA-195 promotes angiogenesis induced by
cerebral infarction via targeting VEGFA. Mol Med Rep. 16:5434–5440.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou H, Wang J, Zhu P, Hu S and Ren J:
Ripk3 regulates cardiac microvascular reperfusion injury: The role
of IP3R-dependent calcium overload, XO-mediated oxidative stress
and F-action/filopodia-based cellular migration. Cell Signal.
45:12–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dai Y, Zhang J, Xiang J, Li Y, Wu D and Xu
J: Calcitriol inhibits ROS-NLRP3-IL-1beta signaling axis via
activation of Nrf2-antioxidant signaling in hyperosmotic stress
stimulated human corneal epithelial cells. Redox Biol.
21:1010932018. View Article : Google Scholar
|
|
35
|
Park SJ, Ahmad F, Um JH, Brown AL, Xu X,
Kang H, Ke H, Feng X, Ryall J, Philp A, et al: Specific Sirt1
activator-mediated improvement in glucose homeostasis requires
Sirt1-independent activation of AMPK. EBioMedicine. 18:128–138.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu J, Jiang C, Ma X and Wang J:
Notoginsenoside Fc attenuates high glucose-induced vascular
endothelial cell injury via upregulation of PPAR-gamma in diabetic
spraguedawley rats. Vascul Pharmacol. 109:27–35. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kojima H, Otani A, Oishi A, Makiyama Y,
Nakagawa S and Yoshimura N: Granulocyte colony-stimulating factor
attenuates oxidative stress-induced apoptosis in vascular
endothelial cells and exhibits functional and morphologic
protective effect in oxygen-induced retinopathy. Blood.
117:1091–1100. 2011. View Article : Google Scholar
|
|
38
|
Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu
P, Ma Q, Tian F and Chen Y: Mff-dependent mitochondrial fission
contributes to the pathogenesis of cardiac microvasculature
ischemia/reperfusion injury via induction of mROS-mediated
cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP
opening. J Am Heart Assoc. 6:e0053282017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang Y, Subramanian M, Yurdagul A Jr,
Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, Ryan TA, Nomura M,
Maxfield FR and Tabas I: Mitochondrial fission promotes the
continued clearance of apoptotic cells by macrophages. Cell.
171:331–345.e22. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu P, Zhang G, Sha L and Hou S: DUSP1
alleviates cerebral ischaemia reperfusion injury via inactivating
JNK-Mff pathways and repressing mitochondrial fission. Life Sci.
210:251–262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ji K, Lin K, Wang Y, Du L, Xu C, He N,
Wang J, Liu Y and Liu Q: TAZ inhibition promotes IL-2-induced
apoptosis of hepatocellular carcinoma cells by activating the
JNK/F-actin/mitochondrial fission pathway. Cancer Cell Int.
18:1172018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hubbard BP and Sinclair DA: Small molecule
SIRT1 activators for the treatment of aging and age-related
diseases. Trends Pharmacol Sci. 35:146–154. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang Q, Deng Q, Zhang J, Ke J, Zhu Y, Wen
RW, Ye Z, Peng H, Su ZZ, Wang C and Lou T: Activation of the
Nrf2-ARE pathway ameliorates hyperglycemia-mediated mitochondrial
dysfunction in podocytes partly through Sirt1. Cell Physiol
Biochem. 48:1–15. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mishra M, Duraisamy AJ and Kowluru RA:
Sirt1: A guardian of the development of diabetic retinopathy.
Diabetes. 67:745–754. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ayanga BA, Badal SS, Wang Y, Galvan DL,
Chang BH, Schumacker PT and Danesh FR: Dynamin-related protein 1
deficiency improves mitochondrial fitness and protects against
progression of diabetic nephropathy. J Am Soc Nephrol.
27:2733–2747. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Galloway CA and Yoon Y: Mitochondrial
dynamics in diabetic cardiomyopathy. Antioxid Redox Signal.
22:1545–1562. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sheng J, Li H, Dai Q, Lu C, Xu M, Zhang J
and Feng J: DUSP1 recuses diabetic nephropathy via repressing
JNK-Mff-mitochondrial fission pathways. J Cell Physiol.
234:3043–3057. 2019. View Article : Google Scholar
|
|
48
|
Becatti M, Barygina V, Mannucci A, Emmi G,
Prisco D, Lotti T, Fiorillo C and Taddei N: Sirt1 protects against
oxidative stress-induced apoptosis in fibroblasts from psoriatic
patients: A new insight into the pathogenetic mechanisms of
psoriasis. Int J Mol Sci. 19:E15722018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang L, Bao D, Li P, Lu Z, Pang L, Chen
Z, Guo H, Gao Z and Jin Q: Particle-induced SIRT1 downregulation
promotes osteoclastogenesis and osteolysis through ER stress
regulation. Biomed Pharmacother. 104:300–306. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Suzuki M, Bandoski C and Bartlett JD:
Fluoride induces oxidative damage and SIRT1/autophagy through
ROS-mediated JNK signaling. Free Radic Biol Med. 89:369–378. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chae HD and Broxmeyer HE: SIRT1 deficiency
downregulates PTEN/JNK/FOXO1 pathway to block reactive oxygen
species-induced apoptosis in mouse embryonic stem cells. Stem Cells
Dev. 20:1277–1285. 2011. View Article : Google Scholar :
|
|
52
|
Yamamoto K, Imamura H and Ando J: Shear
stress augments mitochondrial ATP generation that triggers ATP
release and Ca2+ signaling in vascular endothelial
cells. Am J Physiol Heart Circ Physiol. 315:H1477–H1485. 2018.
View Article : Google Scholar
|
|
53
|
Li X, Fang P, Li Y, Kuo YM, Andrews AJ,
Nanayakkara G, Johnson C, Fu H, Shan H, Du F, et al: Mitochondrial
reactive oxygen species mediate lysophosphatidylcholine-induced
endothelial cell activation. Arterioscler Thromb Vasc Biol.
36:1090–1100. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Placido AI, Pereira CM, Correira SC,
Carvalho C, Oliveira CR and Moreira PI: Phosphatase 2a inhibition
affects endoplasmic reticulum and mitochondria homeostasis via
cytoskeletal alterations in brain endothelial cells. Mol Neurobiol.
5:154–168. 2017. View Article : Google Scholar
|
|
55
|
Park SJ, Lee H, Jo DS, Jo YK, Shin JH, Kim
HB, Seo HM, Rubinsztein DC, Koh JY, Lee EK and Cho DH:
Heterogeneous nuclear ribonucleoprotein A1 post-transcriptionally
regulates Drp1 expression in neuroblastoma cells. Biochim Biophys
Acta. 1849:1423–1431. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kashatus DF: Restraining the divider: A
drp1-phospholipid interaction inhibits drp1 activity and shifts the
balance from mitochondrial fission to fusion. Mol Cell. 63:913–915.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zheng D, Yu Y, Li M, Wang G, Chen R, Fan
GC, Martin C, Xiong S and Peng T: Inhibition of MicroRNA 195
prevents apoptosis and multiple-organ injury in mouse models of
sepsis. J Infect Dis. 213:1661–1670. 2016. View Article : Google Scholar :
|
|
58
|
Mortuza R, Feng B and Chakrabarti S:
miR-195 regulates sirt1-mediated changes in diabetic retinopathy.
Diabetologia. 57:1037–1046. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhu H, Yang Y, Wang Y, Li J, Schiller PW
and Peng T: MicroRNA- 195 p romotes palmitate-induced apoptosis in
cardiomyocytes by down-regulating sirt1. Cardiovasc Res. 92:75–84.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Khader A, Yang WL, Hansen LW, Rajayer SR,
Prince JM, Nicastro JM, Coppa GF and Wang P: SRT1720, a sirtuin 1
activator, attenuates organ injury and inflammation in sepsis. J
Surg Res. 219:288–295. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Guo S, Liao H, Liu J, Liu J, Tang F, He Z,
Li Y and Yang Q: Resveratrol activated sonic hedgehog signaling to
enhance viability of NIH3T3 cells in vitro via regulation of sirt1.
Cell Physiol Biochem. 50:1346–1360. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Pacholec M, Bleasdale JE, Chrunyk B,
Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis
P, Pabst B, et al: SRT1720, SRT2183, SRT1460, and resveratrol are
not direct activators of SIRT1. J Biol Chem. 285:8340–8351. 2010.
View Article : Google Scholar : PubMed/NCBI
|