Introduction
Myocardial ischemia and reperfusion (I/R) injury is
a primary risk factor for causing myocardial infarctions (MI),
which is the most common cause of mortality in developed countries
(1-3). It has been estimated that ~15.9
million individuals worldwide suffered from MI in 2015 (4). Although reperfusion therapy has been
demonstrated to be able to restore impaired cardiac function and to
mitigate the infarct size following ischemic events, the additional
tissue injury caused by reperfusion is almost inevitable (5).
Multiple mechanisms are hypothesized to be involved
in I/R injury (1). The
inflammatory response to I/R is common in myocardial I/R injury
(6). Various cytokines will be
released during the inflammation reactions (7). Reactive oxygen species (ROS)
generation also occurs in the myocardium; however, if ROS cannot be
cleared promptly, intracellular oxidative stress will be initiated,
finally leading to cell apoptosis. Cellular apoptosis is known to
be triggered shortly following MI, and it is evidently increased
during reperfusion (8,9). Apoptosis has been recognized as a
crucial factor in the progression of I/R injury in cultured
myocardial cells (10,11). Therefore, a decrease in
inflammation, ROS generation and/or apoptosis may be therapeutic
targets against I/R injury. Various apop-tosis pathways, including
the mitochondrial pathway and the tumor necrosis factor receptor
superfamily member 6 (Fas)/Fas ligand (FasL) death
receptor-mediated pathway, have been demonstrated to be essential
for the apoptosis observed in the myocardium following I/R
(11-14). Therefore, depressing apoptotic
signaling may be a promising strategy to decrease I/R injury.
Carthamus tinctorius L. has been applied
extensively in the treatment of cerebrovascular and cardiovascular
diseases. Hydroxysafflor Yellow A (HSYA) is the primary active
component of this compound, commonly used in Chinese medicine. The
antioxidant effect of HSYA has been validated (15), and it has been demonstrated that
HSYA may improve I/R injury by decreasing oxidative stress in brain
tissue (16). Nevertheless, the
mechanisms of HSYA-mediated protection from I/R injury are not
completely understood.
The Janus kinase (JAK)/signal transducers and
activators of transcription (STAT) pathway is able to modulate
stress-responsive gene expression (17,18). The phosphorylation of STATs
mediated by JAK triggers alteration of gene transcription (19,20). In addition, STATs have been
demonstrated to regulate apoptosis in multiple cell types (21). Previous studies identified that
STATs may modulate the opening of mitochondrial permeability
transition pores (22,23). In addition, activation of JAK/STAT
signaling was identified in the development of renal I/R injury, in
which the release of inflammatory promotion factors was enhanced
(24). Previous data have also
demonstrated that STAT1 is able to induce the expression levels of
pro-apoptotic genes including Fas and FasL (25), and to repress the expression
levels of anti-apoptotic gene including B-cell lymphoma 2 (Bcl-2)
(26).
The present study was undertaken to examine the
potential role of HSYA in myocardial I/R injury in vivo and
in vitro. Furthermore, considering the crucial role of
JAK/STAT1 in tissue protection, the potential role of this
signaling pathway in HSYA-mediated protection against myocardial
I/R injury was also examined.
Materials and methods
Chemical preparation
HSYA powder (95-99.0% purity, determined by high
performance liquid chromatography) was obtained from Shanghai
Yuanye Biotechnology Co., Ltd. (Shanghai, China). Sodium
pentobarbital was obtained from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany). Cardiac troponin I (cTnI; cat. no. ml059498)
and interleukin-6 (IL-6; cat. no. ml064292) ELISA kits were
obtained from Shanghai Enzyme-linked Biotechnology Co., Ltd.
(Shanghai, China). The superoxide dismutase (SOD; cat. no. S0101),
lactate dehydrogenase (LDH; cat. no. C0016) and malondialdehyde
(MDA; cat. no. S0131) kits were purchased from Beyotime Institute
of Biotechnology (Haimen, China). Dulbecco's modified Eagle's
medium (DMEM) and other cell culture reagents were purchased from
Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Evans
blue and 2,3,5-triphenyltetrazolium chloride (TTC) were obtained
from Sigma-Aldrich; Merck KGaA. The specific inhibitors of JAK2 and
STAT1 (named AG490 and S1491, respectively) were purchased from
MedChemExpress (Monmouth Junction, NJ, USA). The antibodies used
were purchased from Abcam (Cambridge, MA, USA) and Cell Signaling
Technology, Inc. (CST, Inc.; Danvers, MA, USA), and were as
follows: Anti-cleaved caspase (cat. no. ab49822; dilution, 1:500;
Abcam), anti-Fas (cat. no. ab82419; dilution, 1:1,000; Abcam),
anti-FasL (cat. no. ab15285; dilution, 1:500; Abcam), anti-Bcl-2
(cat. no. ab59348; dilution, 1:1,000; Abcam), anti-Bcl-2-associated
X protein (Bax; cat. no. ab32503; 1:3,000; Abcam), anti-
phosphorylated (p)-JAK2 (cat. no. 4406; dilution, 1:1,000; CST,
Inc.), anti-p-STAT1 (cat. no. 7649; dilution, 1:1,000; CST, Inc.),
anti-β-actin (cat. no. ab8226; dilution, 1:5,000; Abcam),
anti-GAPDH (cat. no. ab9385; dilution, 1:1,000; Abcam) and
horseradish peroxidase (HRP)-conjugated secondary antibodies (cat.
no. ab205718; dilution, 1:5,000; Abcam).
I/R model in vivo
The present study was performed, according to the
Guide for the Care and Use of Laboratory Animals, 8th Edition
(27) and approved by the Animal
Subjects Committee of the Affiliated Hospital of Hangzhou Normal
University (Hangzhou, China). The experiments were performed on 40
healthy adult male Sprague-Dawley rats (weighing 250±20 g; 2 months
old) obtained from Guangdong Medical Laboratory Animal Center
(Foshan, China). The animals were housed in a pathogen-free
environment and maintained following standard laboratory animal
feeding protocols. The animals had ad libitum access to food
and water in a light/dark cycle (12/12 h). The animals were given 2
weeks to acclimate prior to the experiments. The local myocardial
I/R model was constructed as follows: The animals were anesthetized
by intraperitoneal administration of sodium pentobarbital (60
mg/kg); ligation of the anterior descending thoracic branch of the
coronary artery was performed for 30 min prior to 2 h of perfusion.
The animals were randomly divided into four groups, which were:
Control (control), which comprised rats without I/R treatment; the
Sham operated group (sham), in which open heart surgery was
performed but the anterior descending branch of the coronary artery
was not ligated; the I/R group (I/R), which included rats that
underwent I/R treatment; and the I/R+HSYA group (I/R+H), which
included rats that underwent I/R treatment, but 5 mg/kg HSYA was
intraperitoneally injected 30 min prior to ischemia. As previously
described and following our preliminary experiments, concentration
of HSYA was determined (28,29). Following perfusion, the
anesthetized animals were prepared for subsequent experiments.
Finally, the animals were sacrificed by intraperitoneal injection
of sodium pentobarbital (200 mg/kg). Mortality was verified by lack
of visible signs of respiration and measurable heartbeat.
I/R model in vitro
H9c2 cells (American Type Culture Collection,
Manassas, VA, USA) were cultured in DMEM medium containing fetal
bovine serum (10%) and streptomycin/penicillin (1%) in an incubator
with 5% CO2. The cells were seeded at a density of
2.5×103 cell/well. The cells were randomly grouped as
follows: i) Control group (control); ii) the hypoxia/reoxygenation
group (H/R); iii) the H/R +20 µM HSYA group (H/R + H); iv)
the H/R +10 µM AG490 group (H/R + A); v) the H/R +10
µM AG490 +20 µM HSYA (H/R + H + A) group; and vi) the
H/R +5 µM S1491 +20 µM HSYA (H/R + H + S1491) group.
As previously described and following our preliminary experiments,
the doses of each agent were selected (29-32). The cells were collected for the
following experiments.
Infarct size measurement
As described previously, the risk area and infarct
size was calculated using a double-staining method with TCC and
Evans blue stains (33). In
brief, following perfusion, the heart was perfused with 3% Evans
blue to indicate the area at risk (AAR). Then, the tissues were
incubated with 2% TTC solution in the dark at 37°C for 15 min, and
then were then stored in 4% paraformaldehyde overnight at 4°C to
demarcate the infarct area. Areas with non-perfusion and blue
staining were considered AAR, while areas with non-perfusion with
TTC staining were labeled as the infarct area. The AAR and infarct
area were calculated using Image-Pro Plus software V6.0 (Media
Cybernetics, Inc., Rockville, MD, USA). The infarct size was
measured as a percentage of the infarct area over the AAR.
Analysis of myocardial damage and
oxidative stress markers
Following reperfusion, blood samples in rats and
cells in the culture medium were harvested. The blood and cells
were then centrifuged at 3,000 × g for 15 min at room temperature
to obtain sera and cell supernatant. The levels of cTnI, IL-6 and
LDH release were examined to estimate myocardial damage. The levels
were measured using commercially available ELISA kits according to
the manufacturer's protocol. The absorbance was read on a
microplate reader (Thermo Fisher Scientific, Inc.). The serum and
the cell culture supernatant were also used to estimate oxidative
stress markers, including the content of MDA and the activity of
SOD, which were detected with commercial assay kits using
fluorescence spectrophotometry (F-7100; Hitachi, Ltd., Tokyo,
Japan) following the manufacturer's protocols.
Detection of myocardial apoptosis
A terminal deoxynucleotidyl transferase-mediated
dUTP nick-end labeling (TUNEL) assay and flow cytometry analysis
were performed to detect the apoptosis levels in heart tissue and
cardiomyocytes, respectively. The heart tissue was fixed in 4%
paraformaldehyde for 10 min at room temperature and embedded in
paraffin. The tissue was subsequently cut into 4-5 µm
sections, and the slides were incubated with TUNEL mixture reagent
(Sigma-Aldrich; Merck KGaA) at 37°C for 1 h. The cells were also
incubated with DAPI (1 µg/ml) for 30 min at room temperature
to stain the nuclei. ProLong™ Gold Antifade Mountant (cat. no.
P36930; Thermo Fisher Scientific, Inc.) was used to prevent
photo-bleaching. The apoptotic cells in ≥6 fields were randomly
selected. The percentage of apop-tosis was calculated as the number
of TUNEL-positive cells within the total populations of
cardiomyocytes. The images and the ratios were obtained using an
Olympus fluorescence microscope (BX51; Olympus Corporation, Tokyo,
Japan) and ImageJ software (version 2.0.0; National Institutes of
Health, Bethesda, MD, USA), respectively. An Annexin V-fluorescein
isothiocyanate (FITC) and propidium iodide (PI) staining kit (BD
Biosciences, San Jose, CA, USA) was used to assess cell apoptosis
in vitro according to the manufacturer's protocols. The
collected cells were stained with 5 µl Annexin V-FITC and 10
µl PI in the dark at room temperature for 10-15 min.
Apoptosis was determined by a FACSCalibur flow cytometer (BD
Biosciences) and analysis software BD CellQuest™ Pro Software
version 1.2 (BD Biosciences).
Cell viability and caspase-3
activity
The cells (5×103 per well) were seeded in
a 96-well plate and treated as described. Next, the cells were
incubated with Cell Counting Kit-8 (CCK-8; Beyotime Institute of
Biotechnology) reagent (10 µl) at 37°C in an incubator for 3
h. The absorbance was determined on a microplate reader (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) at 450 nm. Caspase-3
activity (cat. no. 69-21755; MSK Laboratory, Wuhan, China) was
determined in the cell lysate by conducting an ELISA assay. The
treated cells were collected and plated in a 96-well plate. Cell
lysates were subject to protein quantitation using a Bicinchoninic
Acid Protein Assay kit (Bio-Rad Laboratories, Inc.) prior to
detection. Each well was incubated with substrate solution. Then,
the 96-well plate was maintained at 37°C for 30 min. Finally, the
reaction was terminated with a stop solution. A microplate reader
was used to read the absorbance at a wavelength of 405 nm.
Determination of mitochondrial membrane
potential (MMP) and ROS generation
The alteration of MMP was measured by Rhodamine-123
(Rho-123) dye (Sigma-Aldrich; Merck KGaA). Following reperfusion,
the cells were incubated with 10 µg/ml Rho-123 working
buffer at 37°C for 30 min. The cells were then washed, and analyzed
using a flow cytometer and BD Accuri™ C6 software (BD Biosciences).
Subsequently, 2,7-dichlorofluorescein diacetate (DCFH-DA) dye
(Sigma-Aldrich; Merck KGaA) was used to detect the ROS level.
Briefly, prior to fixation with 4% paraformaldehyde at room
temperature for 15 min, the cells were stained with 20 µM
DCFH-DA at 37°C for 30 min. ROS generation was then determined at
wavelength 488 nm using a fluorescence plate reader (BioTek
Instruments, Inc., Winooski, VT, USA).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Cells were pretreated as described. The total RNA
was isolated with TRIzol® reagent (Thermo Fisher
Scientific, Inc.). DNaseI was used to digest the genomic DNA.
Reverse transcription was completed with random hexamer primers
using a Moloney-Murine Leukemia Virus Enzyme (Promega Corporation,
Madison, WI, USA). RT-qPCR was performed on ABI Prism 7500 using
the SYBR Premix Ex Taq II (Takara Bio, Inc., Otsu, Japan). The qPCR
conditions were set as follows: 10 min pretreatment at 95°C, 96°C
for 15 sec, 63°C for 45 sec (35 cycles) and a final extension at
75°C for 10 min. The specific PCR primers used were as follows: Fas
forward, 5′-CCAGCCACAAAGAGAGGAGA-3′; Fas reverse,
5′-AACGGTTCCTCTCAACACCT-3′; FasL forward,
5′-TGCTGTGTGACAATGCAGAG-3′, FasL reverse,
5′-GAGCCTCCTTTCTCACCCTT-3′; Bcl-2 forward,
5′-CCTGGCATCTTCTCCTTCCA-3′; Bcl-2 reverse,
5′-GGACATCTCTGCAAAGTCGC-3′; Bax forward,
5′-TGGCCTCCTTTCCTACTTCG-3′; Bax reverse,
5′-AAAATGCCTTTCCCCGTTCC-3′; β-actin forward,
5′-TCTATGAGGGTTACGCGCTC-3′; β-actin reverse,
5′-GCTGTGGTGGTGAAGCTGTA-3′. The 2−ΔΔCq method was used
to quantify gene expression levels (34).
Western blot analysis
The harvested cells were treated with
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology). A Bicinchoninic Acid Protein Assay kit (Bio-Rad
Laboratories, Inc.) was used to assess protein concentration.
Following denaturation in boiling water for 5 min, the proteins (20
µg/lane) were separated by electrophoresis on 10% SDS-PAGE
gels. Then, the cells were transferred onto a polyvinylidene
fluoride membrane. Subsequently, 5% skimmed milk was incubated with
the membrane to block the non-specific antigens at room temperature
for 2 h. The primary antibodies were then added, to interact with
the target proteins, at 4°C overnight. Next, the membrane was
incubated with HRP-coupled secondary antibodies at 4°C for 1 h.
Blots were visualized using enhanced chemiluminescence reagent
(Thermo Fisher Scientific, Inc.) An enhanced chemiluminescence
system (GE Healthcare, Chicago, IL, USA) was adopted to detect the
bands. The protein gray intensity was calculated using Quantity one
software (4.6.2; Bio-Rad Laboratories, Inc.).
Statistical analysis
GraphPad Prism Software 6 (GraphPad Software, Inc.,
La Jolla, CA, USA) was used for statistical analysis. Statistical
analysis was conducted by one-way analysis of variance with
Dunnett's post-test comparison. Data are presented as the mean ±
standard deviation from three independent experiments. P<0.05
was considered to indicate a statistically significant
difference.
Results
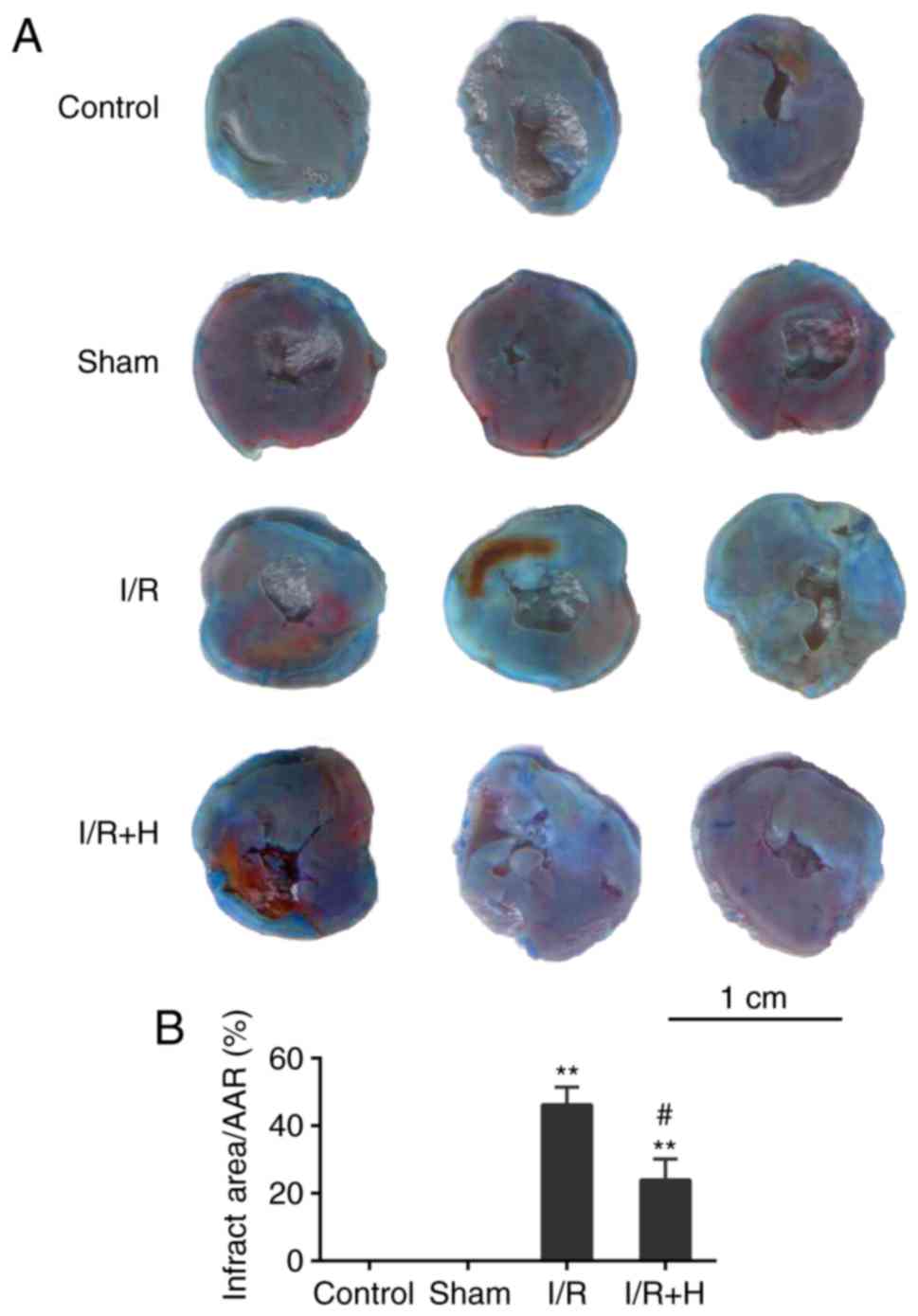
HSYA ameliorates myocardial I/R injury
and oxidant stress in vivo
Infarct size and the release of cTnI and IL-6 were
measured to estimate the effect of HSYA on I/R injury. In contrast
to the Sham group, animals in the I/R group exhibited a significant
increase in myocardial infarct size. The infarct size was markedly
decreased in the HSYA group in comparison with that in the I/R
group (Fig. 1A and B). In
addition, animals in the I/R group exhibited a marked increase in
serum cTnI and IL-6 levels, compared with those rats in the Sham
group. Notably, the treatment with HSYA was associated with a minor
increase in cTnI and IL-6 levels compared with the I/R group
(Fig. 2A and B). Furthermore, as
a marker of tissue damage, the increased activity of LDH induced by
I/R injury was decreased by the treatment with HSYA (Fig. 2C). In addition, the animals in the
I/R group exhibited a noticeable increase in the content of MDA and
a significant decrease in SOD activity compared with the Sham
group. The administration of HSYA in animals with I/R injury
exhibited cardio-protective effects by alleviating oxidative stress
(Fig. 2D and E).
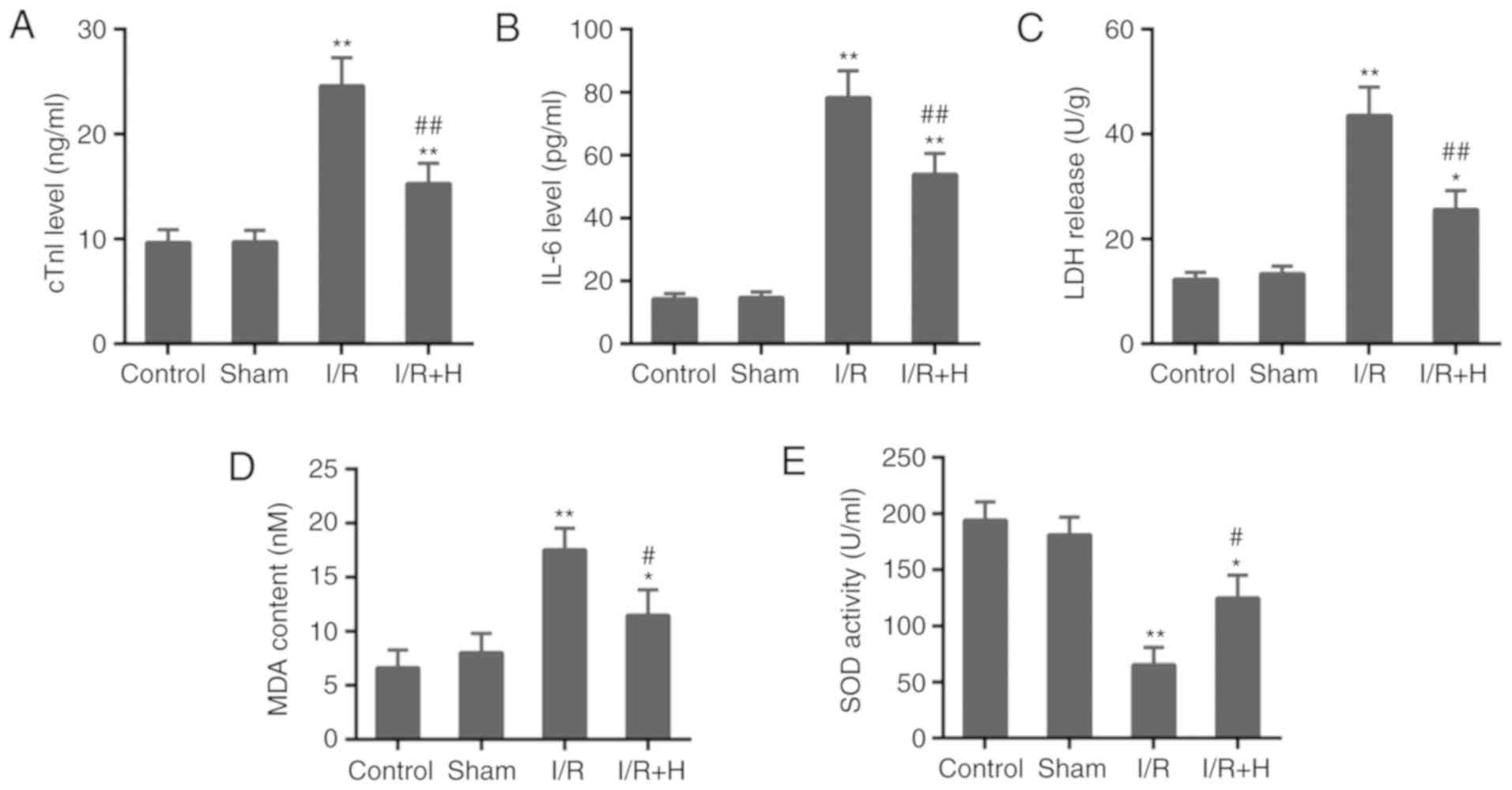 | Figure 2Effect of HSYA on indicators for
myocardial injury and oxidative stress. The serum levels of (A)
cTnI, (B) IL-6, (C) LDH, (D) MDA and (E) SOD in rats.
*P<0.05 and **P<0.01 vs. control group;
#P<0.05 and ##P<0.01 vs. I/R group.
I/R, ischemia/reperfusion; H/HSYA, Hydroxysafflor Yellow A; cTnI,
cardiac troponin; IL-6, interleukin 6; LDH, lactate dehydrogenase;
MDA, malondialdehyde; SOD, superoxide dismutase. |
HSYA decreases myocardial apoptosis in
vivo
To estimate the effect of HSYA on apoptosis, the
TUNEL-positive cardiomyocytes and the expression levels of
apoptosis-associated proteins were determined in vivo. The
percentage of TUNEL-positive cells was elevated in the I/R group in
comparison with the Sham group. Compared with the I/R group, HSYA
significantly decreased the number of TUNEL-positive cardiomyocytes
(Fig. 3A and B). In addition, the
expression levels of pro-apoptotic proteins, including cleaved
caspase-3, Fas, FasL and Bax, were markedly induced by I/R.
However, the administration of HSYA decreased their expression
levels. Concomitantly, the level of anti-apoptotic protein Bcl-2
was decreased in the I/R group, but it was recovered in the I/R +
HSYA group (Fig. 3C and D).
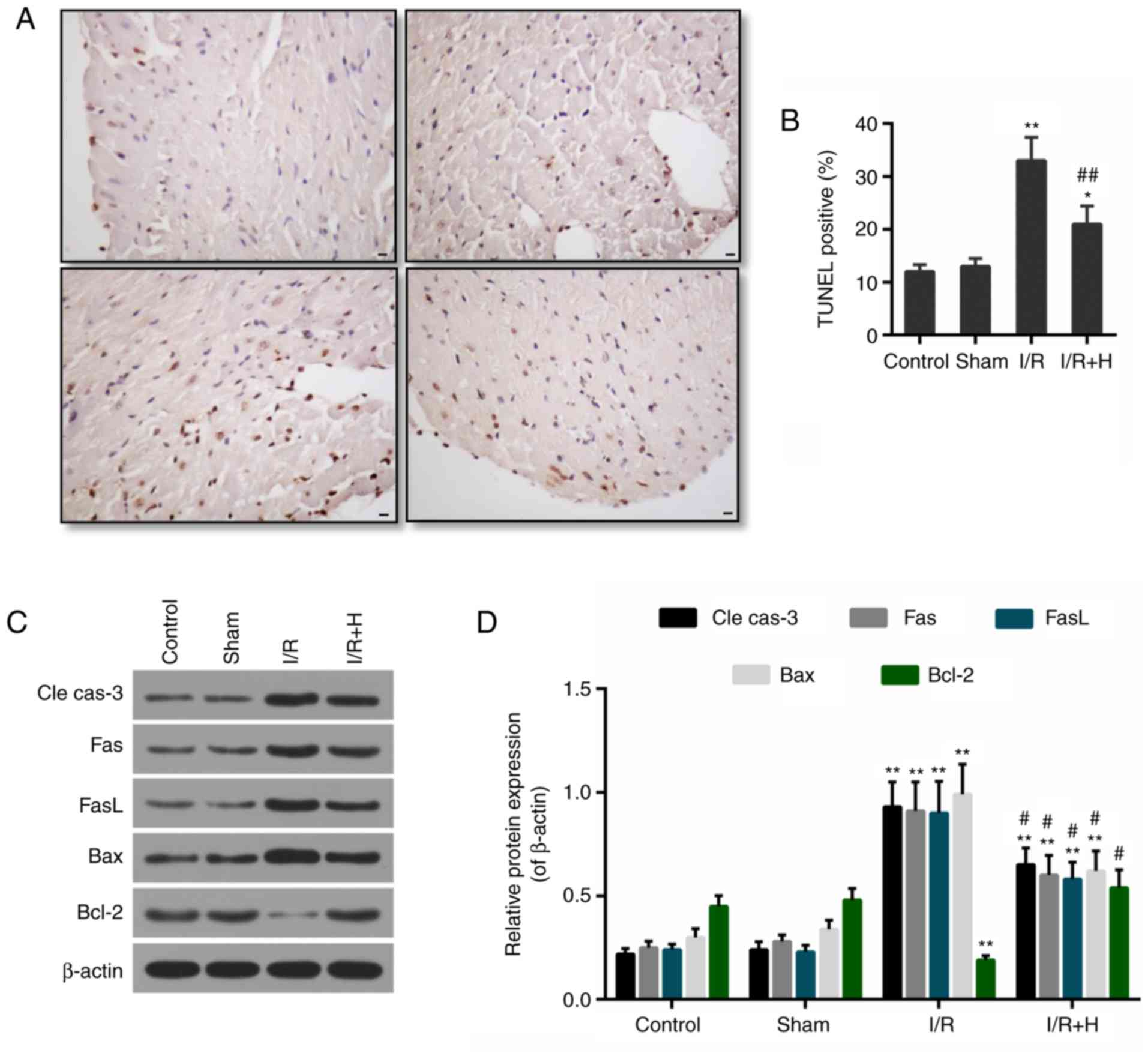 | Figure 3Effect of HSYA on myocardial
apoptosis. (A) Representative TUNEL staining images in each group.
Scale bar=100 µM. (B) Apoptosis was determined by the number
of TUNEL-positive cells. (C) Western blot analysis for the protein
expression levels of cle cas-3, Fas, FasL, Bax and Bcl-2. (D)
Densitometric analysis of the western blot analysis data.
*P<0.05 and **P<0.01 vs. control group;
#P<0.05 and ##P<0.01 vs. I/R group.
I/R, ischemia/reperfusion; H/HSYA, Hydroxysafflor Yellow A; TUNEL,
terminal deoxynucleotidyl transferase-mediated dUTP nick-end
labeling; cle cas-3, cleaved caspase-3; Fas, tumor necrosis factor
receptor superfamily member 6; FasL, Fas ligand; Bcl-2, B-cell
lymphoma 2; Bax, Bcl-2-associated X protein. |
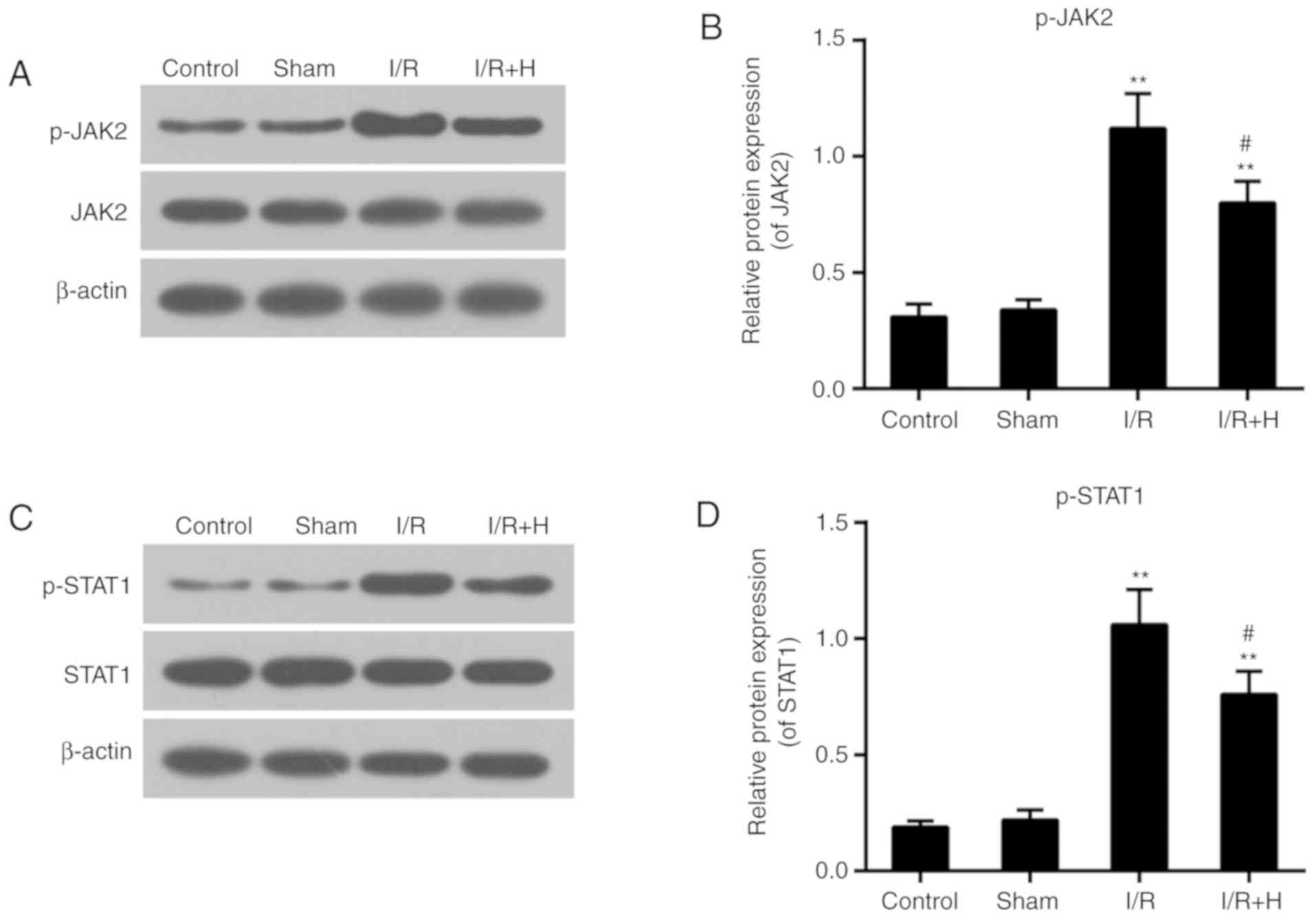
HSYA suppresses myocardial p-JAK2 and
p-STAT1 protein expression levels
The expression levels of p-JAK2 and p-STAT1 were
markedly increased in animals with I/R injury. No significant
difference in the expression levels of total JAK2 and STAT1 between
Sham and I/R group was identified. The administration of HSYA
decreased the levels of p-JAK2 and p-STAT1 (Fig. 4A-D).
Inhibition of JAK2/STAT1 signaling HSYA
prevents H9c2 cardiomyocytes against I/R injury
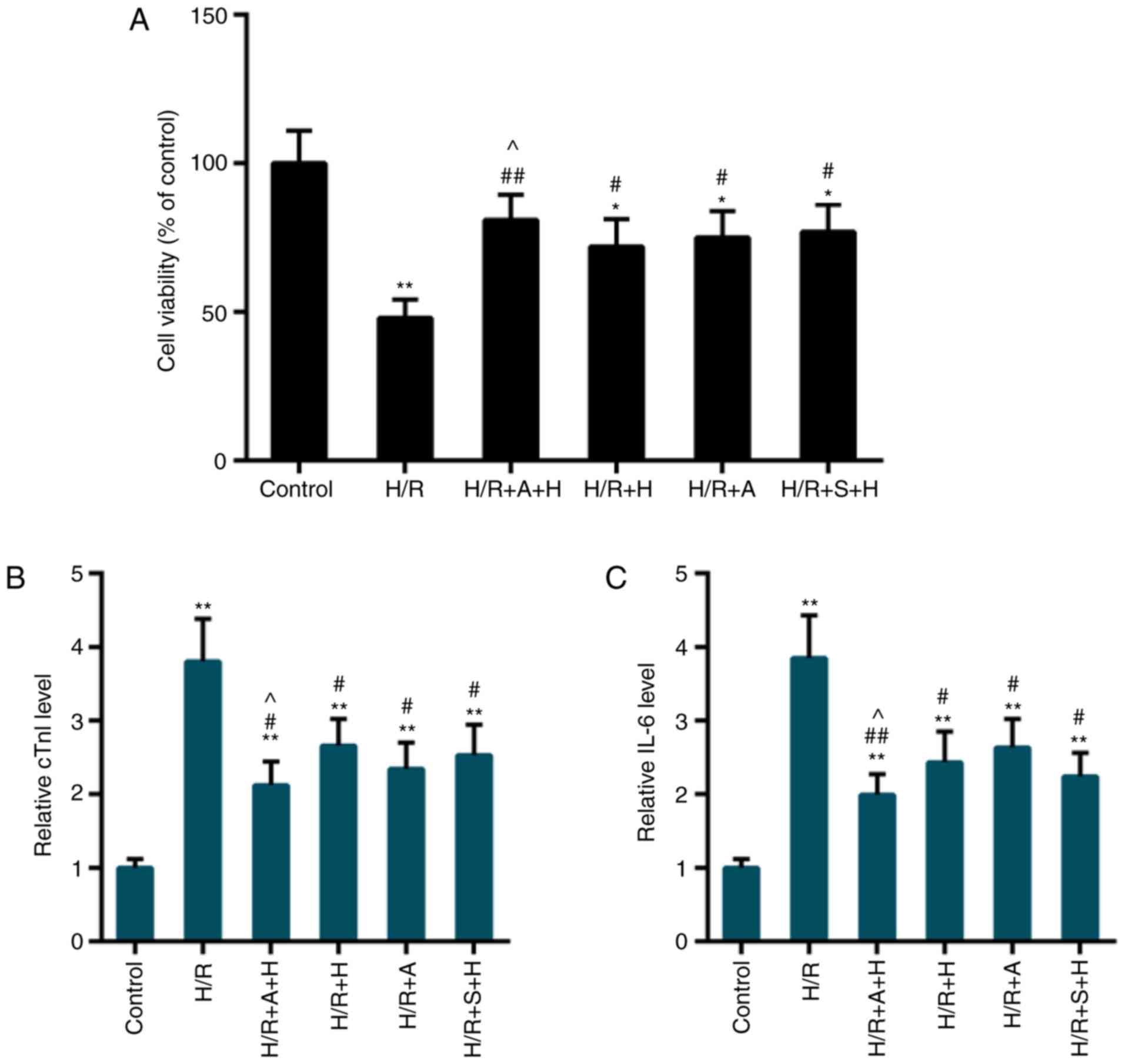
CCK-8 data indicated that compared with the control
group, the cell viability was significantly inhibited following I/R
injury, while pretreatment with HSYA or inhibitors of JAK2/STAT1
(AG490 or S1491) significantly rescued the cell viability (Fig. 5A). In addition, the release of
cTnI and IL-6 was decreased in the H/R group compared with the
control group, while such a release was decreased by the
pretreatment of HSYA, AG490 or S1491 (Fig. 5B and C). The protective effect
appeared more apparent in the HR + A + H or H/R + S + H group
compared with the H/R + H group.
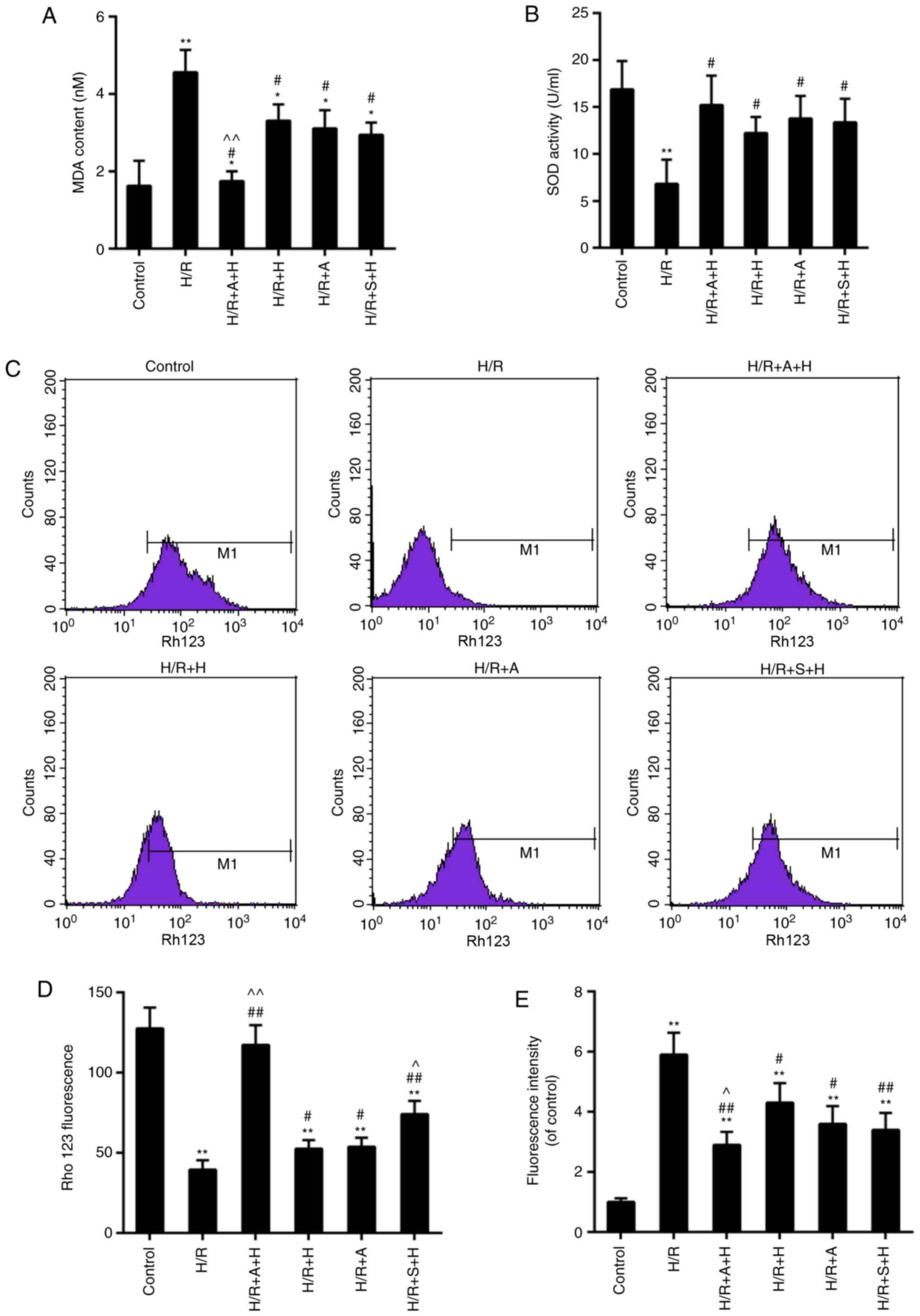
Inhibition of JAK2/STAT1 signaling
strengthens the anti- oxidant effects of HSYA in H9c2
Data from the assays for MDA content and SOD
activity demonstrated that I/R injury accelerated the rate of
oxidative stress compared with the control. The MDA content and SOD
activity were markedly decreased and recovered, respectively, in
the group pretreated with HSYA. Although no significant difference
was identified in the MDA content and SOD activity in H/R + S + H
group when compared with those in the H/R + H group, the protective
effects of HSYA were more significantly enhanced by the combined
use with AG490 or S1491 (Fig. 6A and
B). Loss of MMP may be associated with the disruption of redox
status, which then leads to ROS-triggered oxidative stress. The
results indicated that the loss of MMP was all partially alleviated
in H/R + A + H, H/R + H, H/R + A and H/R + S + H groups, compared
with the control group. Consistently, the ROS contents in these
groups were decreased compared with those in the control group. The
ROS level was significantly decreased in the H/R + A + H and H/R +
S + H groups, compared with the H/R + H group. Although no
significant difference was identified in the H/R + S + H group in
comparison with that in the H/R + H group, the MMP level was also
decreased in the H/R + A + H and H/R + S + H groups, compared with
the H/R + H group (Fig.
6C-E).
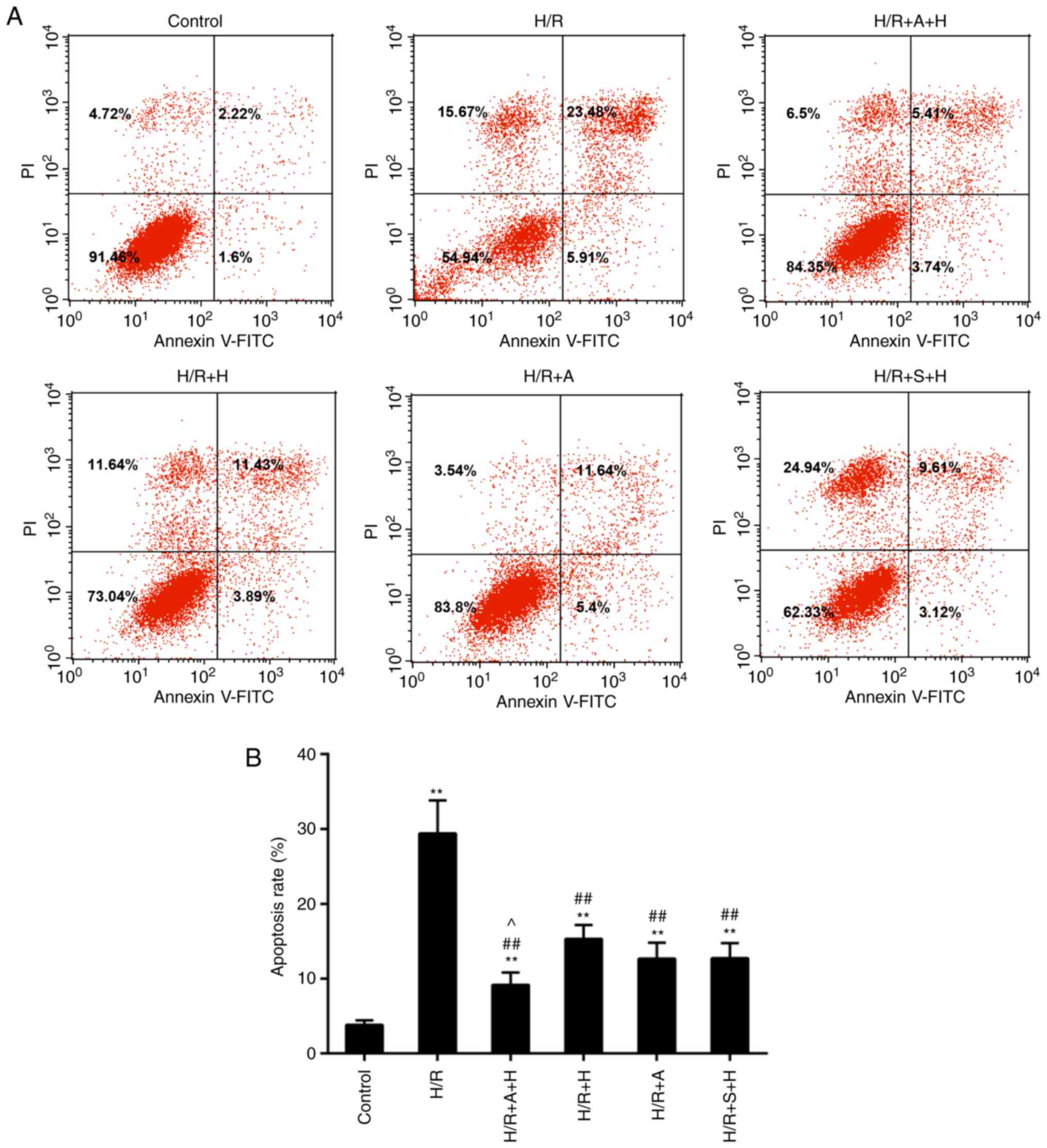
Inhibition of JAK2/STAT1 signaling
enhances the anti- apoptotic effect of HSYA in H9c2
To confirm the effect of HSYA on apoptosis, the
levels of apoptosis were detected by flow cytometry. The results
revealed that the apoptosis rate was accelerated in the H/R group,
while decreased cell apoptosis was identified in the H/R + A + H,
H/R + H, HR + A and H/R + S + H groups compared with the H/R group
(Fig. 7A and B). Furthermore, the
caspase-3 activity was decreased in the H/R + A + H, H/R + H, HR +
A and H/R + S + H groups compared with the H/R group (Fig. 8A). Additionally, the expression
levels of apoptosis-associated factors were evaluated. The data
from the in vitro analyses were compatible with those from
the in vivo experiments, which demonstrated that the
expression levels of pro-apoptotic proteins (cleaved caspase-3,
Fas, FasL and Bax) were decreased and anti-apoptotic protein Bcl-2
was increased in the H/R + A + H, H/R + H, H/R + A and H/R + S + H
groups compared with that in the H/R group (Fig. 8B and C). Similar results were
obtained with regards to the mRNA expression levels of these
apoptosis-associated factors (Fig.
8D). Therefore, the treatment with AG490/S1491 enhanced the
protective effect of HSYA.
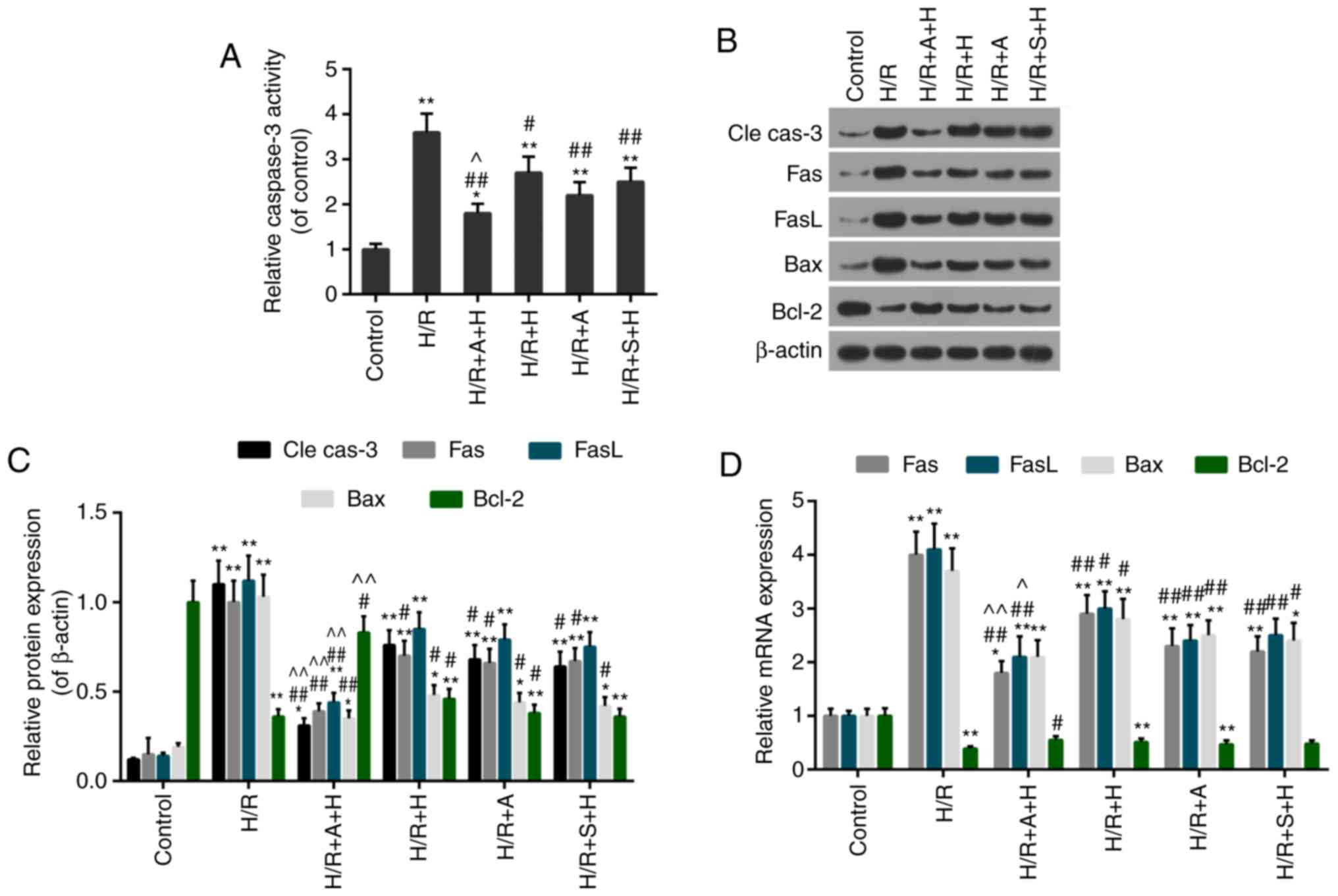 | Figure 8Effect of inhibition of janus kinase
2/signal transducer and activator of transcription 1 on the
activity of apoptosis-associated proteins. (A) Caspase-3 activity
was estimated using an ELISA kit. (B) Western blot analysis gel
examining the protein levels of cle cas-3, Fas, FasL, Bax and
Bcl-2. (C) Quantification of the western blot analysis data. (D)
Reverse transcription quantitative polymerase chain reaction assay
of the expression levels of Fas, FasL, Bax and Bcl-2.
*P<0.05 and **P<0.01 vs. control group;
#P<0.05 and ##P<0.01 vs. HR group;
^P<0.05 and ^^P<0.01 vs. H/R+H group.
cle cas-3, cleaved caspase-3; H/R, hypoxia/reoxygenation; H,
Hydroxysafflor Yellow A; A, AG490; S, S1491; Fas, tumor necrosis
factor receptor superfamily member 6; FasL, Fas ligand; Bcl-2,
B-cell lymphoma 2; Bax, Bcl-2-associated X protein. |
Discussion
I/R injuries are one of the primary causes of MI
(3). HSYA, the primary active
component of Carthamus tinctorius L., has been suggested to
be able to protect against multiple types of cell injuries
(35,36). In the present study, the
protective role of HSYA was demonstrated, following I/R injury in
an in vivo animal model and in vitro experiments
using cultured H9c2 cells. The signaling pathway by which HSYA
delivers its protective effect was also explored.
Infarct size is considered an important index in
determining the severity of I/R injury in heart muscle (37). Therefore, the present study first
determined the effects of HSYA on infarct size. With the
administration of HSYA, the infarct size was decreased compared
with the Sham group. In addition, the levels of cTnI and IL-6,
sensitive markers of cardiac injury (38), were decreased in the I/R+HSYA
treatment group compared with the I/R group. Furthermore, the
release of LDH, which may reflect the degree of I/R damage, was
also decreased following pretreatment with HSYA, indicating that
HSYA is capable of decreasing myocardial injury caused by I/R in
vivo.
The mechanism by which HSYA realized its protective
effects was then investigated. The increased production of free
radicals and the decreased activities of antioxidant enzymes are
closely associated with myocardial I/R injury (39). The disrupted redox status will
trigger oxidative stress (OS). SOD and MDA are common indicators of
OS. The former is responsible for decomposing O2 and
H2O2, while the latter is the product of
lipid peroxidation (40,41). The results from the present study
revealed that these indicators of oxidative stress were alleviated
by HSYA, indicating an antioxidant scavenger capacity of HSYA.
Apoptosis is involved in the destruction of
cardiomyocytes following I/R injury (29,42). It has been recognized that Bcl-2
and Bax are essential proteins in apoptosis (29). The Bax/Bcl-2 ratio regulates the
cell sensitivity death signals by modulating the function of the
mitochondria. Caspase-3 is a key gene of the apoptotic pathways and
may be processed into cleaved caspase-3 (43). Additionally, the Fas/FasL death
receptor-mediated pathway may be activated in I/R injury (11-14). In the present study, HSYA
decreased cleaved caspase-3, Fas and FasL levels, increased the
Bcl-2/Bax ratio and mitigated TUNEL-positive staining in comparison
with the I/R group. Taken together, the results demonstrated an
anti-apoptotic effect of HSYA following I/R injury.
Subsequently, the present study aimed to explore the
molecular signaling methods by which HSYA exerted its effective
role. The JAK/STAT pathway is important in signal transduction and
is involved in mediating cell growth, proliferation and
differentiation, and apoptosis. JAK and STAT family members are
located in myocardial cells and are closely associated with
myocardial I/R injury (44).
STAT-1, a member of the STATs, is able to regulate intracellular
signaling. It has been demonstrated that STAT1 was activated
following myocardial ischemia (45). Therefore, the present study
focused particularly on the potential role of JAK2/STAT1 in
HSYA-mediated anti-MI activity. The activity of the JAK2/STAT1
pathway in I/R injury was determined. HSYA treatment was observed
to prevent the phosphorylation of JAK2, and it also decreased the
phosphorylation of downstream STAT1. Altogether, these results
suggested that the protective effect of HSYA was, at least,
partially dependent on suppressing the JAK2/STAT1 pathway.
JAK/STAT pathways are important signaling pathways
in response to stress, including ischemia, hypoxia and oxidative
stress (46,47). JAK2 may be activated by ROS and
hypoxic conditions (48). STAT-1
is phosphorylated by JAK2, and it serves a critical role in
cerebral I/R injury in vivo (49). To additionally confirm the
potential role of the JAK2/STAT1 signaling pathway in the
HSYA-mediated protective effect, in vitro experiments using
H9c2 cells were performed in the present study. The data indicated
that the inhibition of JAK2 (by AG490) or STAT1 (by S1491) enhanced
the protective role of HSYA by improving cell viability and
decreasing the release of cTnI, IL-6 and LDH. In addition, the
HSYA-derived antioxidant capacity was strengthened by the
inhibition of JAK2 or STAT1. The imbalance between the generation
and removal of free radicals will lead to ROS accumulation.
Mitochondria, the primary sites for ROS production, are associated
with the loss of MMP (50).
Therefore, the MMP and ROS content in H9c2 was measured, and it was
identified that the loss of MMP and the ROS generation were
additionally deteriorated by H/R injury, while the HSYA treatment
reversed this phenomenon. Notably, AG490 or S1491 increased the
protective abilities of HSYA. Furthermore, the caspase-3 activity
and the expression levels of cleaved caspase-3, Fas, FasL and Bax
were additionally decreased in cells treated with AG490 or S1491,
compared with the HSYA treatment alone. By contrast, the expression
levels of Bcl-2 were increased in the groups treated with AG490 or
S1491 compared with that in the H/R+H group. Additionally,
examination of the apoptosis rate demonstrated that AG490 or S1491
treatment markedly enhanced the anti-apoptotic effect of HSYA,
suggesting that HSYA and JAK2/STAT1 signaling have similar
mechanisms of action. Previous studies suggested that the JAK/STAT
signaling pathway may regulate apoptotic signals by decreasing
caspase 3 protein expression and increasing anti-apoptotic Bcl-2
levels (51,52). It has also been identified that
JAK2 and STAT1 were activated following reperfusion, and that
AG-490 may abrogate the activation of JAK2 (47,53). The results from the present study
were in concordance with these conclusions. The combined effects of
HSYA and AG490/S1491 in an in vivo I/R rat model would
provide additional supporting evidence for the involvement of the
JAK/STAT pathway in I/R injury. Altogether, the results from the
present study indicated that JAK2/STAT1 may serve as therapeutic
targets to mitigate I/R injury. However, a previous study revealed
that activating the JAK2/STAT3 signaling pathway may decrease
HR-induced mitochondrial oxidative damage (54). We hypothesized that the
discrepancy may be caused by distinct cell context and study model.
In addition, previous studies have demonstrated that JAK2 may
activate STAT3, STAT5a, STAT5b and STAT6 (55-57), therefore the role of these
proteins in mediating the protective effects of HSYA in I/R injury
should be additionally investigated in future studies.
Certain previous studies have documented the role of
HSYA in myocardial IR injury (58-60). These studies suggested that HSYA
exerted its cardio-protective effect by decreasing inflammation or
oxidative stress via toll-like receptor 4 signaling or the
extracellular signal-regulated kinase/glycogen synthase kinase 3β
pathway. This was a limitation of the present study. However, the
mechanism of the protective effect of HSYA demonstrated in the
present study was different from that in these previous studies. We
hypoth-esized that these results together confirm the protective
effects of HSAY on myocardial I/R injury and potentially provide
insight for future clinical studies into HSYA.
In conclusion, HSYA attenuated myocardial injury
in vitro and in vivo by inhibiting oxidative stress
and apoptosis, in which the JAK2/STAT1 signaling pathway was partly
involved. The results from the present study offered insight into
the effects and the mechanisms of HSYA. It may provide effective
therapeutic strategies to combat MI.
Acknowledgments
Not applicable.
Funding
The present study was supported by The Natural
Science Foundation of Zhejiang Province, China (grant no.
LY17H020001).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
DZ and TD made substantial contributions to the
conception and design of the study. BL, YJ and SL contributed to
data acquisition, and data analysis and interpretation. DZ and TD
drafted the article and critically revised it for important
intellectual content. All authors agreed to be accountable for all
aspects of the work in ensuring that questions related to the
accuracy or integrity of the work are appropriately investigate and
resolved. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was performed according to the
Guide for the Care and Use of Laboratory Animals, 8th Edition and
approved by Animal Subjects Committee of the Affiliated Hospital of
Hangzhou Normal University (Hangzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bainey KR and Armstrong PW: Clinical
perspectives on reperfusion injury in acute myocardial infarction.
Am Heart J. 167:637–645. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maxwell SR and Lip GY: Reperfusion injury:
A review of the pathophysiology, clinical manifestations and
therapeutic options. Int J Cardiol. 58:95–117. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heusch G, Boengler K and Schulz R:
Inhibition of mitochondrial permeability transition pore opening:
The Holy Grail of cardio-protection. Basic Res Cardiol.
105:151–154. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
GBD 2015 Disease and Injury Incidence and
Prevalence Collaborators: Global, regional, and national incidence,
prevalence, and years lived with disability for 310 diseases and
injuries, 1990-2015: A systematic analysis for the Global Burden of
Disease Study, 2015. Lancet. 388:1545–1602. 2016. View Article : Google Scholar
|
|
5
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiong J, Xue FS, Yuan YJ, Wang Q, Liao X
and Wang WL: Cholinergic anti-inflammatory pathway: A possible
approach to protect against myocardial ischemia reperfusion injury.
Chin Med J (Engl). 123:2720–2726. 2010.
|
|
7
|
Naidu BV, Farivar AS, Woolley SM, Grainger
D, Verrier ED and Mulligan MS: Novel broad-spectrum chemokine
inhibitor protects against lung ischemia-reperfusion injury. J
Heart Lung Transplant. 23:128–134. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Olivetti G, Quaini F, Sala R, Lagrasta C,
Corradi D, Bonacina E, Gambert SR, Cigola E and Anversa P: Acute
myocardial infarction in humans is associated with activation of
programmed myocyte cell death in the surviving portion of the
heart. J Mol Cell Cardiol. 28:2005–2016. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao ZQ, Nakamura M, Wang NP, Wilcox JN,
Shearer S, Ronson RS, Guyton RA and Vinten-Johansen J: Reperfusion
induces myocardial apoptotic cell death. Cardiovasc Res.
45:651–660. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Orogo AM and Gustafsson AB: Cell death in
the myocardium: My heart won't go on. IUBMB Life. 65:651–656. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stephanou A, Brar B, Liao Z, Scarabelli T,
Knight RA and Latchman DS: Distinct initiator caspases are required
for the induction of apoptosis in cardiac myocytes during ischaemia
versus reperfusion injury. Cell Death Differ. 8:434–435. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Scarabelli TM, Stephanou A, Pasini E,
Comini L, Raddino R, Knight RA and Latchman DS: Different signaling
pathways induce apoptosis in endothelial cells and cardiac myocytes
during ischemia/reperfusion injury. Circ Res. 90:745–748. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Scarabelli T, Stephanou A, Rayment N,
Pasini E, Comini L, Curello S, Ferrari R, Knight R and Latchman D:
Apoptosis of endothelial cells precedes myocyte cell apoptosis in
ischemia/reperfusion injury. Circulation. 104:253–256. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jeremias I, Kupatt C, Martin-Villalba A,
Habazettl H, Schenkel J, Boekstegers P and Debatin KM: Involvement
of CD95/Apo1/Fas in cell death after myocardial ischemia.
Circulation. 102:915–920. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jin M, Li JR and Wu W: Study on the
antioxidative effect of safflor yellow. Zhongguo Zhong Yao Za Zhi.
29:447–449. 2004.In Chinese.
|
|
16
|
Wei X, Liu H, Sun X, Fu F, Zhang X, Wang
J, An J and Ding H: Hydroxysafflor yellow A protects rat brains
against ischemia-reperfusion injury by antioxidant action. Neurosci
Lett. 386:58–62. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Darnell JE Jr: STATs and gene regulation.
Science. 277:1630–1635. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Imada K and Leonard WJ: The Jak-STAT
pathway. Mol Immunol. 37:1–11. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Negoro S, Kunisada K, Tone E, Funamoto M,
Oh H, Kishimoto T and Yamauchi-Takihara K: Activation of JAK/STAT
pathway transduces cytoprotective signal in rat acute myocardial
infarction. Cardiovasc Res. 47:797–805. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Das A, Salloum FN, Durrant D, Ockaili R
and Kukreja RC: Rapamycin protects against myocardial
ischemia-reperfusion injury through JAK2-STAT3 signaling pathway. J
Mol Cell Cardiol. 53:858–869. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Battle TE and Frank DA: The role of STATs
in apoptosis. Curr Mol Med. 2:381–392. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boengler K, Hilfiker-Kleiner D, Heusch G
and Schulz R: Inhibition of permeability transition pore opening by
mitochondrial STAT3 and its role in myocardial
ischemia/reperfusion. Basic Res Cardiol. 105:771–785. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Heusch G, Musiolik J, Gedik N and
Skyschally A: Mitochondrial STAT3 activation and cardioprotection
by ischemic postconditioning in pigs with regional myocardial
ischemia/reperfusion. Circ Res. 109:1302–1308. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang N, Luo M, Li R, Huang Y, Zhang R, Wu
Q, Wang F, Li Y and Yu X: Blockage of JAK/STAT signalling
attenuates renal ischaemia-reperfusion injury in rat. Nephrol Dial
Transplant. 23:91–100. 2008. View Article : Google Scholar
|
|
25
|
Stephanou A, Brar BK, Knight RA and
Latchman DS: Opposing actions of STAT-1 and STAT-3 on the Bcl-2 and
Bcl-x promoters. Cell Death Differ. 7:329–330. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stephanou A, Scarabelli TM, Townsend PA,
Bell R, Yellon D, Knight RA and Latchman DS: The carboxyl-terminal
activation domain of the STAT-1 transcription factor enhances
ischemia/reperfusion-induced apoptosis in cardiac myocytes. FASEB
J. 16:1841–1843. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press (US); Washington, DC: 2011
|
|
28
|
Zhu HB, Zhang L, Wang ZH, Tian JW, Fu FH,
Liu K and Li CL: Therapeutic effects of hydroxysafflor yellow A on
focal cerebral ischemic injury in rats and its primary mechanisms.
J Asian Nat Prod Res. 7:607–613. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu SX, Zhang Y, Wang YF, Li XC, Xiang MX,
Bian C and Chen P: Upregulation of heme oxygenase-1 expression by
hydroxysafflor yellow A conferring protection from
anoxia/reoxygenation-induced apoptosis in H9c2 cardiomyocytes. Int
J Cardiol. 160:95–101. 2012. View Article : Google Scholar
|
|
30
|
Wu YX, Gao CZ, Fan KL, Yang LM and Mei XF:
STAT1 inhibitor alleviates spinal cord injury by decreasing
apoptosis. Genet Mol Res. 15:2016.
|
|
31
|
Gorina R, Petegnief V, Chamorro A and
Planas AM: AG490 prevents cell death after exposure of rat
astrocytes to hydrogen peroxide or proinflammatory cytokines:
Involvement of the Jak2/STAT pathway. J Neurochem. 92:505–518.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Smith CC, Dixon RA, Wynne AM, Theodorou L,
Ong SG, Subrayan S, Davidson SM, Hausenloy DJ and Yellon DM:
Leptin-induced cardioprotection involves JAK/STAT signaling that
may be linked to the mitochondrial permeability transition pore. Am
J Physiol Heart Circ Physiol. 299:H1265–H1270. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sachdeva J, Dai W, Gerczuk PZ and Kloner
RA: Combined remote perconditioning and postconditioning failed to
attenuate infarct size and contractile dysfunction in a rat model
of coronary artery occlusion. J Cardiovasc Pharmacol Ther.
19:567–573. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
35
|
Liu F, Wei Y, Yang XZ, Li FG, Hu J and
Cheng RF: Hypotensive effects of safflower yellow in spontaneously
hypertensive rats and influence on plasma renin activity and
angiotensin II level. Yao Xue Xue Bao. 27:785–787. 1992.In
Chinese.
|
|
36
|
Ji DB, Zhang LY, Li CL, Ye J and Zhu HB:
Effect of Hydroxysafflor yellow A on human umbilical vein
endothelial cells under hypoxia. Vascul Pharmacol. 50:137–145.
2009. View Article : Google Scholar
|
|
37
|
Panteghini M, Cuccia C, Bonetti G,
Giubbini R, Pagani F and Bonini E: Single-point cardiac troponin T
at coronary care unit discharge after myocardial infarction
correlates with infarct size and ejection fraction. Clin Chem.
48:1432–1436. 2002.PubMed/NCBI
|
|
38
|
Kowalewski M, Urban M, Mroczko B and
Szmitkowski M: Proinflammatory cytokines (IL-6, TNF-alpha) and
cardiac troponin I (cTnI) in serum of young people with ventricular
arrhythmias. Pol Arch Med Wewn. 108:647–651. 2002.In Polish.
PubMed/NCBI
|
|
39
|
Das DK, Engelman RM, Rousou JA, Breyer RH,
Otani H and Lemeshow S: Pathophysiology of superoxide radical as
potential mediator of reperfusion injury in pig heart. Basic Res
Cardiol. 81:155–166. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu J, Hecker JG and Chiamvimonvat N:
Antioxidant enzyme gene transfer for ischemic diseases. Adv Drug
Deliv Rev. 61:351–363. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Taiwo T and Goldstein S: Drug use and its
association with deviant behaviour among rural adolescent students
in South Africa. East Afr Med J. 83:500–506. 2006.
|
|
42
|
Scarabelli TM, Knight R, Stephanou A,
Townsend P, Chen-Scarabelli C, Lawrence K, Gottlieb R, Latchman D
and Narula J: Clinical implications of apoptosis in ischemic
myocardium. Curr Probl Cardiol. 31:181–264. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Salvesen GS: Caspases and apoptosis.
Essays Biochem. 38:9–19. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Barry SP, Townsend PA, Latchman DS and
Stephanou A: Role of the JAK-STAT pathway in myocardial injury.
Trends Mol Med. 13:82–89. 2007. View Article : Google Scholar
|
|
45
|
Shinmura K, Tang XL, Wang Y, Xuan YT, Liu
SQ, Takano H, Bhatnagar A and Bolli R: Cyclooxygenase-2 mediates
the cardio-protective effects of the late phase of ischemic
preconditioning in conscious rabbits. Proc Natl Acad Sci USA.
97:10197–10202. 2000. View Article : Google Scholar
|
|
46
|
Fuglesteg BN, Suleman N, Tiron C, Kanhema
T, Lacerda L, Andreasen TV, Sack MN, Jonassen AK, Mjøs OD, Opie LH
and Lecour S: Signal transducer and activator of transcription 3 is
involved in the cardioprotective signalling pathway activated by
insulin therapy at reperfusion. Basic Res Cardiol. 103:444–453.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mascareno E, El-Shafei M, Maulik N, Sato
M, Guo Y, Das DK and Siddiqui MA: JAK/STAT signaling is associated
with cardiac dysfunction during ischemia and reperfusion.
Circulation. 104:325–329. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Simon AR, Rai U, Fanburg BL and Cochran
BH: Activation of the JAK-STAT pathway by reactive oxygen species.
Am J Physiol. 275:C1640–C1652. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Takagi Y, Harada J, Chiarugi A and
Moskowitz MA: STAT1 is activated in neurons after ischemia and
contributes to ischemic brain injury. J Cereb Blood Flow Metab.
22:1311–1318. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang J, Ouyang C, Chen X, Fu B, Lu Y and
Hong Q: Effect of Jak2 kinase inhibition on Stat1 and Stat3
activation and apoptosis of tubular epithelial cells induced by ATP
depletion/recovery. J Nephrol. 21:919–923. 2008.PubMed/NCBI
|
|
52
|
Dhingra S, Bagchi AK, Ludke AL, Sharma AK
and Singal PK: Akt regulates IL-10 mediated suppression of
TNFα-induced cardiomyocyte apoptosis by upregulating Stat3
phosphorylation. PLoS One. 6:e250092011. View Article : Google Scholar
|
|
53
|
Stephanou A, Brar BK, Scarabelli TM,
Jonassen AK, Yellon DM, Marber MS, Knight RA and Latchman DS:
Ischemia-induced STAT-1 expression and activation play a critical
role in cardio-myocyte apoptosis. J Biol Chem. 275:10002–10008.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bolli R, Dawn B and Xuan YT: Role of the
JAK-STAT pathway in protection against myocardial
ischemia/reperfusion injury. Trends Cardiovasc Med. 13:72–79. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sheng M, Huang Z, Pan L, Yu M, Yi C, Teng
L, He L, Gu C, Xu C and Li J: SOCS2 exacerbates myocardial injury
induced by ischemia/reperfusion in diabetic mice and H9c2 cells
through inhibiting the JAK-STAT-IGF-1 pathway. Life Sci.
188:101–109. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yamaura G, Turoczi T, Yamamoto F, Siddqui
MA, Maulik N and Das DK: STAT signaling in ischemic heart: A role
of STAT5A in ischemic preconditioning. Am J Physiol Heart Circ
Physiol. 285:H476–H482. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jung YY, Lee JH, Nam D, Narula AS,
Namjoshi OA, Blough BE, Um JY, Sethi G and Ahn KS: Anti-myeloma
effects of icariin are mediated through the attenuation of
JAK/STAT3-dependent signaling cascade. Front Pharmacol. 9:5312018.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Han D, Wei J, Zhang R, Ma W, Shen C, Feng
Y, Xia N, Xu D, Cai D, Li Y and Fang W: Hydroxysafflor yellow A
alleviates myocardial ischemia/reperfusion in hyperlipidemic
animals through the suppression of TLR4 signaling. Sci Rep.
6:353192016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Min J and Wei C: Hydroxysafflor yellow A
cardioprotection in ischemia-reperfusion (I/R) injury mainly via
Akt/hexokinase II independent of ERK/GSK-3β pathway. Biomed
Pharmacother. 87:419–426. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Xu AB, Liu JG, Zhang J and LI JX:
Hydroxysafflor yellow A for ameliorating injury of myocardial
ischemia/reperfusion in SD rats. Chin J Evidence Based
Cardiovascular Med. 6:733–736. 2014.In Chinese.
|