Introduction
Glaucoma is a severe eye disease and has been
recognized as one of the leading causes of irreversible blindness
worldwide (1). Glaucoma has been
predicted to affect ~76.0 million people in 2020 globally (2). Despite advancements in the clinical
treatment of this disease, few improvements have been made in
prognosis. It has been reported that oxidative stress and the
apoptosis of retinal ganglion cells (RGCs) are the main hallmarks
of the pathogenesis of glaucoma (3-5).
Therefore, preventing oxidative stress and apoptosis in RGCs may be
an effective therapeutic strategy for treating glaucoma.
Tetramethylpyrazine (TMP), the main active component
of Ligusticum wallichii Franchat, has been used extensively
for the treatment of neurovascular diseases in China for centuries
(6,7). As previously reported, TMP possesses
therapeutic potential in a variety of diseases by mechanisms
mediated by antioxidation and anti-apoptosis (8-10).
Additionally, the neuroprotective effects of TMP in retinal
diseases have been confirmed in vitro and in vivo
(11,12). For example, Yang et al
(10) revealed that TMP could
preserve neuronal morphology and promote survival in retinal cell
cultures, and protected cells against the cytotoxicity of high
doses of hydrogen peroxide. Luo et al (13) suggested that TMP protects RGCs
against N-methyl-D-aspartate-induced excitotoxicity. However, to
the best of our knowledge, the effects of TMP on glaucoma remain
unknown.
MicroRNAs (miRNAs), a class of endogenous small
noncoding RNAs of 18-22 nucleotides, negatively modulate gene
expression at the post-transcription level by inhibiting
translation or inducing RNA degradation (14,15). Several studies have demonstrated
an important role for miRNAs and their target genes in retinal cell
apoptosis (16,17). For example, Li et al
(18) reported that miR-137 was
downregulated under hypoxic conditions and inhibition of miR-137
could protect RGCs against hypoxia-induced apoptosis through
targeting the Notch1 pathway. Zhang et al (19) revealed that miR-187 inhibited the
oxidative stress-induced apoptosis of RGCs by negatively regulating
P2X purino receptor 7. Cheng et al (20) demonstrated that miR-141 attenuated
UV-induced oxidative stress via activating Keap1-Nrf2 signaling in
retinal ganglion cells; however, whether TMP exerts its protective
effect by regulating miRNA in glaucoma remains unclear.
In the present study, we evaluated the protective
effects of TMP against H2O2-induced damage in
primary RGCs (PRGCs) and an in vitro model of oxidative
stress injury; the potential role of the miR-182/mitochondrial
pathway associated with the neuroprotective effects of TMP was
investigated.
Materials and methods
PRGC culture
PRGCs were isolated using Thy1.2-conjugated magnetic
beads from the retinas of 20 male BALB/c mice (weighing 20-30 g,
8-weeks old) obtained from the Laboratory Animal Center of The
First Affiliated Hospital of Xinxiang Medical University (Xinxiang,
China) as previously described (21). All mice were maintained in a
temperature-controlled room (22±2°C) with a 12-h light/dark cycle
and a relative humidity of 40-60%, and had free access to food and
water. For PRGC culture, tissue culture plates were coated with
poly-D-lysine (10 µg/ml; EMD Millipore) and laminin (10
µg/ml; Sigma-Aldrich; Merck KGaA) at 37°C in a humidified
tissue culture incubator with 5% CO2. Primary cells were
identified by immunofluorescence staining with neurofilament-L
(NF-L; cat. no. 2835; Cell Signaling Technology, Inc.) and reverse
transcription-quantitative PCR (RT-qPCR). Western blotting for the
expression of the RGC-specific markers, including Brn3a (cat. no.
MAB1585 clone 5A3.2; EMD Millipore), thymus cell antigen 1 (Thy-1;
cat. no. MAB1406; EMD Millipore) and NF-L was conducted as
described previously (22,23).
The use of animals and the experimental protocols performed were
approved by the Animal Care Committee of The First Affiliated
Hospital of Xinxiang Medical University (approval no. 2017-0163) in
accordance with institutional guidelines. TMP hydrochloride (100
µM) was purchased from Harbin Medisan Pharmaceutical Co.,
dissolved in normal saline and diluted immediately prior to each
experiment.
Establishment of a model of oxidative
stress injury in PRGCs and TMP treatment
The PRGCs were cultured in DMEM supplemented with
10% fetal bovine serum (HyClone; GE Healthcare Life Sciences), 100
U/ml penicillin and streptomycin at 37°C in a humidified atmosphere
with 5% CO2. The PRGCs were treated with different
concentrations H2O2 (0, 100 200, 400 and 800
µM) at 37°C for 16 h. Cell viability was tested to
investigate cell injury induced by H2O2 in
PRGCs, and 400 µM H2O2 was selected to
establish the model of oxidative stress injury in PRGCs.
PRGCs were pre-treated with TMP at different dosages
(25, 50 and 100 µM) at 37°C for 24 h. The cells were then
subjected to oxidative insult with H2O2 (400
µM) at 37°C for 16 h and collected for subsequent
experiments. Blank cells comprised untreated cells and the control
or vehicle was that of cells treated with
H2O2 or H2O2 + DMSO (1%
v/v), respectively.
Cell viability
PRGCs (1×104 cells) were seeded into
96-well plates and cultured overnight at 37°C in a humidified
tissue culture incubator with 5% CO2. At the end of
treatment, cell viability was determined using Cell Counting Kit-8
(CCK-8) assay (Dojindo Molecular Technologies, Inc.). Briefly, 10
µl CCK-8 reagent (Dojindo Molecular Technologies, Inc.) was
added to each well, then the cells were incubated for 3 h at 37°C.
The absorbance of the samples was read at 450 nm with a microplate
reader (Sunrise™; Tecan Group, Ltd.).
Caspase-3 activity assay
At the end of TMP treatment, the activity of
caspase-3 was measured with a caspase-3 assay kit (Abcam) according
to the manufacturer's protocols. The samples were analyzed using a
microplate reader (Model 680, Bio-Rad Laboratories, Inc.) at 405
nm.
Detection of apoptosis by flow
cytometry
PRGCs were seeded in 6-well plates at a density of
1.0×106 cell/well. Following treatment with TMP, the
cells were washed twice with PBS, and then fixed in 70% ice-cold
ethanol in PBS at 4°C for 30 min. Subsequently, the cells were
stained with 5 µl Annexin V-fluorescein isothiocyanate and 1
µl of propidium iodide (Bio-Science, Co. Ltd.). After
incubation for 15 min at room temperature in the dark, cell
apoptosis was analyzed with a FACScan flow cytometer (FCM; Beckman
Coulter, Inc.). FlowJo software version 7.6.1 (FlowJo LLC, USA) was
used to analyze flow cytometry data.
Reactive oxygen species (ROS)
detection
ROS production in PRGCs was analyzed using
2′,7′-dichlorofluorescin-diacetate (DCHF-DA; cat. no. D6883,
Sigma-Aldrich; Merck KGaA). Briefly, PRGCs were incubated with 10
µM DCHF-DA for 30 min at 37°C, followed by two washes with
PBS. Then, the DCFH-DA staining for the detection of ROS production
was observed using a fluorescence microscope (Nikon Corporation).
Fluorescence was read at 485 nm for excitation and 530 nm for
emission with an Infinite M200 Microplate Reader (Tecan Group,
Ltd.).
ELISA
The concentrations of malondialdehyde (MDA) and
superoxide dismutase (SOD) in the conditioned media were analyzed
by an MDA Assay kit (cat. no. MAK085) and SOD Assay kit (cat. no.
19160) from Sigma-Aldrich (Merck KGaA) according to the
manufacturer's protocols.
RT-qPCR
Following treatment, total RNA was extracted from
retinal tissues or cultured cells using a miRNAeasy mini kit
(Qiagen, Inc.) according to the manufacturer's protocols. A total
of 200 ng of RNA was reverse-transcribed with a miRNA reverse
transcription kit (Qiagen, Inc.) and an mRNA RT kit (Invitrogen;
Thermo Fisher Scientific, Inc.), respectively. qPCR was performed
using the iTaqTM Universal SYBR Green Supermix (Bio-Rad
Laboratories, Inc.) on a 7500HT Real-Time PCR System (Thermo Fisher
Scientific, Inc.). Relative expression levels were quantified via
normalization to small nuclear (sno)RNA202. The primers employed
for qPCR were as follows: miR-182 5′-TGC GCT TTG GCA ATG GTA GAA
CTC-3′ (forward) 5′-CCA GTG CAG GGT CCG AGG TAT T-3′ (reverse);
miR-34a 5′-ACA CTC CAG CTG GGT GGC AGT GTC TTA GCT-3′ (forward),
5′-CTC AAC TGG TG TCG TGG A-3′ (reverse); miR-150 5′-TCT CCC AAC
CCT TGT ACC AGT G-3′ (forward) 5′-CTC AAC TGG TGT CGT G G TA-3′
(reverse); miR-214 5′-ATA GAA TTC TTT CT CCC TTT CCC CTT ACT CTC
C-3′ (forward) 5′-CCA GGA TCC TTT CAT AGG CAC CAC TCA CTT TAC-3′
(reverse); miR-137 5′-GCA GCA AGA GTT CTG GTG GC-3′ (forward),
5′-TGG AAC CAG TGC GAA AAC AC-3′ (reverse); miR-210 5′-GTG CAG GGT
CCG AGG T-3′ (forward), 5′-CTG TGC GTG TGA CAG CGG CT GA-3′
(reverse); snoRNA202 5′-GTC GTA TCC AGT GCA GGG TCC GAG GTA TTC GCA
CTG GAT ACG ACC ATC AG-3′ (RT), 5′-GCA GGG TCC GAG GTA TTC-3′
(forward) and 5′-CTT GAT GAA AGT ACT TTT GA-3′ (reverse). Brn3a
5′-TGG CGT CCA TCT GCG ATC-3′ (forward); 5′-CTC AGG TTG TTC ATT TTT
C-3′ (reverse). Thy-1 5′-CGC TTT ATC AAG GTC CTT ACT C-3′ (forward)
5′-GCG TTT TGA GAT ATT TGA A G GT-3′ (reverse). NF-L 5′-ATG CTC AGA
TCT CCG TGG AG A TG-3′ (forward); 5′-GCT TCG CAG CTC ATT CTC CAG
TT-3′ (reverse). and GAPDH 5′-CAT CAA GAA GGT GGT GAA GCA GG-3′
(forward); 5′-CCA CCA CCC TGT TGC TGT AG C CA-3′ (reverse). The
reaction conditions were as follows: 94°C for 5 min, followed by 40
cycles of 94°C for 1 min, 56°C for 1 min and 72°C for 1 min.
RT-qPCR assays were performed in triplicate and the changes in
expression levels were calculated using the 2−ΔΔCq
method (24).
Transfection
PRGCs (5×103) were seeded into each well
of a 6-well plate for 24 h, and the cells were subsequently
transfected with miR-182 mimics, miR-182 inhibitor or the
corresponding control vectors synthesized by Shanghai GenePharma
Co., Ltd., at a final concentration of 50 nM using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) following the manufacturer′s instructions. The
sequences are as follows: miR-182 mimics, 5′-UUU GGC AAU GGU AGA
ACU CAC ACU-3′; mimics negative control (NC), 5′-UUC UCC GAA CGU
GUC ACG UTT-3′; miR-182 inhibitor, 5′-AGU GUG AGU UCU ACC AUU GCC
AAA-3′ and inhibitor NC, 5′-CAG UAC UUU UGU GUA GUA CAA-3′. After
24 h post-transfection, the cells were pre-treated with 100
µM TMP for 24 h, then subjected to oxidative insult with
H2O2 (400 µM) at 37°C for 16 h and
collected for subsequent experiments.
Bioinformatics
TargetScan (version 7.0; www.targetscan.org/) and PicTar (release 2006;
https://pictar.mdc-berlin.de) target
gene prediction software were used to select MIF as a target gene
of miR-182.
Luciferase reporter assay
The Bcl-2 interacting protein 3 (BNIP3)
3′-untranslated region (UTR) containing complementary sequences or
the mutated sequence for the seed sequence of miR-182 was amplified
by PCR as described previously (25), and cloned into the firefly
luciferase expressing vector, pGL3 (Promega Corporation); the
vectors were denoted as wild-type (wt) pGL3-BNIP3-3′-UTR and mutant
(mut) BNIP3-3′-UTR, respectively. For the luciferase reporter
assay, 293T cells (American Type Culture Collection) were seeded
into a 24-well plate (5.0×105/well) and transfected with
the wt or mut reporter vector, together with miR-182 mimics or
miR-182 inhibitor using Lipofectamine 2000. At 48 h after
transfection, the luciferase activity was determined by using a
dual-luciferase reporter assay system (Promega Corporation). The
pRL-TK plasmid (Promega Corporation) was used as a normalizing
control. All experiments were performed in triplicate.
Western blotting
Total protein was extracted from PRGCs or retinal
tissues using radioimmunoprecipitation assay lysis (Sigma-Aldrich;
Merck KGaA) after treatments. The protein concentration was
determined using a BCA protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Total protein samples (30 µg) were
analyzed by 8% SDS-PAGE and transferred to a polyvinylidene
fluoride membrane (EMD Millipore). β-actin was used for the
normalization of protein expression. The membranes were blocked
with 5% non-fat milk at 4°C overnight, and incubated with primary
antibodies overnight at 4°C. Primary antibodies against
Bcl-2-assoiated X protein (Bax; cat. no. sc-4239, 1:1,000), Bcl-2
(cat. no. sc-176463, 1:1,000), BNIP3 (cat. no. sc-1715, 1:1,000)
and β-actin (cat. no. sc-8432, 1:2,000) were purchased from Santa
Cruz Biotechnology, Inc., while cleaved (cle)-caspase-3 (cat. no.
9661, 1:1,000), cle-poly(ADP-ribose)polymerase (PARP; cat. no.
5625, 1:1,000), Cytochrome c (cyto c cat. no. 4280, 1:1,000)
and Complex IV (cat. no. 4850, 1:1,000) were purchased from Cell
Signaling Technology, Inc. After washing with PBS, the membrane was
incubated with horseradish peroxidase-conjugated antibody (cat. no.
ab205718; 1:2,000, Abcam) for 1 h at room temperature, and the
bands were detected with an ECL Advance reagent (GE Healthcare).
The intensity of the bands of interest was analyzed with ImageJ
software version 1.46 (National Institutes of Health).
Mitochondrial membrane potential
assay
After treatments, a commercial Mitochondrial
Membrane Potential Assay kit with JC-1 (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to determine the mitochondrial membrane
potential (ΔΨm) according to the manufacturer's instructions.
Briefly, after washing twice with PBS, PRGCs were stained with JC-1
(5 µg/ml) for 15 min at 37°C. The red and green fluorescence
intensities were detected (excitation wavelength of 488 nm and
emission wavelength at 500 nm) using a Bio-Tek fluorescent
microplate reader (BioTek Instruments, Inc.). The ΔΨm of the PRGCs
in each treatment group was calculated as the ratio of red to green
fluorescence and expressed as a multiple of the level in the
control group.
Statistical analyses
SPSS 13.0 software (SPSS, Inc.) was used to analyze
the data. Data were expressed as the mean ± standard deviation of
three independent experiments. A Student's t-test was used to
analyze differences between two groups. Differences between
multiple groups were analyzed by one-way analysis of variance,
followed by a Tukey's post-hoc test for multiple comparisons.
P<0.05 was determined to indicate a statistically significant
difference.
Results
TMP promotes cell viability and
suppresses cell apoptosis in H2O2-treated
PRGCs
At days 3 and 7 of the first passage, PRGCs were
collected for immunofluorescence analysis of an RGC marker.
Membranous staining of NF-L was observed in the soma and neurites
(Fig. 1A). To verify the purity
of the isolated PRGC population, the expression levels of NF-L,
Thy-1 and Brn3a (RGC markers) were detected by RT-qPCR. As
presented in Fig. 1B-D, the
expression levels of these markers were significantly increased in
the isolated PRGCs compared with in retinal tissue. Similar results
were observed via western blotting (Fig. 1E). These data demonstrated that
the isolated neoplastic stromal cell populations were not
contaminated by histiocytes or giant cells.
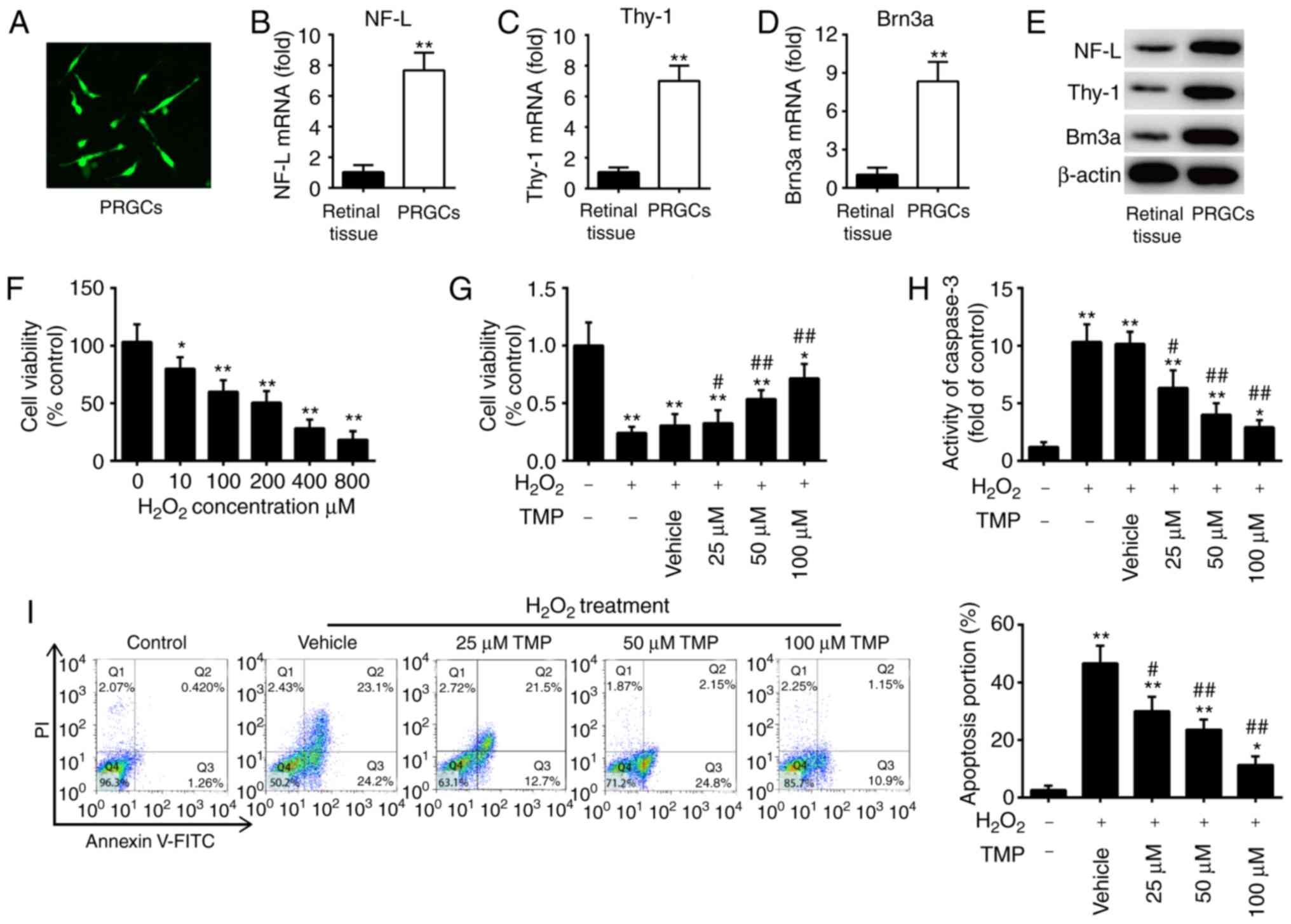 | Figure 1TMP promotes cell viability and
suppresses cell apoptosis in H2O2-treated
PRGCs. (A) NF-L expression was detected by immunofluorescence
(magnification, ×200). (B-D) The mRNA expression levels of NF-L,
Thy-1 and Brn3a were measured by reverse transcription-quantitative
PCR. (E) Protein expression of NF-L, Thy-1 and Brn3a as determined
by western blotting. β-actin was used as a loading control. (F)
PRGCs were treated with 10, 100, 200, 400 and 800 µM of
H2O2 for 24 h, and then cell viability was
determined by a CCK-8 assay. PRGCs were pre-treated with 25, 50 and
100 µM TMP for 24 h prior to exposure with 400 µM of
H2O2. (G) Cell viability was determined by a
CCK-8 assay. (H) The activity of caspase-3 was determined using a
caspase-3 assay kit. (I) Apoptosis was detected via flow cytometric
analysis. Data are presented as mean of three replicates ± standard
deviation. *P<0.05, **P<0.01 vs.
Control group, #P<0.05, ##P<0.01 vs.
Vehicle + H2O2 group. CCK-8, Cell Counting
Kit-8; FITC, fluorescein isothiocyanate; NF-L, neurofilament-L; PI,
propidium iodide; PRGCs, primary retinal ganglion cells; Thy-1,
thymus cell antigen 1; TMP, tetramethylpyrazine. |
H2O2 is widely used to
establish an oxidative injury model using RGCs to mimic the
development of glaucoma in vitro (26,27). As presented in Fig. 1F, H2O2
significantly suppressed the viability of PRGCs in a dose-dependent
manner; however, no significant difference in viability between 400
and 800 µM was observed. Based on previous reports (17,28,29), we used 400 µM
H2O2 to generate a cell model of oxidative
injury. To determine whether TMP promotes the growth of PRGCs, we
treated cells with 25, 50 and 100 µM TMP for 24 h, followed
by 400 µM H2O2. Subsequently, cell
viability was evaluated by a CCK-8 assay. The results showed that
TMP alleviated H2O2-induced suppression of
cell viability, and this effect was does-dependent (Fig. 1G). Further investigation revealed
that H2O2 significantly increased the
activity of caspase-3 compared with in the control group, whereas
TMP attenuated the promoting effects of H2O2
on caspase-3 activity (Fig. 1H).
Consistent with these results, TMP also significantly attenuated
H2O2-induced cell apoptosis (Fig. 1I). Of note, 100 µM TMP was
used for subsequent experiments and this concentration has also
been used previously (17,28,29).
Collectively, these results indicated that the oxidative injury
model of PRGCs was successfully established and TMP treatment could
improve H2O2-induced cell damage.
TMP attenuates
H2O2-induced oxidative damage in PRGCs
It has been acknowledged that glaucoma is associated
with oxidative stress, which can lead to the apoptosis of RGCs
(4,5). MDA is a biomarker of oxidative
stress of cells; SOD is the main antioxidative enzyme and can
resist the damage of oxygen-free radicals to cells (30). Therefore, in the present study,
the levels of ROS, MDA and SOD were examined to assess the
protective effects of TMP in H2O2-treated
PRGCs. As presented in Fig. 2A and
B, H2O2 treatment significantly increased
intracellular ROS levels compared with in Blank cells. However,
pretreatment with TMP significantly decreased ROS levels in a
dose-dependent manner. It was also observed that
H2O2 treatment significantly increased the
levels of MDA and decreased the levels of SOD compared with that in
Blank cells, but these effects were significantly attenuated by TMP
(Fig. 2C and D). Our findings
suggested that TMP attenuated H2O2-induced
cell damage by reducing oxidative stress.
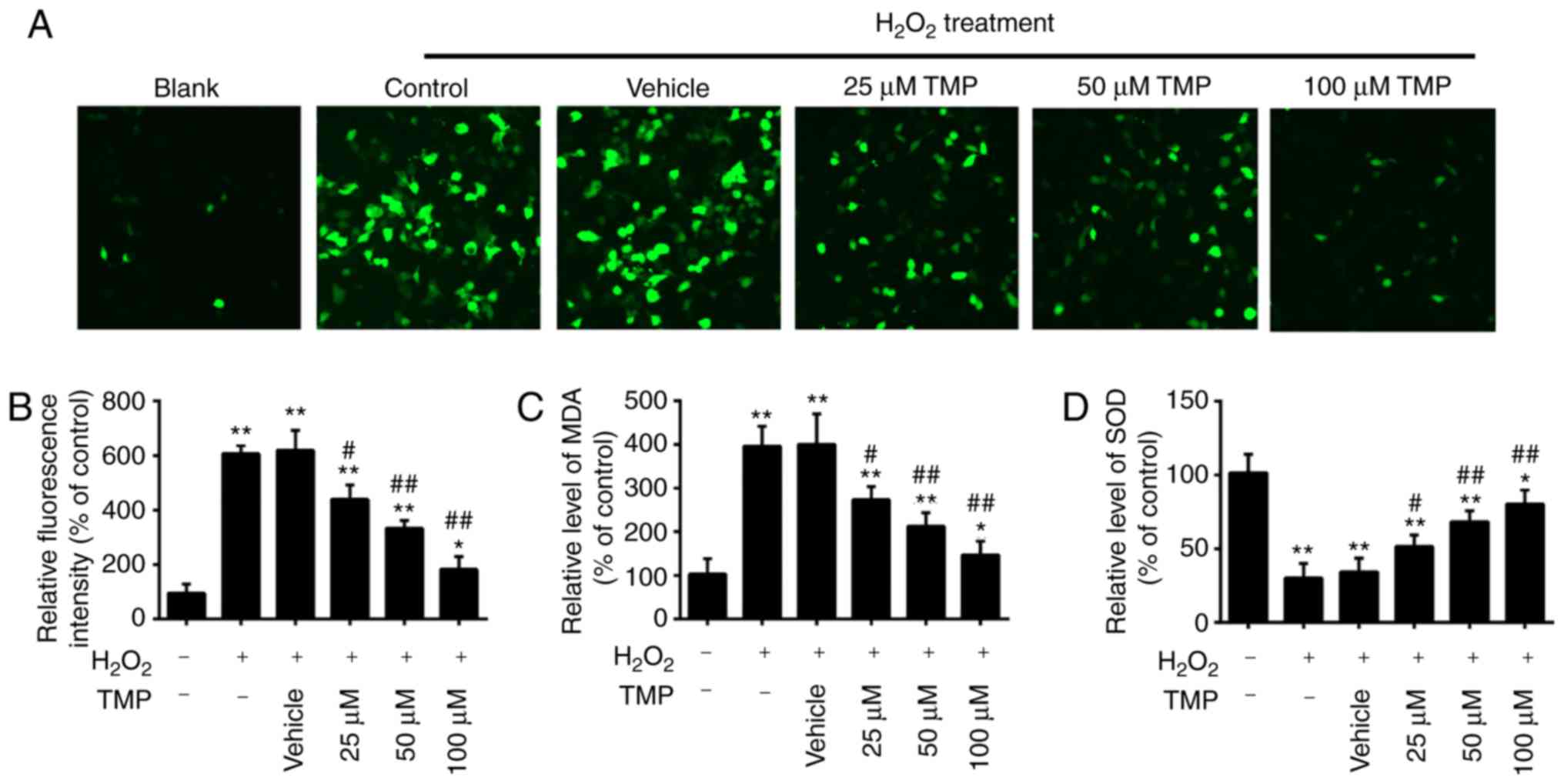 | Figure 2TMP attenuates
H2O2-induced oxidative damage in PRGCs. PRGCs
were pre-treated with 25, 50 and 100 µM of TMP for 24 h
prior to being exposed to 400 µM of
H2O2. (A and B) ROS production was measured
by a 2′,7′-dichlorofluorescin-diacetate assay (magnification,
×200). (C and D) The levels of MDA and SOD were detected by an
ELISA assay. Data are presented as the mean of three replicates ±
standard deviation. *P<0.05, **P<0.01
vs. Control group, #P<0.05, ##P<0.01
vs. Vehicle + H2O2 group. PRGCs, primary
retinal ganglion cells; MDA, malondialdehyde; SOD, superoxide
dismutase; TMP, tetramethylpyrazine. |
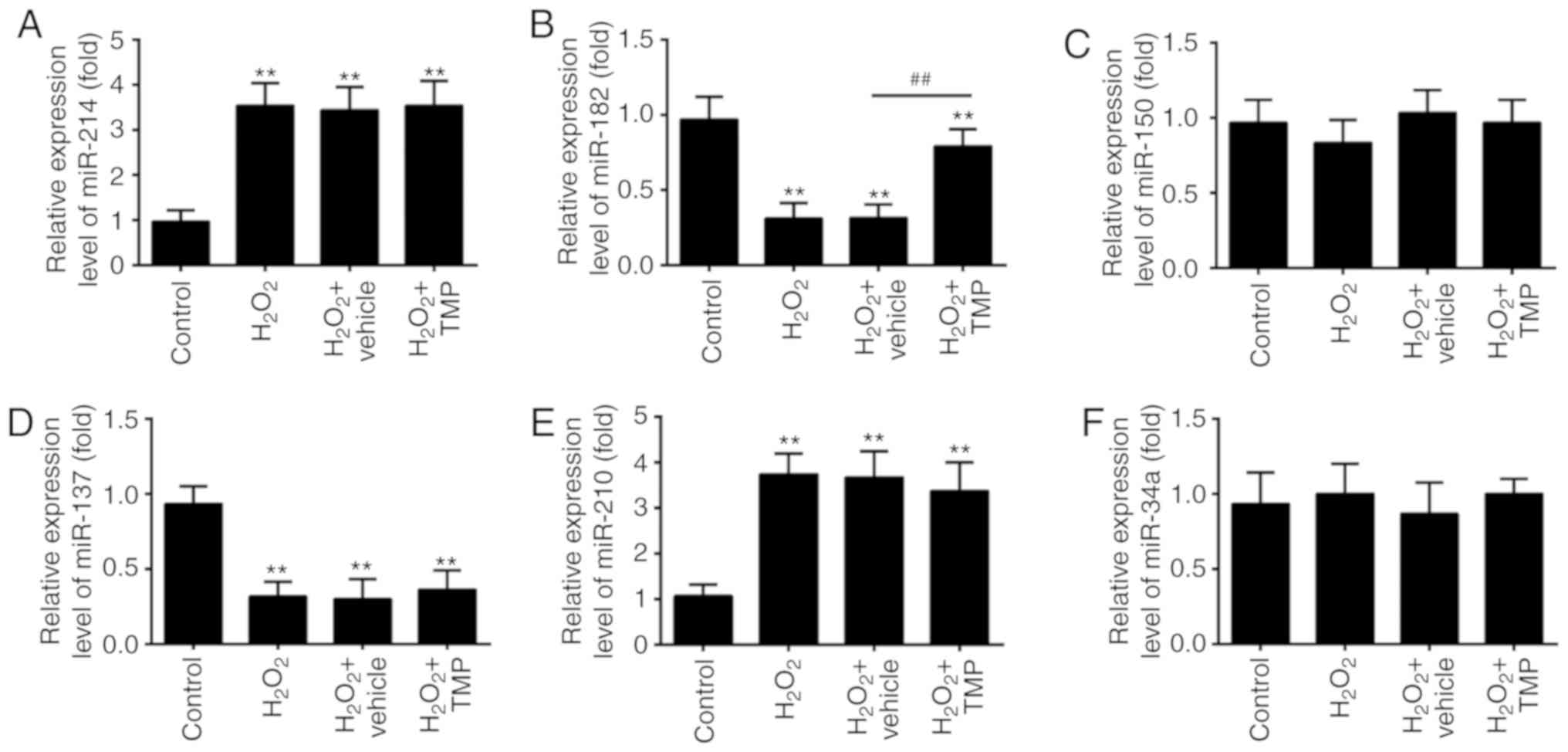
TMP upregulated the expression of
miR-182
Recently, ROS has been reported to alter the
expression of certain miRNAs (31,32). Given TMP suppressed the
accumulation of ROS, we hypothesized that TMP exerts its protective
effects against H2O2-induced cell injury via
the regulation of miRNAs. Thus, we investigated the regulatory
effects of TMP on the changes in the expression of miRNAs, which
have been reported be altered in response to ROS exposure
previously (33). The results of
RT-qPCR showed that H2O2 significantly
increased the expression of miR-214 and miR-210, and decreased the
expression of miR-182 and miR-137, compared with control group,
which corresponds with previous studies (34-37). Interestingly, the expression
levels of miR-150 and miR-34a were markedly unaltered after
H2O2 treatment, which differs with previous
reports (38,39); this may be due to the use of
different cell lines. Of note, the expression of miR-182 in
H2O2-treated PRGCs was significantly
downregulated compared with the control, but increased in response
to TMP; however, no significant changes were observed in the
expression of miR-214, miR-137 and miR-210 following TMP treatment
(Fig. 3A-F). These findings
prompted us to investigate the role of miR-182 in TMP-mediated
protective effects against H2O2-induced cell
damage.
Knockdown of miR-182 inhibits the
protective effects of TMP in H2O2-induced
cell damage
Previous studies have been reported that miR-182
possesses potent antioxidative and antiapoptotic activity in a
variety of diseases (33,35). To investigate whether ectopic
expression of miR-182 involves the protective effects of TMP on
H2O2-induced cell damage, miR-182 inhibitor
or miR-182 mimics were transfected into PRGCs prior to
H2O2 and TMP treatment. As presented in
Fig. 4A, miR-182 expression was
significantly decreased and increased after miR-182 inhibitor or
miR-182 mimics transfection in PRGCs, respectively. It was observed
that miR-182 knockdown significantly alleviated the protective
effects of TMP in H2O2-treated PRGCs, by
reducing cell viability and inducting apoptosis (Fig. 4B and C). The intracellular ROS
levels in the H2O2 + TMP + miR-182 inhibitor
group were significantly higher than H2O2 +
TMP group (Fig. 4D). In addition,
as shown in Fig. 4E and F, the
levels of MDA were significantly increased and the levels of SOD
were decreased in the H2O2 + TMP + miR-182
inhibitor group, compared with that of the
H2O2 + TMP group. These findings suggested
that miR-182 knockdown suppressed the protective effects of TMP in
H2O2-induced injury.
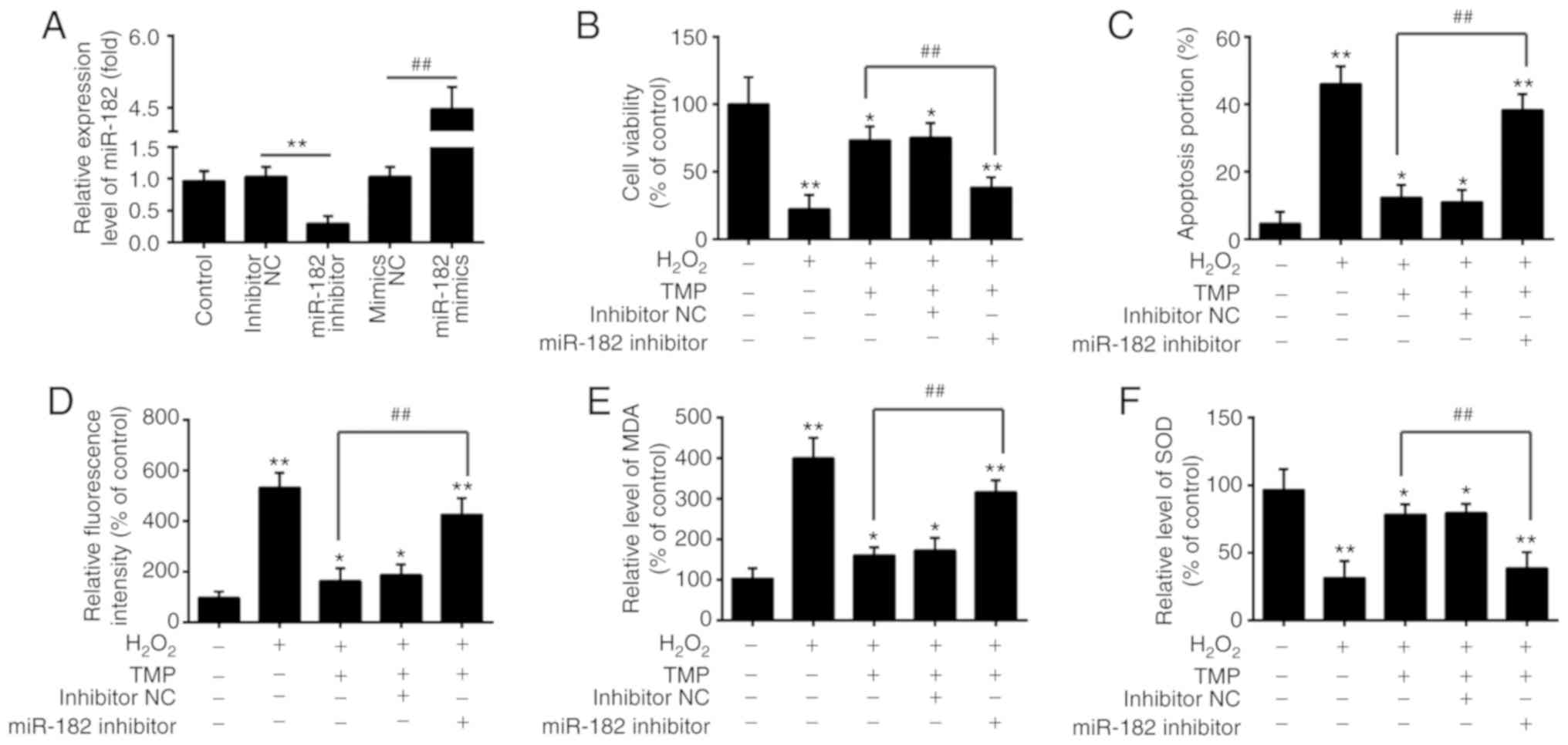 | Figure 4Knockdown of miR-182 inhibits the
protective effects of TMP in H2O2-induced
cell damage. (A) The expression of miR-182 was measured by reverse
transcription-quantitative PCR after miR-182 inhibitor or miR-182
mimics transfection. Data are presented as the mean of three
replicates ± standard deviation. **P<0.01 vs.
inhibitor NC group, ##P<0.01 vs. mimics NC group.
Cells were transfected with miR-182 inhibitor or inhibitor NC
before treatment with TMP for 24 h, and then exposed to
H2O2. (B) Cell viability was then determined
by a Cell Counting Kit-8 assay. (C) Apoptosis was detected by flow
cytometric analysis. (D) ROS production was measured by a
2′,7′-dichlorofluorescin-diacetate assay. (E and F) The levels of
MDA and SOD were detected by an ELISA assay. Data are presented as
the mean of three replicates ± standard deviation.
*P<0.05, **P<0.01 vs. Control group,
##P<0.01 vs. H2O2 + TMP group.
MDA, malondialdehyde; miR, microRNA; NC, negative control; ROS,
reactive oxygen species; SOD, superoxide dismutase; TMP,
tetramethylpyrazine. |
BINP3 is a direct target of miR-182
To explore the molecular mechanism by which miR-182
functions in the protective effects of TMP in
H2O2-induced injury, we performed TargetsScan
and PicTar analyses to predict the target genes of miR-182 and
identified BNIP3 as a potential target of miR-182, with the target
site located in the 3′-UTR (Fig.
5A). To validate this bioinformatic predication, we established
the luciferase reporter plasmids containing the wt or mut 3′-UTR
segments of BNIP3. The luciferase reporter assay revealed that
miR-182 mimics significantly inhibited the luciferase activity
compared with the mimic NC, while miR-182 inhibitor significantly
enhanced the luciferase activity compared with the inhibitor NC
(Fig. 5B). Additionally, miR-182
mimics or inhibitor did not markedly affect luciferase activity
when the targeted BNIP3 sequence was mutated in the miR-182-binding
site (Fig. 5B). To further
confirm that BNIP3 expression is regulated by miR-182, BNIP3
protein expression was analyzed by western blotting. We found that
the expression levels of BNIP3 were notably downregulated by
miR-182 mimics, but markedly enhanced by miR-182 inhibitor
(Fig. 5C). Taken together, these
findings indicated that miR-182 inhibit the expression of BNIP3,
further suggesting that the miR-182/BNIP3 axis may be involved in
the protective effects of TMP in H2O2-induced
injury.
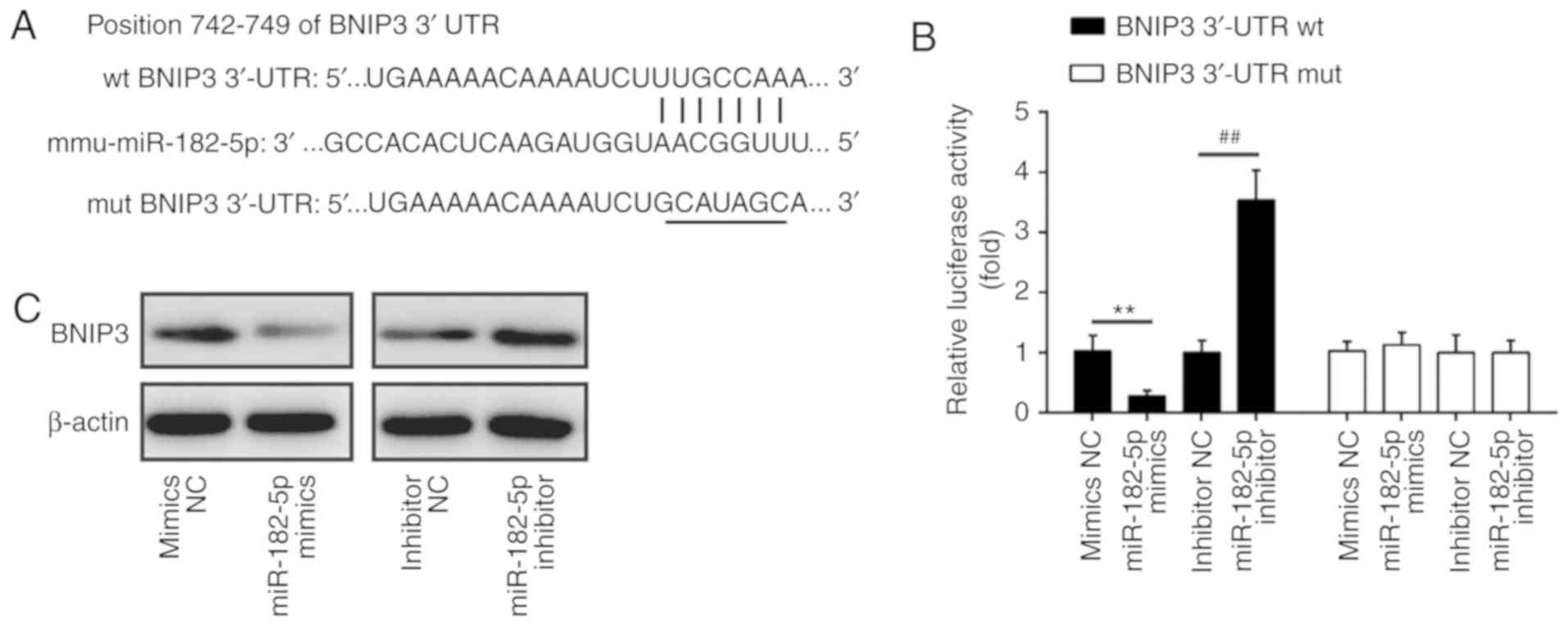 | Figure 5BNIP3 is a direct target of miR-182.
(A) The putative binding site of miR-182 and BNIP3; (B) a
luciferase assay of 293 co-transfected with firefly luciferase
constructs containing the BNIP3 wt or mut 3′-UTR and miR-182
mimics, mimic NC, miR-182 inhibitor or inhibitor NC (n=3). Data are
presented as the mean of three replicates ± standard deviation.
**P<0.01 vs. mimics NC, ##P<0.01 vs.
inhibitor NC. (C) The expression of BNIP3 protein after
transfection with miR-182 mimic or miR-182 inhibitor, as measured
by western blotting. β-actin was used as a loading control. BNIP3,
Bcl-2 interacting protein 3; miR, microRNA; mut, mutated; NC,
negative control; UTR, untranslated region; wt, wild-type. |
TMP attenuates
H2O2-induced PRGC apoptosis via the
miR-182/mitochondrial apoptotic pathway
BNIP3 is a well-known effector of the
mitochondria-mediated apoptosis, which induces the formation of the
pathological mitochondrial permeability transition pore (40,41). Thus, we sought to determine
whether TMP could regulate the mitochondrial apoptotic pathway via
the miR-182/BNIP3 axis. Western blotting was performed to detect
the expression levels of apoptosis-related proteins in PRGCs
transfected with miR-182 inhibitor prior to
H2O2 and TMP treatment. As presented in
Fig. 6A,
H2O2 treatment significantly increased the
expression of pro-apoptotic proteins (BNIP3, Bax, cle-caspase-3 and
cle-PARP) and decreased that of anti-apoptotic Bcl-2, compared with
the control group. Of note, the levels of these pro-apoptotic
proteins were suppressed, whereas the anti-apoptotic protein was
upregulated after TMP pretreatment. However, these effects of TMP
were attenuated by miR-182 inhibitor. cyto c release from
mitochondria into the cytosol is a critical event in apoptosis
(42). Therefore, we further
detected the effects of TMP on cyto c release. The data showed that
H2O2 treatment induced the release of cyto c
from the mitochondria, which was attenuated by TMP. However, the
inhibitory effects of TMP were significantly reversed by knockdown
of miR-182 (Fig. 6B). In
addition, whether TMP could reduce the
H2O2-induced Δψm loss was investigated as
dysregulated mitochondrial function causes the loss of Δψm. As
presented in Fig. 6C,
H2O2 treatment resulted in decreased
red/green fluorescence ratio, which indicated that Δψm had
dissipated, whereas TMP pretreatment attenuated
H2O2-induced Δψm loss. However, knockdown of
miR-182 significantly suppressed the promoting effects of TMP on
Δψm. These results suggest that TMP may attenuate
H2O2-induced PRGC apoptosis via the
miR-182/mitochondrial apoptotic pathway.
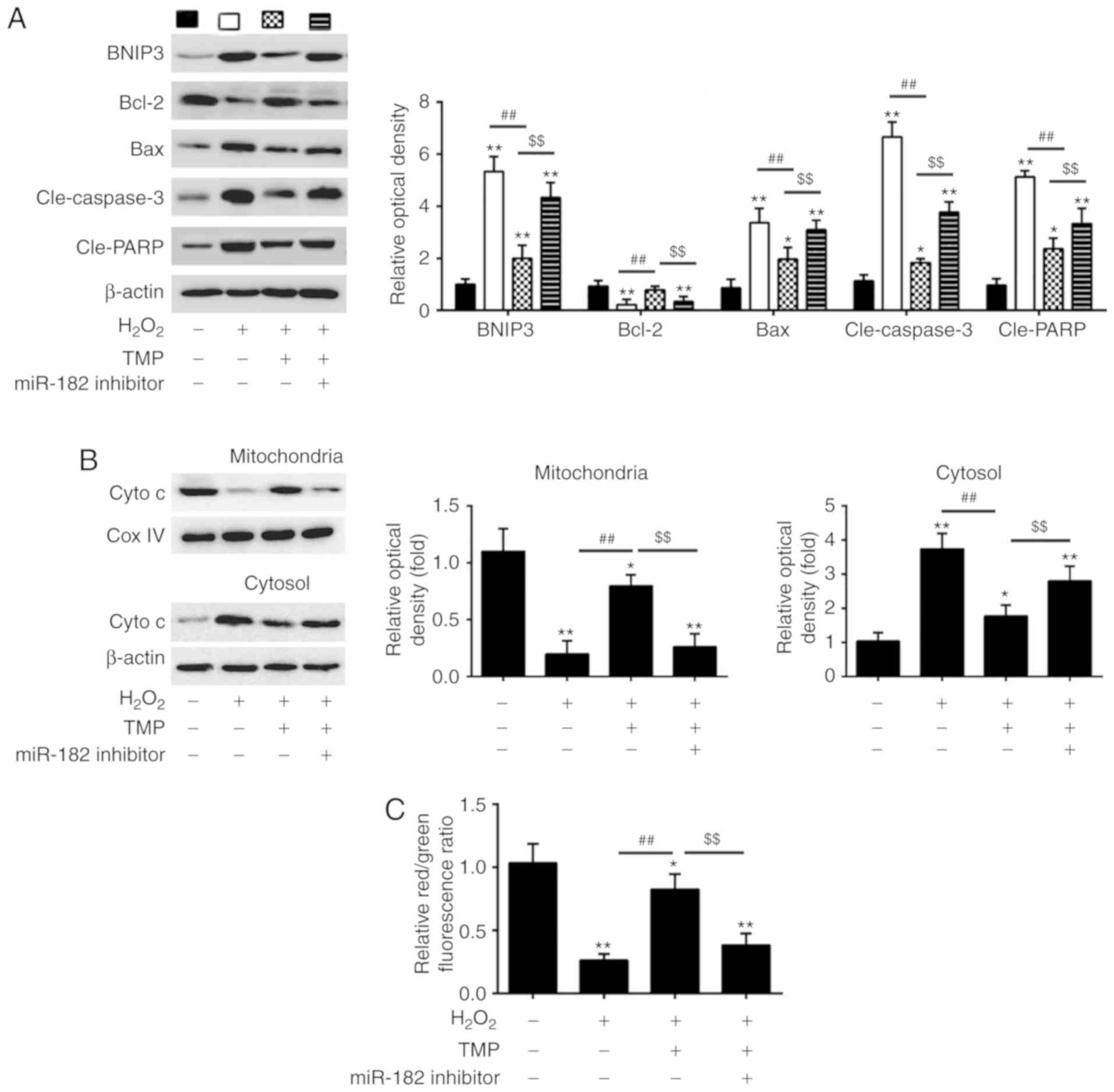 | Figure 6TMP attenuates
H2O2-induced PRGC apoptosis via
miR-182/mitochondrial apoptotic pathway. Cells were transfected
with miR-182 inhibitor or inhibitor NC prior to treatment with TMP
for 24 h and then exposed to H2O2. (A)
Western blot analysis was conducted to determine the expression of
apoptosis-related proteins (BNIP3, Bcl-2, cle-caspase-3, Bax and
cle-PARP and Bcl-2). (B) Protein levels of cyto c in mitochondria
and cytosol was measured using western blot analysis. β-actin and
Cox IV were used as loading controls for the cytosolic and
mitochondrial fractions, respectively. (C) Mitochondrial membrane
potential levels in the treated PRGCs were analyzed using the JC-1
fluorescent probe. Data are presented as the mean of three
replicates ± standard deviation. *P<0.05,
**P<0.01 vs. Control group, ##P<0.01
vs. H2O2 group, $$P<0.01 vs.
H2O2 + TMP group. Bcl-2, B-cell lymphoma 2;
Bax, Bcl-2-assoiated X protein; BNIP3, Bcl-2 interacting protein 3;
cle, cleaved; Cox, IV, Complex IV; Cyto c, cytochrome c;
miR, microRNA; PARP, poly(ADP-ribose)polymerase; PRGCs, primary
retinal ganglion cells; TMP, tetramethylpyrazine. |
Discussion
To the best of our knowledge, the present study is
the first to demonstrate that TMP could attenuate
H2O2-induced damage in PRGCs by suppressing
apoptosis and oxidative stress. Additionally, we reported that TMP
increased the expression of miR-182 and that miR-182 mediated the
protective functions of TMP in H2O2-induced
damage by targeting BNIP3 via inhibiting the mitochondrial
apoptotic pathway. Therefore, our findings suggest that TMP may be
a potential therapeutic agent for the treatment of glaucoma.
Previously, TMP has been reported to exhibit
neuropro-tective effects against retinal diseases in a number of
in vitro and in vivo studies (43-45). Wang et al (46) found that the protective effects of
TMP against all-trans-retinal toxicity in differentiated Y-79
cells, an in vitro model of photoreceptors, were mediated
via upregulation of interphotoreceptor retinoid-binding protein
expression. Yu et al (47)
revealed that TMP attenuated transforming growth factor-β-induced
pathological changes in the trabecular meshwork through the C-X-C
chemokine receptor type 4 pathway, indicating that TMP is a
potential therapeutic method for treating primary open-angle
glaucoma; the roles of TMP in glaucoma require further
investigation. The present study extensively evaluated the
protective effects of TMP against
H2O2-induced damage in PRGCs, an in
vitro model of glaucoma. The results showed that TMP
pre-treatment attenuated H2O2 induced PRGC
injury, as suggested by increases in cell viability, reductions in
the activity of caspase-3 and cell apoptosis, as well as the low
levels of ROS, the high levels of MDA and the low level of SOD.
Collectively, these results indicated that TMP treatment exerts its
protective effects against H2O2-induced
damage through suppressing apoptosis and oxidative stress.
It has been proposed that oxidative stress is an
important mechanism involved in triggering RGC apoptosis in
glaucoma (48); however, the
precise nature of RGC damage caused by oxidative stress remains
unclear. Emerging evidence suggests that ROS can modulate the
expression of some miRNAs in many diseases (49,50). For example, miR-181a has been
shown to be upregulated upon treatment with 600 µM
H2O2 in rat bone marrow mesenchymal stem
cells and this increased expression induced cell death (51). In this study, TMP could inhibit
ROS production; thus, the protective effects of TMP in
H2O2-induced injury may be mediated by miRNAs
induced by ROS. In the present study, a total of six differentially
expressed miRNAs that were associated with ROS were screened out,
in which the levels of miR-182 were significantly downregulated
upon exposure to H2O2, but were upregulated
following treatment with TMP. Interestingly, the literature shows
miR-182 to be involved in anti-apoptotic and antioxidative
processes (52,53). For example, miR-182 inhibited
oxidative stress and apoptosis induced by oxidized low-density
lipoprotein via targeting Toll-like receptor 4 in RAW264.7 cells
(35). Thus, we investigated
whether TMP attenuated H2O2-induced damage
via regulating miR-182. As expected, suppression of miR-182
abolished the protective effects of TMP on
H2O2-induced damage; however, how miR-182
functions in the protective effects of TMP remains unknown.
We explored the underlying molecular mechanism
responsible for the protective effects of TMP in
H2O2-induced damage. Based on bioinformatics
analysis and the dual-lucif-erase reporter assay, our results
showed that miR-182 could directly target BNIP3, an effector of
mitochondria-mediated apoptosis (54-56). Therefore, the protective effects
of TMP on H2O2-induced damage may be mediated
by the miR-182/mitochondria apoptotic pathway. In this study, our
results demonstrated that TMP downregulated BNIP3,
cleaved-caspase-3, Bax and cleaved-PARP, and increased Bcl-2
expression in the H2O2-treated PRGCs.
However, these effects were attenuated by miR-182 inhibition.
Furthermore, TMP attenuated H2O2-induced
release of cyto c and alleviated dissipated Δψm. Similarly, these
effects were attenuated by miR-182 inhibition. Taken together,
these findings indicated that TMP exerts its protective effects on
H2O2-induced damage via modulating the
miR-182/mitochondria apoptotic pathway.
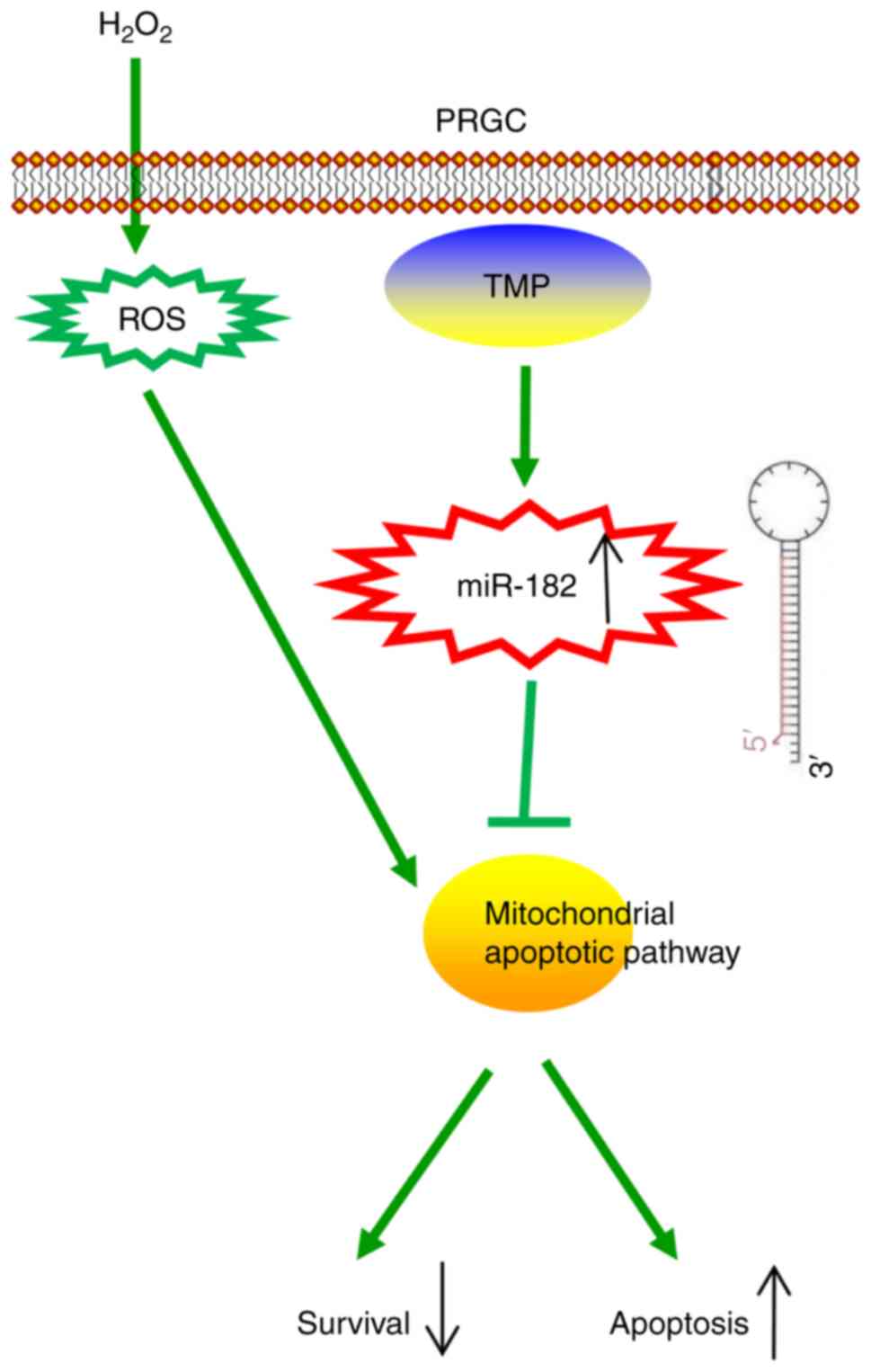
In conclusion, the present study revealed that TMP
improved H2O2-induced damage in PRGCs via the
miR-182/mitochondria apoptotic pathway (Fig. 7). Our findings suggest that TMP
may be considered as a candidate therapeutic agent for the
prevention and treatment of glaucoma. However, the application and
efficacy of TMP in clinical practice requires further
investigation.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study
are included in this published article.
Authors' contributions
XL, QW, YR, XW, HC, HY and BW performed the
experiments, contributed to data analysis and wrote the paper. HY
and BW made substantial contributions to the design of the present
study, contributed to data analysis and acquired experimental
materials. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The use of animals and the experimental protocols
performed were approved by the Animal Care Committee of The First
Affiliated Hospital of Xinxiang Medical University (approval no.
2017-0163) in accordance with institutional guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Koriyama Y, Ohno M, Kimura T and Kato S:
Neuroprotective effects of 5-S-GAD against oxidative stress-induced
apoptosis in RGC-5 cells. Brain Res. 1296:187–195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tham YC, Li X, Wong TY, Quigley HA, Aung T
and Cheng CY: Global prevalence of glaucoma and projections of
glaucoma burden through 2040: A systematic review and
meta-analysis. Ophthalmology. 121:2081–2090. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee D, Shim MS, Kim KY, Noh YH, Kim H, Kim
SY, Weinreb RN and Ju WK: Coenzyme Q10 inhibits glutamate
excitotoxicity and oxidative stress-mediated mitochondrial
alteration in a mouse model of glaucoma. Invest Ophthalmol Vis Sci.
55:993–1005. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sancho P, Fernández C, Yuste VJ, Amrán D,
Ramos AM, de Blas E, Susin SA and Aller P: Regulation of
apoptosis/necrosis execution in cadmium-treated human promonocytic
cells under different forms of oxidative stress. Apoptosis.
11:673–686. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Almasieh M, Wilson AM, Morquette B, Cueva
Vargas JL and Di Polo A: The molecular basis of retinal ganglion
cell death in glaucoma. Prog Retin Eye Res. 31:152–181. 2012.
View Article : Google Scholar
|
|
6
|
Zhai L, Zhang P, Sun RY, Liu XY, Liu WG
and Guo XL: Cytoprotective effects of CSTMP, a novel stilbene
derivative, against H2O2-induced oxidative stress in human
endothelial cells. Pharmacol Rep. 63:1469–1480. 2011. View Article : Google Scholar
|
|
7
|
Wu J, Song R, Song W, Li Y, Zhang Q, Chen
Y, Fu Y, Fang W, Wang J, Zhong Z, et al: Chlorpromazine protects
against apoptosis induced by exogenous stimuli in the developing
rat brain. PLoS One. 6:e219662011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tang Z, Wang Q, Xu H and Zhang W:
Microdialysis sampling for investigations of tetramethylpyrazine
following transdermal and intraperitoneal administration. Eur J
Pharm Sci. 50:454–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gong X, Wang Q, Tang X, Wang Y, Fu D, Lu
H, Wang G and Norgren S: Tetramethylpyrazine prevents
contrast-induced nephropathy by inhibiting p38 MAPK and FoxO1
signaling pathways. Am J Nephrol. 37:199–207. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang G, Qian C, Wang N, Lin C, Wang Y,
Wang G and Piao X: Tetramethylpyrazine protects against
oxygen-glucose deprivation-induced brain microvascular endothelial
cells injury via Rho/Rho-kinase signaling pathway. Cell Mol
Neurobiol. 37:619–633. 2017. View Article : Google Scholar
|
|
11
|
Lu C, Zhang J, Shi X, Miao S, Bi L, Zhang
S, Yang Q, Zhou X, Zhang M, Xie Y, et al: Neuroprotective effects
of tetramethyl-pyrazine against dopaminergic neuron injury in a rat
model of Parkinson's disease induced by MPTP. Int J Biol Sci.
10:350–357. 2014. View Article : Google Scholar :
|
|
12
|
Zhong M, Ma W, Zhang X, Wang Y and Gao X:
Tetramethyl pyrazine protects hippocampal neurons against
anoxia/reoxygenation injury through inhibiting apoptosis mediated
by JNK/MARK signal pathway. Med Sci Monit. 22:5082–5090. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Luo X, Yu Y, Xiang Z, Wu H, Ramakrishna S,
Wang Y, So KF, Zhang Z and Xu Y: Tetramethylpyrazine nitrone
protects retinal ganglion cells against
N-methyl-d-aspartate-induced excitotoxicity. J Neurochem.
141:373–386. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo R, Shen W, Su C, Jiang S and Wang J:
Relationship between the Pathogenesis of Glaucoma and miRNA.
Ophthalmic Res. 57:194–199. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kong N, Lu X and Li B: Downregulation of
microRNA-100 protects apoptosis and promotes neuronal growth in
retinal ganglion cells. BMC Mol Biol. 15:252014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li H, Zhu Z, Liu J, Wang J and Qu C:
MicroRNA-137 regulates hypoxia-induced retinal ganglion cell
apoptosis through Notch1. Int J Mol Med. 41:1774–1782. 2018.
|
|
19
|
Zhang QL, Wang W, Alatantuya, Dongmei, Lu
ZJ, Li LL and Zhang TZ: Down-regulated miR-187 promotes oxidative
stress-induced retinal cell apoptosis through P2X7 receptor. Int J
Biol Macromol. 120:801–810. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cheng LB, Li KR, Yi N, Li XM, Wang F, Xue
B, Pan YS, Yao J, Jiang Q and Wu ZF: miRNA-141 attenuates
UV-induced oxida-tive stress via activating Keap1-Nrf2 signaling in
human retinal pigment epithelium cells and retinal ganglion cells.
Oncotarget. 8:13186–13194. 2017.PubMed/NCBI
|
|
21
|
Jiao J, Huang X, Feit-Leithman RA, Neve
RL, Snider W, Dartt DA and Chen DF: Bcl-2 enhances Ca(2+) signaling
to support the intrinsic regenerative capacity of CNS axons. EMBO
J. 24:1068–1078. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rodriguez AR, de Sevilla Müller LP and
Brecha NC: The RNA binding protein RBPMS is a selective marker of
ganglion cells in the mammalian retina. J Comp Neurol.
522:1411–1443. 2014. View Article : Google Scholar :
|
|
23
|
Zhang XM, Li Liu DT, Chiang SW, Choy KW,
Pang CP, Lam DS and Yam GH: Immunopanning purification and
long-term culture of human retinal ganglion cells. Mol Vis.
16:2867–2872. 2010.
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Lee SY, Lee S, Choi E, Ham O, Lee CY, Lee
J, Seo HH, Cha MJ, Mun B, Lee Y, et al: Small molecule-mediated
up-regulation of microRNA targeting a key cell death modulator
BNIP3 improves cardiac function following ischemic injury. Sci Rep.
6:234722016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ju WK, Liu Q, Kim KY, Crowston JG, Lindsey
JD, Agarwal N, Ellisman MH, Perkins GA and Weinreb RN: Elevated
hydrostatic pressure triggers mitochondrial fission and decreases
cellular ATP in differentiated RGC-5 cells. Invest Ophthalmol Vis
Sci. 48:2145–2151. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu Q, Ju WK, Crowston JG, Xie F, Perry G,
Smith MA, Lindsey JD and Weinreb RN: Oxidative stress is an early
event in hydrostatic pressure induced retinal ganglion cell damage.
Invest Ophthalmol Vis Sci. 48:4580–4589. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lv B, Chen T, Xu Z, Huo F, Wei Y and Yang
X: Crocin protects retinal ganglion cells against H2O2-induced
damage through the mitochondrial pathway and activation of NF-κB.
Int J Mol Med. 37:225–232. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang QL, Wang W, Jiang Y, A-Tuya,
Dongmei, Li LL, Lu ZJ, Chang H and Zhang TZ: GRGM-13 comprising 13
plant and animal products, inhibited oxidative stress induced
apoptosis in retinal ganglion cells by inhibiting P2RX7/p38 MAPK
signaling pathway. Biomed Pharmacother. 101:494–500. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen H, Chow PH, Cheng SK, Cheung AL,
Cheng LY and O WS: Male genital tract antioxidant enzymes: Their
source, function in the female, and ability to preserve sperm DNA
integrity in the golden hamster. J Androl. 24:704–711. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin J, Chuang CC and Zuo L: Potential
roles of microRNAs and ROS in colorectal cancer: Diagnostic
biomarkers and therapeutic targets. Oncotarget. 8:17328–17346.
2017.PubMed/NCBI
|
|
32
|
Lan J, Huang Z, Han J, Shao J and Huang C:
Redox regulation of microRNAs in cancer. Cancer Lett. 418:250–259.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu Y, Qiang W, Xu X, Dong R, Karst AM,
Liu Z, Kong B, Drapkin RI and Wei JJ: Role of miR-182 in response
to oxidative stress in the cell fate of human fallopian tube
epithelial cells. Oncotarget. 6:38983–38998. 2015.PubMed/NCBI
|
|
34
|
Lv G, Shao S, Dong H, Bian X, Yang X and
Dong S: MicroRNA-214 protects cardiac myocytes against H2O2-induced
injury. J Cell Biochem. 115:93–101. 2014. View Article : Google Scholar
|
|
35
|
Qin SB, Peng DY, Lu JM and Ke ZP:
MiR-182-5p inhibited oxidative stress and apoptosis triggered by
oxidized low-density lipoprotein via targeting toll-like receptor
4. J Cell Physiol. 233:6630–6637. 2018. View Article : Google Scholar
|
|
36
|
Li J and Li J, Wei T and Li J:
Down-regulation of MicroRNA-137 improves high glucose-induced
oxidative stress injury in human umbilical vein endothelial cells
by up-regulation of AMPKα1. Cell Physiol Biochem. 39:847–859. 2016.
View Article : Google Scholar
|
|
37
|
Shi YF, Liu N, Li YX, Song CL, Song XJ,
Zhao Z and Liu B: Insulin protects H9c2 rat cardiomyoblast cells
against hydrogen peroxide-induced injury through upregulation of
microRNA-210. Free Radic Res. 49:1147–1155. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li X, Kong M, Jiang D, Qian J, Duan Q and
Dong A: MicroRNA-150 aggravates H2O2-induced cardiac myocyte injury
by down-regulating c-myb gene. Acta Biochim Biophys Sin (Shanghai).
45:734–741. 2013. View Article : Google Scholar
|
|
39
|
Li QC, Xu H, Wang X, Wang T and Wu J:
miR-34a increases cisplatin sensitivity of osteosarcoma cells in
vitro through up-regulation of c-Myc and Bim signal. Cancer
Biomark. 21:135–144. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lomonosova E and Chinnadurai G: BH3-only
proteins in apop-tosis and beyond: An overview. Oncogene. 27(Suppl
1): S2–S19. 2008. View Article : Google Scholar
|
|
41
|
Quinsay MN, Lee Y, Rikka S, Sayen MR,
Molkentin JD, Gottlieb RA and Gustafsson AB: Bnip3 mediates
permeabilization of mitochondria and release of cytochrome c via a
novel mechanism. J Mol Cell Cardiol. 48:1146–1156. 2010. View Article : Google Scholar :
|
|
42
|
Circu ML and Aw TY: Reactive oxygen
species, cellular redox systems, and apoptosis. Free Radic Biol
Med. 48:749–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cai X, Chen Z, Pan X, Xia L, Chen P, Yang
Y, Hu H, Zhang J, Li K, Ge J, et al: Inhibition of angiogenesis,
fibrosis and thrombosis by tetramethylpyrazine: Mechanisms
contributing to the SDF-1/CXCR4 axis. PLoS One. 9:e881762014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang Z, Zhang Q, Ge J and Tan Z:
Protective effects of tetra-methylpyrazine on rat retinal cell
cultures. Neurochem Int. 52:1176–1187. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fu YS, Lin YY, Chou SC, Tsai TH, Kao LS,
Hsu SY, Cheng FC, Shih YH, Cheng H, Fu YY and Wang JY:
Tetramethylpyrazine inhibits activities of glioma cells and
glutamate neuro-excitotox-icity: Potential therapeutic application
for treatment of gliomas. Neuro Oncol. 10:139–152. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang K, Zhu X, Zhang K, Zhou F and Zhu L:
Neuroprotective effect of tetramethylpyrazine against
all-trans-retinal toxicity in the differentiated Y-79 cells via
upregulation of IRBP expression. Exp Cell Res. 359:120–128. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yu N, Zhang Z, Chen P, Zhong Y, Cai X, Hu
H, Yang Y, Zhang J, Li K, Ge J, et al: Tetramethylpyrazine (TMP),
an active ingredient of chinese herb medicine chuanxiong,
attenuates the degeneration of trabecular meshwork through
SDF-1/CXCR4 axis. PLoS One. 10:e01330552015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Izzotti A, Sacca SC, Cartiglia C and De
Flora S: Oxidative deoxyribonucleic acid damage in the eyes of
glaucoma patients. Am J Med. 114:638–646. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou B, Li C, Qi W, Zhang Y, Zhang F, Wu
JX, Hu YN, Wu DM, Liu Y, Yan TT, et al: Downregulation of miR-181a
upregulates sirtuin-1 (SIRT1) and improves hepatic insulin
sensitivity. Diabetologia. 55:2032–2043. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Muratsu-Ikeda S, Nangaku M, Ikeda Y,
Tanaka T, Wada T and Inagi R: Downregulation of miR-205 modulates
cell susceptibility to oxidative and endoplasmic reticulum stresses
in renal tubular cells. PLoS One. 7:e414622012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lee S, Yun I, Ham O, Lee SY, Lee CY, Park
JH, Lee J, Seo HH, Choi E and Hwang KC: Suppression of miR-181a
attenuates H2O2-induced death of mesenchymal stem cells by
maintaining hexokinase II expression. Biol Res. 48:452015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cao MQ, You AB, Zhu XD, Zhang W, Zhang YY,
Zhang SZ, Zhang KW, Cai H, Shi WK, Li XL, et al: miR-182-5p
promotes hepatocellular carcinoma progression by repressing FOXO3a.
J Hematol Oncol. 11:122018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang D, Lu G, Shao Y and Xu D: MiR-182
promotes prostate cancer progression through activating
Wnt/β-catenin signal pathway. Biomed Pharmacother. 99:334–339.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Crow MT: Hypoxia, BNip3 proteins, and the
mitochondrial death pathway in cardiomyocytes. Circ Res.
91:183–185. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Regula KM, Ens K and Kirshenbaum LA:
Inducible expression of BNIP3 provokes mitochondrial defects and
hypoxia-mediated cell death of ventricular myocytes. Circ Res.
91:226–231. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang J, Ye J, Altafaj A, Cardona M, Bahi
N, Llovera M, Cañas X, Cook SA, Comella JX and Sanchis D: EndoG
links Bnip3-induced mitochondrial damage and caspase-independent
DNA fragmentation in ischemic cardiomyocytes. PLoS One.
6:e179982011. View Article : Google Scholar : PubMed/NCBI
|