Introduction
Acute respiratory distress syndrome (ARDS)
represents the most severe form of acute noncardiogenic refractory
respiratory failure and is associated with high morbidity and
mortality in critically ill patients (1,2).
Numerous studies have emphasized the importance of uncontrolled
inflammation in the pathogenesis of ARDS; however, the fundamental
cellular mechanisms that regulate acute lung inflammation and
injury in ARDS still require to be clarified (3,4).
Classical dendritic cells (cDCs), which exist in the
lung in relatively small numbers, are ideally positioned to serve a
priming and central role in the immune response during
infection/inflammation (5,6).
cDCs are pivotal antigen-presenting cells (APCs); the involvement
of pulmonary cDCs in the pathogenesis of ARDS has been revealed in
several recent studies, wherein cDCs were found to act as
pro-inflammatory initiators and mediators (7,8).
However, the precise mechanism by which pulmonary cDCs affect acute
lung inflammation remains to be clarified.
Fms-like tyrosine kinase 3 (FLT3), to which
FLT3-ligand (FLT3L) binds, represents an important hematopoietic
factor that is abundantly expressed on hematopoietic stem cells and
DC progenitors (9,10). FLT3 signaling is one of the most
important pathways that manage the entire lifespan of DCs. Once
FLT3L binds to FLT3, the FLT3 signaling is activated, leading to
the aggregation of functional DCs, stimulation of DC maturation and
decreased apoptosis of mature DCs (9,11).
By contrast, inhibition of FLT3 signaling with lestaurtinib is
associated with decreased aggregation and maturation of cDCs
(9,12). Our previous pilot study also
indicated that FLT3 signaling controls the accumulation and
maturation of cDCs in mouse models of acute lung injury (13). Therefore, FLT3 signaling may
represent a reliable approach for the manipulation of cDCs in
vivo.
Although the mechanisms responsible for the
pathogenesis of ARDS remain controversial, previous studies have
suggested that cDCs may contribute to the pathology of ARDS by
regulating several processes, such as polarizing the T helper cell
(Th) 1 response and regulating neutrophil infiltration (1,9).
Generally, Th cells are the primary target of cDC manipulation,
leading to a shifted balance between Th1 and Th2 responses
(14). Previous studies have
reported that aggregation and maturation of cDCs results in a
Th1-skewed cytokine pattern, thus aggravating the inflammatory
response, whereas inhibited expansion and maturation of cDCs was
associated with a Th2-skewed cytokine pattern (9,15).
Recently, cDCs have also been implicated in crosstalk with
neutrophils, such as reinforcing the recruitment of neutrophils,
prolonging neutrophil survival and inducing the upregulation of the
innate immune response (16,17). In summary, favoring the Th1
response and enhancing neutrophil infiltration may be two
underlying mechanisms by which cDCs participate in the development
of acute lung inflammation during lipopolysaccharide (LPS)-induced
ARDS.
The purpose of the present study was to explore
whether pulmonary cDCs manage lung inflammation and injury during
LPS-induced ARDS and the mechanisms by which pulmonary cDCs affect
lung inflammation and injury in vivo.
Materials and methods
Animals
Seventy-five specific-pathogen-free male C57BL/6
mice (age, 6-8 weeks; weight, 20-25 g) were purchased from the
Laboratory Animal Center, Academy of Military Medical Sciences of
People's Liberation Army (Beijing, China). Mice were maintained
under specific pathogen-free conditions with a 12-h light/dark
cycle (temperature, 18-23°C; humidity, 40-60%) and fed standard
rodent food and water ad libitum. All animal experiments
were conducted in accordance with the National Institutes of Health
Guide for the Care and Use of Laboratory Animals and with the
approval of the Institutional Animal Care and Use Committee of
Nanjing Medical University.
Murine ARDS model
The murine ARDS model was induced as previously
described with minor modifications (18). Briefly, mice were anesthetized by
an intraperitoneal injection of 50 mg/kg pentobarbital, and a
midline incision was made in the neck to expose the trachea. ARDS
was generated by a direct intratracheal instillation of LPS (2
mg/kg; Escherichia coli 0111:B4; Sigma-Aldrich; Merck KGaA)
through a tracheostomy, and the incision was sutured. Mice were
returned to the cage until fully awake.
Experimental groups and sample
acquisition
Mice were randomly allocated to one of the following
groups (n=15 mice per group): Control group, mice received
intratracheal administration of 0.9% normal saline (NS); ARDS
group, mice received 2 mg/kg LPS to establish the ARDS model;
FLT3L+ARDS group, mice received 10 μg/d FLT3L for 5 days
followed by 2 mg/kg LPS; lestaurtinib+ARDS group, mice received 40
mg/kg/d lestaurtinib for 5 days followed by 2 mg/kg LPS; and
DMSO+ARDS group, mice were pretreated with an equal volume of 10%
DMSO for 5 days (DMSO was the solvent for lestaurtinib, and
therefore used here as a vehicle control) followed by 2 mg/kg LPS.
FLT3L (LC Laboratories), lestaurtinib (Miltenyi Biotec, GmbH) and
DMSO (Sigma-Aldrich; Meck KGaA) were all administered
subcutaneously. Mice were euthanized by barbital overdose at 6 or
24 h following LPS challenge. These two post-insult time-points
were selected because they are critical points for the aggregation
and maturation of cDCs (19,20), and they could pathophysiologically
represent the early-phase and late-phase of ARDS, respectively
(21,22). The whole lung was removed and
perfused with saline/EDTA to wipe out the intravascular blood
cells. Specimens were snap-frozen in liquid nitrogen and stored at
−80°C for subsequent measurements.
Measurement of the aggregation and
maturation of pulmonary cDCs by flow cytometry
The aggregation of pulmonary cDCs was quantified
using the relative percentage of cDCs among total lung cells
(23,24). The maturation of pulmonary cDCs
was quantified using the percentage of expression of major
histocompatibility complex class II (MHC II) and CD80 in all
pulmonary cDCs (5). Briefly, the
entire left lung was collagenase-digested into single-cell
suspension, as previously described (25). After red blood cell lysis and Fc
receptor blockade, cells were stained with monoclonal antibodies or
their corresponding isotype controls, according to the
manufacturer's recommendations: Cells were incubated with
FITC-labeled anti-CD11c, Percp-cy5.5-labeled anti-CD11b, PE-labeled
anti-MHC II and APC-labeled anti-CD80 (all at 1:100 dilution; cat.
nos. 11-0114-82, 45-0112-82, 12-5321-82, 17-0801-82; eBioscience;
Thermo Fisher Scientific, Inc.) at 4°C for 30 min, then washed and
resuspended with PBS. The cells were fixed in 1% paraformaldehyde
at 25°C for 1 h and kept in the dark at 4°C until analysis.
CD11c/CD11b double-positive cells represent pulmonary cDCs
(23,26). CD11c/CD11b/MHC II or
CD11c/CD11b/CD80 triple-positive cells represent mature cDCs
(26,27). Flow cytometry analysis was
conducted using a FACSCanto (BD Biosciences) and CellQuest software
(BD Biosciences). For each analysis, 20,000 events were
recorded.
Pulmonary myeloperoxidase (MPO) activity
assay
MPO activity, an index of neutrophil infiltration in
the lung, was measured by chromometry, as previously described,
using a commercially available kit (Nanjing Jiancheng
Bioengineering Institute) (28).
In brief, MPO was extracted from homogenized lung tissue (10 mg per
assay) by suspending the sample in 0.5% hexadecyl trimethyl
ammonium bromide in 0.5 ml of potassium phosphate buffer (10
mmol/l, pH 7.0). A 0.2 ml aliquot of homogenate was mixed with a
1.6 mmol/l solution of tetramethyl benzidine and 0.1 mmol/l
H2O2. The rate of change in absorbance at 460
nm was measured by an MK3 spectrophotometer (Thermo Fisher
Scientific, Inc.). MPO activity was expressed as units per gram of
the sample.
Tumor necrosis factor (TNF)-α,
interleukin (IL)-6, interferon (IFN)-γ, IL-1β, IL-4 and IL-10
assay
The right lower lobe was weighted and homogenized.
The homogenate was then centrifuged at 1,000 × g for 5 min at 4°C
and the supernatant was harvested. The concentrations of TNF-α,
IL-6 IFN-γ, IL-1β, IL-4 and IL-10 in the supernatant were measured
using commercial ELISA kits (cat. nos. EM008-96, EM004-96,
EM007-96, EM001-96, EM003-96 and EM005-96; ExCell Biology, Inc.),
according to the manufacturer's protocols. The concentrations were
expressed as picograms per milligram of the sample.
Determination of mRNA expression levels
of Akt, ERK1/2, STAT5, T-box-expressed-in-T-cells (T-bet) and GATA
binding protein 3 (GATA-3)
mRNA expression in lung tissues was measured by
reverse transcription-quantitative PCR (RT-qPCR). Briefly, lung
tissues (10 mg per assay) were harvested and homogenized, and the
total RNA was extracted using TRIzol reagent (Takara Biotechnology
Co., Ltd.). A total of 5 μg of total RNA was subsequently
reverse transcribed to cDNA, using the High-Capacity cDNA Reverse
Transcription kit (Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. An ABI PRISM 7300 Sequence Detection
System (Applied Biosystems; Thermo Fisher Scientific, Inc.) was
used for PCR amplification. Each sample was analyzed in triplicate
with the following thermocycling conditions: 40 cycles, including a
denaturation step at 95°C for 15 sec, an annealing step at 56°C for
20 sec and an extension step at 72°C for 40 sec. SYBR Green I
(Thermo Fisher Scientific, Inc.) was selected as the fluorophore
dye. The 2−ΔΔCq method was used to calculate the
relative expression of mRNA, and β-actin was used as the internal
reference gene (29). The primers
used were as follows: Akt, forward 5′-TCTATGGCGCTGAGATTGTG-3′ and
reverse 5′-CTTAATGTGCCCGTCCTTGT-3′; ERK1, forward
5′-TCCGCCATGAGAATGTTATAGGC-3′ and reverse
5′-GGTGGTGTTGATAAGCAGATTGG-3′; ERK2, forward
5′-GGTTGTTCCCAAATGCTGACT-3′ and reverse
5′-CAACTTCAATCCTCTTGTGAGGG-3′; STAT5, forward
5′-AGTATTACACTCCTGTACTTGCGAAAG-3′ and reverse
5′-GGAGCTTCTAGCGGAGGTGAAGAGACC-3′; T-bet, forward
5′-ACCACCTGTTGTGGTCCAAG-3′ and reverse 5′-CACCAAGACCACATCCACAA-3′;
GATA-3, forward 5′-ACCGGGTTCGGATGTAAGTC-3′ and reverse
5′-AGGCATTGCAAAGGTAGTGC-3′; and β-actin, forward
5′-CCTCTATGCCAACACAGTGC-3′ and reverse
5′-GTACTCCTGCTTGCTGATCC-3′.
Determination of protein expression
levels of Akt, ERK1/2, STAT5, T-bet, GATA-3 and FLT3L
Protein expression in lung tissue was measured by
western blotting. Briefly, the ReadyPrep™ Protein Extraction kit
(cat. no. 1632086; Bio-Rad Laboratories, Inc.) was used to extract
the total protein. A BCA assay was used to measure the protein
concentration. Proteins were denatured and added to the wells in
aliquots of 25 μg per well. After SDS-PAGE (10%), the
proteins were transferred to a polyvinylidene fluoride membrane,
blocked with Tris-buffered saline with Tween-20 (TBST) containing
5% skim milk powder at 25°C for 1 h, and incubated at 4°C overnight
with primary antibodies against phosphorylated (p-) Akt, total Akt,
p-ERK1/2, total ERK1/2, p-STAT5, total STAT5 (all at 1:400
dilution; cat. nos. ab38449, ab8805, ab201015, ab17942, ab32364,
ab16276; Abcam), T-bet, GATA-3, FLT3L and β-actin (all at 1:400
dilution; cat. nos. sc-21763, sc-130057, sc-365266, sc-47778; Santa
Cruz Biotechnology, Inc.). β-actin was used as an internal
reference protein. The membrane was washed three times with TBST
and incubated with horseradish peroxidase-conjugated secondary
antibody (1:1,000 dilution; cat. no. A0216; Beyotime Institute of
Biotechnology) at 25°C for 1 h. The membrane was washed four times
with TBST, developed with the Pierce™ Enhanced Chemiluminescence
reagent (Thermo Fisher Scientific, Inc.) and placed on an X-ray
film for imaging. Quantity One 4.6.7 software (Bio-Rad
Laboratories, Inc.) was used for quantitative analysis of the
grayscale values of the bands.
Evaluation of lung edema
Lung edema was assessed by the ratio of lung wet
weight to body weight (LWW/BW) (30). In brief, the intact lung was
harvested and trimmed to remove extra-pulmonary tissues, and the
LWW/BW was calculated based on the recorded lung wet weight and
body weight.
Lung histopathological analysis
The right upper lobe was fixed in 10% neutral
formaldehyde at 4°C for 24 h and embedded in paraffin. After
consecutively transverse slicing into 5-μm-thick sections
and sequentially staining with hematoxylin for 5 min and eosin for
2 min at 25°C, 10 high-magnification (×400; light microscopy)
visual fields were randomly selected for semi-quantitative
evaluation of lung injury based on the method reported by Smith
et al (31), and the mean
sum of each field score was determined.
Statistical analysis
Statistical analyses were conducted using the SPSS
16.0 software package (SPSS, Inc.) All data were presented as the
means ± standard deviation. For multiple group comparison, a
one-way analysis of variance followed by Bonferroni's post hoc test
was used. For two-group comparison, the Mann-Whitney U test was
applied. P<0.05 was considered to indicate a statistically
significant difference.
Results
Regulation of FLT3 signaling in lung
tissue by FLT3L and lestaurtinib
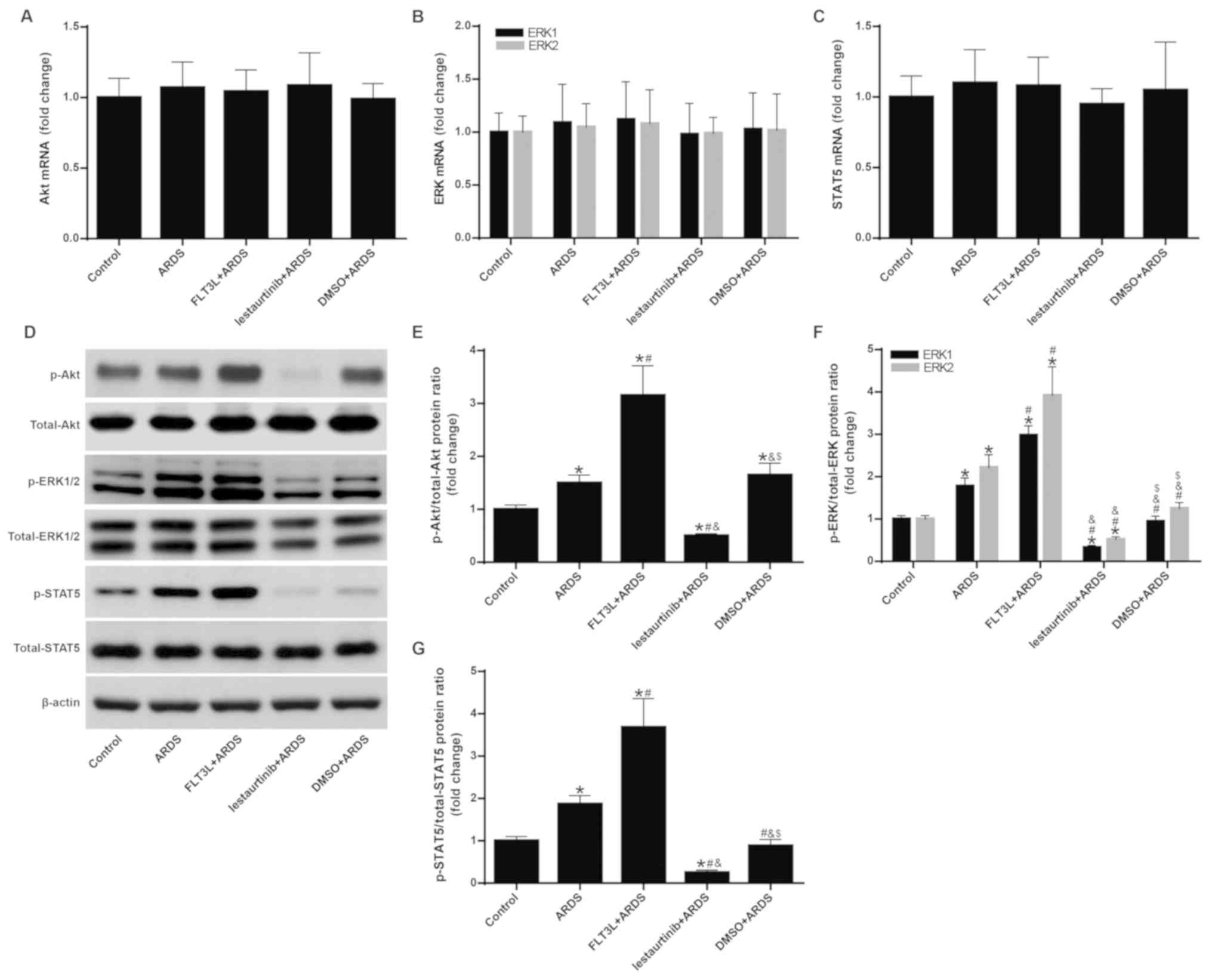
The mRNA and protein expression levels of Akt,
ERK1/2 and STAT5 were measured to confirm the regulation of FLT3
signaling by FLT3L and lestaurtinib. The results demonstrated that
the mRNA expression levels of Akt, ERK1/2 and STAT5 exhibited no
significant difference among the groups (Fig. 1A-C). However, FLT3L pretreatment
in the FLT3L+ARDS group significantly increased the phosphorylation
of Akt, ERK1/2 and STAT5 compared with the ARDS group alone
(P<0.05; Fig. 1D-G), and
lestaurtinib pretreatment in the lestaurtinib + ARDS group
significantly decreased the phosphorylation of Akt, ERK1/2 and
STAT5 compared with the DMSO+ARDS vehicle control group (P<0.05;
Fig. 1D-G). These results
indicated that FLT3L and lestaurtinib could effectively activate
and suppress FLT3 signaling, respectively.
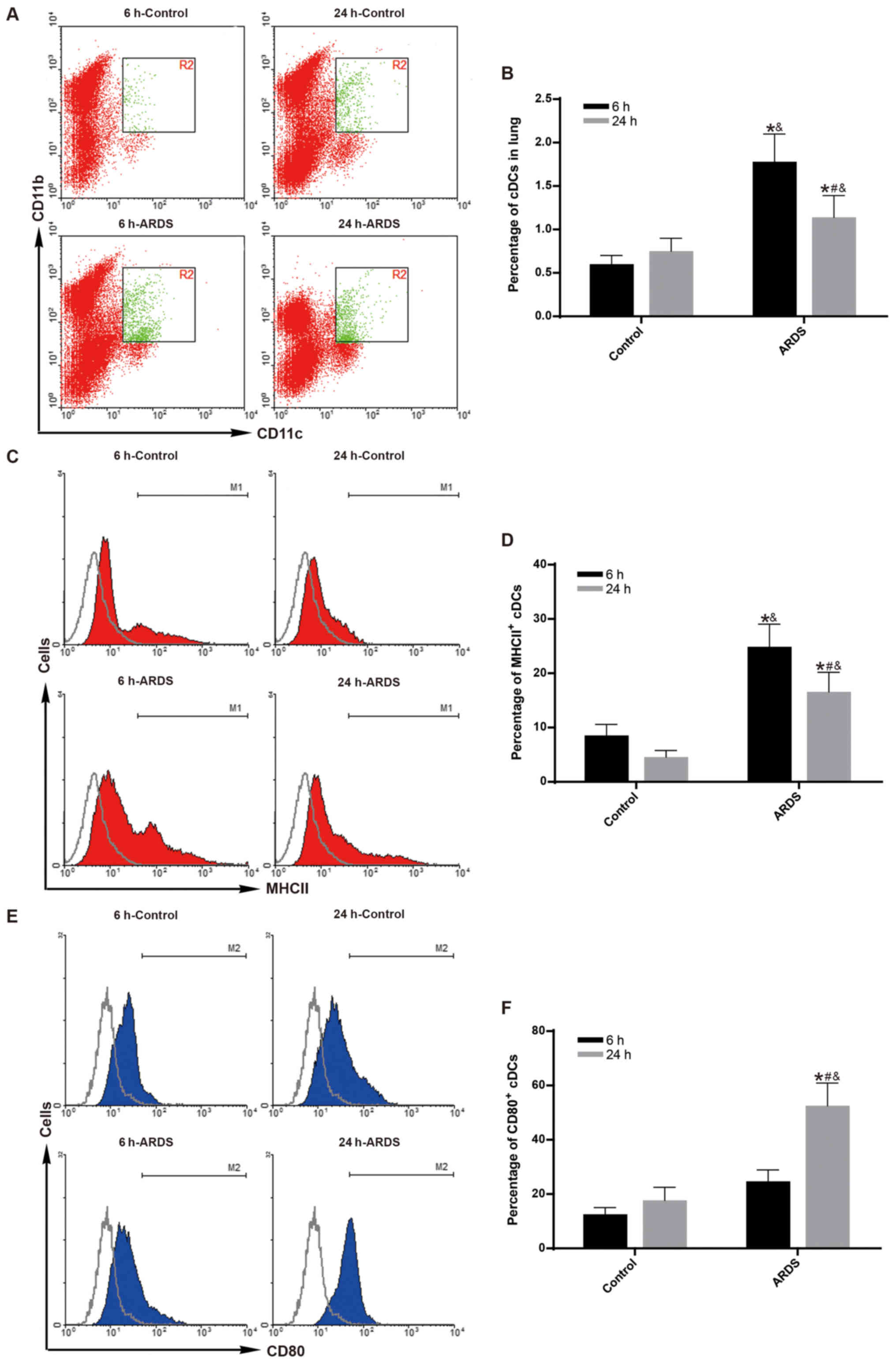
Aggregation and maturation of cDCs peaks
as early as 6 h following LPS challenge
To observe the maximum effect of FLT3 signaling on
the aggregation and maturation of cDCs at early-phase of ARDS
(19,21), the aggregation and maturation of
cDCs was measured at 6 h following LPS challenge. Compared with the
control group, both the percentage of pulmonary cDCs (Fig. 2A and B) and the percentage of MHC
II-expressing cDCs (Fig. 2C and
D) were significantly increased in the ARDS group just 6 h
after LPS challenge (P<0.05).
To observe the effect of FLT3 signaling on the
aggregation and maturation of cDCs at late-phase of ARDS (20,22), the aggregation and maturation of
cDCs was measure at 24 h following LPS challenge. Compared with the
control group, both the percentage of pulmonary cDCs (Fig. 2A and B) and the percentage of MHC
II-expressing cDCs (Fig. 2C and
D) remained higher in the ARDS group at 24 h after LPS
challenge (P<0.05). However, the effect was gradually reduced at
24 h compared to 6 h for the ARDS group (Fig. 2B and D; P<0.05). Notably, the
expression levels of CD80 on pulmonary cDCs remained unchanged at 6
h after LPS challenge between the control group and the ARDS group
(Fig. 2E and F); however, a
subsequent sharp increase was observed at 24 h after LPS challenge
(Fig. 2E and F; P<0.05).
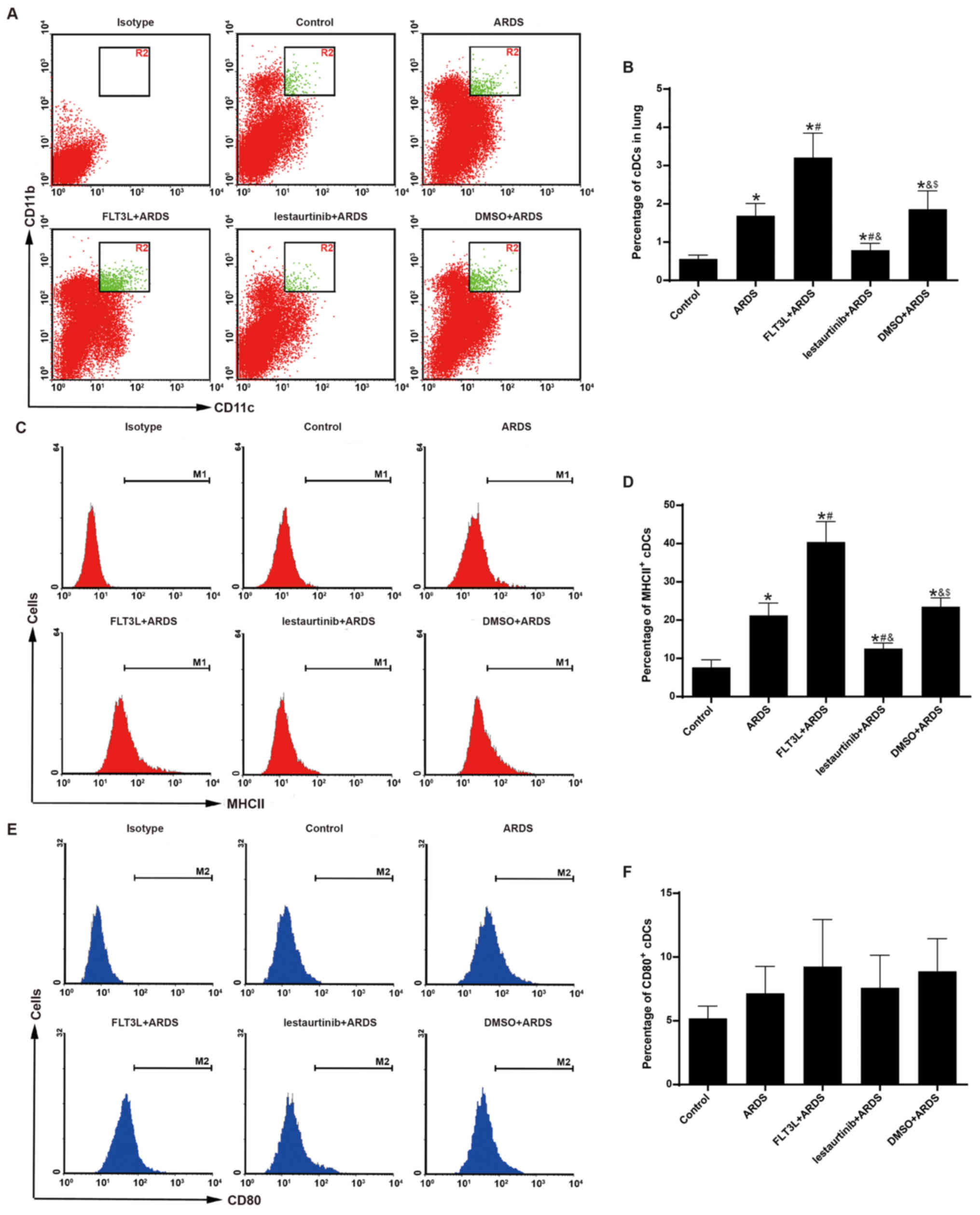
Aggregation and maturation of cDCs is
modulated by FLT3 signaling
The aggregation and maturation of pulmonary cDCs was
assessed at 6 h following LPS challenge to confirm the DC-targeting
property of FLT3 signaling in vivo. As expected, FLT3L
pretreatment in the FLT3L+ARDS group was associated with a
significant increase in the percentage of pulmonary cDCs just 6 h
after LPS challenge compared with the ARDS alone group (Fig. 3A and B). Furthermore, lestaurtinib
pretreatment in the lestaurtinib+ARDS group significantly reduced
the percentage of pulmonary cDCs at 6 h after LPS challenge
compared with the vehicle control DMSO+ARDS group (Fig. 3A and B).
The expression of MHC II and CD80 was further
examined in the pulmonary cDCs, in order to evaluate their
maturation. FLT3L pretreatment in the FLT3L+ARDS group
significantly increased the expression of MHC II in pulmonary cDCs
compared with the ARDS alone group (Fig. 3C and D). Furthermore, compared
with the DMSO+ARDS group, the lestaurtinib+ARDS group exhibited
decreased expression of MHC II in pulmonary cDCs (Fig. 3C and D). However, the expression
of CD80 in pulmonary cDCs exibited no significant difference among
the groups (Fig. 3E and F).
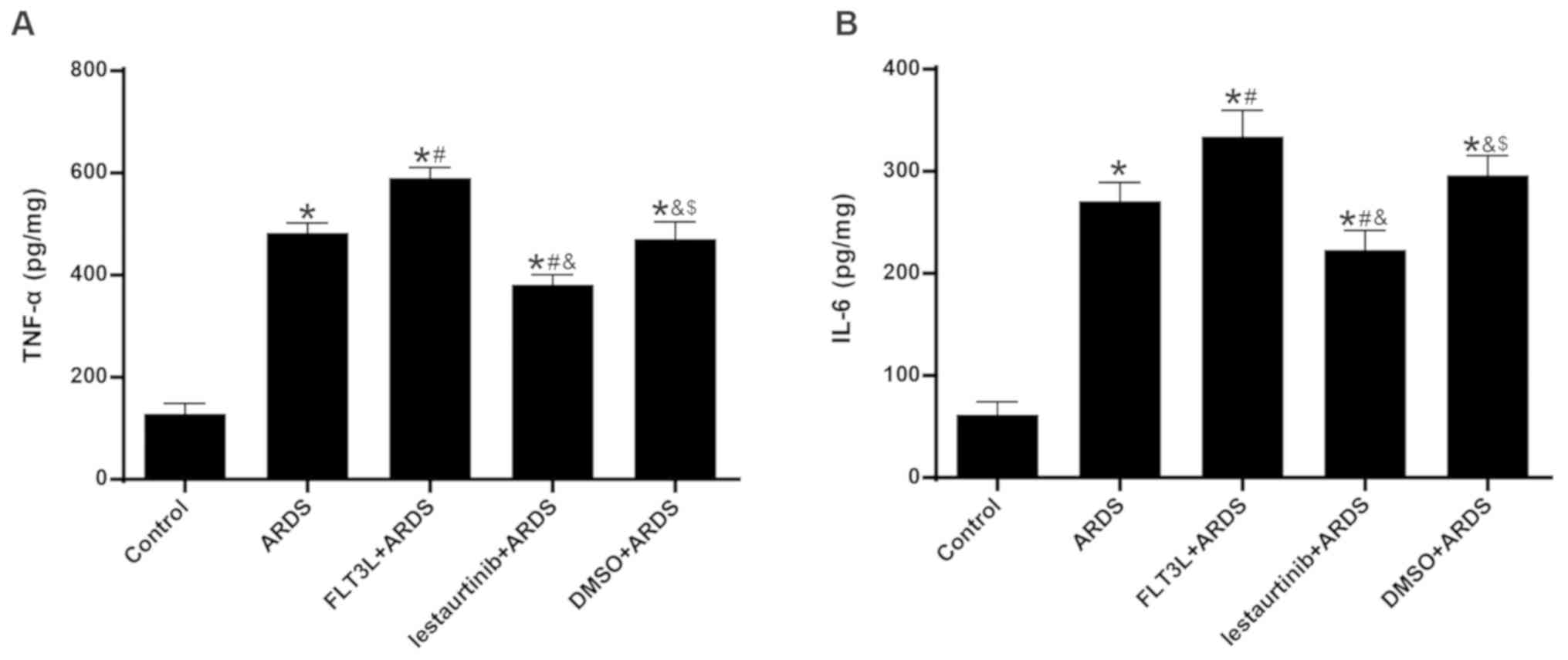
Effect of cDC manipulation on acute lung
inflammation
The levels of TNF-α and IL-6 in the lungs at 6 h
following LPS challenge were measured, in order to evaluate acute
lung inflammation. The results demonstrated that the levels of both
TNF-α and IL-6 were significantly increased in the ARDS group
compared with the control group (Fig.
4). FLT3L pretreatment in the FLT3L+ARDS group significantly
increased the TNF-α and IL-6 levels in the lung compared with the
ARDS alone group (Fig. 4).
Additionally, lestaurtinib pretreatment in the lestaurtinib+ARDS
group significantly decreased the TNF-α and IL-6 levels compared
with the DMSO+ARDS group (Fig.
4).
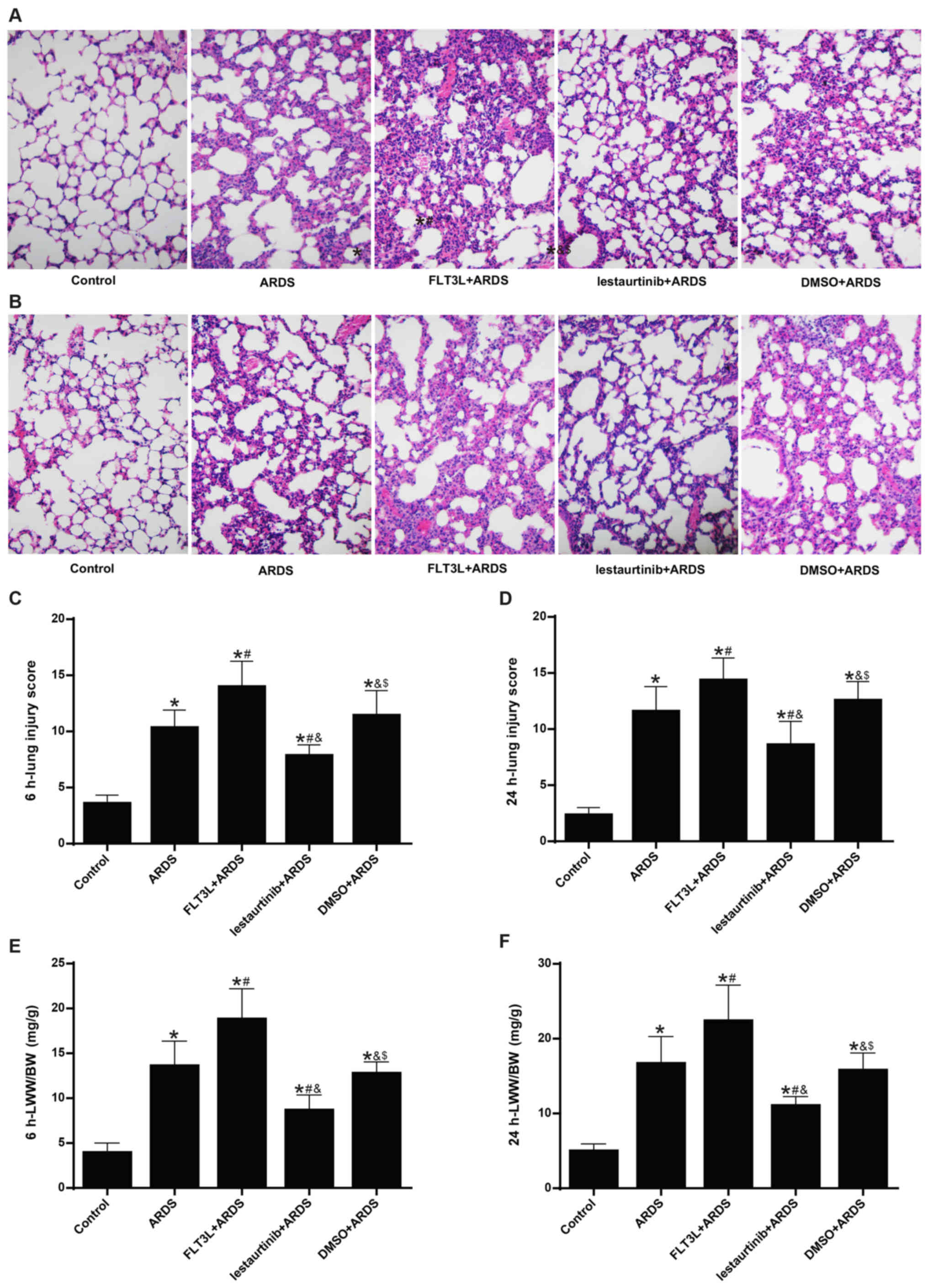
Effect of cDC manipulation on lung
injuries and lung edema
The lung injury score was calculated next. The
histopatho-logical analysis showed alveolar wall thickening,
diffused infiltration of inflammatory cells, hemorrhage,
intra-alveolar exudates and edema in the ARDS group at 6 h and 24 h
following LPS challenge (Fig. 5A and
B), with an elevated lung injury score (Fig. 5C and D). The pathologic changes
were markedly aggravated (Fig. 5A and
B) and the lung injury was significantly increased (Fig. 5C and D) in the FLT3L+ARDS group
compared with the ARDS alone group. By contrast, lestaurtinib
pretreatment in the lestaurtinib+ARDS group greatly alleviated the
pathologic changes (Fig. 5A and
B) and significantly decreased the lung injury score (Fig. 5C and B) compared with the
DMSO+ARDS group.
The LWW/BW ratio was calculated to evaluate lung
edema. The LWW/BW ratio in the ARDS group was significantly
increased at 6 h and 24 h following LPS challenge compared with the
control group (Fig. 5E and F).
The LWW/BW ratio in the FLT3L+ARDS group was further increased
compared with the ARDS alone group (Fig. 5E and F). By contrast, the LWW/BW
ratio in the lestaurtinib+ARDS group was significantly decreased
compared with the DMSO+ARDS group (Fig. 5E and F).
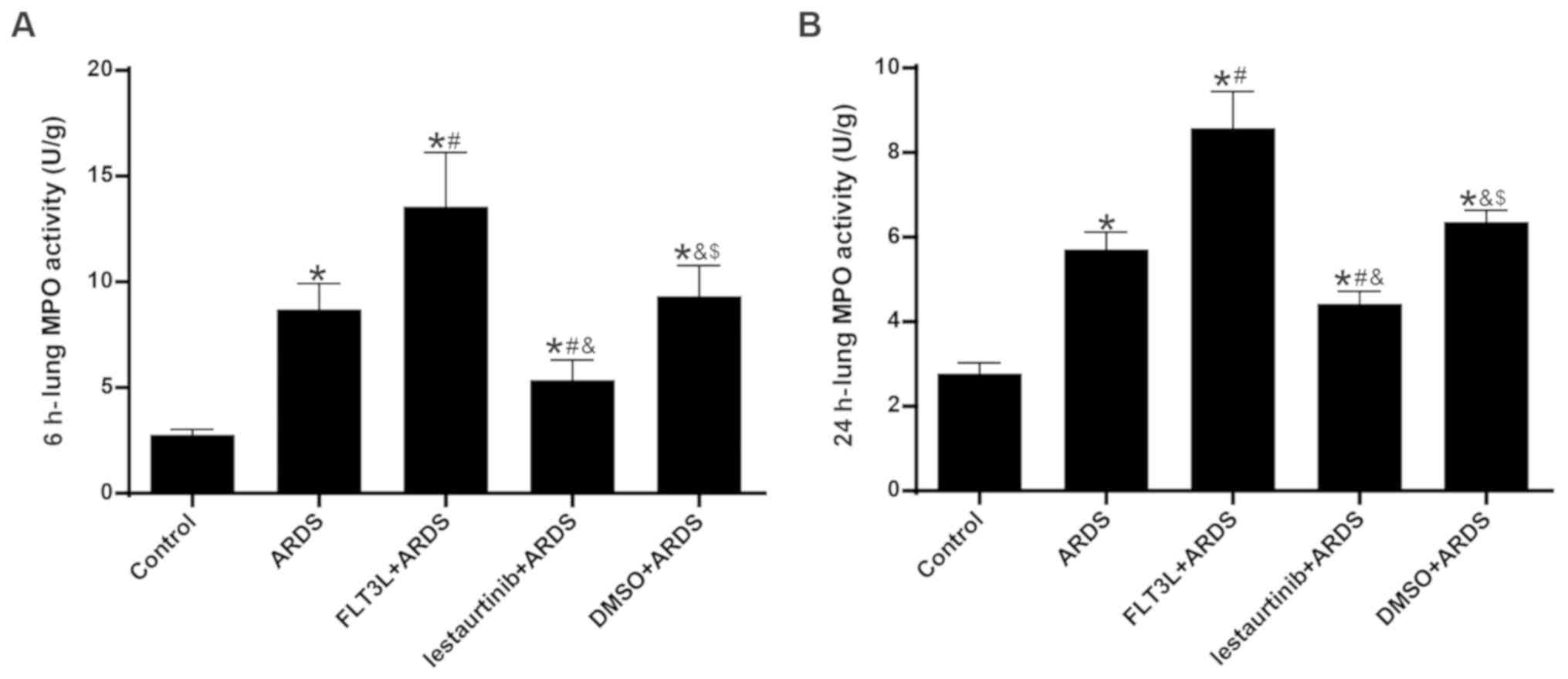
Effect of cDC manipulation on neutrophil
infiltration
MPO activity was assessed to evaluate neutrophil
infiltration in the lung. The results demonstrated that, compared
with the control group, lung MPO activity in the ARDS group was
increased significantly at 6 h and 24 h following LPS challenge
(Fig. 6). FLT3L pretreatment in
the FLT3L+ARDS group further augmented MPO activity compared with
the ARDS alone group (Fig. 6). By
contrast, lestaurtinib pretreatment in the lestaurtinib+ARDS group
significantly decreased MPO activity compared with the DMSO+ARDS
group (Fig. 6).
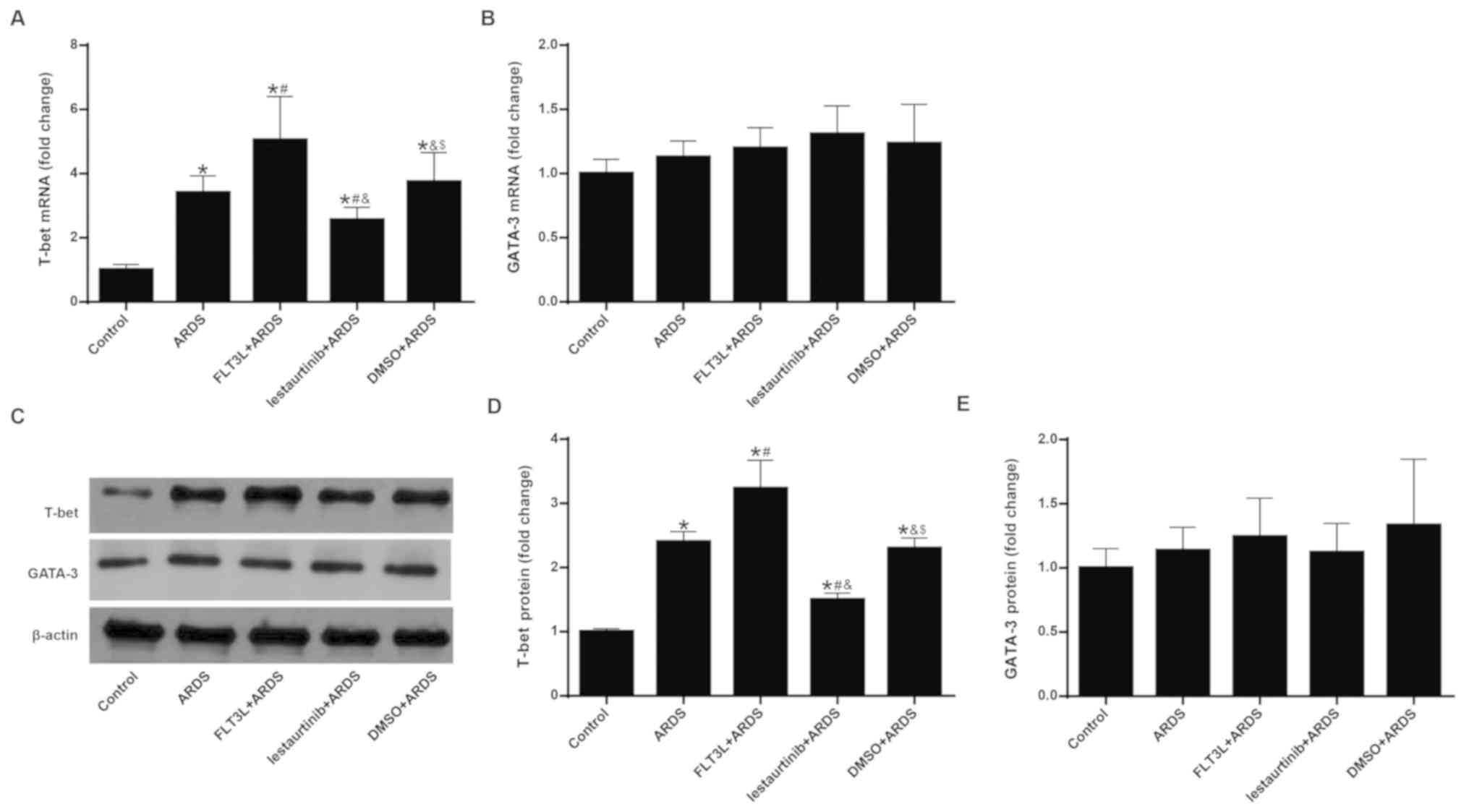
Effect of cDC manipulation on the balance
of the Th1/Th2 response
The mRNA and protein expression levels of T-bet and
GATA-3 were measured in order to evaluate the balance of the
Th1/Th2 response. Since it has been demonstrated that mobilized DCs
appear in draining lymph nodes at least 6 h after the invasion of
antigens (27), mRNA and protein
expression of T-bet and GATA-3 were measured at 24 h following LPS
challenge, instead of at 6 h. The results demonstrated that LPS
challenge in the ARDS group led to significant elevation of mRNA
and protein expression of T-bet (Fig.
7A, C and D), but not GATA-3 (Fig. 7B, C and E) in the lung, indicating
a Th1-biased response. FLT3L pretreatment in the FLT3L+ARDS group
further amplified this effect (Fig.
7A-E). By contrast, lestaurtinib pretreatment in the
lestaurtinib+ARDS group significantly decreased the mRNA and
protein expression levels of T-bet, but not GATA-3, in the lung
compared with the DMSO+ARDS group (Fig. 7A-E), indicating a Th2-biased
response.
Effect of cDC manipulation on Th1/Th2
cytokine production
The concentrations of IFN-γ/IL-1β and IL-4/IL-10 in
the lungs at 24 h following LPS challenge were measured in order to
evaluate the Th1 and Th2 cytokine production, respectively. The
results demonstrated that the concentration of IFN-γ and IL-1β in
the lungs was significantly increased in the ARDS group compared
with the control group (Fig. 8A and
B), indicating an enhanced Th1-skewed cytokine pattern. FLT3L
pretreatment in the FLT3L+ARDS group further increased the
concentration of IFN-γ and IL-1β compared with the ARDS alone group
(Fig. 8A and B). By contrast,
lestaurtinib pretreatment in the lestaurtinib+ARDS group
significantly decreased the concentration of IFN-γ and IL-1β in the
lung compared with the DMSO+ARDS group, indicating a repression of
the Th1-skewed cytokine pattern (Fig.
8A and B). Unexpectedly, neither LPS challenge nor cDC
manipulation affected the concentration of IL-4 and IL-10 in the
lungs among all groups (Fig. 8C and
D), indicating a steady Th2 cytokine pattern (Fig. 8B).
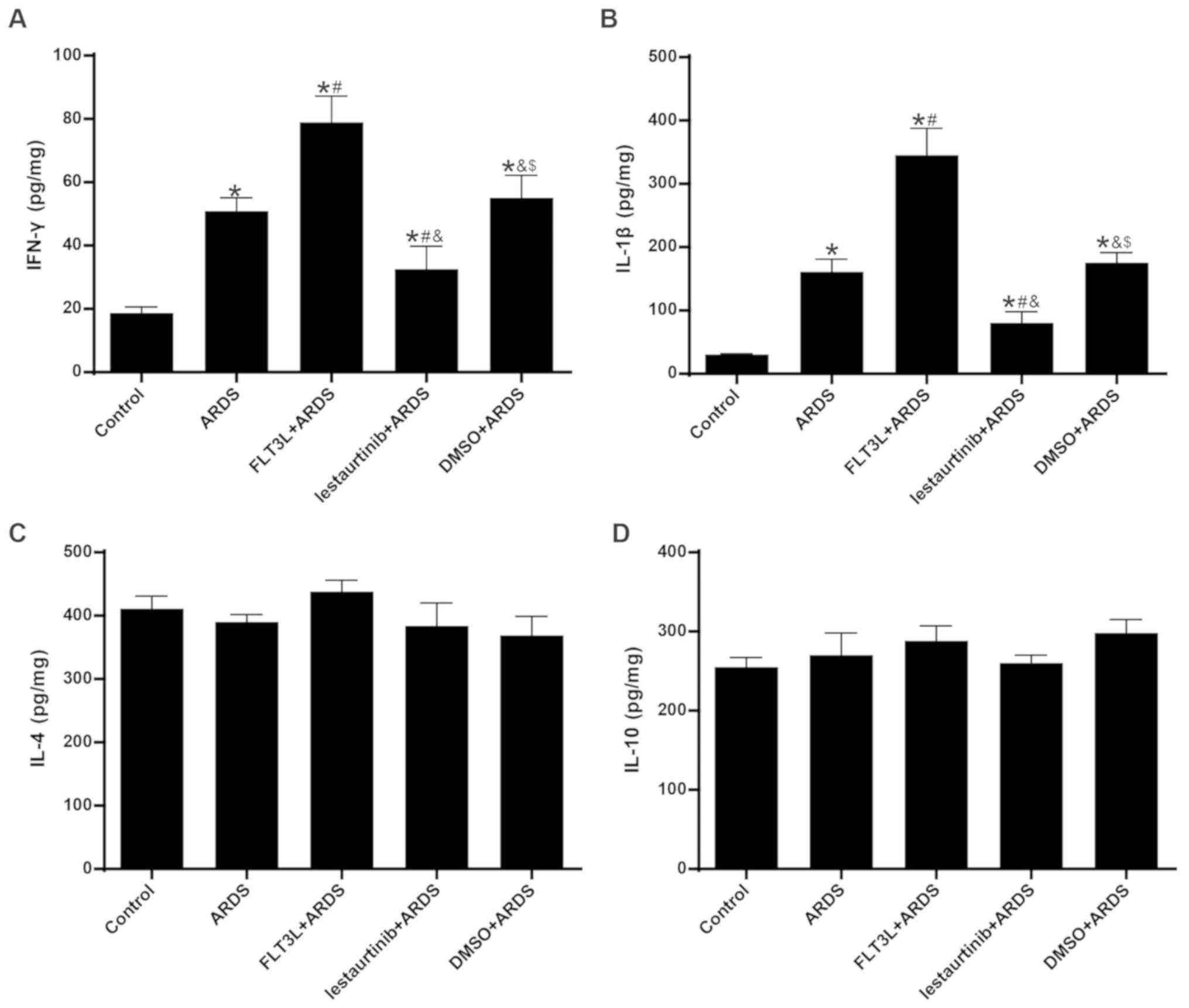 | Figure 8Effect of cDC manipulation on Th1/Th2
cytokine production. (A) Levels of IFN-γ, (B) IL-1β, (C) IL-4 and
(D) IL-10 were measured in the lungs by ELISA at 24 h post-LPS
challenge, in order to evaluate the Th1/Th2 cytokine production.
Data are presented as the mean ± standard deviation (n=6).
*P<0.05 vs. Control; #P<0.05 vs. ARDS;
&P<0.05 vs. FLT3L+ARDS; and $P<0.05
vs. lestaurtinib+ARDS. cDC, classical dendritic cell; Th, T helper
cell; IFN, interferon; IL, interleukin; LPS, lipopolysaccharide;
ARDS, acute respiratory distress syndrome; FLT3L, Fms-like tyrosine
kinase 3-ligand. |
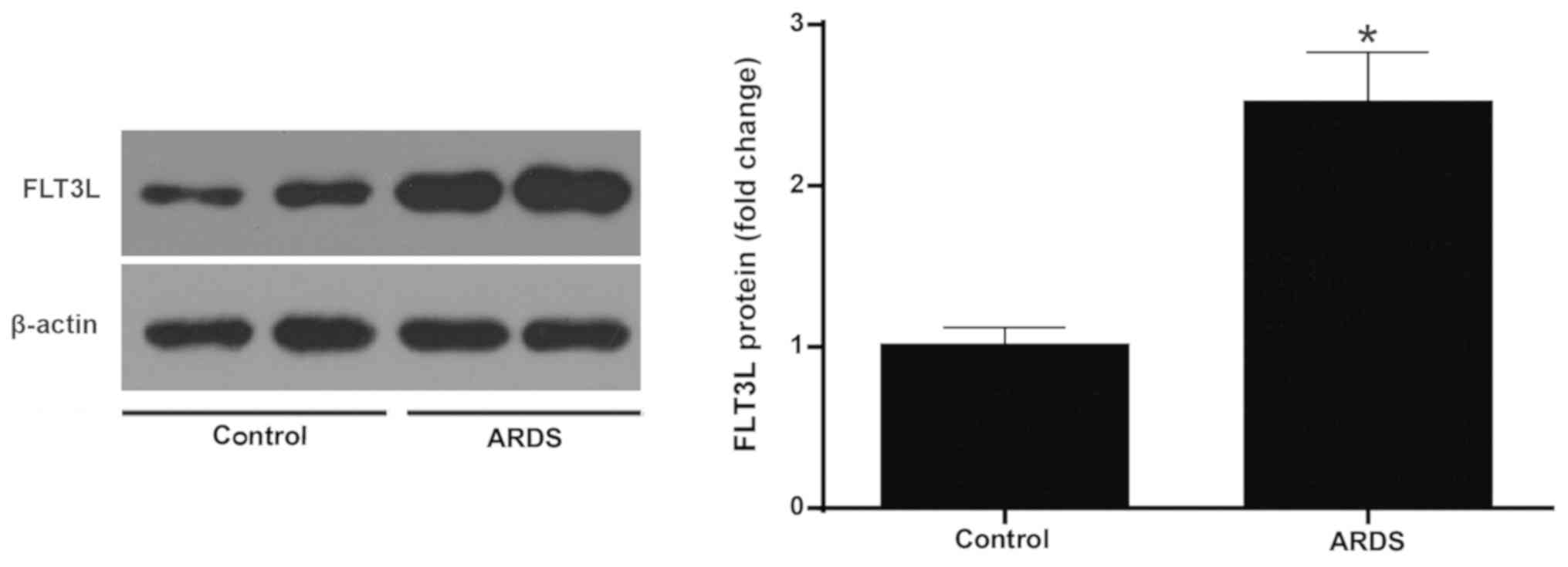
Increased expression of FLT3L in ARDS
lung tissue
The protein expression of FLT3L was measured in
normal and ARDS lung tissue at 6 h following LPS challenge. The
results demonstrated that the LPS challenge led to a signifi-cantly
higher expression of FLT3L in the ARDS lung tissues compared with
the control normal lung tissues (Fig.
9).
Discussion
The development of ARDS is characterized by the
sequestration of activated neutrophils and monocytes/macrophages,
inducing delayed apoptosis and an increased respiratory burst,
which results in the devastation of the pulmonary epithelium and
disruption of the blood-air barrier (1). The present study described the
involvement of pulmonary cDCs in the pathogenesis of ARDS. The
results demonstrated that pulmonary cDCs regulated acute lung
inflammation and injury in an ARDS model, favoring the Th1 response
and enhancing neutrophil infiltration, which may constitute the
underlying mechanisms for the contribution of pulmonary cDCs to
acute lung inflammation and injury in LPS-induced ARDS.
It has been generally accepted that ARDS primarily
represents uncontrolled inflammation of the lung (1,3).
Pulmonary cDCs act as sentinels and key elements in the immune
network of the lung, and cDCs have recently been reported to
modulate the progression of inflammation in several pulmonary
inflammatory disorders (5,6);
however, their distinctive role in the pathogenesis of ARDS has yet
to be elucidated. Therefore, the present study investigated the
aggregation and maturation status of pulmonary cDCs during the
initial 24 h period of LPS-induced ARDS. Although several studies
have reported that pulmonary cDCs aggregate and mature in the case
of ARDS (26,32), the present results highlighted
that the aggregation and maturation of pulmonary cDCs peaked as
early as 6 h following LPS challenge. Both the concentration of
TNF-α and IL-6, two indicators of acute lung inflammation, were
elevated in parallel with the aggregation and maturation of
pulmonary DCs. Additionally, the present results demonstrated that
the aggregation and maturation of pulmonary cDCs decreased sharply
to a level almost comparable to that of normal control mice just 24
h following LPS challenge, implying that pulmonary cDCs mainly
function during the early stage of ARDS. Together, these results
suggested that pulmonary cDCs may, at least in part, initiate acute
lung inflammation in ARDS.
cDCs have also been reported to manage the
progression of inflammation in multiple pulmonary and non-pulmonary
diseases (23,33). Consistent with observations from
other inflammatory disorders (34), the present study demonstrated
that, along with the increased aggregation and maturation of
pulmonary cDCs, acute lung inflammation and histopathologic injury
were also exacerbated. By contrast, inhibited aggregation and
maturation of pulmonary cDCs by lestaurtinib was associated with an
improvement in acute lung inflammation and injury. Therefore, the
present findings indicated the regulatory role of pulmonary cDCs in
the case of ARDS.
Since FLT3 signaling is one of the primary pathways
that controls the development and behavior of cDCs, including
differentiation, migration, aggregation, maturation and even
apoptosis (35), FLT3 signaling
was recently employed as a specific and efficient tool for cDC
manipulation under several inflammatory conditions (9,13,36,37). The present study also took
advantage of the cDC-targeting property of FLT3 signaling in ARDS.
As expected, the results revealed that FLT3L pretreatment resulted
in significantly increased aggregation and maturation of cDCs,
while blocking FLT3 signaling with lestaurtinib inhibited
aggregation and maturation of cDCs. These observations suggested
that FLT3 signaling may be a promising pathway for the manipulation
of cDCs in vivo.
As one of the most important antigen-presenting
cells (APCs), cDCs are generally recognized as a bridge linking
innate and adaptive immune responses (5). Previous studies have suggested that
cDCs determine the ensuing immune response via multiple mechanisms,
such as shifting the balance of the Th1/Th2 response, secreting
pro-inflammatory cytokines, and affecting innate immune elements
(16,38,39). Thus, the present study
investigated the mechanisms through which cDCs may affect
LPS-induced acute lung inflammation and injury. The present results
demonstrated that modulation of FLT3 signaling was associated with
altered aggregation and maturation of pulmonary DCs, as well as a
shifted the balance of the Th1/Th2 response and ensuing cytokine
production. Emerging data also suggest an underlying role of FLT3
signaling in the modulation of the inflammatory process (40,41). Furthermore, in vitro
administration of lestaurtinib only decreases the cDC population
and activation, but not T cell aggregation or activation (9). Together, these results suggest that
FLT3 signaling directly regulates the aggregation and maturation of
pulmonary cDCs, which in turn shifts the balance of the Th1/Th2
response and ensuing cytokine production.
Regulating neutrophil infiltration may be another
mechanism by which cDCs regulate LPS-induced acute lung
inflammation and injury. Neutrophils are the key elements of innate
immunity, and neutrophil infiltration serves a critical role in the
development of ARDS. Neutrophils are considered to be the first
mobilized cellular element in the pathogenesis of ARDS (42,43). However, recent studies have
demonstrated that pulmonary cDCs may aggregate before the
infiltration of neutrophils (44). In vitro studies have
demonstrated that cDCs not only enhance the chemotaxis of
neutrophils, but also protect neutrophils from apoptosis (45,46). The results from the present study
are consistent with these previous results.
Finally, the present results demonstrated that the
expression of FLT3L, a dedicated activator for FLT3 signaling, was
higher in ARDS lung tissue compared with normal lung tissue. These
findings suggested that the FLT3L-induced activation of FLT3
signaling during ARDS may be protective or self-reparative
(44), which provided a rationale
for DC-targeting therapy in ARDS.
It may be argued that cDCs may not be the only APCs
responsible for immunity or tolerance in the lung. Indeed,
plasmacytoid
CD11c+CD11b−B220+Gr-1+
DCs (pDCs) have been demonstrated to downregulate the production of
inflammatory cytokines in the lung, thus attenuating lung injury in
experimental ARDS (26). However,
pDCs account for <5% of the total pulmonary DC population and
are only found within the bronchial epithelium (5); their effect on the pulmonary immune
microenvironment is negligible compared with that of cDCs. However,
the distinctive role of pDCs in the pathogenesis of ARDS deserves
further investigation.
Of note, the results on CD80 expression on pulmonary
cDCs in the present study were unexpected. Neither LPS nor FLT3
signaling altered the cell-surface expression of CD80 in pulmonary
cDCs at 6 h following LPS challenge. One possible explanation is
that in most APC populations, the peak expression of both
costimulatory molecules (CD80 and CD86) occurs at least 18 h
following LPS challenge, and CD80 is expressed only inducibly and
later than CD86 (47,48). Therefore, the 6 h observation
period used in the present study may have been too short to detect
changes in CD80 expression.
Another limitation arises from the imbalance of the
Th1/Th2 skewed cytokine profiles in the lung. Although the
production of IFN-γ and IL-1β varied consistently with the relative
expression of T-bet, the concentration was fairly low at 24 h
following LPS challenge. Since the strong antigen-presenting
activity between DCs and T cells in draining lymph nodes occurs at
least 24 h after insult, it is reasonable that the concentration of
IFN-γ and IL-1β in the lung fluctuated in the low range at 24 h
after LPS challenge (49). In
addition, pulmonary IL-4 and IL-10 exhibited comparable
concentrations, which were much higher than those of IFN-γ and
IL-1β. Previous studies have proposed that under steady-state
conditions, an inherently Th2-biased response predominates in lung
parenchyma, providing a counteraction to potentially
tissue-damaging Th1 responses induced by innocuous inhaled antigens
(5). The increased concentrations
of IL-4 and IL-10 may thus reflect the inherently Th2-biased
response.
In conclusion, the present results highlighted that
pulmonary cDCs regulated acute lung inflammation and injury in
LPS-induced ARDS and revealed that pulmonary cDCs modulated
LPS-induced acute lung inflammation and injury through the
manipulation of neutrophil infiltration and balance of the Th1/Th2
response. The present study demonstrated that inhibition of FLT3
signaling by lestaurtinib attenuated acute lung injury, providing a
novel perspective for the prevention and treatment of ARDS.
However, clinical trials are required for the confirmation and
translation of the present research results in patients.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81400054), the
Natural Science Foundation of Jiangsu Province (grant no.
BK20140122), the Talented Youth Program of Jiangsu Province (grant
no. QNRC2017179) and the 333 High Level Talent Training Project of
Jiangsu Province (grant no. 2018III-0299).
Availability of data and materials
All data generated or analyzed during this study are
included in this article.
Authors' contributions
LL and LD conceived and designed the study. LL, LD,
DZ and FG performed the experiments, analyzed the data, and
interpreted the results. LD, FG and JY generated the figures and
drafted the manuscript. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
The experimental protocols were approved by the
Institutional Animal Care and Use Committee at Nanjing Medical
University (approval no. LLSP201607155). All animal experiments
were conducted in accordance with the National Institutes of Health
Guidance for the Care and Use of Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Thompson BT, Chambers RC and Liu KD: Acute
respiratory distress syndrome. N Engl J Med. 377:562–572. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fan E, Brodie D and Slutsky AS: Acute
respiratory distress syndrome: Advances in diagnosis and treatment.
JAMA. 319:698–710. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reilly JP, Christie JD and Meyer NJ: Fifty
years of research in ARDS. Genomic contributions and opportunities.
Am J Respir Crit Care Med. 196:1113–1121. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
ARDS Definition Task Force; Ranieri VM,
Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E,
Camporota L and Slutsky AS: Acute respiratory distress syndrome:
The Berlin Definition. JAMA. 307:2526–2533. 2012.PubMed/NCBI
|
|
5
|
Kumar V: Dendritic cells in sepsis:
Potential immunoregulatory cells with therapeutic potential. Mol
Immunol. 101:615–626. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lambrecht BN and Hammad H: The role of
dendritic and epithelial cells as master regulators of allergic
airway inflammation. Lancet. 376:835–843. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Buttignol M, Pires-Neto RC, Rossi E Silva
RC, Albino MB, Dolhnikoff M and Mauad T: Airway and parenchyma
immune cells in influenza A(H1N1)pdm09 viral and non-viral diffuse
alveolar damage. Respir Res. 18:1472017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
von Wulffen W, Steinmueller M, Herold S,
Marsh LM, Bulau P, Seeger W, Welte T, Lohmeyer J and Maus UA: Lung
dendritic cells elicited by Fms-like tyrosine 3-kinase ligand
amplify the lung inflammatory response to lipopolysaccharide. Am J
Respir Crit Care Med. 176:892–901. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Whartenby KA, Calabresi PA, McCadden E,
Nguyen B, Kardian D, Wang T, Mosse C, Pardoll DM and Small D:
Inhibition of FLT3 signaling targets DCs to ameliorate autoimmune
disease. Proc Natl Acad Sci USA. 102:16741–16746. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lau CM, Nish SA, Yogev N, Waisman A,
Reiner SL and Reizis B: Leukemia-associated activating mutation of
Flt3 expands dendritic cells and alters T cell responses. J Exp
Med. 213:415–431. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim SW, Choi SM, Choo YS, Kim IK, Song BW
and Kim HS: Flt3 ligand induces monocyte proliferation and enhances
the function of monocyte-derived dendritic cells in vitro. J Cell
Physiol. 230:1740–1749. 2015. View Article : Google Scholar
|
|
12
|
Sato T, Yang X, Knapper S, White P, Smith
BD, Galkin S, Small D, Burnett A and Levis M: FLT3 ligand impedes
the efficacy of FLT3 inhibitors in vitro and in vivo. Blood.
117:3286–3293. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dong L, He H, Liu J, Liu L, Yang Y and Qiu
H: The control effects of FLT3 signaling-dependent pulmonary
conventional dendritic cells on the initiation of acute lung
inflammation response to lipopolysaccharide induced acute lung
injury in mice. Zhonghua Ji Zhen Yi Xue Za Zhi. 25:1412–1417.
2016.In Chinese.
|
|
14
|
Mellman I: Dendritic cells: Master
regulators of the immune response. Cancer Immunol Res. 1:145–149.
2013. View Article : Google Scholar
|
|
15
|
Shao Z, Bharadwaj AS, McGee HS, Makinde TO
and Agrawal DK: Fms-like tyrosine kinase 3 ligand increases a lung
DC subset with regulatory properties in allergic airway
inflammation. J Allergy Clin Immunol. 123:917–924.e2. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ludwig IS, Geijtenbeek TB and van Kooyk Y:
Two way communication between neutrophils and dendritic cells. Curr
Opin Pharmacol. 6:408–413. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dieker J, Tel J, Pieterse E, Thielen A,
Rother N, Bakker M, Fransen J, Dijkman HB, Berden JH, de Vries JM,
et al: Circulating apoptotic microparticles in systemic lupus
erythematosus patients drive the activation of dendritic cell
subsets and prime neutrophils for NETosis. Arthritis Rheumatol.
68:462–472. 2016. View Article : Google Scholar
|
|
18
|
Baudiß K, de Paula Vieira R, Cicko S,
Ayata K, Hossfeld M, Ehrat N, Gómez-Muñoz A, Eltzschig HK and Idzko
M: C1P attenuates lipopolysaccharide-induced acute lung injury by
preventing NF-κB activation in neutrophils. J Immunol.
196:2319–2326. 2016. View Article : Google Scholar
|
|
19
|
McWilliam AS, Nelson D, Thomas JA and Holt
PG: Rapid dendritic cell recruitment is a hallmark of the acute
inflammatory response at mucosal surfaces. J Exp Med.
179:1331–1336. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Olguín-Alor R, de la Fuente-Granada M,
Bonifaz LC, Antonio-Herrera L, García-Zepeda EA and Soldevila G: A
key role for inhibins in dendritic cell maturation and function.
PLoS One. 11:e01678132016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schlosser K, Taha M, Deng Y, McIntyre LA,
Mei SHJ and Stewart DJ: High circulating angiopoietin-2 levels
exacerbate pulmonary inflammation but not vascular leak or
mortality in endotoxin-induced lung injury in mice. Thorax.
73:248–261. 2018. View Article : Google Scholar :
|
|
22
|
Aggarwal NR, King LS and D'Alessio FR:
Diverse macrophage populations mediate acute lung inflammation and
resolution. Am J Physiol Lung Cell Mol Physiol. 306:L709–L725.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wen H, Hogaboam CM, Gauldie J and Kunkel
SL: Severe sepsis exacerbates cell-mediated immunity in the lung
due to an altered dendritic cell cytokine profile. Am J Pathol.
168:1940–1950. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Smit JJ, Lindell DM, Boon L, Kool M,
Lambrecht BN and Lukacs NW: The balance between plasmacytoid DC
versus conventional DC determines pulmonary immunity to virus
infections. PLoS One. 3:e17202008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vermaelen K and Pauwel R: Accurate and
simple discrimination of mouse pulmonary dendritic cell and
macrophage populations by flow cytometry: Methodology and new
insights. Cytometry A. 61:170–177. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Venet F, Huang X, Chung CS, Chen Y and
Ayala A: Plasmacytoid dendritic cells control lung inflammation and
monocyte recruitment in indirect acute lung injury in mice. Am J
Pathol. 176:764–773. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Masten BJ and Lipscomb MF: Comparison of
lung dendritic cells and B cells in stimulating naive
antigen-specific T cells. J Immunol. 162:1310–1317. 1999.PubMed/NCBI
|
|
28
|
Liu H, Wu J, Yao JY, Wang H and Li ST: The
role of oxidative stress in decreased acetylcholinesterase activity
at the neuromuscular junction of the diaphragm during sepsis. Oxid
Med Cell Longev. 2017:97186152017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li L, Dong L, Wang Y, Zhang X and Yan J:
Lats1/2-mediated alteration of hippo signaling pathway regulates
the fate of bone marrow-derived mesenchymal stem cells. Biomed Res
Int. 2018:43879322018. View Article : Google Scholar
|
|
30
|
Li L, Dong L, Zhang J, Gao F, Hui J and
Yan J: Mesenchymal stem cells with downregulated Hippo signaling
attenuate lung injury in mice with lipopolysaccharide-induced acute
respiratory distress syndrome. Int J Mol Med. 43:1241–1252.
2019.PubMed/NCBI
|
|
31
|
Smith KM, Mrozek JD, Simonton SC, Bing DR,
Meyers PA, Connett JE and Mammel MC: Prolonged partial liquid
ventilation using conventional and high-frequency ventilatory
techniques: Gas exchange and lung pathology in an animal model of
respiratory distress syndrome. Crit Care Med. 25:1888–1897. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hubbell JA, Thomas SN and Swartz MA:
Materials engineering for immunomodulation. Nature. 462:449–460.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lambrecht BN, Salomon B, Klatzmann D and
Pauwels RA: Dendritic cells are required for the development of
chronic eosinophilic airway inflammation in response to inhaled
antigen in sensitized mice. J Immunol. 160:4090–4097.
1998.PubMed/NCBI
|
|
34
|
Riccardi F, Della Porta MG, Rovati B,
Casazza A, Radolovich D, De Amici M, Danova M and Langer M: Flow
cytometric analysis of peripheral blood dendritic cells in patients
with severe sepsis. Cytometry B Clin Cytom. 80:14–21. 2011.
View Article : Google Scholar
|
|
35
|
Schlitzer A, Zhang W, Song M and Ma X:
Recent advances in understanding dendritic cell development,
classification, and phenotype. F1000Res. 7:2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Agrawal DK, Hopfenspirger MT, Chavez J and
Talmadge JE: Flt3 ligand: A novel cytokine prevents allergic asthma
in a mouse model. Int Immunopharmacol. 1:2081–2089. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bohannon J, Fang G, Cui W, Sherwood E and
Toliver-Kinsky T: Fms-like tyrosine kinase-3 ligand alters
antigen-specific responses to infections after severe burn injury.
Shock. 32:435–441. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jankovic D, Liu Z and Gause WC: Th1- and
Th2-cell commitment during infectious disease: Asymmetry in
divergent pathways. Trends Immunol. 22:450–457. 2011. View Article : Google Scholar
|
|
39
|
Abe K, Nguyen KP, Fine SD, Mo JH, Shen C,
Shenouda S, Corr M, Jung S, Lee J, Eckmann L and Raz E:
Conventional dendritic cells regulate the outcome of colonic
inflammation independently of T cells. Proc Natl Acad Sci USA.
104:17022–17027. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Beshara R, Sencio V, Soulard D, Barthélémy
A, Fontaine J, Pinteau T, Deruyter L, Ismail MB, Paget C, Sirard
JC, et al: Alteration of Flt3-Ligand-dependent de novo generation
of conventional dendritic cells during influenza infection
contributes to respiratory bacterial superinfection. PLoS Pathog.
14:e10073602018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Madan B, Goh KC, Hart S, William AD,
Jayaraman R, Ethirajulu K, Dymock BW and Wood JM: SB1578, a novel
inhibitor of JAK2, FLT3, and c-Fms for the treatment of rheumatoid
arthritis. J Immunol. 189:4123–4134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kangelaris KN, Prakash A, Liu KD,
Aouizerat B, Woodruff PG, Erle DJ, Rogers A, Seeley EJ, Chu J, Liu
T, et al: Increased expression of neutrophil-related genes in
patients with early sepsis-induced ARDS. Am J Physiol Lung Cell Mol
Physiol. 308:L1102–L1113. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li H, Zhou X, Tan H, Hu Y, Zhang L, Liu S,
Dai M, Li Y, Li Q, Mao Z, et al: Neutrophil extracellular traps
contribute to the pathogenesis of acid-aspiration-induced ALI/ARDS.
Oncotarget. 9:1772–1784. 2017.
|
|
44
|
Udayanga KG, Nakamura Y, Nakahashi-Oda C
and Shibuya A: Immunoreceptor CD300a on mast cells and dendritic
cells regulates neutrophil recruitment in a murine model of sepsis.
Int Immunol. 28:611–615. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sallusto F, Palermo B, Lenig D, Miettinen
M, Matikainen S, Julkunen I, Forster R, Burgstahler R, Lipp M and
Lanzavecchia A: Distinct patterns and kinetics of chemokine
production regulate dendritic cell function. Eur J Immunol.
29:1617–1625. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dransfield I and Rossi AG: Granulocyte
apoptosis: Who would work with a 'real' inflammatory cell? Biochem
Soc Trans. 32:447–451. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sharpe AH and Freeman GJ: The B7-CD28
superfamily. Nat Rev Immunol. 2:116–126. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hathcock KS, Laszlo G, Pucillo C, Linsley
P and Hodes RJ: Comparative analysis of B7-1 and B7-2 costimulatory
ligands: Expression and function. J Exp Med. 180:631–640. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xia W, Pinto CE and Kradin RL: The
antigen-presenting activities of Ia+ dendritic cells
shift dynamically from lung to lymph node after an airway challenge
with soluble antigen. J Exp Med. 181:1275–1283. 1995. View Article : Google Scholar : PubMed/NCBI
|