Introduction
There are different degrees of apoptosis in
myocardial ischemia/reperfusion (I/R) injury. Reducing myocardial
apoptosis has become a focus of research on myocardial protection.
In 2003, Wang et al (1)
first reported that calcium-sensing receptor (CaSR) was expressed
in myocardial tissue, leading to the hypothesis that CaSRs
participated in myocardial I/R injury induced by calcium overload
(2); however, the specific
mechanism remains unknown.
With I/R, CaSR activation of the phospholipase C
(PLC) pathway causes calcium release from the endoplasmic reticulum
(ER) (1,3-5),
resulting in excessive ER stress (6). Multiple studies have demonstrated
that the protein kinase C (PKC) family participates in the
protection of cardiomyocytes or damage from myocardial I/R injury
(5-7). The promotion or inhibition of
apoptosis depends on the PKC subtype (8,9).
PKCδ, one of the PKC subtypes, is an important signal in multiple
types of cells, including myocardial infarction (10), lung inflammation (11), development of atherosclerosis
(12) and the salivary gland
(13). Previous studies have
confirmed that myocardial I/R injury results in the transfer of
PKCδ from the cell cytoplasm to the mitochondria and ER (5,7,14,15). During I/R, CaSR induces
phosphorylation of PKCδ and migrates to mitochondria to induce
cardiomyocyte apoptosis (5);
however, the mechanism by which PKCδ participates in cell apoptosis
has not been fully elucidated. The aim of the present study was to
confirm that CaSR could activate PKCδ to induce cardiomyocyte
apoptosis through ER stress-associated apoptotic pathways during
I/R in vivo.
Materials and methods
Animals
Male Wistar rats, weighing 200-250 g, were obtained
from the Animal Center of Harbin Medical University. All animals
were treated according to the Guidelines for the Care and Use of
Laboratory Animals and all protocols were approved by the Ethics
Committee of Basic Medical College of Harbin Medical University.
The associated permit number is SCXK (Hei) 2013-001.
Materials
NPS-2390, GdCl3, verapamil, rottlerin and
amiloride were obtained from Sigma-Aldrich (Merck KGaA). The
terminal deoxynucleotidyl transferase (TdT)-mediated-UTP nick end
labeling (TUNEL) kit, and the
5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocar
bocyanine iodide (JC-1) kit were from Roche Diagnostics GmbH.
Primary antibodies targeting PKCδ (cat. no. sc-213),
glucose-regulated protein 78 (GRP78; cat. no. sc-13968), Caspase-12
(cat. no. sc-5627), c-Jun N-terminal kinase (JNK; cat. no. sc-474),
phosphorylated (p-) JNK (cat. no. sc-6254) and Caspase-3 (cat. no.
sc-7148) were obtained from Santa Cruz Biotechnology, Inc. The
primary antibody targeting CaSR was from Alpha Diagnostic
International Inc. (cat. no. ACR-004). β-actin (cat no. M01263-2)
and Calnexin (cat. no. A03372) were obtained from Boster Biological
Technology.
Experimental protocol
Heparinized rats [2,000 U/kg, by intraperitoneal
(i.p.) injection] were anesthetized with 2% pentobarbital sodium
(40 mg/kg; i.p.). Following endotracheal intubation, an animal
breathing machine was provided with a rodent respirator at a rate
of 50 breaths/min and a tidal volume of 20 ml/kg (16). Between the third and fourth ribs,
a left parasternal incision was made to expose the heart. The left
anterior descending artery (LAD) was ligated using a 6-0 silk
suture. Myocardial I/R was induced by the compression of the LAD
(17). The changes in the ST-T
segment, caused by tightening or loosing of the silk suture, were
monitored by ECG. After coronary artery ligation for 30 min, the
ligation was removed to restore myocardial blood perfusion. At 2 h
after reperfusion, the rats were sacrificed by exsanguination under
anesthesia, and aortic inverse irrigation with PBS was performed to
the hearts in order to remove residual blood. Subsequently, a
portion of the tissue of the anterior wall of the left ventricle
near the apex was obtained for further analysis.
The animals were randomly divided into five groups:
i) Control group (n=10), thread only, without ligation; ii) I/R
group (I/R; n=10), coronary artery ligation for 30 min, and
loosening of the silk suture to restore myocardial blood perfusion
for 2 h; iii) I/R+Amiloride+Verapamil+GdCl3 group
(I/R+A+V+Gd; n=10), 20 µM amiloride (inhibitor of
Na+-Ca2+ exchanger), 1 µM verapamil
(L-type calcium channel blocker) and 30 µM GdCl3
(activator of CaSR) were added by injection through the femoral
vein 10 min prior to reperfusion; iv)
I/R+Amiloride+Verapamil+GdCl3+NPS-2390 group
(I/R+A+V+Gd+NPS; n=10), NPS-2390 (inhibitor of CaSR) was added by
injection through the femoral vein 10 min prior to reperfusion,
while all other procedures were the same as those performed in the
I/R+Amiloride+Verapamil+GdCl3 group; and v)
I/R+Amiloride+Verapamil+GdCl3+Rottlerin group
(I/R+A+V+Gd+PKCδI; n=10), amiloride (20 µM), verapamil (1
µM), GdCl3 (30 µM) and 10 µM
rottlerin (specific inhibitor of PKCδ) were added by injection
through the femoral vein 10 min before reperfusion, and all other
procedures were identical to those performed in the
I/R+Amiloride+Verapamil+Gd Cl3+NPS-2390 group.
ER isolation
Frozen heart specimens were minced and ground by
pestle on ice and then homogenized using a tissue homogenizer
thirty times in 3 ml of cool lysis buffer (250 mM sucrose, 20
mmol/l Tris-HCl pH 7.2, 10 mmol/l KCl, 1.5 mmol/l MgCl2,
1 mmol/l EDTA, 1:300 protease inhibitor cocktail, 1:300 phosphatase
inhibitor cocktail). The muscle cells were then collected in a
microcentrifuge tube and incubated for 30 min at 4°C. The
homogenates were centrifuged at 800 × g for 10 min at 4°C, and the
resulting supernatant was centrifuged at 10,000 × g for 20 min at
4°C. The new supernatants were centrifuged at 100,000 × g for 1 h
(4°C) to obtain the ER (pellet) and cytosolic (supernatant)
extracts. The ER pellet was resuspended in lysis buffer containing
1% Triton X-100 (14). The ER
extract was stored at −70°C.
TUNEL staining
Identification of apoptotic cells was performed
using an in situ cell death detection kit (Roche Diagnostics
GmbH), according to the manufacturer's instructions. After rinsing
with PBS, the heart specimens were fixed with 4% formaldehyde
solution at room temperature for 24 h. Following routine protocols,
the heart specimens were paraffin-embedded, sectioned at 4
µm thickness and dewaxed. After rinsing with PBS, the slides
were soaked in a solution containing 0.1% Triton X-100 and 0.1%
sodium citrate for 2 min to increase the cell membrane
permeability. The slides were incubated with 50 µl of TUNEL
reaction mixture for 60 min at 37°C. 3,3′-Diaminobenzidine (DAB;
0.05%) was added to the slide for 30 min and then incubated in the
substrate solution (100 µl) for an additional 10-15 min.
Labeled myocytes were analyzed using a light microscope (XSZ-D2;
Olympus Corporation).
Transmission electron microscopy
Heart samples were fixed with 2.5% glutaraldehyde
for 24 h at 4°C, washed with PBS, and fixed with 1% osmium
tetroxide for 2 h. Samples were then dehydrated with fractionated
acetone, embedded in Epon-Araldite resin, and cut into ultrathin
sections of 50-70 nm. Ultrathin sections were placed on copper
grids and stained with uranyl acetate and lead citrate. Samples
were examined under a JEM-2000EX transmission electron microscope
(16,18). The images were captured using a
Hamamatsu camera (Hamamatsu Photonics, Ltd.).
Western blot analysis
Protein extracts were prepared as aforementioned and
subjected to 10% SDS-PAGE. Each lane contained 60 µg of
protein. All samples were transferred to nitrocellulose membranes
and blocked for 1 h at room temperature with 5% BSA in
Tris-buffered saline/Tween-20 (TBST) buffer. The membranes were
then exposed to the rabbit monoclonal anti-rat CaSR antibody, the
rabbit polyclonal serum against rat total PKCδ, and GRP78,
Calnexin, Caspase-12, p-JNK, JNK and Caspase-3 antibodies at a
concentration of 1:1,000 overnight at 4°C. The secondary antibody
alkaline phosphatase IgG (1:2,000; cat. no. S3731; Promega
Corporation) was then added for 2 h at room temperature. The final
signals were quantified using a Bio-Rad Chemi Doc EQ densitometer
and Bio-Rad Quantity One software (Bio-Rad Laboratories, Inc.).
β-actin was used as internal reference for the total cytoplasmic
extract. Calnexin was used as internal reference for the ER
extract. All band intensities were normalized to β-actin or
Calnexin and expressed as a percentage of the control sample.
Isolation of acute myocardial cells
Rats without any treatment were anesthetized with 2%
pentobarbital (100 mg/kg, i.p.) and the hearts were quickly
removed. Then the hearts were perfused (5 min) through the aorta
with standard Tyrode's solution at 37°C until the residue was clear
(19). The composition of the
standard Tyrode's solution was: 136 mM NaCl, 5.4 mM KCl, 0.35 mM
NaH2PO4, 1.0 mM MgCl2, 2.0 mM
CaCl2, 10 mM dextrose and 10 mM HEPES (pH adjusted to
7.4 with NaOH). The solution was equilibrated with 95%
O2 and 5% CO2 at room temperature prior to
use. Then the hearts were sequentially perfused with
Ca2+-free Tyrode's solution for 10 min, and a
Ca2+-free Tyrode's solution containing 0.015 g/l
collagenase for 30 min. The ventricular tissue (2-3 mm in diameter)
was excised and placed in KB solution. The composition of the KB
solution was: 70 mM glutamic acid, 15 mM taurin, 30 mM KCl, 10 mM
KH2PO4, 10 mM HEPES, 0.5 mM MgCl2,
0.5 mM ethylene glycol tetraacetic acid and 10 mM glucose (pH
adjusted to 7.3-7.4 with KOH). Myocardial cells were isolated and
kept at room temperature (5).
The acutely isolated myocardial cells were then
divided into five treatment groups: i) Control group, myocardial
cells were incubated in Tyrode's solution without any treatment;
ii) I/R group, myocardial cells were incubated with Tyrode's
solution for 8 min, then incubated in simulated ischemic solution
(NaCl 136 mM, KCl 5.4 mM, NaH2PO4 0.35 mM,
MgCl2 1.0 mM and Hepes 10 mM, pH 6.8) at 37°C for 30
min, and finally incubated with Tyrode's solution for another 8
min; iii) I/R+A+V+Gd, myocardial cells were incubated with
simulated ischemia solution for 30 min, followed by reperfusion for
8 min in Tyrode solution immediately after addition of 20 µM
Amiloride, 1 µM Verapamil and 30 µM GdCl3;
iv) I/R+A+V+Gd+NPS, 20 µM Amiloride, 1 µM Verapamil,
30 µM GdCl3 and 10 µM NPS-2390 were added to the
myocardial cells before reperfusion with Tyrode's solution for 8
min; and v) I/R+A+V+Gd+PKCδI, 20 µM Amiloride, 1 µM
Verapamil and 30 µM GdCl3 and 10 µM
Rottlerin were added to the myocardial cells before reperfusion
with Tyrode's solution for 8 min. Following treatments, cells were
analyzed as indicated.
JC-1 fluorescence staining
The decrease of mitochondrial membrane potential is
a landmark event in the early stages of apoptosis. This method can
identify apoptotic cells earlier than other methods. Mitochondrial
membrane potential was measured by JC-1 fluorescence staining. JC-1
is an ideal fluorescent probe for detecting mitochondrial membrane
potential. When the mitochondrial membrane potential is high, JC-1
aggregates in the matrix of the mitochondria to form a polymer,
which can produce red fluorescence. When the mitochondrial membrane
potential is low, JC-1 cannot accumulate in the matrix of
mitochondria, and therefore JC-1 exists as a monomer which can
produce green fluorescence. Therefore, changes in mitochondrial
membrane potential can be easily detected through the change of
fluorescence color. The relative ratio of green and red
fluorescence is commonly used to measure the proportion of
mitochondrial depolarization. The appearance of green fluorescence
indicates a decrease in mitochondrial membrane potential, and the
cell is likely to be in the early stages of apoptosis. Red
fluorescence indicates that the mitochondrial membrane potential is
normal and the state of the cells is normal.
In the present study, isolated cardiomyocytes were
stained with JC-1 (1 µg/ml) for 15 min at 37°C and washed
with Tyrode's solution three times. The changes of mitochondrial
membrane potential were observed using confocal laser microscopy.
The excitation light was set to 488 nm when the JC-1 monomer
(green) was detected, and the emission light was set to 530 nm.
When the JC-1 polymer (red) was detected, the excitation light was
set to 525 nm, and the emission light was set to 590 nm. Multiple
fields of view (>10) were captured from each sample and the
average intensity of each field was quantified. The intensity ratio
of JC-1 polymer to JC-1 monomer in each field was calculated. A
decrease of the ratio indicated a decrease in the membrane
potential, and an increase of the ratio indicated an increase in
the membrane potential.
Statistical analysis
Data were expressed as the mean ± SD from three
independent experiments. Data were analyzed using one-way ANOVA
followed by Bonferroni post hoc tests. All statistical analyses
were performed using Prism 5 (GraphPad Software, Inc.). P<0.05
was considered to indicate a statistically significant
difference.
Results
CaSR involved in cardiomyocyte apoptosis
during I/R
Previous studies have confirmed that myocardial I/R
results in the translocation of PKCδ from the cell cytoplasm to the
ER (7,14,15), and activation of CaSR by
myocardial I/R can induce cardiomyocyte apoptosis through the ER
stress-related apoptosis pathway (6). However, the interaction of the CaSR
with PKCδ to induce cardiomyocyte apoptosis in vivo has not
been fully elucidated. Thus, the following experimental methods
were used to confirm the relationship between CaSR and PKCδ in
cardiomyocyte apoptosis during I/R in vivo.
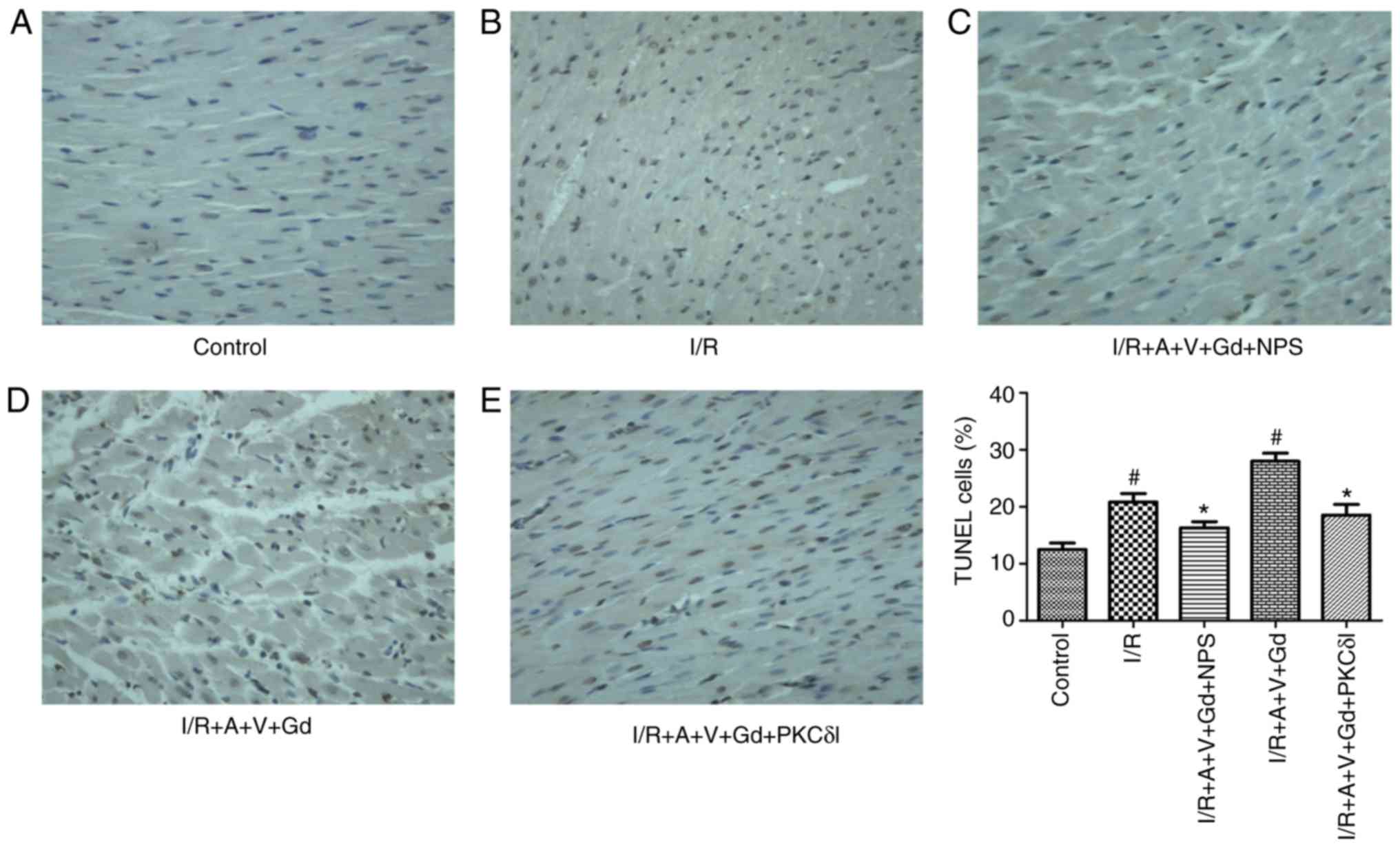
Cardiomyocyte apoptosis was detected by TUNEL
staining and JC-1 fluorescence staining. In the TUNEL experiment,
normal cardiomyocyte nuclei were stained blue, while apoptotic cell
nuclei were stained brown. The ratio of TUNEL-positive cells was
increased significantly in the I/R and I/R+A+V+Gd groups compared
with the Control group (P<0.05; Fig. 1). In the I/R+A+V+Gd+NPS group and
I/R+A+V+Gd+PKCδI group, the ratio of apoptotic cells was lower
compared with the I/R+A+V+Gd group (P<0.05; Fig. 1). The results of cardiomyocyte
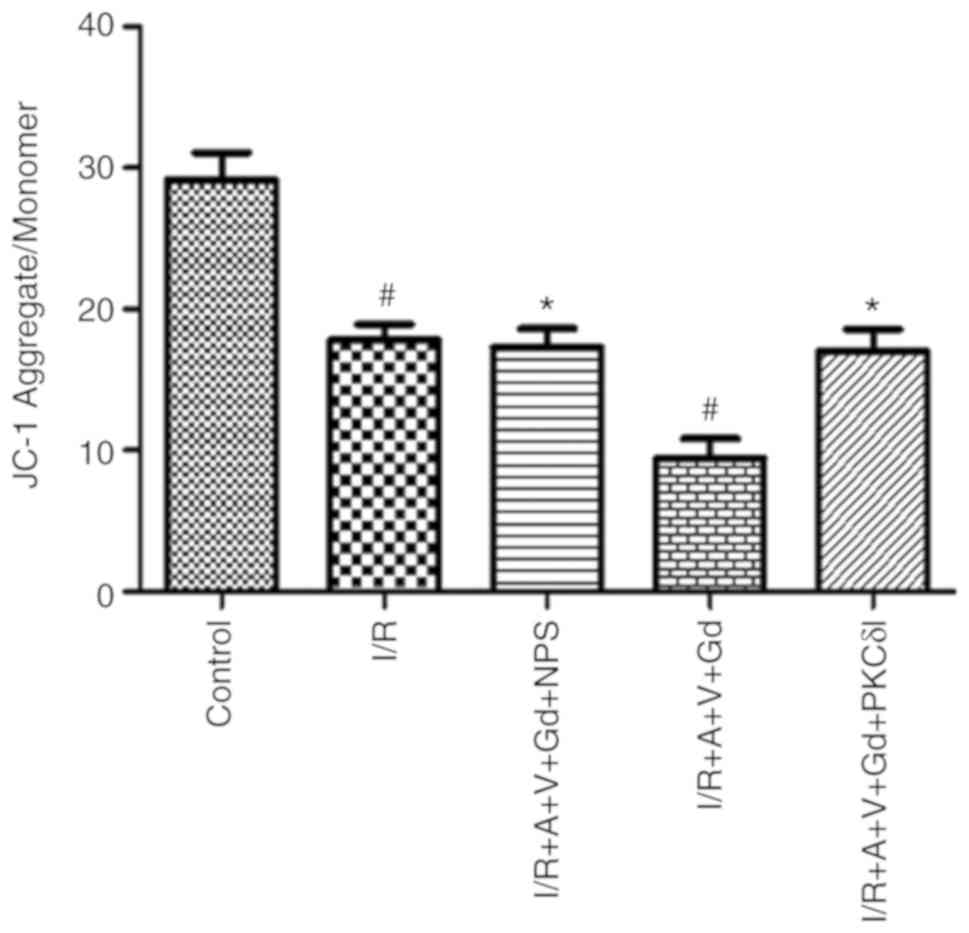
JC-1 staining revealed that the intensity ratio of JC-1
polymer/monomer decreased significantly in the I/R and I/R+A+V+Gd
groups compared with the Control group (P<0.05; Fig. 2). However, compared with the
I/R+A+V+Gd group, the ratio of JC-1 polymer/monomer in the
I/R+A+V+Gd+NPS and I/R+A+V+Gd+PKCδI groups remained at a high level
(P<0.05; Fig. 2).
Morphological characterization of
cardiomyocyte apoptosis
In the present study, ultrastructural changes in
cardiomyocytes were examined by transmission electron microscopy.
Morphological characteristics indicating apoptosis include
condensation, aggregation and margination of nuclear chromatin, ER
expansion, capsule bubble and mitochondrial swelling, and
mitochondrial crest fracture. These apoptotic characteristics were
observed in the I/R+A+V+Gd and I/R groups, but not in the Control
group (Fig. 3). By contrast, in
the I/R+A+V+Gd+NPS and I/R+A+V+Gd+PKCδI groups, only a slight
margination of the nuclear chromatin, expansion of the ER and
swelling of the mitochondrion were observed (Fig. 3).
CaSR protein expression in myocardial
cells
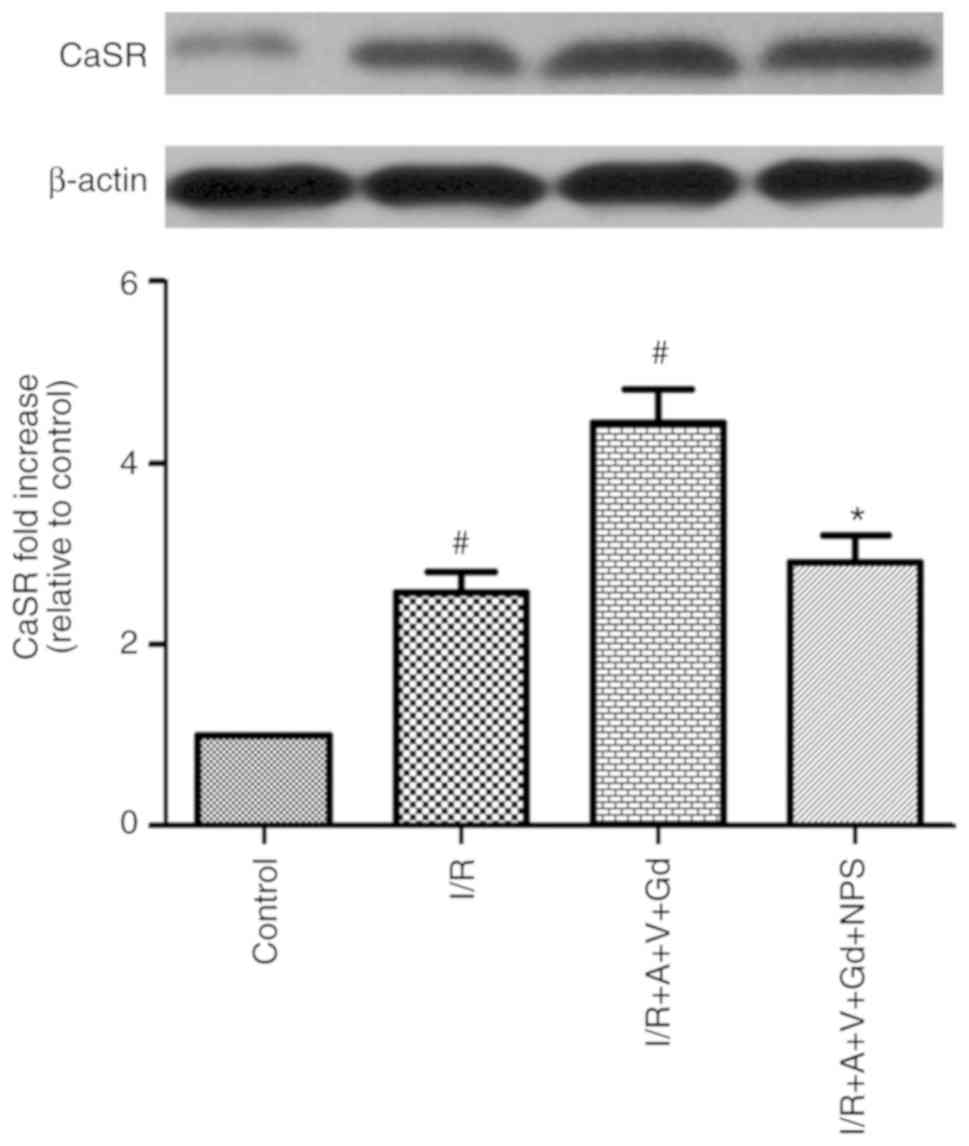
Since CaSR serves a key role in the elevation of
[Ca2+]i in cardiomyocytes (5), the expression levels of the CaSR
protein, with a relative molecular mass of 130 kDa, were determined
by western blot analysis. Compared with the Control group, the
protein expression levels of CaSR in the I/R and I/R+A+V+Gd groups
were increased (P<0.05; Fig.
4). Additionally, the CaSR protein expression levels in the
I/R+A+V+Gd+NPS group was decreased compared with the I/R+A+V+Gd
group (P<0.05; Fig. 4). These
results demonstrated that GdCl3 could upregulate the
expression of CaSR (Fig. 4).
Effect of CaSR in myocardial I/R on the
expression of ER stress-related proteins in vivo
The ER environment can be stimulated by various
stimuli, such as a perturbation of Ca2+ homeostasis,
which leads to initiation of the unfolded protein response (UPR).
The UPR is a protective mechanism against ER stress, which is
conducive to the stability of the cellular environment. If
excessive ER stress is not resolved, it will induce cell apoptosis
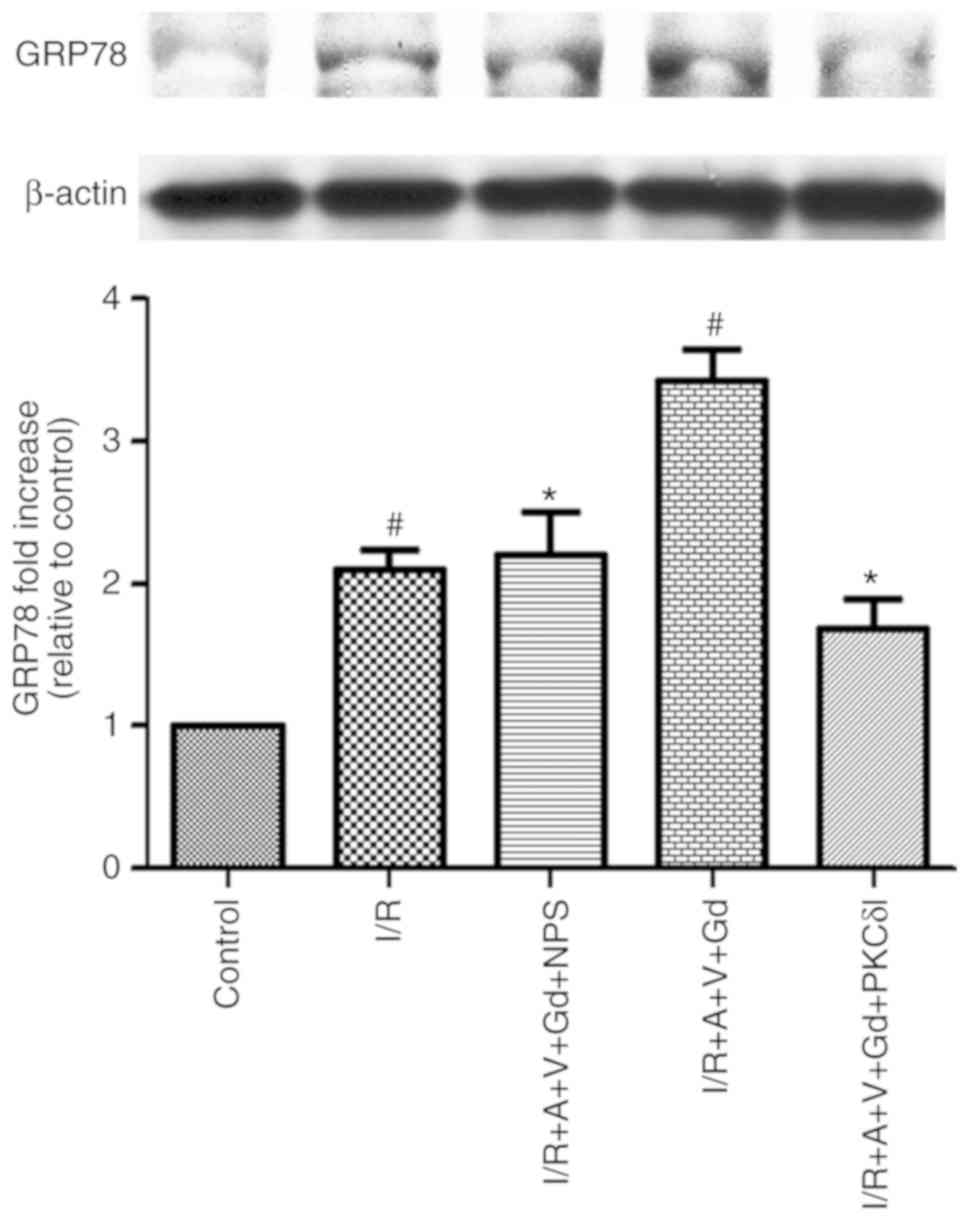
(20). To detect the occurrence
of ER stress, protein expression levels of ER stress-related
proteins, such as GRP78, can be detected, which is resistant to the
UPR and has been extensively used as a marker for ER stress and UPR
(21). In the present study, the
protein expression levels of GRP78 were detected by western blot
analysis. The results demonstrated that GRP78 expression in the I/R
and I/R+A+V+Gd groups was significantly higher compared with the
Control group, but in the I/R+A+V+Gd+NPS and I/R+A+V+Gd+PKCδI
groups, GRP78 expression was significantly lower compared with the
I/R+A+V+Gd group (P<0.05; Fig.
5). The present results demonstrated that activated CaSR in I/R
induced ER stress in vivo. These results also determined
that inhibition of PKCδ may protect the myocardium from ER stress
in an in vivo model of I/R myocardial injury.
CaSR activates PKCδ induction of the ER
stress-induced apoptosis pathway in vivo
The translocation of PKCδ to the ER, to participate
in ER stress-induced apoptosis, has been previously documented
(14). Next, the present study
investigated whether CaSR activating PKCδ participated in the
apoptotic pathway in vivo. Previous studies have shown that
several molecules, including JNK, CCAAT/enhancer-binding
protein-homologous protein (CHOP) and Caspase-12, are involved in
the ER stress-induced apoptosis pathway (22). In the current study, the protein
expression of Caspase-12, p-JNK and Caspase-3 in cardiomyocytes was
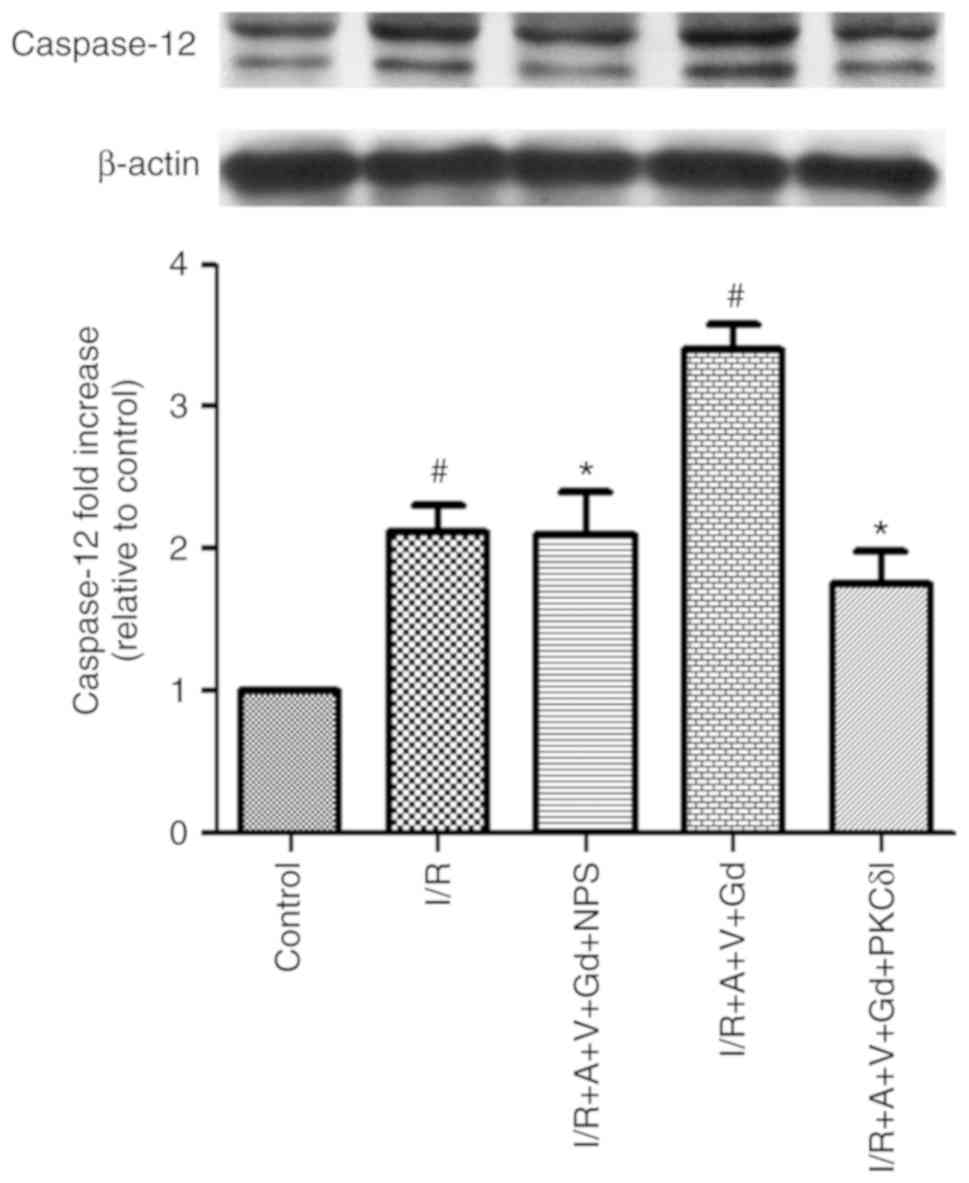
examined by western blot analysis. Cleaved Caspase-12 (the 42 kDa
proteolytic fragment of Caspase-12) was significantly increased in
the I/R and I/R+A+V+Gd groups compared with the Control group
(P<0.05; Fig. 6). The
expression levels of cleaved Caspase 12 were decreased in the
I/R+A+V+Gd+NPS and I/R+A+V+Gd+PKCδI groups compared with the
I/R+A+V+Gd group (P<0.05, Fig.
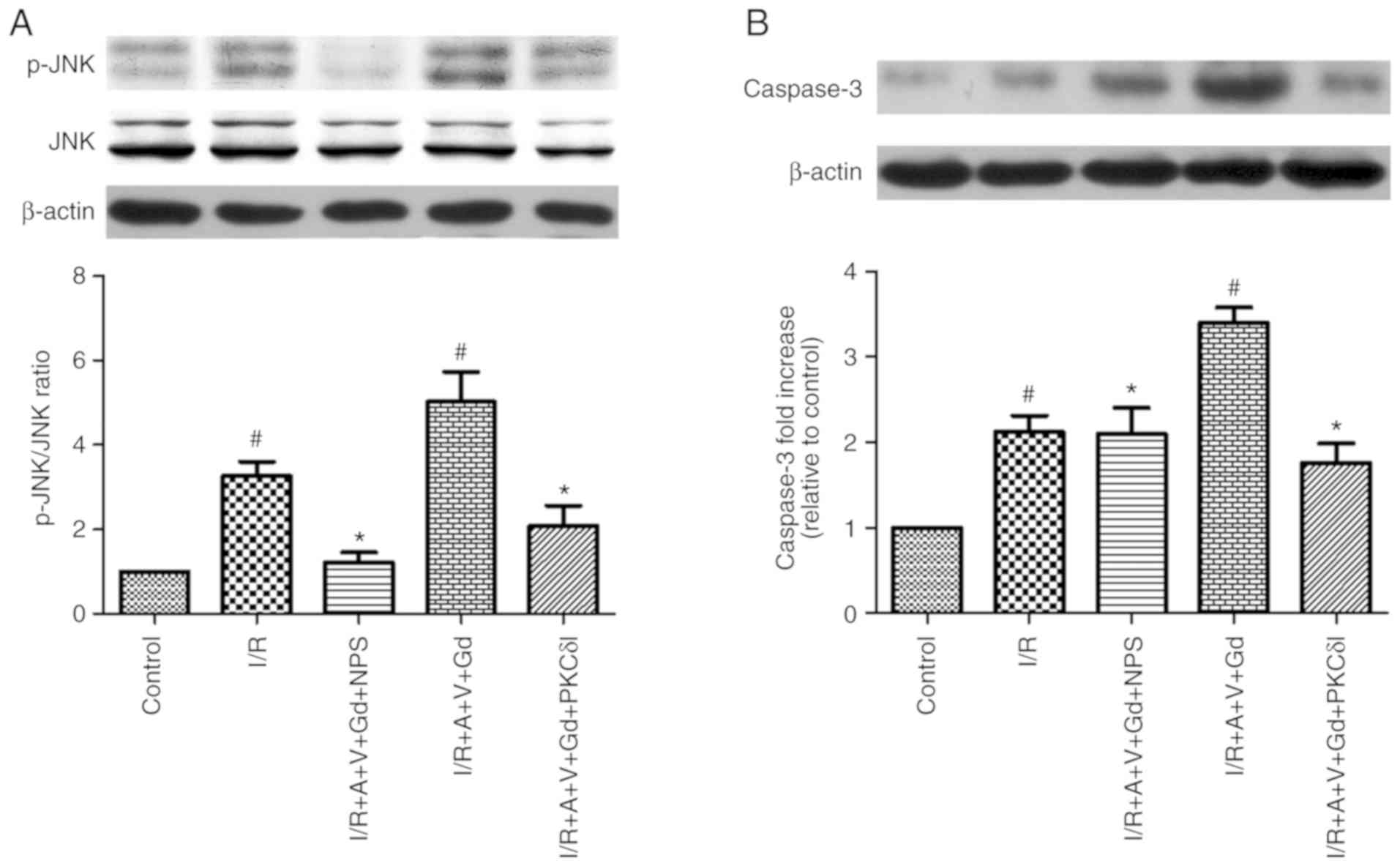
6). As presented in Fig. 7,
the protein expression levels of Caspase-3 and the ratio of
p-JNK/JNK in the I/R and I/R+A+V+Gd groups were higher compared
with the Control group. The levels of Caspase-3 and the p-JNK/JNK
ration in the I/R+A+V+Gd+NPS and I/R+A+V+Gd+PKCδI groups were
decreased compared wih the I/R+A+V+Gd group (P<0.05; Fig. 7). The protein expression levels of
Caspase-12, p-JNK and Caspase-3 are related to the rate of
apoptosis of cardiomyocytes. Therefore, the present results
revealed that CaSR activated PKCδ to participate in apoptosis
through the ER stress-induced apoptosis pathway.
CaSR induces PKCδ translocation to the ER
following I/R and participates in the ER stress-induced apoptosis
pathway
In the myocardium, inhibition of PKCδ activation
could significantly inhibit the ER stress response at the beginning
of reperfusion (14). In the
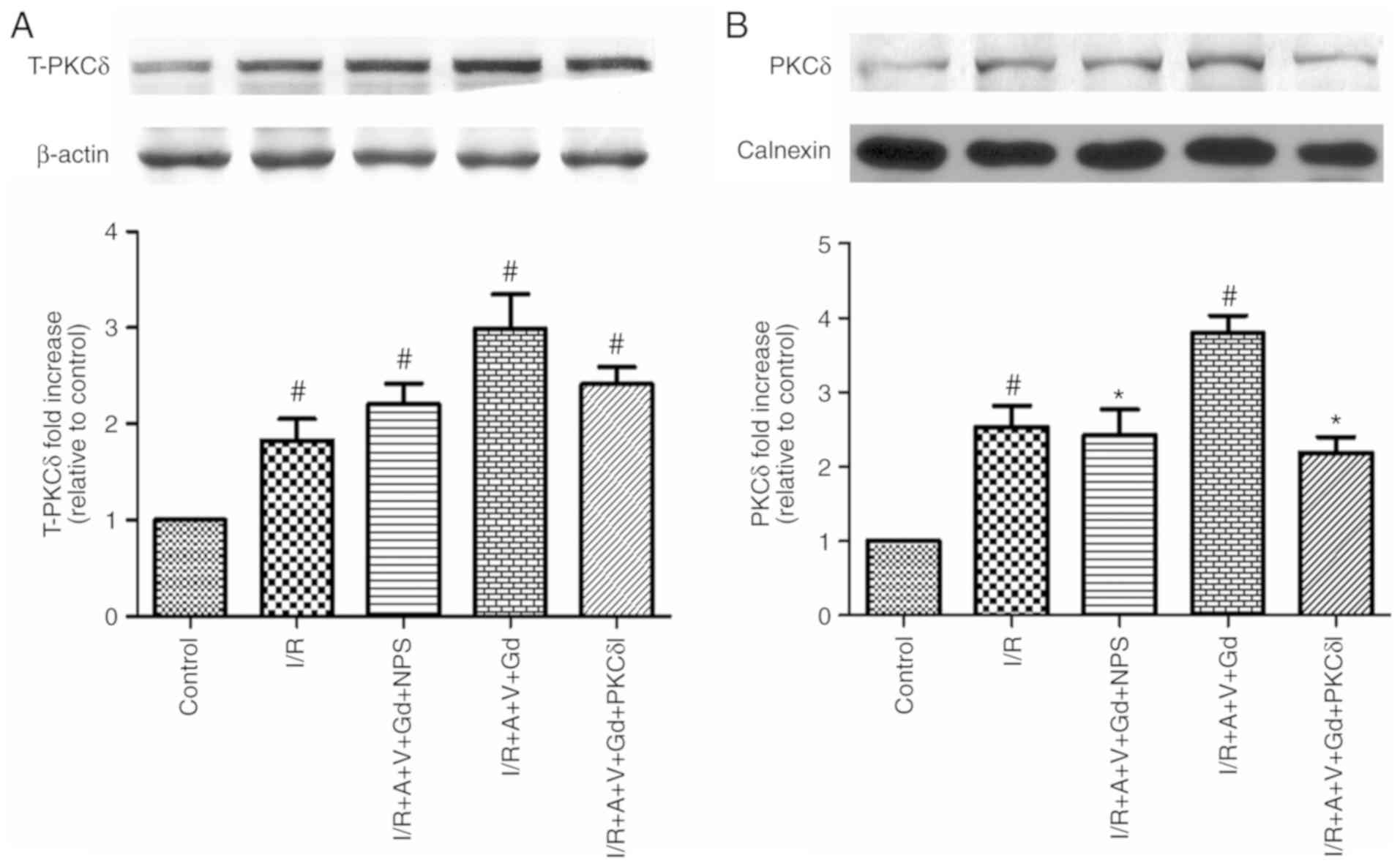
present study, expression of total-PKCδ in the cytoplasm of
myocardial cells was detected by western blotting in each group
(Fig. 8A). In the I/R,
I/R+A+V+Gd, I/R+A+V+Gd+NPS and I/R+A+V+Gd+PKCδI groups, total-PKCδ
protein expression levels were significantly higher compared with
the Control group (P<0.05; Fig.
8A). The present study also quantitatively analyzed the
expression of PKCδ in the ER fractions (Fig. 8B). The PKCδ expression levels of
the I/R and I/R+A+V+Gd groups were significantly higher compared
with the Control group (P<0.05; Fig. 8B), while the levels of PKCδ from
the ER fractions were significantly inhibited in the I/R+A+V+Gd+NPS
and I/R+A+V+Gd+PKCδI groups compared with the I/R+A+V+Gd group
(P<0.05; Fig. 8B). These
results demonstrated that CaSR induced PKCδ translocation to the ER
following I/R and participated in the ER stress-induced apoptosis
pathway.
Discussion
The results of the present study confirmed that the
activation of CaSR induced PKCδ translocation to the ER following
I/R and participated in cardiomyocyte apoptosis via the ER
stress-associated apoptotic pathway in vivo.
It is well-established that I/R injury can lead to
activation of PKC. PKC, as a kind of Ca2+ and
phospholipid-dependent protein kinase, is involved in the apoptosis
of cardiac muscle cells by regulating [Ca2+]i
homeostasis. The PKC family contains 13 subtypes, and the PKCε and
PKCδ subtypes have been well-studied. The former mainly serevs a
role in the inhibition of apoptosis, while the latter mainly acts
in the promotion of apoptosis. CaSR acts as a protein-coupled
receptor, causing the accumulation of inositol phosphate (IP),
which leads to an increase in intracellular calcium. However, the
PKCδ inhibitor, rottlerin, can reverse the occurrence of this
phenomenon by inhibiting CaSR (5,23).
The links between PKCδ, CaSR and ER stress during I/R remain
unclear. To investigate the effect of activation of PKCδ by CaSR on
myocardial I/R, GdCl3 (an activator of CaSR), NPS-2390
(an inhibitor of CaSR), amiloride (an inhibitor of the
Na+-Ca2+ exchanger), verapamil (an L-type
calcium channel blocker) and rottlerin (a PKCδ inhibitor) were used
in the present study. The experimental animals were divided into
five groups: Control, I/R, I/R+A+V+Gd, I/R+A+V+Gd+NPS and
I/R+A+V+Gd+PKCδI groups.
First, the expression of CaSR in myocardial I/R was
detected by western blot analysis. CaSR, also known as the
parathyroid cell CaSR, is an integral membrane protein belonging to
the G-protein coupled receptor 3 family. The molecular weights of
CaSR are 110, 130 and 150 kDa, of which the 150 kDa form is the
mature form of CaSR. CaSR is present on the cell membrane. In the
present study, the 130 kDa CaSR protein was detected in the cardiac
tissues. Compared with the Control group, the expression of CaSR in
the I/R and I/R+A+V+Gd groups increased. However, in the
I/R+A+V+Gd+NPS group, the CaSR protein expression levels was lower
compared with the I/R+A+V+Gd group. Thus, I/R and GdCl3
were demonstrated to increase CaSR expression.
Multiple studies have indicated that PKC can
regulate the intracellular Ca2+ concentration (24,25) and participate in the protection or
damage of cardiac muscle cells by CaSR-mediated intracellular
calcium release. In the present study, cardiomyocyte apoptosis was
examined by JC-1 fluorescence staining and TUNEL staining. The
decrease of mitochondrial membrane potential is a landmark event in
the early stages of apoptosis. JC-1 is an ideal fluorescent probe
for detecting mitochondrial membrane potential. Therefore, the JC-1
method was used to detect early apoptosis following myocardial I/R.
The sensitivity of the TUNEL method for detecting apoptosis is very
high, and it is also possible to quantitatively analyze apoptotic
cells of both the middle or late stages. The results of
cardiomyocyte JC-1 straining analysis revealed that the intensity
ratio of JC-1 polymer/monomer decreased significantly in the I/R
and I/R+A+V+Gd groups compared with the Control group. Similarly,
the ratio of TUNEL-positive cells was significantly higher in the
I/R and I/R+A+V+Gd groups compared with the Control group. In
addition, the ultrastructural changes of the myocardial cells were
observed by transmission electron microscopy. In the I/R and
I/R+A+V+Gd groups, the myocardial cell structure was disordered,
and the nuclear chromatin was condensed and concentrated. By
contrast, in the I/R+A+V+Gd+NPS and I/R+A+V+Gd+PKCδI groups, only a
slight margination of the nuclear chromatin, expansion of the ER
and swelling of the mitochondrion were observed. Based on the
aforementioned results, the experiments revealed that
GdCl3 significantly increased the occurrence of
cardiomyocyte apoptosis following I/R. The data also demonstrated
that inhibition of PKCδ or CaSR significantly reduced the rate of
cardiomyocyte apoptosis and protected cardiomyocytes from
myocardial I/R injury.
The ER environment can be impaired by various
stimuli, such as a perturbation of Ca2+ homeostasis,
which leads to the UPR. Excessive UPR can stimulate ER stress
(20). Previous studies have
demonstrated that CaSR-induced Ca2+ release from
internal stores is a PLC-mediated/IP3-dependent process. The
enhancement of [Ca2+]i may be related to the release of
Ca2+ by the ER (6,26).
To examine the occurrence of ER stress, the protein levels of GRP78
were detected. GRP78 is an ER molecular chaperone protein that
promotes protein folding and the internal environment of calcium
ions, protecting cells against oxidative stress and apoptosis, and
has been extensively used as a marker for ER stress and the onset
of the UPR (21,27). Protein expression of GRP78 can
thus be used as a marker of ER stress (28-30). In the present study, the
expression of GRP78 was detected by western blot analysis. The
results demonstrated that GRP78 protein expression levels in the
I/R and I/R+A+V+Gd groups were significantly higher compared with
the Control group; however, the levels of the I/R+A+V+Gd+NPS and
I/R+A+V+Gd+PKCδI groups were decreased compared with the I/R+A+V+Gd
group. Based on the present results, activated CaSR in I/R was
demonstrated to induce ER stress in vivo. These data also
determined that inhibition of PKCδ may protect the myocardium from
ER stress in an in vivo model of I/R injury.
Next, the current study investigated whether
activation of PKCδ by CaSR participated in ER stress-initiated
apoptotic signaling. Studies have confirmed that several molecules,
including Caspase-12, CHOP and JNK, are involved in the ER
stress-induced apoptosis pathway (22). Caspase-12, an ER membrane-resident
apoptotic molecule, can lead to cardiomyocyte apoptosis and cardiac
dysfunction (31,32). The JNK signaling pathway is one of
the important mitogen-activated protein kinase pathways, and serves
an important regulatory role in a variety of physiological and
pathological programmed cell death processes (33). Both in vitro and in
vivo experiments have demonstrated that high-intensity ER
overstress activates inositol dependent enzyme 1 (IRE1) and its
downstream tumor necrosis factor receptor-associated factor 2
(TRAF2)/apoptosis signal-regulated kinase (ASK1)/JNK apoptotic
signaling pathway (34,35). JNK exerts its pro-apoptotic effect
by activating the downstream caspase-9 and caspase-3 (36). In the present study, the protein
expression levels of Caspase-12, p-JNK and Caspase-3 in
cardiomyocytes were measured by western blot analysis. The results
demonstrated that the activation of Caspase-12, p-JNK and Caspase-3
were significantly higher following I/R injury. Therefore,
CaSR-activated PKCδ induced endoplasmic reticulum Ca2+
depletion and activated the ER stress-associated pathway to
participate in the process of cardiomyocyte apoptosis during I/R
injury.
Several studies have indicated that inhibition of
PKCδ activation could significantly inhibit the ER stress response
at the beginning of reperfusion in the myocardium (14). In the present study, the
expression of total-PKCδ in the cytoplasm of myocardial cells was
detected by western blotting. Compared with the Control group, the
expression levels of total-PKCδ protein in the I/R, I/R+A+V+Gd,
I/R+A+V+Gd+NPS and I/R+A+V+Gd+PKCδI groups were significantly
increased. Expression of PKCδ was also quantitatively analyzed in
the ER fractions. The levels of PKCδ in the I/R and I/R+A+V+Gd
groups were significantly higher compared with the Control group,
while the levels of PKCδ in the ER fractions was significantly
lower in the I/R+A+V+Gd+NPS and I/R+A+V+Gd+PKCδI groups compared
with the I/R+A+V+Gd group. These results revealed that CaSR induced
PKCδ translocation to the ER, which then participated in
cardiomyocyte apoptosis via ER stress-associated pathways.
In summary, the present results confirmed that
activation of CaSR induced the translocation of PKCδ to the ER
following I/R and participated in cardiomyocyte apoptosis via ER
stress-associated apoptotic pathways in vivo.
Acknowledgments
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant nos. 81670344, 81370421 and
81370330), the Natural Science Foundation of Heilongjiang (grant
no. D201070) and the High-Priority Health Projects of Tianjin
(grant no. 16KG146).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
WZ conceived and designed the experiments. CL, MZ,
HZ, TF, FL, HL and SD performed the experiments and analyzed the
data. CL and HL wrote the paper. HZ, MZ and HL contributed to the
manuscript revisions. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Basic Medical College of Harbin Medical University
[permit no. SCXK (Hei) 2013-001].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang R, Xu C, Zhao W, Zhang J, Cao K, Yang
B and Wu L: Calcium and polyamine regulated calcium-sensing
receptors in cardiac tissues. Eur J Biochem. 270:2680–2688. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lu FH, Tian Z, Zhang WH, Zhao YJ, Li HL,
Ren H, Zheng HS, Liu C, Hu GX, Tian Y, et al: Calcium-sensing
receptors regulate cardiomyocyte Ca2+ signaling via the
sarcoplasmic reticulum-mitochondrion interface during
hypoxia/reoxygenation. J Biomed Sci. 17:502010. View Article : Google Scholar
|
|
3
|
Di Lisa F and Bernardi P: Mitochondria and
ischemia-reperfusion injury of the heart: Fixing a hole. Cardiovasc
Res. 70:191–199. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Grover GJ: Mitochondrial ATP-sensitive
potassium channels and mitochondrial protein kinase C: Sometimes
it's good to have a close neighbor. Am J Physiol Heart Circ
Physiol. 290:H1752–H1753. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zheng H, Liu J, Liu C, Lu F, Zhao Y, Jin
Z, Ren H, Leng X, Jia J, Hu G, et al: Calcium-sensing receptor
activating phosphorylation of PKCδ translocation on mitochondria to
induce cardiomyocyte apoptosis during ischemia/reperfusion. Mol
Cell Biochem. 358:335–343. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lu F, Tian Z, Zhang W, Zhao Y, Bai S, Ren
H, Chen H, Yu X, Wang J, Wang L, et al: Calcium-sensing receptors
induce apoptosis in rat cardiomyocytes via the endo(sarco)plasmic
reticulum pathway during hypoxia/reoxygenation. Basic Clin
Pharmacol Toxicol. 106:396–405. 2010.
|
|
7
|
Murriel CL, Churchill E, Inagaki K, Szweda
LI and Mochly-Rosen D: Protein kinase Cdelta activation induces
apoptosis in response to cardiac ischemia and reperfusion damage: A
mechanism involving BAD and the mitochondria. J Biol Chem.
279:47985–47991. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dong S, Teng Z, Lu FH, Zhao YJ, Li H, Ren
H, Chen H, Pan ZW, Lv YJ, Yang BF, et al: Post-conditioning
protects cardiomyocytes from apoptosis via PKC(epsilon)-interacting
with calcium-sensing receptors to inhibit endo(sarco)plasmic
reticulum-mitochondria crosstalk. Mol Cell Biochem. 341:195–206.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Churchill EN and Mochly-Rosen D: The roles
of PKCdelta and epsilon isoenzymes in the regulation of myocardial
ischaemia/reperfusion injury. Biochem Soc Trans. 35:1040–1042.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kostyak JC, Hunter JC and Korzick DH:
Acute PKCdelta inhibition limits ischaemia-reperfusion injury in
the aged rat heart: Role of GSK-3beta. Cardiovasc Res. 70:325–334.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Parihar SP, Ozturk M, Marakalala MJ, Loots
DT, Hurdayal R, Beukes D, Van Reenen M, Zak DE, Mbandi SK, Darboe
F, et al: Protein kinase C-delta (PKCδ), a marker of inflammation
and tuberculosis disease progression in humans, is important for
optimal macrophage killing effector functions and survival in mice.
Mucosal Immunol. 11:579–580. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Q, Park K, Xia Y, Matsumoto M, Qi W, Fu
J, Yokomizo H, Khamaisi M, Wang X, Rask-Madsen C and King GL:
Regulation of macrophage apoptosis and atherosclerosis by lipid
induced PKCδ isoform activation. Circ Res. 121:1153–1167. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wie SM, Wellberg E, Karam SD and Reyland
ME: Tyrosine kinase inhibitors protect the salivary gland from
radiation damage by inhibiting activation of protein kinase C-δ.
Mol Cancer Ther. 16:1989–1998. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qi X, Vallentin A, Churchill E and
Mochly-Rosen D: deltaPKC participates in the endoplasmic reticulum
stress-induced response in cultured cardiac myocytes and ischemic
heart. J Mol Cell Cardiol. 43:420–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qi X and Mochly-Rosen D: The PKCdelta-Abl
complex communicates ER stress to the mitochondria-an essential
step in subsequent apoptosis. J Cell Sci. 121:804–813. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mayhew TM, Lucocq JM and Griffiths G:
Relative labelling index: A novel stereological approach to test
for non-random immunogold labelling of organelles and membranes on
transmission electron microscopy thin sections. J Microsc.
205:153–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chimenti S, Carlo E, Masson S, Bai A and
Latini R: Myocardial infarction: Animal models. Methods Mol Med.
98:217–226. 2004.PubMed/NCBI
|
|
18
|
Davey KA, Garlick PB, Warley A and
Southworth R: Immunogold labeling study of the distribution of
GLUT-1 and GLUT-4 in cardiac tissue following stimulation by
insulin or ischemia. Am J Physiol Heart Circ Physiol.
292:H2009–H2019. 2007. View Article : Google Scholar
|
|
19
|
Ding JW, Tong XH, Yang J, Liu ZQ, Zhang Y,
Yang J, Li S and Li L: Activated protein C protects myocardium via
activation of anti-apoptotic pathways of survival in
ischemia-reperfused rat heart. J Korean Med Sci. 25:1609–1615.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao GL, Yu LM, Gao WL, Duan WX, Jiang B,
Liu XD, Zhang B, Liu ZH, Zhai ME, Jin ZX, et al: Berberine protects
rat heart from ischemia/reperfusion injury via activating
JAK2/STAT3 signaling and attenuating endoplasmic reticulum stress.
Acta Pharmacol Sin. 37:354–367. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kockskämper J, Zima AV, Roderick HL,
Pieske B, Blatter LA and Bootman MD: Emerging roles of inositol
1,4,5-trisphosphate signaling in cardiac myocytes. J Mol Cell
Cardiol. 45:128–147. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mao W, Iwai C, Qin F and Liang CS:
Norepinephrine induces endoplasmic reticulum stress and
downregulation of norepinephrine transporter density in PC12 cells
via oxidative stress. Am J Physiol Heart Circ Physiol.
288:H2381–H2389. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kaur K, Singh M, Singh N and Jaggi AS:
Possible mechanism of rottlerin induced modulation of ischemia
reperfusion injury in isolated rat hearts. Biol Pharm Bull.
31:1745–1748. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hernández-Bedolla MA, González-Domínguez
E, Zavala- Barrera C, Gutiérrez-López TY, Hidalgo-Moyle JJ,
Vázquez- Prado J, Sánchez-Torres C and Reyes-Cruz G:
Calcium-sensing-receptor (CaSR) controls IL-6 secretion in
metastatic breast cancer MDA-MB-231 cells by a dual mechanism
revealed by agonist and inverse-agonist modulators. Mol Cell
Endocrinol. 436:159–168. 2016. View Article : Google Scholar
|
|
25
|
Sano R and Reed JC: ER stress-induced cell
death mechanisms. Biochim Biophys Acta. 1833:3460–3470. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lv Z, Liu C, Zhai M, Zhang Q, Li J, Zheng
F and Peng M: LPS pretreatment attenuates cerebral
ischaemia/reperfusion injury by inhibiting inflammation and
apoptosis. Cell Physiol Biochem. 45:2246–2256. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu C, Fu Q, Mu R, Wang F, Zhou C, Zhang
L, Yu B, Zhang Y, Fang T and Tian F: Dexmedetomidine alleviates
cerebral ischemia-reperfusion injury by inhibiting endoplasmic
reticulum stress dependent apoptosis through the
PERK-CHOP-Caspase-11 pathway. Brain Res. 1701:246–254. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang M, Wu A, Shen Y, Chen H, Tu J and
Zhai C: Effects of L-carnitine and bisoprolol on endoplasmic
reticulum stress-mediated myocardial injury after cardiopulmonary
resuscitation in rats. Zhonghua Yi Xue Za Zhi. 95:1475–1478.
2015.In Chinese. PubMed/NCBI
|
|
29
|
Zhang L, Zhang H, Lv M, Jia J, Fan Y, Tian
X, Li X, Li B, Ji J, Wang L, et al: Increased expression of 78 kD
glucose-regulated protein promotes cardiomyocyte apoptosis in a rat
model of liver cirrhosis. Int J Clin Exp Pathol. 8:9256–9263.
2015.PubMed/NCBI
|
|
30
|
Xuan LY, Tao XX, Zhao YJ, Ge HY, Bao LH,
Wang DP and Zhao M: Effect of total flavonoids of astragalus on
endoplasmic reticulum chaperone, calumenin and connecxin 43 in
suckling mouse myocardium with myocarditis caused by coxsackievirus
B3. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 32:51–54. 2016.In
Chinese. PubMed/NCBI
|
|
31
|
Fu HY, Sanada S, Matsuzaki T, Liao Y,
Okuda K, Yamato M, Tsuchida S, Araki R, Asano Y, Asanuma H, et al:
Chemical endoplasmic reticulum chaperone alleviates
doxorubicin-induced cardiac dysfunction. Circ Res. 118:798–809.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Iurlaro R and Muñoz-Pinedo C: Cell death
induced by endoplasmic reticulum stress. FEBS J. 283:2640–2652.
2016. View Article : Google Scholar
|
|
34
|
Chen H, Yang H, Pan L, Wang W, Liu X, Ren
X, Liu Y, Liu W, Zhang Y, Jiang L, et al: The molecular mechanisms
of XBP-1 gene silencing on IRE1α-TRAF2-ASK1-JNK pathways in oral
squamous cell carcinoma under endoplasmic reticulum stress. Biomed
Pharmacother. 77:108–113. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Brandt B, Abou-Eladab EF, Tiedge M and
Walzel H: Role of the JNK/c-Jun/AP-1 signaling pathway in
galectin-1-induced T-cell death. Cell Death Dis. 1:e232010.
View Article : Google Scholar
|