Introduction
Peroxisomes are ubiquitous organelles in eukaryotic
cells with multiple metabolic functions (1,2).
The three established functions of peroxisomes are α-oxidation
(release of one carbon of a 3-methyl-branched or 2-hydroxyfatty
acid), β-oxidation (release of two carbons of the backbone of
diverse substrates) and ether lipid synthesis. Almost all diseases
caused by peroxisome dysfunction are manifested by a variety of
neurological abnormalities, underscoring the importance of
peroxisomes in the nervous system.
X-linked adrenoleukodystrophy (X-ALD), the most
frequent peroxisomal disorder, is caused by inactivity of a
transmembrane ATP binding cassette (ABC) transporter, which imports
very long chain fatty acid (VLCFA)-CoA into the peroxisome for
β-oxidation (3). Up to 60% of
male patients develop a catastrophic form of cerebral inflammatory
demyelination, cerebral ALD (CALD), characterized by increased
blood-brain barrier (BBB) permeability to monocytes with marked
perivascular microglial/monocyte activation at the lesion edge
(4-7).
Multifunctional protein 2 (MFP2) deficiency, another
peroxisomal disorder, is caused by mutations in the hydroxysteroid
17-β dehydrogenase 4 (HSD17B4) gene, which encodes D-bifunctional
protein, the central enzyme in the second step of the peroxisomal
β-oxidation pathway that is essential for the shortening of
branched-chain and straight-chain compounds (8). Defects of MFP2 are associated with a
spectrum of neurological disorders, encompassing developmental and
degenerative pathologies, and marked monocytic activation and
neuroinflammation have been demonstrated in a mouse model lacking
MFP2 (9,10). While the pathogenetic mechanisms
underlying cerebral degeneration following peroxisomal dysfunction
remain largely elusive, previous studies on X-ALD suggest that
abnormal BBB function precedes inflammatory demyelination (11,12). As the key component of the BBB,
brain microvascular endothelial cells (BMECs) strictly control the
flow of substances into and out of the brain, as well as the entry
of leukocytes through regulation of adhesion molecules and
intercellular tight-junction protein expression (13-15). Abnormal microvascular function in
peroxisomal disorders has been previously demonstrated in CALD: i)
Ex vivo histopathology demonstrated distorted microvascular
permeability beyond the edge of the demyelinating lesions (12) and ii) in vitro experiments
indicated that lack of ABC subfamily D member 1 (ABCD1) in human
BMECs causes dysregulation of adhesion molecules and tight-junction
proteins, leading to an increase in permeability to leukocytes
(12). However, how loss of
peroxisomal function secondary to ABCD1 or MFP2 deficiency leads to
endothelial dysfunction has remained largely elusive.
As the most conserved mammalian sirtuin (SIRT)
family NAD+ dependent protein deacetylase, SIRT1 has
been demonstrated to be the key metabolic sensor across different
tissue types, regulating numerous transcriptional factors and
co-factors involved in systemic metabolic homeostasis, including
peroxisomal disorders (16,17). Endothelial cell dysfunction is
markedly associated with activation of nuclear factor (NF)-κB
signaling, a pathway that is strictly regulated by SIRT1 (11,12,18). Reduced SIRT1 expression has been
observed in numerous abnormalities of vascular endothelial
function, including diabetic vasculopathy, whereas improvement of
SIRT1 activity by either overexpression or replacement treatment
markedly ameliorated the dysfunction and had significant beneficial
effects (19-21). Given the master regulator role of
SIRT1 in energy metabolism homeostasis and its protective role in
endothelial cells (22,23), understanding whether SIRT1 is also
able to modulate endothelial dysfunction in peroxisomal disorders
may lead to novel therapeutic and less toxic interventions.
In the present study, an in vitro model of
the human BBB was utilized to investigate the effects of ABCD1 and
MFP2 deficiency on the BME and assess whether modulation of SIRT1
levels can ameliorate the endothelial dysfunction and normalize its
interactions with monocytes.
Materials and methods
Cell cultures
Primary human brain microvascular endothelial cells
(HBMECs) from CSC systems were purchased from ScienCell Research
Laboratories, Inc. They were maintained in EGM-2 Endothelial Cell
Growth Medium-2 Bullel kit (Lonza Group, Ltd.) on a Collagen type 1
(Corning, Inc.)-coated 10 cm diameter plate in a 37°C humidified
atmosphere of 95% air and 5% CO2 in incubator and used
for experiments at 80% confluency.
THP-1 was purchased from ZQ Cell Research and
cultured in RPMI1640 (GE Healthcare) medium supplemented with 0.05
mM β-Mercaptoethanol (Sigma-Aldrich; Merck KGaA), 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.) and 1% penicillin streptomycin
(Thermo Fisher Scientific, Inc.). Cells were grown in a humidified
atmosphere of 5% CO2 at 37°C and used for experiments at
>80% confluency.
Chemical treatment
Resveratrol was purchased from Sigma-Aldrich; Merck
KGaA and a 50 mM storage solution in DMSO was prepared. Per
experimental needs, cells were treated with 20 µM
Resveratrol for 48 h before further analysis. Sirtinol
(Sigma-Aldrich; Merck KGaA) was made into 20 mM storage solution by
DMSO and diluted into a 20 µM working solution for cell
treatment. For tumor necrosis factor (TNF)-α treatment, cells were
treated with 10 ng/ml TNF-α (eBioscience; Thermo Fisher Scientific,
Inc.) for 6 h before different assays.
ABCD1, HSD17B4 and Krüppel-like factor 4
(KLF4) gene silencing
SiGENOME Human ABCD1 (cat. no. M009605010005) small
interfering (si)RNA-SMARTpool and siGENOME non-targeting siRNA
(cat. no. D0012100105) were purchased from GE Healthcare Dharmacon,
Inc. HSD17B4siRNA-Homo-427/990/1870 and KLF4 siRNA were purchased
from Shanghai GenePharma Co., Ltd. Silencing was performed
according to the protocol provided by the manufacturer. Briefly,
1.5-2×105 HBMECs were seeded in 6-well plates for 24 h.
siRNA (5 µM) dissolved in serum free medium was mixed with
DharmaFECT transfection reagent (GE Healthcare Dharmacon, Inc.) and
incubated for 20 min at room temperature. Cells were then replaced
with fresh medium containing 25 nM siRNA and then cultured for 48 h
for further analysis.
Reverse transcription
quantitative-PCR
Total RNA was extracted with Qiagen RNeasy kit.
First-strand cDNA was synthesized (25°C, 10 min; 42°C, 30 min;
85°C, 5 min) using Hiscript QRT SuperMix (Thermo Fisher Scientific,
Inc.) in 20 µl reaction. Thermocycling conditions were 95°C
for 10 min and 40 PCR cycles (95°C, 20 sec; 55°C, 30 sec; 72°C, 20
sec). After the amplification reaction, the temperature was 95°C
for 5 sec and 65°C for 60 sec. PCRs were performed using SYBR
master mix (Thermo Fisher Scientific, Inc.) and the data were
analyzed by calculating the ∆Cq value (24). The primers used were listed as
below: Claudin 5 forward: 5′-CTC TGC TGG TTC GCC AAC AT-3′, and
Claudin 5 reverse: 5′-CAG CTC GTA CTT CTG CGA CA-3′; β-actin
forward: 5′-CGA GCA CAG AGC CTC GCC TTT GCC-3′ and β-actin reverse:
5′-TGT CGA CGA CGA GCG CGG CGA TAT-3′.
Western blotting protein analysis
HBMEC protein was extracted with
radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris-HCl pH
7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate and 0.1% SDS).
After lysis on an ice for 0.5 h, samples were collected and
centrifuged at 4°C at 20,817 g for 0.5 h. The supernatant was
transferred into fresh tubes and protein concentration was
determined by the bicinchoninic acid (BCA) method. Nuclear Protein
was extracted using Nucleoprotein Extraction kit from Beyotime
Institute of Biotechnology according to the manufacturer's
protocol. Briefly, cells were lysed with RIPA buffer on ice for 0.5
h, which were collected and centrifuged at 4°C at 20,817 × g for
0.5 h. The supernatant was removed. After adding 100 µl
buffer C, the precipitation was centrifuged at 4°C at 20,817 × g
for 0.5 h. The supernatant was transferred into fresh tubes for
next step.
Equal amounts of protein (60 µg/lane) were
separated by 10% SDS-PAGE and transferred onto nitrocellulose
filter membrane (EMD Millipore). Membranes were blocked with 5%
non-fat milk in phosphate-buffered saline containing 0.05% Tween-20
at room temperature for 2 h and probed with antibodies at 4°C for
14 h including Anti-ABCD1 (1:1,000; OriGene Technologies, Inc.;
cat. no. TA803208), and anti-hydroxysteroid (17B4) dehydrogenase 4
(1:2,000; Abcam; cat. no. ab128565), anti-SIRT1 (1:1,000; Santa
Cruz Biotechnology, Inc.; cat. no. sc15404), anti-intracellular
adhesion molecular (ICAM1; 1:1,000; Santa Cruz Biotechnology, Inc.;
cat. no. sc7891), anti-vascular cellular adhesion molecule (VCAM1;
1:1,000; Santa Cruz Biotechnology, Inc.; cat. no. sc13160),
anti-Claudin 5 (1:2,000; Abcam; cat. no. ab15106), anti-KLF4
(1:2,000; Proteintech; cat. no. 11880), anti-phosphorylated NF-κB
inhibitor (p-IκBα; 1:2,000; Abcam; cat. no. ab133462), anti-NF-κB
(1:2,000; Abcam; cat. no. ab16502) and anti-Acetyl-Lysine (1:2,000;
ABclonal; cat. no. A2391). Anti-β-actin (1:5,000; Santa Cruz
Biotechnology, Inc.; cat. no. sc47778) was used as a protein
loading control while anti-Lamin b (1:5,000; Santa Cruz
Biotechnology, Inc. cat. no. sc56143) was elected as nuclear
protein loading control. Membranes were developed with Odyssey
Infrared laser scanning image-forming system after being washed and
incubated with appropriate secondary antibodies (1:2,000;
horseradish peroxidase, Goat anti mouse or Goat anti rabbit, Vicmed
Company; cat. no. C30409) at room temperature for 2 h.
Immunoprecipitation (IP)
The protein lysates, which obtained as
aforementioned, were incubated with 1 µg IgG (Beyotime
Institute of Biotechnology) and 20 µl resuspended Protein
A/G PLUS-Agarose (Santa Cruz Biotechnology, Inc.) for 30 min at
4°C, then centrifugation was used to remove the non-specific
protein adhering to protein A/G at 4°C at 2,500 × g for 5 min.
After collecting and measuring the concentration of protein by the
bicinchoninic acid method, the supernatants were made up to a total
amount of 500 µl with IP buffer. The corresponding primary
antibody with 5 µl was added to the samples including
anti-KLF4 (ProteinTech Group, Inc.; cat. no. 11880), or anti-SIRT1
(Santa Cruz Biotechnology, Inc.; cat. no. sc-15404) or IgG
(Beyotime Institute of Biotechnology) which as a contrast, followed
by incubation overnight at 4°C. Subsequently, the samples were
incubated with 100 µl Protein A/G PLUS-Agarose at 4°C for 4
h. Finally, the result was produced by western blot analysis.
Human monocyte-endothelial adhesion
assay
For the adhesion assay, HBMECs were plated in 6-well
plates at a density of 5×104 cells/well until 80-90%
confluent, with certain groups treated with tumor necrosis factor
(TNF)-α (10 ng/ml) for 6 h. Furthermore, 1×105 THP-1
cells/well were incubated with 1 µl Calcein AM
(Sigma-Aldrich; Merck KGaA; 4 mM) at a dilution of 1:1,000 for 1 h
at 37°C. Subsequently, the Calcein AM-labeled THP-1 cells were
seeded onto the endothelial monolayer at a density of
1×105 cells/well and incubated with RPMI-1640 medium
plus 10% FBS for 1 h at 37°C. The cells were washed with PBS and
fixed in 4% paraformaldehyde at room temperature for 20 min prior
to imaging. Images were captured in five randomly selected
microscopic fields at a ×10 magnification using an inverted
fluorescence microscope (TH4-200; Olympus Corp.) and fluorescence
was quantified using ImageJ software (Version no. 1.4.3.67,
National Institutes of Health).
Determination of the NADP/NADPH
ratio
The Amplite™ Fluorimetric NADP/NADPH Ratio Assay kit
was purchased from AAT Bioquest. The assay was performed according
to the manufacturer's protocol. In brief, HBMECs were plated at a
density of 1×105 cells/well in 6-well plates and allowed
to attach for 24 h. After removing the medium, 100 µl
NADP/NADPH lysis buffer (AAT Bioquest, Inc.) was added to each
well, followed by incubation at room temperature for 15 min. The
supernatant was collected by centrifugation for subsequent analysis
at room temperature at 2,000 × g for 10 min. The fluorescence
intensity was measured using a microplate reader (Gene Company,
Ltd.) with excitation and emission wavelengths of 530 and 590 nm,
respectively.
Malondialdehyde (MDA) and superoxide
dismutase (SOD) activity detection
The MDA Assay kit and the SOD assay kit were
purchased from NJJC Institute of Bioengineering. First, HBMECs were
plated at a density of 1×105 cells/well in 6-well plates
and allowed to attach for 24 h, followed by treatment with or
without TNF-α (10 ng/ml) for 6 h. Next, the cells were collected
and the corresponding reagents were added according to the
manufacturer's protocols. The absorbance was measured using a
microplate reader (Gene Company, Ltd.) at 550 nm for SOD and 532 nm
for MDA, respectively.
Statistical analysis
All the results in this study were analyzed by SPSS
16.0 statistical software; GraphPad Prism 6.0 software (GraphPad
Software, Inc.) was used for the generation of graphs. Values are
expressed as the mean ± standard deviation. Statistical analyses
were performed using one-way ANOVA with Tukey multiple comparison
test to determine statistical significance between the groups.
P<0.05 was considered to indicate a statistically significance
difference.
Results
ABCD1 or HSD17B4 silencing in HBMECs
leads to reduction of SIRT1 levels and alteration in adhesion
molecules and tight-junction proteins
In the first set of experiments, the direct impact
of ABCD1 and HSD17B4 deficiency in HBMECs was investigated. Several
key protein markers involved in endothelial interactions and
permeability to leukocytes, including tight-junction proteins and
adhesion molecules, as well as changes in SIRT1 expression levels
prior to and after activation with TNF-α (10 ng/ml), were measured.
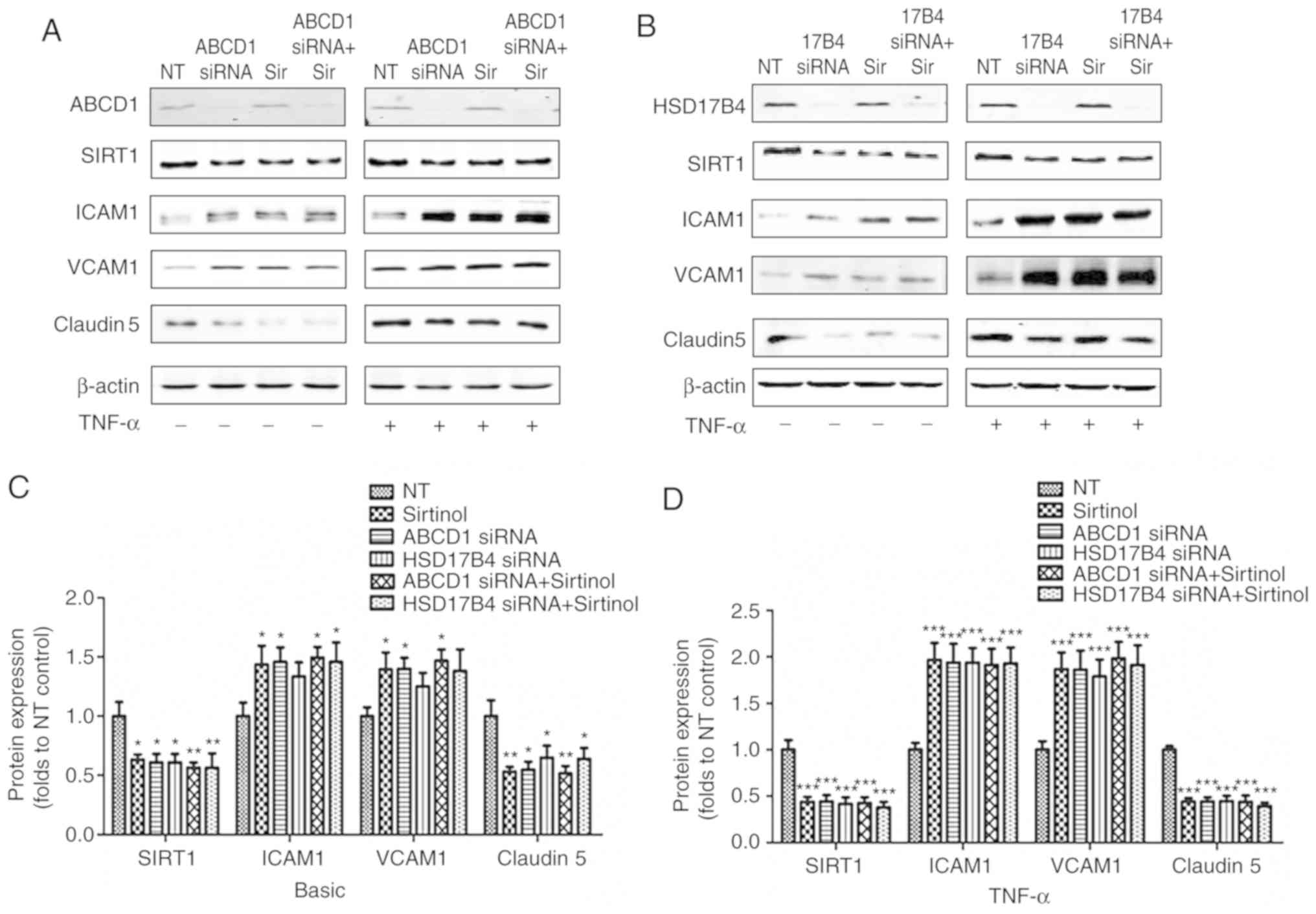
As presented in Fig. 1, silencing
of ABCD1 led to a significant 35% reduction in Claudin 5
(P<0.05), a 38% reduction in SIRT1 (P<0.05) as well as a 41%
increase in ICAM1 (P<0.05) and a 33% increase in VCAM1
(P<0.05), while stimulation with TNF-α lead to further decreases
in Claudin 5 (53%, P<0.001) and SIRT1 (47%, P<0.001), as well
as a 2 and 2.8-fold increase in ICAM1 (P<0.001) and VCAM1
(P<0.001), respectively. Similar results were observed following
HSD17B4 silencing, which led to a ~25% reduction in Claudin 5
(P<0.05), a 36% increase in ICAM1 and a ~25% increase in VCAM1,
accompanied by a 39% reduction in SIRT1 (P<0.05) at the basal
level without TNF-α stimulation. Upon TNF-α stimulation, a 57%
reduction of Claudin 5 (P<0.001), a 58% reduction of SIRT1
(P<0.001), a 2.1-fold increase of ICAM1 (P<0.001) and a
2.2-fold increase of VCAM1 (P<0.001) were observed. Of note,
inhibition of SIRT1 by treatment with Sirtinol (20 µmol/l)
led to comparable alterations of the expression of adhesion
molecules and Claudin 5 prior to and after endothelial activation
(39/58% reduction in Claudin 5; 43/94% increase in ICAM1 and
36/300% increase of VCAM1 expression prior to/after stimulation
with TNF-α, respectively). A increased higher expression of ICAM1
and VCAM1 and decreased Claudin 5 levels were detected in the
groups with ABCD1 silencing plus Sirtinol and HSD17B4 silencing
plus Sirtinol, but this was not statistically significant (Fig. 1).
SIRT1 activation by resveratrol restores
protein changes in HBMECs after ABCD1 or HSD17B4 silencing
It was then investigated whether increasing SIRT1
function using the SIRT1 activator resveratrol (20 µmol/l)
attenuates the protein alterations associated with peroxisomal
dysfunction caused by ABCD1 or HSD17B4 silencing. The targeting
association was demonstrated by significant increases in SIRT1
protein levels after resveratrol treatment at a baseline and
following TNF-α treatment (P<0.05). Resveratrol also partially
normalized SIRT1 levels after ABCD1 and HSD17B4 silencing,
particularly when HBMECs were activated with TNF-α (P<0.01).
HBMECs treated with resveratrol exhibited a reduction of ICAM1 and
VCAM1 and increased Claudin 5 expression under basal conditions
(P<0.05) and following TNF-α treatment (P<0.01). Of note,
resveratrol treatment of HBMECs with silencing of ABCD1 or HSD17B4
attenuated the overexpression of ICAM1 and VCAM1 and significantly
increased Claudin 5 expression after activation with TNF-α
(Fig. 2).
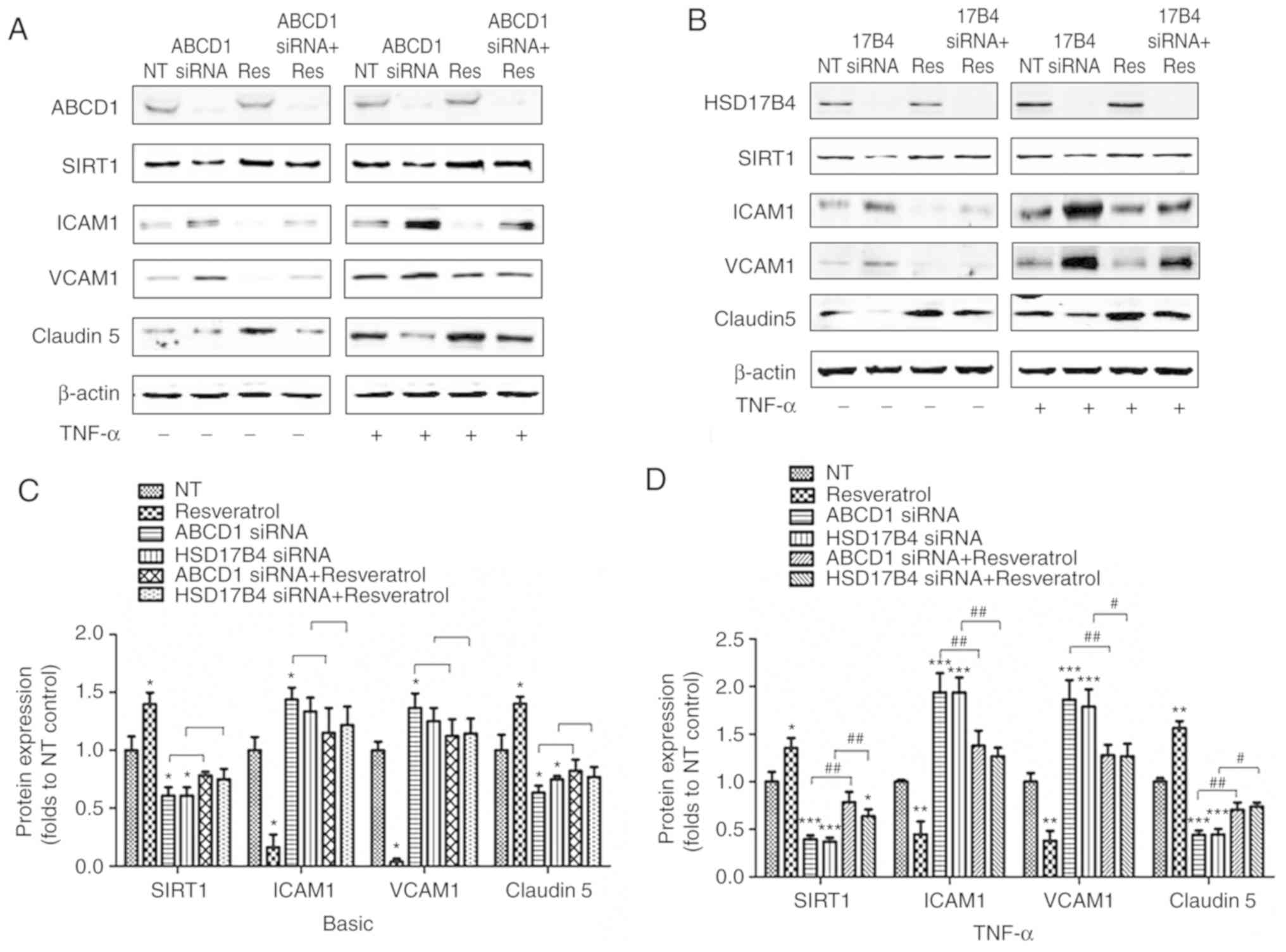 | Figure 2SIRT1 activation by resveratrol
restores protein biomarker changes in HBMECs after ABCD1 or HSD17B4
silencing. Representative western blot images as well as intensity
quantification showing SIRT1, tight-junction protein (Claudin 5)
and adhesion molecule (ICAM1 and VCAM1) changes in (A) ABCD1 or (B)
HSD17B4 silenced HBMECs with or without resveratrol (20 µM)
treatment at (C) basal or (D) TNF-α (10 ng/ml) treated condition.
Data are presented as the mean ± standard deviation of three
different experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. NT control
group. #P<0.05 and ##P<0.01. NT,
non-targeting; ABCD, ATP binding cassette subfamily D member 1;
HBMECs, human brain microvascular endothelial cells; TNF, tumor
necrosis factor; ICAM, intracellular cellular adhesion molecule;
VCAM, vascular cellular adhesion molecule; si, small interfering;
SIRT1, sirtuin1; NT, non-targeting. |
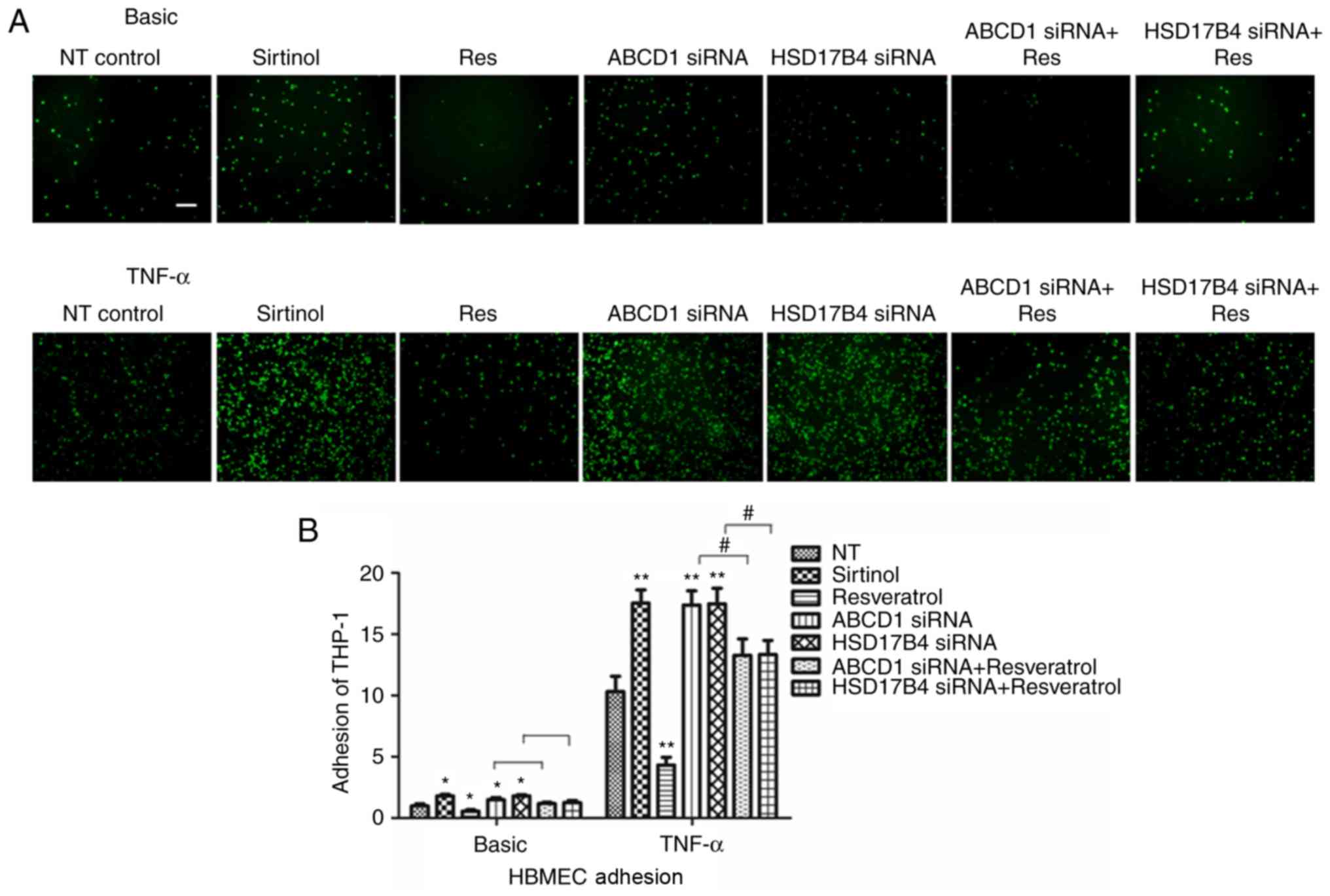
SIRT1 expression levels in HBMECs
modulate endothelial-monocyte interactions
After determining the changes of molecular
biomarkers, the functional consequences of low SIRT1 levels in
HBMECs achieved by inhibition with Sirtinol or silencing of ABCD1
or HSD17B4. As previously described, a significant increase in
adhesion of THP-1 cells was identified after ABCD1 silencing under
basal conditions (P<0.05) and following TNF-α treatment
(P<0.01; Fig. 3). Similarly, a
significant increase in adhesion was observed after HSD17B4
silencing and Sirtinol treatment under basal conditions (P<0.05)
and upon stimulation with TNF-α (P<0.01; Fig. 3).
To further assess whether restoration of SIRT1
levels in ABCD1- and HSD17B4-silenced cells is able to attenuate
their increased adhesion to monocytes that had been observed,
silenced HBMECs were treated with resveratrol. First, resveratrol
treatment alone significantly reduced monocyte adhesion to 48 and
41% under basal conditions (P<0.05) and following TNF-α
treatment (P<0.01), respectively. Furthermore, in ABCD1-silenced
HBMECs, treatment with resveratrol markedly inhibited monocyte
adhesion by 32 and 41% under basal and TNF-α treatment conditions,
respectively (P<0.05). Similarly, in HSD17B4-silenced HBMECs,
treatment with resveratrol also markedly inhibited monocyte
adhesion by 31 and 40% at basal and TNF-α treatment conditions,
respectively (P<0.05), suggesting functional correction by
resveratrol treatment (Fig.
3).
Disruption of NADP/NADPH and increased
oxidative stress in HBMECs after ABCD1 or HSD17B4 silencing
As a NAD+-dependent protein deacetylase
and the key metabolic sensor, SIRT1 tightly regulates numerous
cellular activities involved in systemic metabolic homeostasis
(16). In the present study, it
was revealed that the NADP/NADPH ratio in HBMECs was significantly
increased after ABCD1 or HSD17B4 silencing (P<0.01;
Table I), suggesting a
disturbance of metabolic homeostasis. By measuring oxidative stress
makers MDA and SOD, it was unveiled that ABCD1 and HSD17B4
silencing led to significantly increased MDA levels and decreased
SOD levels in HBMECs, suggesting increased oxidative stress. This
effect was mimicked by sirtinol treatment, while resveratrol
treatment significantly reduced the oxidative stress under basal
and TNF-α treatment conditions (P<0.01; Table II).
 | Table INADP/NADPH ratio in human brain
microvascular endothelial cells after ABCD1 and HSD17B4
silencing. |
Table I
NADP/NADPH ratio in human brain
microvascular endothelial cells after ABCD1 and HSD17B4
silencing.
| NT | ABCD1 SIRNA | HSD17B4 SIRNA |
|---|
| NADP/NADPH | 0.95±0.20 |
1.40±0.28A |
1.41±0.22A |
| Table IIMDA and SOD levels in HBMECs at
different treatment conditions. |
Table II
MDA and SOD levels in HBMECs at
different treatment conditions.
| Basic
| TNF-α
|
|---|
| MDA (nmol/ml) | SOD (U/ml) | MDA (nmol/ml) | SOD (U/ml) |
|---|
| Control | 0.75±0.12 | 64.08±4.38 | 1.54±0.27 | 103.41±5.88 |
| ABCD1 siRNA | 1.12±0.23a | 33.38±5.27a | 3.24±0.29a | 71.72±4.75a |
| Sirtinol | 1.15±0.28a | 34.05±4.75a | 3.03±0.39a | 72.05±8.18a |
| ABCD1
siRNA+sirtinol | 1.23±0.32a | 34.39±4.35a | 3.31±0.40a | 72.42±4.78a |
| Resveratrol | 0.39±0.14a | 83.73±5.57a | 0.60±0.34a | 125.93±5.51a |
| ABCD1
siRNA+resveratrol | 0.94±0.20a,b | 45.48±6.40a,b | 2.23±0.31a,b | 83.43±6.65a,b |
| Control | 0.73±0.18 | 63.62±5.62 | 1.51±0.36 | 101.75±6.33 |
| 17B4 siRNA | 1.13±0.31a | 35.82±6.49a | 3.28±0.39a | 68.72±9.57a |
| Sirtinol | 1.16±0.34a | 34.38±5.12a | 3.21±0.48a | 67.95±7.53a |
| 17B4
siRNA+sirtinol | 1.27±0.31a | 33.71±5.90a | 3.41±0.45a | 70.38±8.39a |
| Resveratrol | 0.40±0.18a | 83.89±6.59a | 0.47±0.56a | 126.93±4.76a |
| 17B4
siRNA+resveratrol | 0.89±0.21a,b | 44.23±5.86a,b | 2.13±0.73a,b | 81.86±8.26a,b |
NF-κB and Krüppel-like factor 4 (KLF4)
mediate SIRT1- dependent transcriptional regulation of ICAM1 and
Claudin 5 in HBMECs following ABCD1 or HSD17B4 silencing
In order to investigate the molecular mechanisms
that mediate the effect of SIRT1 upon upregulation of adhesion
molecules, activation of NF-κB, a transcriptional factor known to
regulate ICAM1 expression, was then assessed. Indeed, SIRT1
depletion by sirtinol or silencing of ABCD1 or HSD17B4 in HBMECs
led to increased IκBα phosphorylation and subsequent NF-κB
accumulation in the nucleus under basal conditions (P<0.05;
Fig. 4A and B) and following
TNF-α treatment (P<0.01; Fig. 5A
and B), suggesting activation of the NF-κB signaling pathway.
When SIRT1 activity was enhanced by resveratrol treatment, a
significant reduction of IκBα phosphorylation and nuclear NF-κB
levels was observed in the control as well as in the ABCD1 or
HSD17B4 gene silencing groups under basal conditions (P<0.01;
Fig. 4C and D) and following
TNF-α treatment (P<0.01; Fig. 5C
and D), indicating that restoration of the function of SIRT1 is
able to reverse NF-κB activation and significantly inhibit the
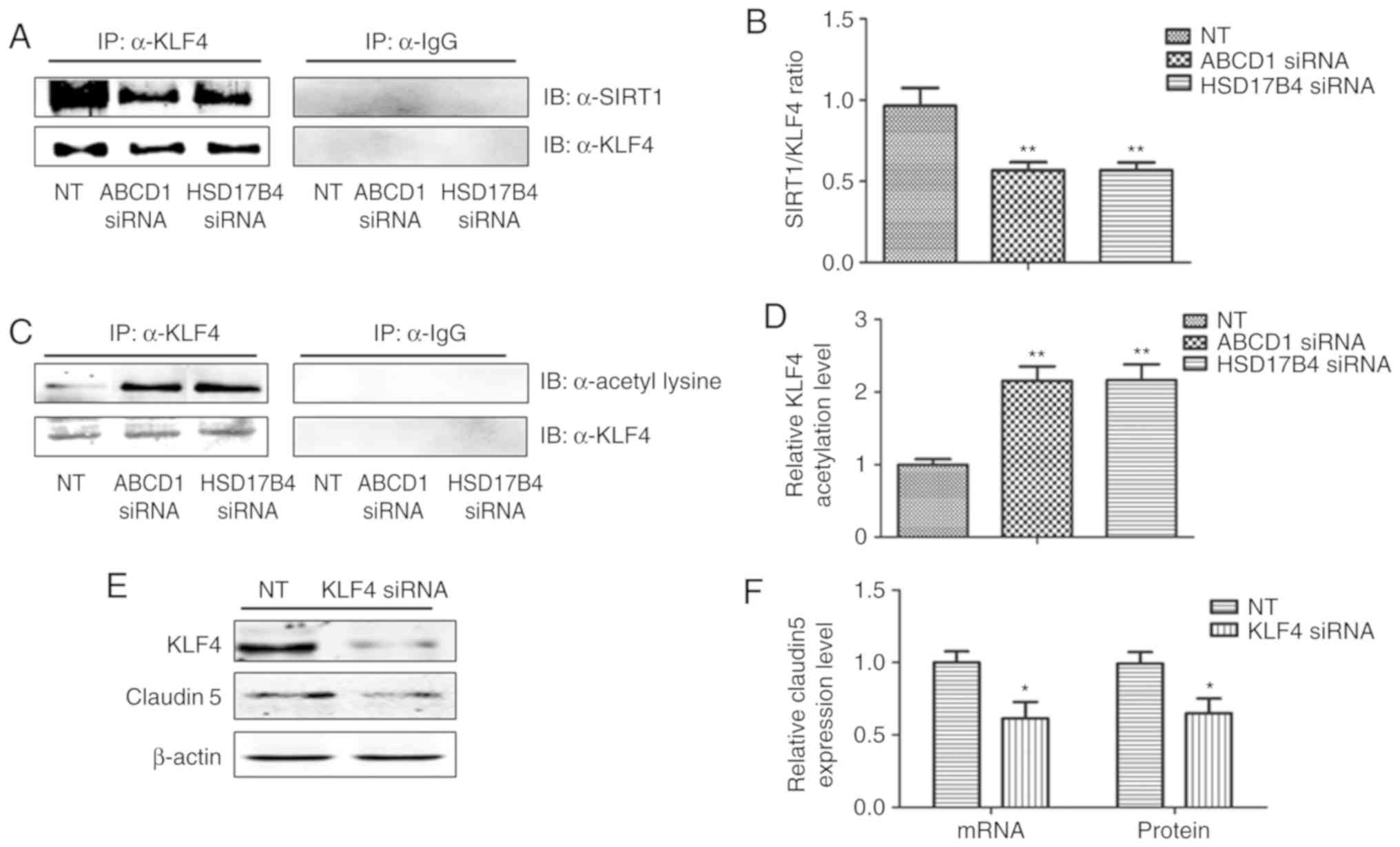
expression of adhesion molecules, including ICAM1. A recent study
reported that SIRT1 deacetylated KLF4 to activate Claudin 5
transcription in ovarian cancer cells (25). The present results indicated that
ABCD1 and HSD17B4 silencing significantly reduced nuclear KLF4
levels under basal (P<0.05) and TNF-α treatment conditions
(P<0.001), and the same effect was observed following SIRT1
inhibition using sirtinol (Figs. 4A
and B; 5A and C). Increased
SIRT1 activation by resveratrol significantly increased nuclear
KLF4 levels in the control group and also restored the levels of
KLF4 that were reduced by ABCD1 and HSD17B4 silencing under basal
(P<0.05; Fig. 4C and D) and
TNF-α treatment conditions (P<0.01; Fig. 5C and D). The direct interaction
between SIRT1 and KLF4 and subsequent regulation of Claudin 5
expression was assessed by IP and silencing of KLF4. Reduction of
SIRT1 binding to KLF4 protein and increase in KLF4 protein
acetylation following ABCD1 and HSD17B4 silencing were confirmed
(P<0.01; Fig. 6A-D). Finally,
KLF4 silencing significantly inhibited Claudin 5 transcription in
HBMECs (P<0.05; Fig. 6E and
F).
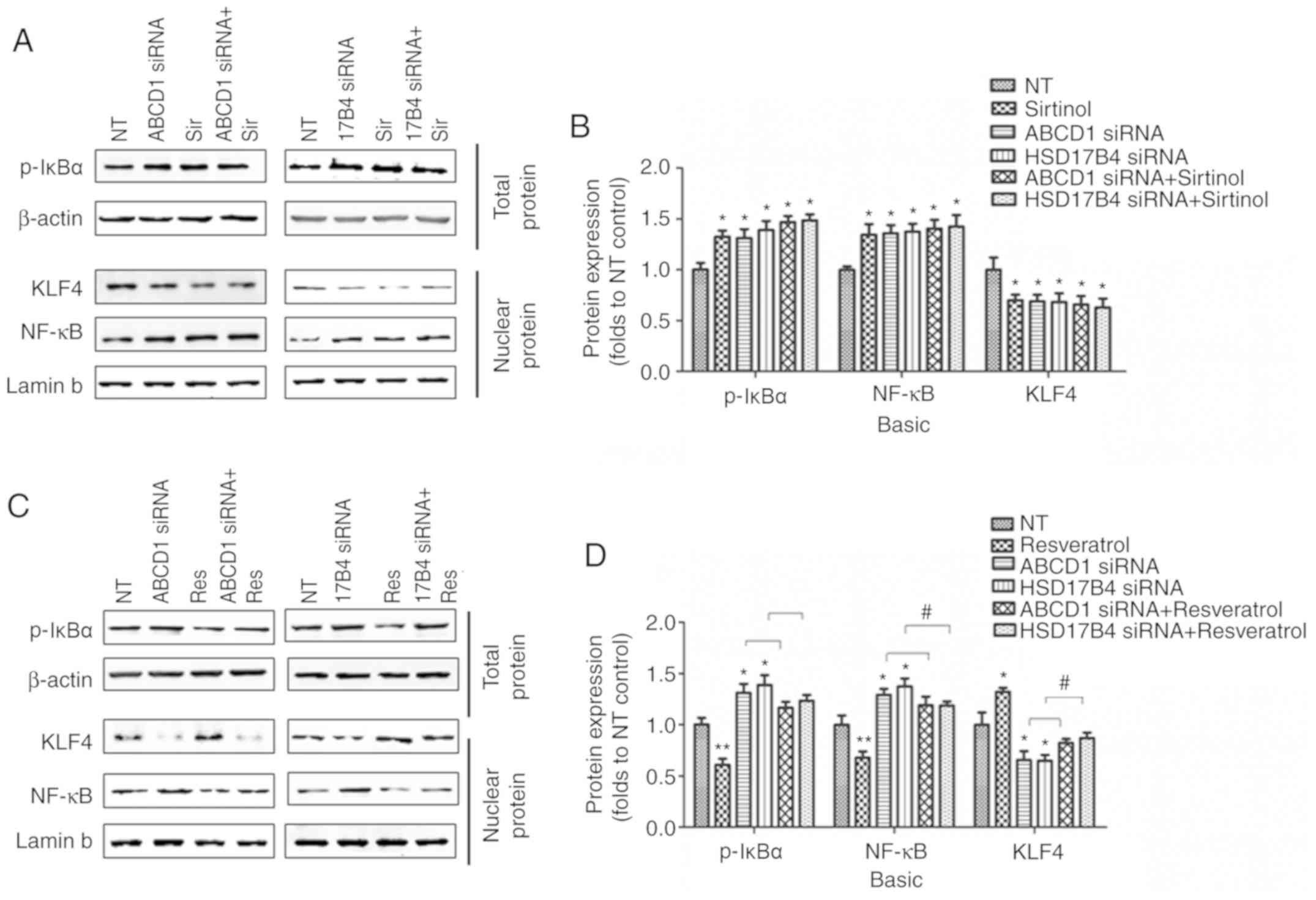 | Figure 4Regulation of SIRT1 on NF-κB and KLF4
following ABCD1 or HSD17B4 silencing. (A) Representative western
blot images as well as (B) intensity quantification of p-IκBα,
nuclear NF-κB and nuclear KLF4 in ABCD1 or HSD17B4 silenced HBMECs
with or without sirtinol (20 µM) treatment at basal
condition. (C) Representative western blot images as well as (D)
intensity quantification of p-IκBα, nuclear NF-κB and nuclear KLF4
in ABCD1 or HSD17B4 silenced HBMECs with or without resveratrol (20
µM) treatment at basal condition. *P<0.05 and
**P<0.01 vs. NT control group. #P<0.05.
NT, non-targeting; ABCD, ATP binding cassette subfamily D member 1;
HBMECs, human brain microvascular endothelial cells; TNF, tumor
necrosis factor; NF, nuclear factor; si, small interfering; KLF4,
Krüppel-like factor 4; NF, nuclear factor; p-IκBα,
phosphorylated-inhibitor of NF-κB; NT, non-targeting. |
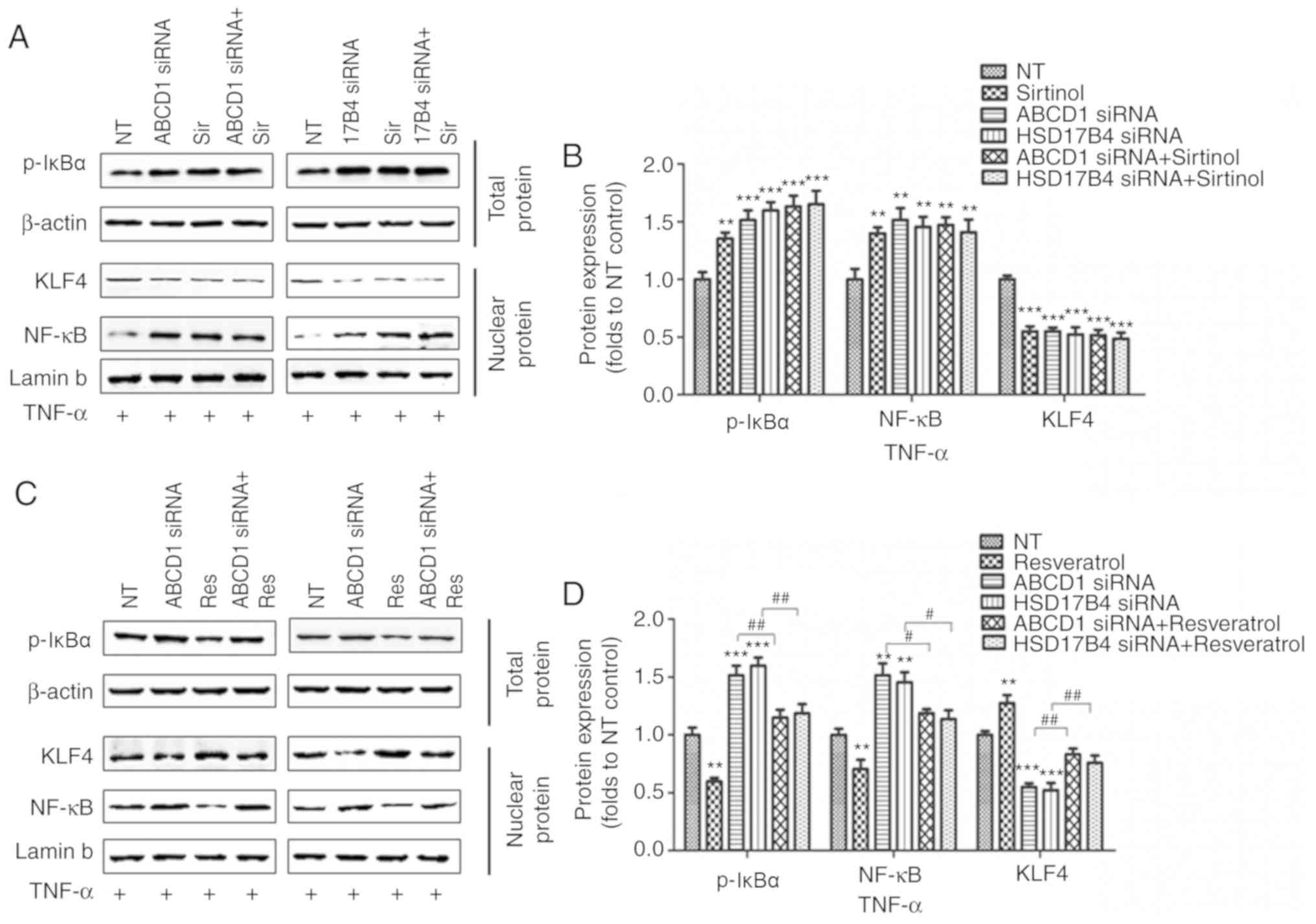 | Figure 5Regulation of SIRT1 on NF-κB and KLF4
following ABCD1 or HSD17B4 silencing at TNF-α treated condition.
(A) Representative western blot images as well as (B) intensity
quantification of p-IκBα, nuclear NF-κB and nuclear KLF4 in ABCD1
or HSD17B4 silenced HBMECs with or without sirtinol (20 µM)
treatment at TNF-α (10 ng/ml) treated condition. (C) Representative
western blot images as well as (D) intensity quantification of
p-IκBα, nuclear NF-κB and nuclear KLF4 in ABCD1 or HSD17B4 silenced
HBMECs with or without resveratrol (20 µM) treatment at
TNF-α (10 ng/ml) treated condition. **P<0.01 and
***P<0.001 vs. the NT control group.
#P<0.05 and ##P<0.05. NT,
non-targeting; ABCD, ATP binding cassette subfamily D member 1;
HBMECs, human brain microvascular endothelial cells; TNF, tumor
necrosis factor; NF, nuclear factor; si, small interfering; KLF4,
Krüppel-like factor 4; p-IκBα, phosphorylated-inhibitor of NF-κB;
NT, non-targeting. |
Discussion
The present study demonstrated that dysfunction of
ABCD1 and HSD17B4, two peroxisomal disorders manifesting with
neurodegeneration, caused brain microvascular endothelium
dysfunction in the in vitro cell model of the BBB. This
dysfunction was correlated with a reduction of SIRT1 levels in
HBMECs. Inhibition of SIRT1 activity by sirtinol mimicked the
molecular and functional changes caused by ABCD1 and HSD17B4
silencing, while improvement of SIRT1 function by resveratrol
ameliorated HBMEC dysfunction.
The BBB constitutes specialized endothelial cells,
pericytes and astrocytes, and tightly regulates the communication
between the immune and nervous systems (26). Leukocyte infiltration is
considered a critical step in the pathogenesis of numerous CNS
diseases (27-30), but under normal conditions, brain
leukocyte traffic into the brain is limited due to the tight
endothelial barrier (30-32).
A leukocyte may cross the BBB and enter the brain
parenchyma through several steps, consisting of the initial rolling
and binding of leukocytes on the endothelium by selectins, and
subsequent adhesion of leukocytes and their transendothelial egress
by ICAM1 and VCAM1 (33,34). Elevated levels of soluble
E-selectin, VCAM1 and ICAM1 have been reported in the sera and
cerebrospinal fluid of patients multiple sclerosis (MS), and are
correlated with disease activity (35). Tight junctions are essential to
the transendothelial egress of leukocytes. It is known that
abnormalities in ZO1 and occludin occur in the brain of subjects
with MS, causing interruption of junctional integrity (36). Dysfunctional tight junctions may
allow for a greater influx of blood-borne cells and cytokines, thus
amplifying inflammation and further parenchymal damage. In the
present study, it was demonstrated that ABCD1 silencing [as
previously reported (12)] and
HSD17B4 silencing increase ICAM1 and VCAM1 expression in HBMECs,
while reducing Claudin 5 expression, which facilitates monocyte
adhesion and subsequent transmigration, a potential mechanism
contributing to neuroinflammation in cerebral ALD and MFP
deficiency.
It is well known that impaired bioenergetics and
mitochondrial metabolism, together with oxidative stress, are
factors underlying a variety of neurodegenerative diseases,
including Parkinson's, Huntington's and Alzheimer's disease, or
amyotrophic lateral sclerosis (37-40). CALD and MFP deficiency are
peroxisomal disorders that lead to a deficit of VLCFA metabolism.
Disrupted energy homeostasis with diminished ATP, NADH and GSH
levels, accompanied by dysregulation of oxidative phosphorylation,
was discovered in patients with X-ALD as well as
ABCD1−/− mouse tissue samples (41-43). Similarly, increased oxidative
stress and disrupted energy homeostasis was also reported in MFP
deficiency (44). As a
NAD+-dependent protein deacetylase and the key metabolic
sensor, SIRT1 tightly regulates numerous cellular activities
involved in systemic metabolic homeostasis. In the present study,
increased NADP/NADPH levels and oxidative stress were detected in
HBMECs after ABCD1 or HSD17B4 silencing. The disturbance of
metabolic homeostasis may be the potential trigger of SIRT1
inhibition upon ABCD1 or HSD17B4 deficiency.
The crucial role of SIRT1 in maintaining normal
vascular endothelial function has been well established in a
variety of vascular disorders (19-21). Jia et al (45) reported the inhibitory effect of
SIRT1 on ICAM1 expression in human umbilical vein endothelial cells
by suppressing its promoter activity. Transcriptional factor NF-κB
activation and SIRT1 are closely regulated by each other and have
an essential role in the maintenance of endothelial function
(11,12). It has been well established that
ICAM1 has a binding site for NF-κB in its promoter region and its
expression is largely controlled by NF-κB activation (45-47). The present results first suggest
that depletion of SIRT1 by ABCD1 or HSD17B4 silencing or sirtinol
causes activation of NF-κB signaling in HBMECs and reciprocally,
increases in SIRT1 expression caused by resveratrol treatment
prevent NF-κB activation, and subsequently ameliorates endothelial
dysfunction. Another protective effect of SIRT1 on barrier function
was via regulation of tight-junction proteins in endothelial cells
(48,49). The present study indicated that
upregulation of SIRT1 increases Claudin 5 expression. In terms of
the mechanism, Zhang et al (25) reported that, in ovarian cancer
cells, activation of Claudin 5 transcription was mediated by
SIRT1-induced deacetylation of KLF4 and subsequent transport of
KLF4 to the nucleus. In the present study, it was proved that ABCD1
and HSD17B4 silencing down-regulates Claudin 5 transcription in
HBMECs via reducing SIRT1 and its binding to KLF4. Furthermore,
resveratrol effectively ameliorates brain endothelial dysfunction,
while pleiotropic effects other than NF-κB and KLF4 signaling may
also occur. Regarding those possible effects, resveratrol is well
known as an anti-oxidant, which is able to inhibit
mitochondria-derived oxidative damage (50,51) and regulate inflammatory and immune
responses in endothelial cells (52). Further studies are necessary to
demonstrate its beneficial effects in more complex model systems of
peroxisomal dysfunction, including in vitro multicellular
BBB models and in vivo mouse models.
In conclusion, the present study demonstrates, for
the first time, that brain endothelial peroxisomal dysfunction
secondary to X-ALD and MFP deficiency causes dysregulation of
SIRT1, which alters the transcriptional regulation of adhesion
molecules and tight junction proteins via the NF-κB and KLF4
signaling pathways. SIRT1 depletion in brain endothelial cells,
which is most likely caused by disruption of β-oxidation and
oxidative stress, may be one of the underlying mechanisms for
monocyte and microglia activation during neuroinflammation in
peroxisomal disorders. Restoration of endothelial function by
normalizing SIRT1 levels with substances including resveratrol or
by targeting its downstream signaling pathways may give rise to
novel therapeutic strategies for these devastating
neurodegenerative disorders.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81501038).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YG and YW were responsible for conception and design
of the study, YZ, YW and YG were responsible for data acquisition
and analysis. YG and YZ were responsible for drafting manuscript
and figures. GC was responsible for providing general guidance. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
X-ALD
|
X-linked adrenoleukodystrophy
|
|
MFP2
|
multifunctional protein 2
|
|
HSD17B4
|
hydroxysteroid 17-β dehydrogenase
4
|
|
ABCD1
|
ATP binding cassette subfamily D
Member 1
|
|
SIRT1
|
sirtuin1
|
|
ATP
|
adenosine triphosphate
|
|
VLCFA
|
very long chain fatty acid
|
|
BBB
|
blood brain barrier
|
|
BMECs
|
brain microvascular endothelial
cells
|
|
HBMECS
|
human brain microvascular endothelial
cells
|
|
NAD
|
nicotinamide adenosine
dinucleotide
|
|
NADP
|
Nicotinamide adenine dinucleotide
phosphate
|
|
TNF-α
|
tumor necrosis factor-α
|
|
ICAM
|
intercellular adhesion molecule 1
|
|
VCAM1
|
vascular cell adhesion molecule 1
|
|
NF-κB
|
nuclear factor κ-light-chain-enhancer
of activated B cells
|
|
KLF4
|
Krüppel-like factor 4
|
|
MDA
|
malondialdehyde
|
|
SOD
|
superoxide dismutase
|
|
RIPA
|
radioimmunoprecipitation assay
|
|
SD
|
standard deviation
|
Acknowledgments
We sincerely thank the help provided by the
Department of Neurobiology, Xuzhou Medical University and The
Affiliated Hospital of Xuzhou Medical University.
References
|
1
|
Trompier D, Vejux A, Zarrouk A, Gondcaille
C, Geillon F, Nury T, Savary S and Lizard G: Brain peroxisomes.
Biochimie. 98:102–110. 2014. View Article : Google Scholar
|
|
2
|
Wanders RJ and Waterham HR: Biochemistry
of mammalian peroxisomes revisited. Annu Rev Biochem. 75:295–332.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mosser J, Douar AM, Sarde CO, Kioschis P,
Feil R, Moser H, Poustka AM, Mandel JL and Aubourg P: Putative
X-linked adrenoleukodystrophy gene shares unexpected homology with
ABC transporters. Nature. 361:726–730. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eichler F and Van Haren K: Immune response
in leukodystrophies. Pediatr Neurol. 37:235–244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van der Voorn JP, Pouwels PJ, Powers JM,
Kamphorst W, Martin JJ, Troost D and Spreeuwenberg MD: Correlating
quantitative MR imaging with histopathology in X-linked
adrenoleukodystrophy. AJNR Am J Neuroradiol. 32:481–489. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Eichler FS, Ren JQ, Cossoy M, Rietsch AM,
Nagpal S, Moser AB, Frosch MP and Ransohoff RM: Is microglial
apoptosis an early pathogenic change in cerebral X-linked
adrenoleukodystrophy? Ann Neurol. 63:729–742. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Musolino PL, Rapalino O, Caruso P,
Caviness VS and Eichler FS: Hypoperfusion predicts lesion
progression in cerebral X-linked adrenoleukodystrophy. Brain.
135:2676–2683. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Berger J, Dorninger F, Forss-Petter S and
Kunze M: Peroxisomes in brain development and function. Biochim
Biophys Acta. 1863:934–955. 2016. View Article : Google Scholar :
|
|
9
|
Verheijden S, Beckers L, De Munter S, Van
Veldhoven PP and Baes M: Central nervous system pathology in MFP2
deficiency: Insights from general and conditional knockout mouse
models. Biochimie. 98:119–126. 2014. View Article : Google Scholar
|
|
10
|
Ferdinandusse S, Denis S, Mooyer PA,
Dekker C, Duran M, Soorani-Lunsing RJ, Boltshauser E, Macaya A,
Gärtner J, Majoie CB, et al: Clinical and biochemical spectrum of
D-bifunctional protein deficiency. Ann Neurol. 59:92–104. 2006.
View Article : Google Scholar
|
|
11
|
Zhang W, Huang Q, Zeng Z, Wu J, Zhang Y
and Chen Z: Sirt1 inhibits oxidative stress in vascular endothelial
cells. Oxid Med Cell Longev. 2017:75439732017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang H, Zhang W, Pan H, Feldser HG, Lainez
E, Miller C, Leung S, Zhong Z, Zhao H, Sweitzer S, et al: SIRT1
activators suppress inflammatory responses through promotion of p65
deacetylation and inhibition of NF-κB activity. PLoS One. 7:pp.
e463642012, View Article : Google Scholar
|
|
13
|
Rubin LL and Staddon JM: The cell biology
of the blood-brain barrier. Annu Rev Neurosci. 22:11–28. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ueno M: Molecular anatomy of the brain
endothelial barrier: An overview of the distributional features.
Curr Med Chem. 14:1199–1206. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pardridge WM: Blood-brain barrier
delivery. Drug Discov Today. 12:54–61. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bordone L and Guarente L: Calorie
restriction, SIRT1 and metabolism: Understanding longevity. Nat Rev
Mol Cell Biol. 6:298–305. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Morató L, Ruiz M, Boada J, Calingasan NY,
Galino J, Guilera C, Jové M, Naudí A, Ferrer I, Pamplona R, et al:
Activation of sirtuin 1 as therapy for the peroxisomal disease
adrenoleukodystrophy. Cell Death Differ. 22:1742–1753. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stamatovic SM, Martinez-Revollar G, Hu A,
Choi J, Keep RF and Andjelkovic AV: Decline in Sirtuin-1 expression
and activity plays a critical role in blood-brain barrier
permeability in aging. Neurobiol Dis. 126:105–116. 2019. View Article : Google Scholar
|
|
19
|
Orimo M, Minamino T, Miyauchi H, Tateno K,
Okada S, Moriya J and Komuro I: Protective role of SIRT1 in
diabetic vascular dysfunction. Arterioscler Thromb Vasc Biol.
29:889–894. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng
W, Liu G, Wei YS, Cai H, Liu DP and Liang CC: Endothelium-specific
overexpression of class III deacetylase SIRT1 decreases
atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res.
80:191–199. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vassallo PF, Simoncini S, Ligi I, Chateau
AL, Bachelier R, Robert S, Morere J, Fernandez S, Guillet B,
Marcelli M, et al: Accelerated senescence of cord blood endothelial
progenitor cells in premature neonates is driven by SIRT1 decreased
expression. Blood. 123:2116–2126. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim D, Nguyen MD, Dobbin MM, Fischer A,
Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, et
al: SIRT1 deacetylase protects against neurodegeneration in models
for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J.
26:3169–3179. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Donmez G and Outeiro TF: SIRT1 and SIRT2:
Emerging targets in neurodegeneration. EMBO Mol Med. 5:344–352.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Zhang X, Chen J, Sun L and Xu Y: SIRT1
deacetylates KLF4 to activate Claudin-5 transcription in ovarian
cancer cells. J Cell Biochem. 119:2418–2426. 2018. View Article : Google Scholar
|
|
26
|
Obermeier B, Daneman R and Ransohoff RM:
Development, maintenance and disruption of the blood-brain barrier.
Nat Med. 19:1584–1596. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wekerle H, Schwab M, Linington C and
Meyermann R: Antigen presentation in the peripheral nervous system:
Schwann cells present endogenous myelin autoantigens to
lymphocytes. Eur J Immunol. 16:1551–1557. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cross AH, Dolich S and Raine CS: Antigen
processing of myelin basic protein is required prior to recognition
by T cells inducing EAE. Cell Immunol. 129:22–31. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Raine CS, Cannella B, Duijvestijn AM and
Cross AH: Homing to central nervous system vasculature by
antigen-specific lymphocytes. II Lymphocyte/endothelial cell
adhesion during the initial stages of autoimmune demyelination Lab
Invest. 63:476–489. 1990.
|
|
30
|
Zlokovic BV: The blood-brain barrier in
health and chronic neurodegenerative disorders. Neuron. 57:178–201.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wilson EH, Weninger W and Hunter CA:
Trafficking of immune cells in the central nervous system. J Clin
Invest. 120:1368–1379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Man S, Ubogu EE, Williams KA, Tucky B,
Callahan MK and Ransohoff RM: Human brain microvascular endothelial
cells and umbilical vein endothelial cells differentially
facilitate leukocyte recruitment and utilize chemokines for T cell
migration. Clin Dev Immunol. 2008:3849822008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Miller DW: Immunobiology of the
blood-brain barrier. J Neurovirol. 5:570–578. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Minagar A and Alexander JS: Blood-brain
barrier disruption in multiple sclerosis. Mult Scler. 9:540–549.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Droogan AG, McMillan SA, Douglas JP and
Hawkins SA: Serum and cerebrospinal fluid levels of soluble
adhesion molecules in multiple sclerosis: Predominant intrathecal
release of vascular cell adhesion molecule-1. J Neuroimmunol.
64:185–191. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Plumb J, McQuaid S, Mirakhur M and Kirk J:
Abnormal endothelial tight junctions in active lesions and
normal-appearing white matter in multiple sclerosis. Brain Pathol.
12:154–169. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Calabrese V, Cornelius C, Dinkova-Kostova
AT, Calabrese EJ and Mattson MP: Cellular stress responses, the
hormesis paradigm, and vitagenes: Novel targets for therapeutic
intervention in neurodegenerative disorders. Antioxid Redox Signal.
13:1763–1811. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Di Domenico F, Perluigi M, Butterfield DA,
Cornelius C and Calabrese V: Oxidative damage in rat brain during
aging: Interplay between energy and metabolic key target proteins.
Neurochem Res. 35:2184–2192. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Leuner K, Hauptmann S, Abdel-Kader R,
Scherping I, Keil U, Strosznajder JB, Eckert A and Müller WE:
Mitochondrial dysfunction: The first domino in brain aging and
Alzheimer's disease? Antioxid Redox Signal. 9:1659–1675. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Martinez A, Portero-Otin M, Pamplona R and
Ferrer I: Protein targets of oxidative damage in human
neurodegenerative diseases with abnormal protein aggregates. Brain
Pathol. 20:281–297. 2010. View Article : Google Scholar
|
|
41
|
Galino J, Ruiz M, Fourcade S, Schlüter A,
López-Erauskin J, Guilera C, Jove M, Naudi A, García-Arumí E,
Andreu AL, et al: Oxidative damage compromises energy metabolism in
the axonal degeneration mouse model of X-adrenoleukodystrophy.
Antioxid Redox Signal. 15:2095–2107. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schlüter A, Espinosa L, Fourcade S, Galino
J, López E, Ilieva E, Morató L, Asheuer M, Cook T, McLaren A, et
al: Functional genomic analysis unravels a metabolic-inflammatory
interplay in adrenoleukodystrophy. Hum Mol Genet. 21:1062–1077.
2012. View Article : Google Scholar :
|
|
43
|
Fourcade S, López-Erauskin J, Galino J,
Duval C, Naudi A, Jove M, Kemp S, Villarroya F, Ferrer I, Pamplona
R, et al: Early oxidative damage underlying neurodegeneration in
X-adrenoleukodystrophy. Hum Mol Genet. 17:1762–1773. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ferdinandusse S, Finckh B, de Hingh YC,
Stroomer LE, Denis S, Kohlschütter A and Wanders RJ: Evidence for
increased oxidative stress in peroxisomal D-bifunctional protein
deficiency. Mol Genet Metab. 79:281–287. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jia Y, Gao P, Chen H, Wan Y, Zhang R,
Zhang Z, Yang R, Wang X, Xu J and Liu D: SIRT1 suppresses PMA and
ionomycin-induced ICAM-1 expression in endothelial cells. Sci China
Life Sci. 56:19–25. 2013. View Article : Google Scholar
|
|
46
|
Melotti P, Nicolis E, Tamanini A, Rolfini
R, Pavirani A and Cabrini G: Activation of NF-kB mediates ICAM-1
induction in respiratory cells exposed to an adenovirus-derived
vector. Gene Ther. 8:1436–1442. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ledebur HC and Parks TP: Transcriptional
regulation of the inter-cellular adhesion molecule-1 gene by
inflammatory cytokines in human endothelial cells. Essential roles
of a variant NF-kappa B site and p65 homodimers. J Biol Chem.
270:933–943. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ma Y, Xu C, Wang W, Sun L, Yang S, Lu D,
Liu Y and Yang H: Role of SIRT1 in the protection of intestinal
epithelial barrier under hypoxia and its mechanism. Zhonghua Wei
Chang Wai Ke Za Zhi. 17:602–606. 2014.In Chinese. PubMed/NCBI
|
|
49
|
Ma J, Wang P, Liu Y, Zhao L, Li Z and Xue
Y: Krüppel-like factor 4 regulates blood-tumor barrier permeability
via ZO-1, occludin and claudin-5. J Cell Physiol. 229:916–926.
2014. View Article : Google Scholar
|
|
50
|
Liu CW, Sung HC, Lin SR, Wu CW, Lee CW,
Lee IT, Yang YF, Yu IS, Lin SW, Chiang MH, et al: Resveratrol
attenuates ICAM-1 expression and monocyte adhesiveness to
TNF-α-treated endothelial cells: Evidence for an anti-inflammatory
cascade mediated by the miR-221/222/AMPK/p38/NF-κB pathway. Sci
Rep. 7:446892017. View Article : Google Scholar
|
|
51
|
Kaisar MA, Prasad S and Cucullo L:
Protecting the BBB endothelium against cigarette smoke-induced
oxidative stress using popular antioxidants: Are they really
beneficial? Brain Res. 1627:90–100. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Schwager J, Richard N, Widmer F and
Raederstorff D: Resveratrol distinctively modulates the
inflammatory profiles of immune and endothelial cells. BMC
Complement Altern Med. 17:3092017. View Article : Google Scholar : PubMed/NCBI
|