Introduction
Autophagy is a genetically and evolutionarily
conserved process that occurs in all eukaryotic cells from yeast to
mammals, which is used for the non-selective removal of long-lived
proteins, large protein aggregates, and subcellular organelles
(1–3). At the beginning of autophagy,
portions of the cytoplasm and intra-cellular organelles are
sequestered in double membrane-bound structures to form
autophagosomes. Subsequently, they fuse with lysosomes to form
autophagolysosomes, where the sequestered content is degraded by
lysosomal hydrolases or recycled (4).
There is increasing evidence describing the role of
autophagy in cancer (5), and
various antineoplastic therapies were reported to induce autophagy
in human cancer cell lines (6–8).
However, fundamental questions regarding whether autophagy kills
cancer cells or protects them from unfavorable conditions have not
yet been clearly answered (9).
Some studies have suggested an inverse relationship between
autophagy and malignant growth, as Beclin 1, a key autophagy
protein regulator, is a haploinsufficient tumor suppressor gene in
mice (10,11) but is frequently absent in human
cancers (12). Also, the
identification of damage-regulated autophagy modulator (DRAM), a
protein necessary to induce p53-dependent autophagy, provides
another link between autophagy and tumor suppression (13). Therefore, these studies suggest
that defects in autophagy may favor carcinogenesis and that the
restoration of autophagy may have promising therapeutic
implications in cancer (14).
However, there is no specific cancer therapy that targets autophagy
in colorectal cancer, consequently a better understanding on the
molecular mechanisms by which carcinogens lead to autophagy may
contribute to the advancement of a rational clinical therapy in
this cancer type.
Autophagy is a multistep process, and various
signaling pathways have been implicated in its upregulation and/or
downregulation (2,3,15).
Nevertheless, independently whether the cell is normal or
malignant, the mammalian target of rapamycin (mTOR) serves as the
main regulator of autophagy (16).
Oncogenic forms of Ras are also implicated in the negative control
of autophagy through the activation of class I PI3K (17), and PTEN, the classic negative
regulator of PI3K, regulates autophagy suppressing Akt activity
leading to autophagy initiation (18). The mitogen-activated kinases
(MAPKs) also were reported to regulate autophagy in cancer cells
(19). Thus, various signaling
pathways may control autophagy, but it is not fully understood if
these pathways induce or suppress autophagy formation depending on
the cell type. In epithelial tumor cells, such as breast, prostate,
and colon cancer cells, it was reported that irradiation induced
autophagosome formation, but apoptosis has little or no role in
cell death (20–22). However, the cell signaling events
underlying the formation of these organelles after IR, particularly
in colon cancer cells has been only slightly explored, and it
remains unclear if autophagy inhibition contributes to cell
death.
Therefore, we hypothesized that IR promotes
autophagosome formation in an event mediated by cell signaling
pathways activated by this cancer therapy and that autophagy
inhibition could lead to cell death. To test this hypothesis, we
used the highly invasive human colon cancer HCT-116 cells, which
are an appropriate model to investigate the IR-induced effects
(23), and blockade of
autophagosomes formation by pharmacological interference. Our
finding clearly demonstrate that blockade of IR-induced
autophagosomes impairs proliferation but do not enhance cell death
in colon cancer cells.
Materials and methods
Antibodies and reagents
The following antibodies were used in this study:
Akt and phospho-Akt antibodies (Cell Signaling Technology, Boston,
MA, USA), v-Src and phospho-Src antibodies (Oncogene Research
Products, Boston, MA, USA), LC3B antibody (Sigma Chemical Co., St.
Louis, MO, USA), α-tubulin antibody (Zymed Lab. Inc., San
Francisco, CA, USA). The following secondary antibodies were used:
Alexa fluor 488-conjugated antibody (Invitrogen Co., Carlsbad, CA,
USA) and horseradish peroxidase-conjugated antibody (Sigma Co.)
Both 2-(4-Morpholinyl)-8-phenyl-4H-1-benzopyran-4-1 (LY294002) and
1-tert-Butyl-3-(4-chlorophenyl)-1H-pyrazolo(3,4-d)pyrimidin-4-amine
(PP2) were purchased from Calbiochem (La Jolla, CA, USA). Annexin V
FITC-conjugated and 3-methyladenine (3-MA) were obtained from Sigma
Co., and Zymed Lab., respectively.
Cell culture, γ irradiation, and
pharmacological inhibition
HCT-116 cells (ATCC, number CCL-247, Manassas, VA,
USA) were maintained in Dulbelcco’s modified Eagle’s medium (DMEM)
containing 10% fetal bovine serum (FBS; Gibco) at 37°C and 5%
CO2. Irradiation was carried out 48 h after plating
(time 0) at 25°C using a 137 Cs Irradiator (IBL 437) with a dose of
8.5 Gy at a dose rate of 2.54 Gy/min. All analyses were carried out
12, 24, 36, 48 and 72 h after irradiation.
For pharmacological interference assays, cells were
incubated for 60 min with the kinase inhibitors: 10 μM LY294002
(PI3K) and 170 nM PP2 (Src). Cells were also treated for 60 min
with 10 mM 3-MA, a well-known autophagy inhibitor. Cells were
rinsed with phosphate buffered saline (PBS), irradiated as
described above, and processed for later analysis.
Annexin V/PI doucble staining
Cells were trypsinized, washed in PBS (pH 7.4), and
resuspended in 1X Annexin binding buffer (10 mM HEPES/NaOH, pH 7.4;
140 mM NaCl; 2.5 mM CaCl2). A cell suspension with
105 cells (95 μl) was then incubated with 5 μl of
Annexin V-FITC for 60 min at 4°C. Prior to flow cytometry analysis,
5 μl of a 20 μg/ml propidium iodide stock solution was added to the
cells, and then analyzed with a FACScalibur flow cytometer and
analyzed in CellQuest software (BD Bioscience, San Jose, CA,
USA).
Cell cycle analysis
Cells were harvested by trypsinization and washed
once with ice-cold PBS. The cells were then stained in the dark
with 75 μM propidium iodide and 20 μg/ml RNase A in PBS for at
least 30 min in the presence of NP-40. At least 10,000 events for
cell cycle were assessed in these experiments using a FACScalibur
flow cytometer and CellQuest software.
Cell proliferation assay
HCT-116 cells (5×106/well) were cultured
in 96-well plates and after 48 h of treatment, cell proliferation
was assayed in triplicate samples. Cells were fixed in ethanol and
stained in a solution of 0.05% violet crystal in 20% ethanol. Then,
washed, treated in methanol and the optical densities were measured
at 595 nm using a Spectra Max 190 spectrophotometer (Molecular
Devices, Sunnyvale, CA, USA).
Supravital cell staining with acridine
orange
Cells were cultured on coverslips for 48 h. After
irradiation, acridine orange (Sigma) was added at a final
concentration of 1 μg/ml. Where indicated, Bafilomycin A1 (Sigma),
a specific inhibitor of the vacuolar H+-ATPase, was
added to the cells at a final concentration of 0.2 μM before the
addition of acridine orange. The coverslips were washed and mounted
using n-propyl gallate. The fluorescence was detected using an
Axiovert S 100 microscope equipped with a KS300 image analyzer
(Carl Zeiss Inc., Germany).
Immunofluorescence analysis
Cell monolayers were grown on coverslips and
subjected to different treatments. Cells were washed, fixed in 4%
freshly prepared formaldehyde, and incubated in NH4Cl.
The cells were then permeabilized with 0.5% Triton X-100 and
blocked with 3% BSA in PBS. Subsequently, they were incubated with
the primary antibody against LC3B (10 μg/ml) followed by incubation
with an Alexa fluor 488-conjugated secondary antibody. The
coverslips were washed and then mounted using n-propyl gallate. The
cell staining was detected using the Axiovert S 100 microscope.
Transmission electron microscopy
(TEM)
Cells were grown on Transwell polycarbonate filters
(0.4-μm-pore size; Costar, Cambridge, MA, USA). After IR, cells
were fixed in 2.5% glutaraldehyde, 1% freshly prepared
formaldehyde, 0.8% sucrose, and 2 mM CaCl2 in 0.1 M
cacodylate buffer (pH 7.4). Post-fixation was carried out with 1%
osmium tetroxide in cacodylate buffer containing 0.8% potassium
ferrocyanide and 5 mM CaCl2. Subsequently, cells were
dehydrated in a graded series of acetone and embedded in epoxy
resin. Ultrathin sections (60 nm) were stained with uranyl acetate
and lead citrate, and examined using a Zeiss CEM-900 transmission
electron microscope.
In order to monitor autophagosome formation, cells
were grown as described above and incubated for 2 h at 37°C with 10
nm BSA-Au. Experiments were also carried out in the presence or
absence of protein kinase inhibitors to monitor the involvement of
cell signaling pathways in organelle formation, and in the presence
or absence of 3-MA. After the treatments, cells were washed,
irradiated, and incubated for 12, 36 or 48 h at 37°C. At the
indicated time periods, cells were processed for routine electron
microscopy as described above. At least 100 cells from each
treatment were randomly analyzed.
Western blot analysis
Total lysates were obtained by homo-genizing cell
samples in RIPA buffer (1% NP-40; 0.5% deoxycholate; 0.2% SDS; 150
mM PBS; and 50 mM Tris-HCl, pH 7.4) containing 20 mM sodium
fluoride, 1 mM sodium orthovanadate, and a cocktail of protease
inhibitors (1:100 dilution). Equal amounts of protein samples were
separated by SDS-PAGE and transferred to nitrocellulose sheets. The
blots were blocked in TBS-T (20 mM Tris-HCl, pH 7.6; 137 mM NaCl;
and 0.1% Tween) containing 1% BSA, incubated overnight with the
indicated antibodies, and then visualized using an enhanced
chemiluminiscence (ECL) detection kit (Amersham Biosciences,
Buckingham, UK). The protein levels of Akt and Src with their
respective phosphorylated forms were quantified by densitometry
using LabWorks 4.6 software (Bio-Rad). In each case, the specific
activity score was calculated using the following equation:
Arbitrary score = amount of phosphorylated protein/amount of total
protein. The protein levels of LC3B were calculated using α-tubulin
as a loading control. The score for non-irradiated cells was
normalized as one in each case.
Statistical analysis
Statistical analysis was performed using two-way
ANOVA with a post hoc Bonferroni or Dunnet test in three
independent experiments. The significantly different values are
indicated in the figure legends as P<0.05 or P<0.01. These
values are presented as means ± standard deviations (SD) from three
independent experiments. Significantly different values relative to
the control group, and the values relative to irradiated cells are
reported.
Results
Effect of IR treatment on the cell
viability and apoptosis of HCT-116 cells
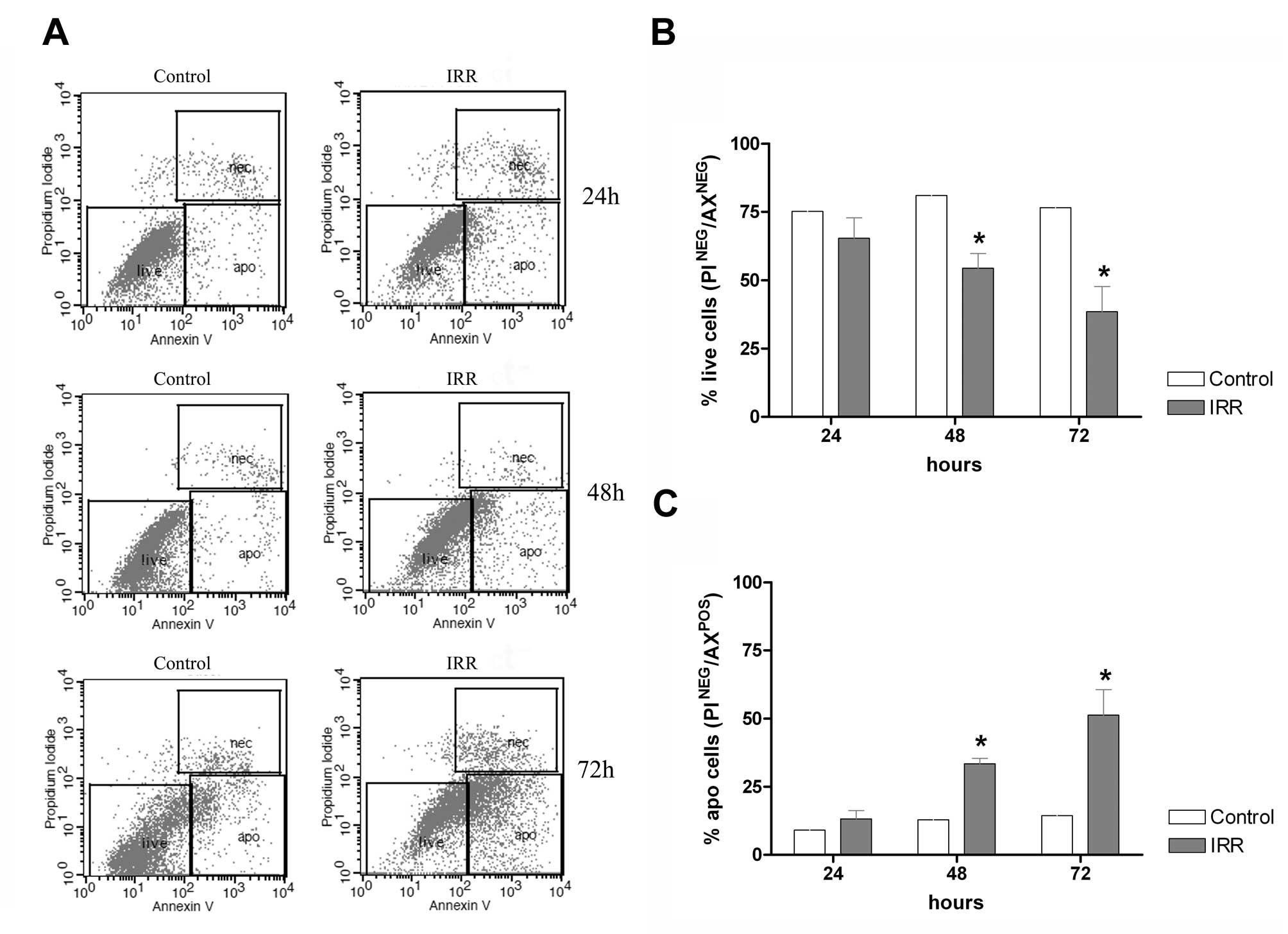
Cells were grown and irradiated with 8.5 Gy. After
24, 48 and 72 h, the cells were subjected to Annexin V/PI staining
and analyzed by flow cytometry (Fig.
1A). We observed that treatment of the HCT-116 cells with 8.5
Gy dose after 24 h did not affect the cell survival, but after 48
and 72 h the cell survival was affected in 15 and 40%,
respectively, as compared to untreated cells (Fig. 1B). We further examined if IR was
able to induce cell death by apoptosis using flow cytometry
analysis of cells stained with Annexin V/PI. We observed that IR
after 24 h did not alter the levels of apoptotic cells as compared
with untreated cells, but after 48 and 72 h a rate of 15 and 60% of
apoptotic cells was observed, respectively (Fig. 1C). Based on these results, we used
the time of 48 h after IR as treatment condition for all subsequent
studies.
IR promotes acidic vacuole formation that
corresponds to autophagosomes
We observed that a major population of cell survived
to IR after 48 h, while a minor amount was induced to die. Thus, we
decided to analyze other events in programmed cell death, such as
the additional type II of programmed death or autophagy, which is
characterized by the presence of acidic vacuole formation (20). These vacuoles are characterized by
labeling with acridine orange, widely known to accumulate in acidic
compartments. The majority of untreated cells had only a few
labeled vesicles (Fig. 2A); in
contrast irradiated cells at 48 h had large fluorescent vacuoles in
the cytoplasm (Fig. 2B). We
confirm the acidic nature of the vacuoles by incubating the cells
with Bafilomicyn A1, a well-known inhibitor of the vacuolar
H+-ATPase responsible for preventing the proper
acidification of lysosomal compartments (24). In the presence of the inhibitor, no
acridine orange-labeled vacuoles were observed in irradiated cells
(Fig. 2C).
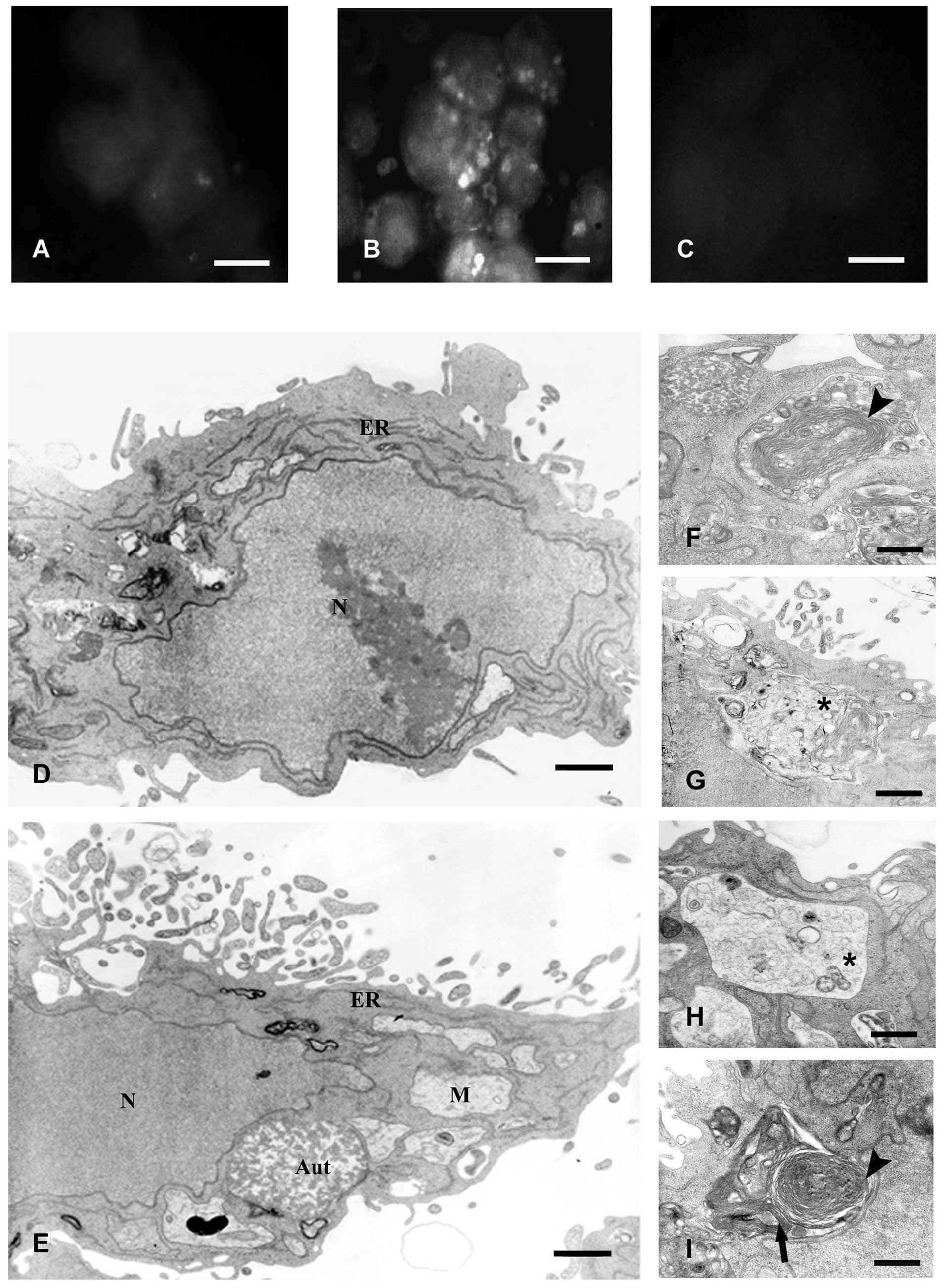 | Figure 2IR promotes acidic vacuole formation
that corresponds to autophagosomes. Vital staining with acridine
orange of non-irradiated cells (A), 48 h after irradiation with 8.5
Gy (B), and incubated with 200 nM Bafilomycin A1 for 30 min before
the addition of acridine orange (C). Bar, 10 μm. (D and E)
Representative electron microscopy micrographs of non-irradiated
and irradiated cells with 8.5 Gy after 48 h, respectively. The
nuclei of irradiated cells exhibited a similar ultrastructure as
that observed in control cells and did not show morphological
characteristics of apoptosis, such as chromatin margination or
nuclear pyknosis. At a higher magnification, it was observed that
most of the autophagic vacuoles arise from newly formed lamellar
structures (arrowhead) (F) and double-membrane autophagosomes that
sequester organelles (arrow) (I) to single-membrane organelles that
contain digested material (asterisks) (G and H). (D and E) Bars,
0.2 μm. (F-I) Bars, 0.3 μm. N, nuclei; ER, endoplasmic reticulum;
M, mitochondria; and Aut, autophagolysosomes. |
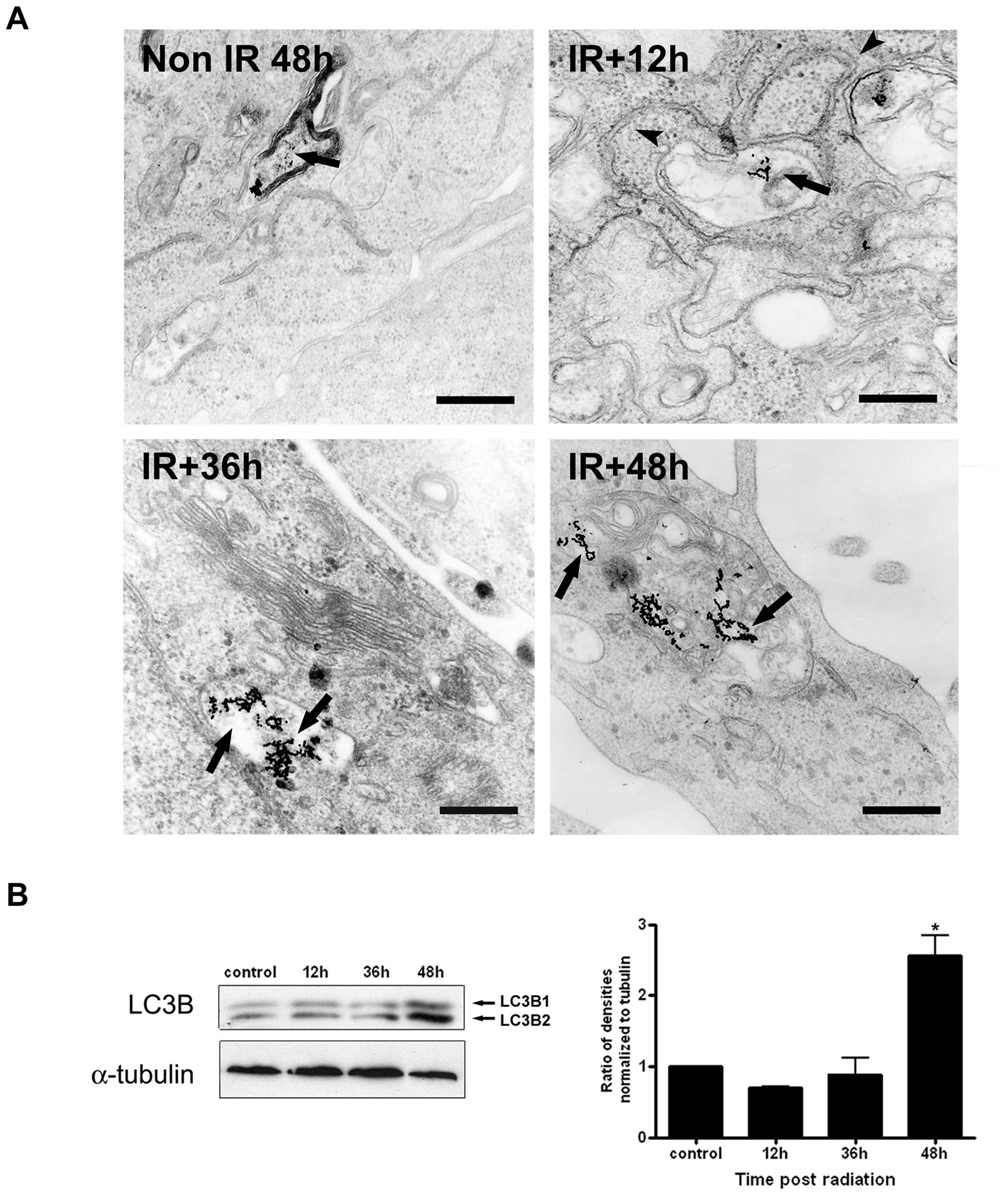
Further analysis by TEM showed that the IR-induced
acidic vacuoles corresponded to large vacuoles containing electron
dense material but the nuclei and organelles had typical morphology
similar to control cells and did not show morphological
characteristics of apoptosis, such as chromatin margination or
nuclear pyknosis (Fig. 2D and E).
Forty-eight hours after IR, accumulation of these organelles was
observed in ~80% of the analyzed cells, which displayed a core
composed of granular, vesicular, or lamellar contents. Furthermore,
the organelles were frequently surrounded by smooth and/or rough
membrane cisternae or in fusion with smooth vesicles of unknown
origin (Fig. 2F-I). Early studies
have characterized the integration of the endocytic and autophagic
pathways using BSA-Au as a marker of fluid-phase endocytosis
(25). Using this approach, we
observed the labeling with the marker of lysosome-like organelles
in unirradiated cells after 48 h. In irradiated cells the marker
was observed in double membranes in apparent sequestration of
organelles (12 h), initial autophagosomes contained non-degraded
material, indicating that the absence of lysosomal hydrolases (36
h), and in single membrane-bound autophagosomes containing
multivesicular material (48 h) (Fig.
3A).
We confirmed these results by Western blot analysis
using anti-LC3B, an antibody that recognizes two forms of LC3B
(LC3B-I and LC3-II). In Fig. 3B,
we show a significant increase in LC3B-II levels 48 h after IR
treatment. The increased LC3B-II level is associated with increased
autophagosome membranes and correlates with the extent of
autophagosomes formation (26).
Irradiation induces PI3K/Akt and Src
activation concomitantly with autophagosome formation at early
stages after irradiation
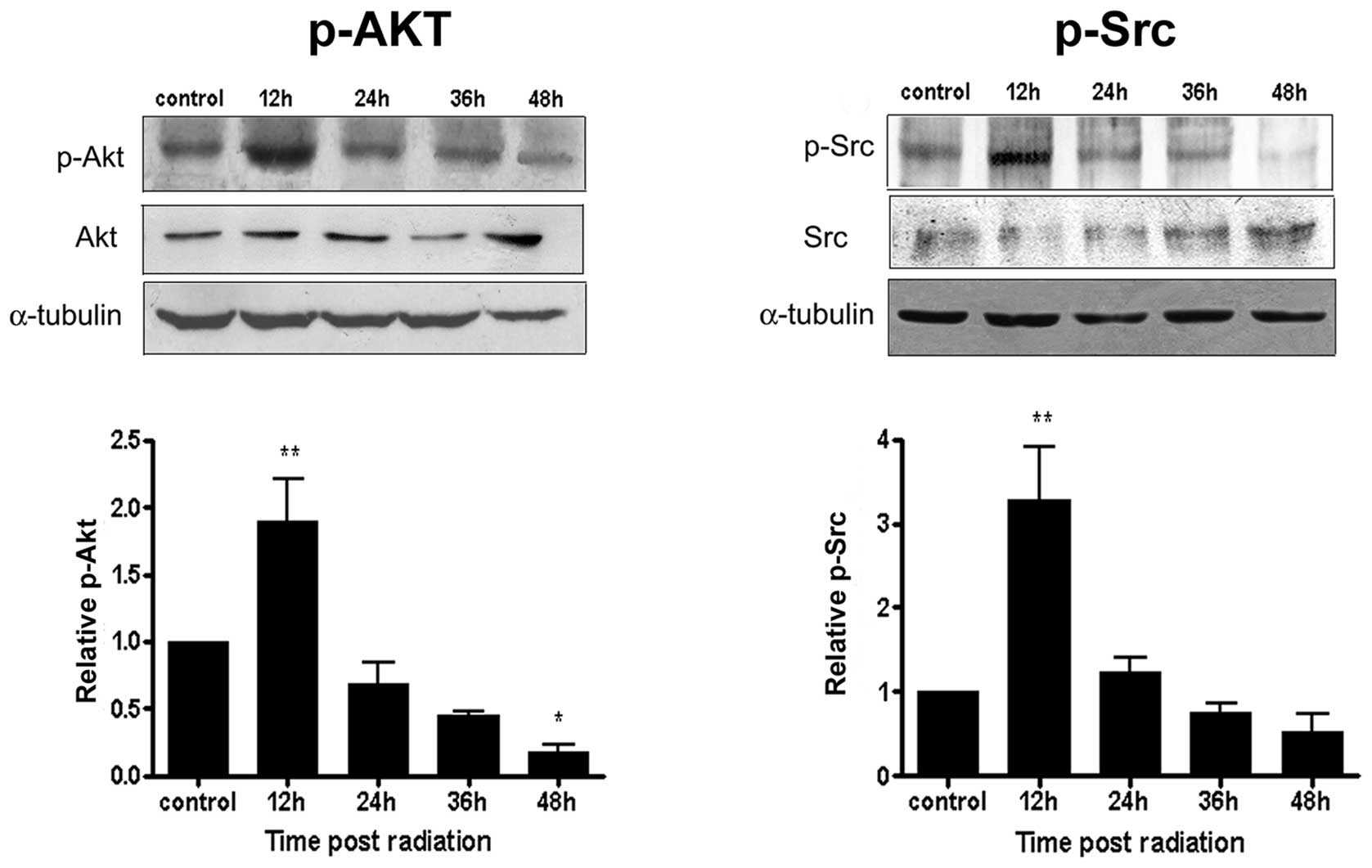
To identify cell signaling pathways involved in the
formation of autophagosomes, we first analyzed by Western blotting
the phosphorylation status of Akt, a downstream target of PI3K, and
of Src after irradiation. We observed a significant increase in the
phosphorylation levels of these kinases 12 h after IR. At
subsequent post-IR times, the phosphorylation of the kinases
decreased when compared to control cells (Fig. 4). These data implicate the
involvement of PI3K/Akt and Src activation at the initial stages of
autophagosome formation, where organelle sequestration and the
initial formation of autophagosomes occur.
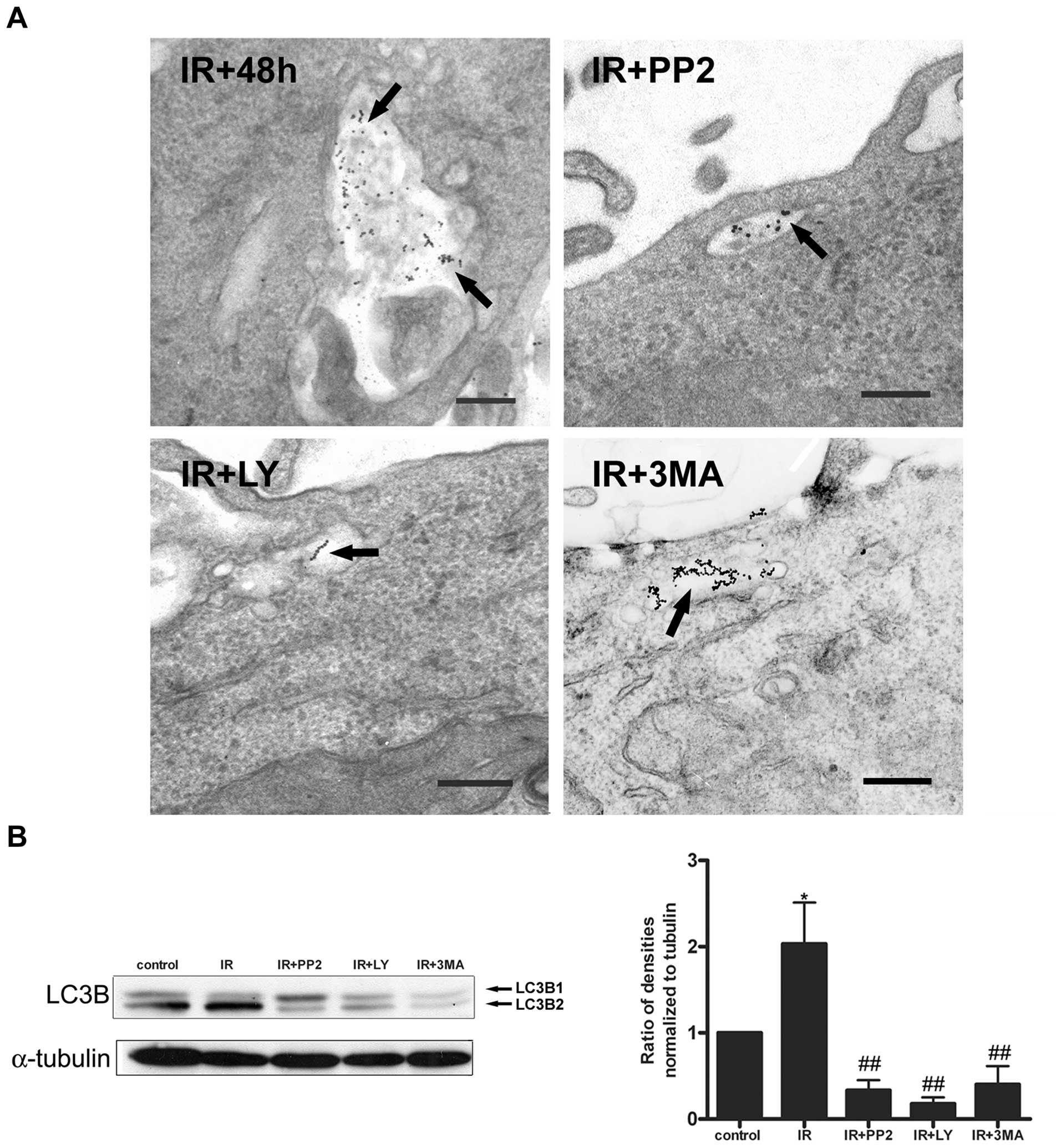
We monitored the autophagosome formation in response
to PI3K and Src inhibition using the specific inhibitors, LY294002
and PP2, respectively, and 3-MA as a control for autophagy
inhibition. TEM analysis showed that ~80% of the irradiated cells
displayed a strong BSA-Au labeling into the autophagosomes, and the
treatment with the inhibitors caused a blockade of autophagosome
formation. Irradiated cells treated with the inhibitors only had
organelles resembling early and late endosomes labeled with the
marker (Fig. 5A). We further
confirmed that the autophagosome formation depends on the early
activation of PI3K/Akt and Src by Western blotting using LC3B.
Fig. 5B shows that pretreatment
with the inhibitors significantly inhibited the expression of LC3B
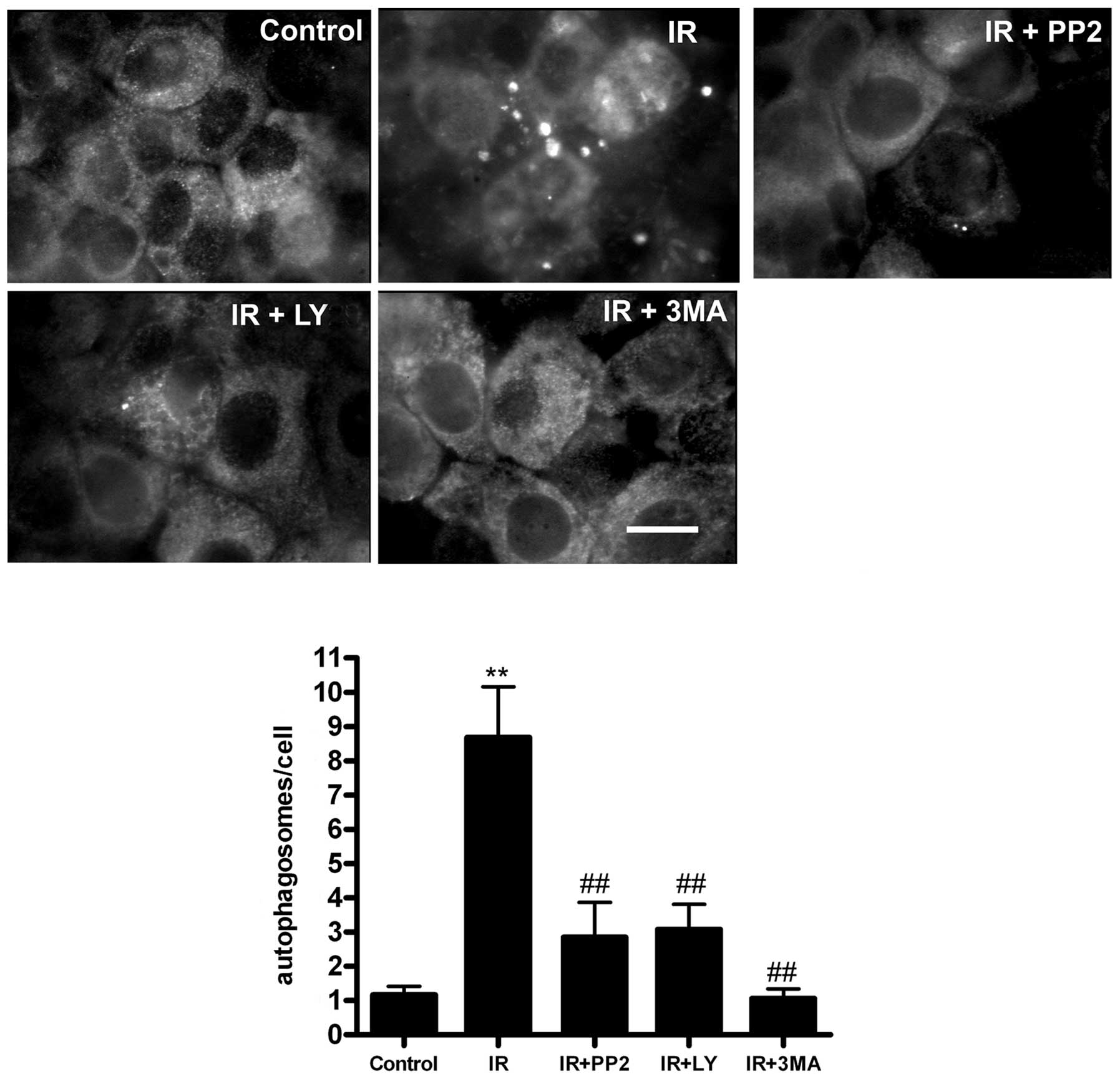
in response to IR. Next, quantitative immunofluorescence using the
LC3B marker demonstrated that pretreatment with the inhibitors
significantly inhibited autophagosome formation (Fig. 6). Together, these data indicate
that the activation of PI3K/Akt and Src during the early stages of
irradiation (12 h) is necessary to induce a cell signaling cascade
that leads to autophagosome formation in HCT-116 cells.
Blockade of IR-induced autophagosome
formation by PI3K/Akt and Src inhibition impairs proliferation but
does not enhance cell death
Various studies suggest that the autophagic response
of cancer cells to radiotherapy is a major pathway that may lead to
cell death or survival in contrast to apoptosis, which only leads
to cell death (27). Autophagy is
a temporary survival mechanism under stress conditions, but its
inhibition may either promote or inhibit cell death depending on
the conditions and agents used (7,28).
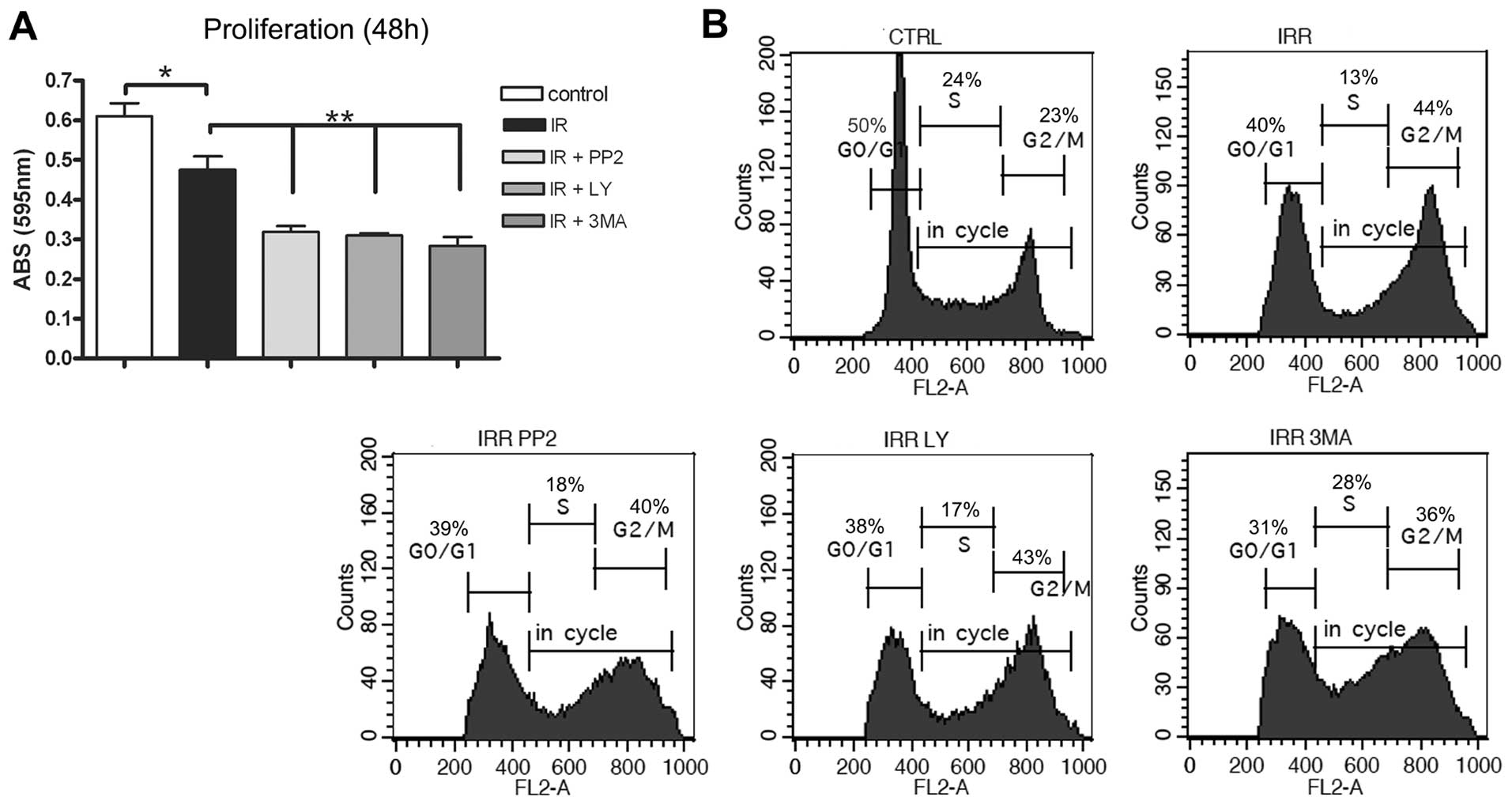
Thus, we decided to analyze cell proliferation measuring crystal
violet incorporation, which correlates with the total cell number,
after the blockade of autophagosome formation with PI3K/Akt, Src,
and 3-MA. Fig. 7A shows that IR
caused a decrease in proliferation after 48 h when compared to
non-irradiated cells, and that the pretreatment with the inhibitors
significantly decreased proliferation relative to cells treated
with IR alone. This suggests that the blockade of IR-induced
autophagosomes causes a synergistic inhibitory effect on the total
number of irradiated cells at the time of 48 h.
In order to determine whether the inhibition of
HCT-116 cell growth caused by the blockade of IR-induced
autophago-some formation was due to alteration of the cell cycle,
we investigated the cycle profiles analyzing the DNA content by
flow cytometry. As shown in Fig.
7B, the phases of cell cycle distribution indicated that IR
promoted arrest in G2/M phase at 48 h, as compared to
untreated cells. Pretreatment with kinase inhibitors and 3-MA
increased the amount of cells in S phase as compared with
irradiated cells. The percentage of pretreated cells in the
Sub-G0 phase was very similar to those irradiated only
(data not shown). At 72 h, the population of cells pretreated with
the inhibitors in G2/M phase was similar to those
irradiated only (data not shown). Taken together, these results
suggest that the growth-inhibitory effect as consequence of the
blockade of autophagosomes formation could be partly due to its
ability to retard S phase of the cell cycle rather than leading
cells to apoptosis-mediated death. To investigate in more detail
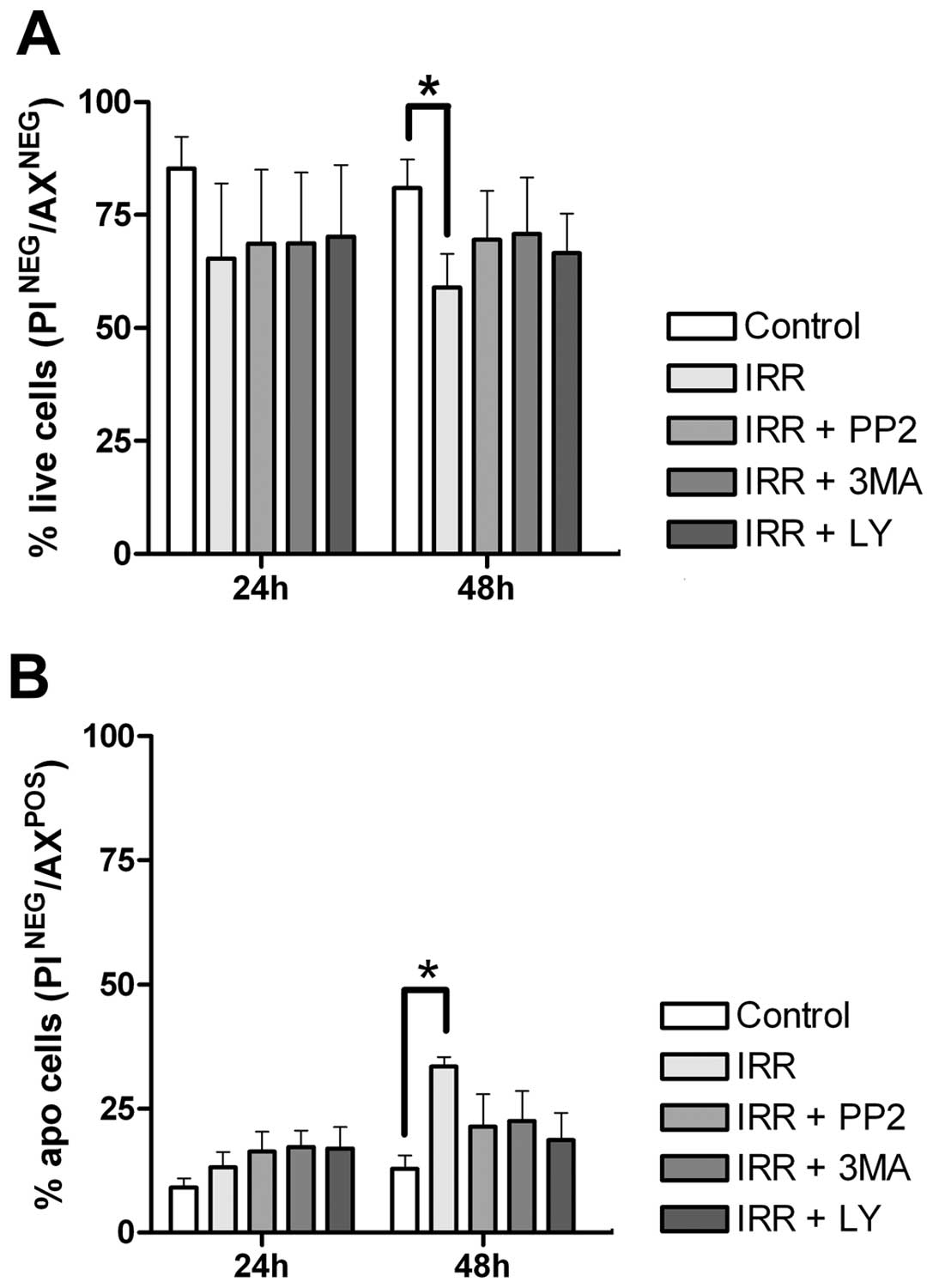
the role of autophagy on the inhibitory effect of proliferation, we
examined whether the autophagy inhibitors could attenuate cell
survival. We observed that pretreatment with the inhibitors did not
alter cell viability (Fig. 8A) or
the cell death by apoptosis (Fig.
8B) in relation to those resulting from 48 h after IR.
Together, these results indicate that autophagy inhibition
decreases cell proliferation but did not enhance the IR-mediated
cell death. Additionally, an accentuated effect on these two
parameters was observed after 72 h, however none of the inhibitors
altered the effects on cell death after IR (data not shown).
Discussion
Colorectal cancer is the second most prevalent
cancer and the third leading cause of cancer deaths worldwide, and
radiotherapy remains the major adjuvant therapy to improve survival
rates and reduce the risk of local recurrence (29). However, this therapy may contribute
to cellular responses related to neoplastic progression. One of
these responses involve autophagosome formation as a cell defense
mechanism (30), but the precise
molecular mechanisms underlying formation of these organelles and
its relation with tumor cell survival in response to radiation
remain to be defined.
To further address this issue, we initially analyzed
whether IR impairs cell viability or induces apoptosis in HCT-116
cells. We observed that IR induced approximately15% of cell death
by apoptosis after 48 h of treatment, but 75% of cells survived
this treatment. These findings corroborate previous studies showing
that irradiated cells develop an adaptive response as a
self-defense mechanism that protects cells rather than causing cell
death (31). It was shown that
depletion or inhibition of p53 could induce autophagy suggesting a
key role for the p53 tumor suppressor in the regulation of
autophagy (32). However, recently
using both WT and p53-null HCT-116 cells that were treated with the
cell wall skeleton of Mycobacterium bovis Bacillus
Calmette-Guerin (BCG/CWS) plus IR, it was shown that p53 was not
involved in autophagy induction (33). Therefore, these findings also
indicate that autophagy and apoptosis use different mechanisms of
control.
Various studies have suggested that anticancer
therapies, such as hormonal agents, chemotherapy and IR, frequently
induce autophagy, in most cases as a pro-survival response
potentially contributing to treatment resistance (34,35).
Thus, we evaluated autophagosome formation, which is the most
distinctive feature of autophagy. Our results showed that IR
induced the accumulation of large vacuoles derived from the
autophagic pathway, as characterized by the following parameters:
a) acidic nature, b) enclosure by a double membrane, c) presence of
degraded material derived from organelles such as the endoplasmic
reticulum or mitochondria in their lumen, and d) labeling with LC3B
(Figs. 2 and 3). As previously reported, these criteria
are well-known characteristics of autophagosome formation (36).
Previous morphological studies have shown a link
between the endocytic and autophagic pathways (37); therefore we used the convergence of
these pathways to monitor the formation of IR-induced
autophagosomes using BSA-Au, a classic fluid-phase endocytosis
marker, and TEM analysis (25).
Using this approach we showed that IR induced the formation of
autophagosomes after 48 h (Fig.
3A), which was confirmed by measuring LC3 expression levels by
Western blotting (Fig. 3B). There
are no previous reports demonstrating the biogenesis of IR-induced
autophagosomes in invasive colon cancer cells. The present study
reinforces a previous report suggesting that the integration of
autophagic vacuoles with vacuoles of the endocytic pathway is a
prerequisite of autophagosome maturation (1), and provide a valuable model to
investigate molecular mechanisms that control autophagosome
biogenesis.
Autophagy is a dynamic process regulated by several
cell signaling pathways (5,38),
however, the molecular control of IR-induced autophagy,
particularly in colon cancer cells, remains poorly understood. In
human breast tumor cells, a link between autophagic responses to IR
and mTOR signaling was reported (31. It was shown that clinically
relevant IR doses increased the phosphorylated forms of mTOR, Akt,
and S6 ribosomal proteins (39).
Therefore, inhibitors of the PI3K/Akt/mTOR pathway have emerged as
important and attractive therapeutic strategies for cancer therapy.
Our results demonstrated that PI3K/Akt and Src signaling were
involved in autophagy control (Figs.
4-6). We suggest that IR,
known to activate growth-factor receptors (40,41),
could increase the activity of Ras family oncoproteins causing
subsequent activation of downstream effectors such as PI3K/Akt and
Src, which in turn, may modulate autophagosome formation. Once
these IR-activated signaling pathways are correlated with cell
survival, we can also speculate that IR-induced autophagy in this
cell type is an attempt to avoid apoptosis in response to this
treatment. It is important to point out that LY294002 is a known
inhibitor of autophagy as well as of PI3K signaling (42) and suppresses the activity of the
downstream Akt. However, this inhibitor also induces rather than
inhibits autophagy in malignant glioma cells. One explication to
this controversy could be that the effects of LY294002 on autophagy
depend on the cell type or treatment conditions, such as
concentration used and exposure time, which can affect the mTOR
pathway (43).
We further analyzed cell proliferation and survival
rates to investigate if the blockade of autophagy could be
sufficient to enhance cell death induced by IR. Interestingly, the
concentration of inhibitors that induced autophagy blockade did not
increase apoptotic cell death but significantly inhibited the
proliferation of irradiated cells (Figs. 7 and 8). This latter observation could indicate
that the decreased proliferation induced by the blockade of
autophagy was due to a delay of the cell cycle in S phase. To
confirm this hypothesis we analyzed the cell cycle progression 72 h
after IR, and observed a restoration of the cell population in
G2/M in the group treated with kinase inhibitors as
compared with the irradiated group only. This suggests that the
presence of autophagosomes is important to progress the cell cycle
from S to G2/M where G2 checkpoint allows the
cell to repair the DNA damage after IR. Recently, it was shown that
IR-induced autophagy promotes post-IR cell survival and contributes
to cellular radioresistance in breast cancer cell lines (44); however experimental evidence to
explain this conclusion was not explored. Of note, the PI3K/Akt
pathway is a survival pathway that via its inhibition does not
always cause substantial apoptosis, therefore, additional treatment
with other agents is necessary to cause abnormal autophagosome
accumulation and leads to accelerated cell death (45).
In conclusion, we showed that IR-induced autophagy
is a initial mechanism of tumor cell survival, and that autophagy
inhibition induces a delay of the cell cycle in S phase rather than
leading cells to apoptosis-mediated death. Therefore, cell death in
response to autophagy inhibition may vary among each type of cancer
due to the different biological characteristics and therapeutic
treatments, as previously reported (46). In the future, the understanding of
the molecular mechanisms underlying IR-induced autophagy as well as
the combination with other drugs that interfere with other defense
programs may increase the efficacy in radiation oncology.
Acknowledgements
This study was sponsored by Conselho Nacional de
Desenvol-vimento Cientifico e Tecnologico (CNPq), Ministério da
Saúde, Brasil, and Fundação Carlos Chagas Filho de Amparo à
Pesquisa do Estado de Rio de Janeiro (FAPERJ). We are grateful to
Barbara DuRocher for her valuable help in the interpretation of the
cell cycle experiments, Livia Goto-Silva for suggestions made at
the beginning of the present study, the Programa de Cooperação
INCA/FIOCRUZ for the use of their facility and the employers of
Departamento de Hemoterapia-INCA for the use of gamma
irradiator.
References
|
1
|
Razi M, Chan EY and Tooze SA: Early
endosomes and endosomal coatomer are required for autophagy. J Cell
Biol. 185:305–321. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Meijer AJ and Codogno P: Regulation and
role of autophagy in mammalian cells. Int J Biochem Cell Biol.
36:2445–2462. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ogier-Denis E and Codogno P: Autophagy: a
barrier or an adaptive response to cancer. Biochim Biophys Acta.
1603:113–128. 2003.PubMed/NCBI
|
|
5
|
Rubinsztein DC, Gestwicki JE, Murphy LO
and Klionsky DJ: Potential therapeutic applications of autophagy.
Nat Rev Drug Discov. 6:304–312. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Apel A, Herr I, Schwarz H, Rodemann HP and
Mayer A: Blocked autophagy sensitizes resistant carcinoma cells to
radiation therapy. Cancer Res. 68:1485–1494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Amaravadi RK, Yu D, Lum JJ, et al:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bursch W, Ellinger A, Kienzl H, et al:
Active cell death induced by the anti-estrogens tamoxifen and ICI
164 384 in human mammary carcinoma cells (MCF-7) in culture: the
role of autophagy. Carcinogenesis. 17:1595–1607. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yue Z, Jin S, Yang C, Levine AJ and Heintz
N: Beclin 1, an autophagy gene essential for early embryonic
development, is a haploinsufficient tumor suppressor. Proc Natl
Acad Sci USA. 100:15077–15082. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qu X, Yu J, Bhagat G, et al: Promotion of
tumorigenesis by heterozygous disruption of the Beclin 1 autophagy
gene. J Clin Invest. 112:1809–1820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aita VM, Liang XH, Murty VV, et al:
Cloning and genomic organization of Beclin 1, a candidate tumor
suppressor gene on chromosome 17q21. Genomics. 59:59–65. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Crighton D, Wilkinson S, O’Prey J, et al:
DRAM, a p53-induced modulator of autophagy, is critical for
apoptosis. Cell. 126:121–134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Corcelle E, Nebout M, Bekri S, et al:
Disruption of autophagy at the maturation step by the carcinogen
lindane is associated with the sustained mitogen-activated protein
kinase/extracellular signal-regulated kinase activity. Cancer Res.
66:6861–6870. 2006. View Article : Google Scholar
|
|
15
|
Klionsky DJ and Erm SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–172.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen S, Rehman SK, Zhang W, Wen A, Yao L
and Zhang J: Autophagy is a therapeutic target in anticancer drug
resistance. Biochim Biophys Acta. 1806:220–229. 2010.PubMed/NCBI
|
|
17
|
Furuta S, Hidaka E, Ogata A, Yokota S and
Kamata T: RAS is involved in the negative control of autophagy
through the class IP3-kinase. Oncogene. 23:3898–3904. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Arico S, Petiot A, Bauvy C, et al: The
tumor suppressor PTEN positively regulates macroautophagy by
inhibiting the phosphatidylinositol 3-kinase/protein kinase B
pathway. J Biol Chem. 276:35243–35246. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ogier-Denis E, Pattingre S, Benna J and
Codogno P: Erk1/2-dependent phosphorylation of Galpha-interacting
protein stimulates its GTPase accelerating activity and autophagy
in human colon cancer cells. J Biol Chem. 275:39090–39095. 2000.
View Article : Google Scholar
|
|
20
|
Paglin S, Hollister T, Delohery T, et al:
A novel response of cancer cells to radiation involves autophagy
and formation of acidic vesicles. Cancer Res. 61:439–444.
2001.PubMed/NCBI
|
|
21
|
Brown M and Wounters BG: Apoptosis, p53,
and tumor cell sensitivity to anticancer agents. Cancer Res.
59:1391–1399. 1999.PubMed/NCBI
|
|
22
|
Finkel E: Does cancer therapy trigger cell
suicide? Science. 286:2256–2258. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Speake WJ, Dean RA, Kumar A, Morris TM,
Scholefield SA and Watson JH: Radiation induced MMP expression from
rectal cancer is short lived but contributes to in vitro invasion.
Eur J Surg Oncol. 31:869–871. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yamamoto A, Tagawa Y, Yoshimori T,
Moriyama Y, Masaki R and Tashiro Y: Bafilomycin A1 prevents
maturation of autophagic vacuoles by inhibiting fusion between
autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E
cells. Cell Struct Funct. 23:33–42. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fengsrud M, Roos N, Berg T, Liou W, Slot
JW and Seglen PO: Ultrastructural and immunocytochemical
characterization of autophagic vacuoles in isolated hepatocytes:
effects of vinblastine and asparagine on vacuole distributions. Exp
Cell Res. 221:504–519. 1995. View Article : Google Scholar
|
|
26
|
Kabeya Y, Mizushima N, Ueno T, et al: LC3,
a mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zois CE and Koukourakis MI:
Radiation-induced autophagy in normal and cancer cells: towards
novel cytoprotection and radio-sensitization policies? Autophagy.
5:442–450. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Levine B and Yuan J: Autophagy in cell
death: an innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li J, Hou N, Faried A, Tsutsumi S and
Kuwano H: Inhibition of autophagy augments 5-fluorouracil
chemotherapy in human colon cancer in vitro and in vivo model. Eur
J Cancer. 46:1900–1909. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kondo Y and Kondo S: Autophagy and cancer
therapy. Autophagy. 2:85–90. 2006. View Article : Google Scholar
|
|
31
|
Paglin S, Lee NY, Nakar C, et al:
Rapamycin-sensitive pathway regulates mitochondrial membrane
potential, autophagy, and survival in irradiated MCF-7 cells.
Cancer Res. 65:11061–110702. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tasdemir E, Maiuri MC, Galluzzi L, et al:
Regulation of autophagy by cytoplasmic p53. Nat Cell Biol.
10:676–687. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yuk JM, Shin DM, Song KS, et al: Lim K,
Kim KH, Lee SH, Kim JM, Lee JS, Paik TH, Kim JS and Jo EK: Bacillus
calmette-guerin cell wall cytoskeleton enhances colon cancer
radiosensitivity through autophagy. Autophagy. 6:46–60. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Al-Ejeh F, Kumar R, Wiegmans A, Lakhani
SR, Brown MP and Khanna KK: Harnessing the complexity of DNA damage
response pathways to improve cancer treatment outcomes. Oncogene.
29:6085–6098. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
John S, Nayvelt I, Hsu HC, Yang P, Liu W
and Das GM: Regulation of estrogenic effects by beclin 1 in breast
cancer cells. Cancer Res. 68:7855–7863. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Eskelinen EL: Maturation of autophagic
vacuoles in mammalian cells. Autophagy. 1:1–10. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hamasaki M and Yoshimori T: Where do they
come from? Insights into autophagosome formation. FEBS Lett.
584:1296–1301. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Corcelle EA, Puustinen P and Jäättlelä M:
Apoptosis and autophagy: targeting autophagy signaling in cancer
cells - ‘trick or treats’? FEBS J. 276:6084–6096. 2009.
|
|
39
|
Albert JM, Cao KW and Lu B: Targeting the
Akt/mammalian target of rapamycin pathway for radiosensitization of
breast cancer. Mol Cancer Ther. 5:1183–1189. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dent P, Reardon DB, Park JS, et al:
Radiation-induced release of transforming growth factor alpha
activates the epidermal growth factor receptor and
mitogen-activated protein kinase pathway in carcinoma cells,
leading to increased proliferation and protection from
radiation-induced cell death. Mol Biol Cell. 10:2493–2506.
1999.
|
|
41
|
Goldkorn T, Balaban N, Shannon M and
Matsukuma K: EGF receptor phosphorylation is affected by ionizing
radiation. Biochim Biophys Acta. 1358:289–299. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Aki T, Yamaguchi K, Fujimiya T and
Mizukami Y: Phosphoinositide 3-kinase accelerates autophagic cell
death during glucose deprivation in the rat cardiomyocyte-derived
cell line H9c2. Oncogene. 22:8529–8535. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Takeuchi H, Kondo Y, Fujiwara K, et al:
Synergistic augmentation of rapamycin-induced autophagy in
malignant glioma cells by phosphatidylinositol 3-kinase/protein
kinase B inhibitors. Cancer Res. 65:3336–3346. 2005.PubMed/NCBI
|
|
44
|
Chaachouay H, Ohneseit P, Toulany M,
Kehlbach R, Multhoff G and Rodemann HP: Autophagy contributes to
resistance of tumor cells to ionizing radiation. Radiother Oncol.
99:287–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Degtyarev M, De Mazi è re A, Orr C, et al:
Akt inhibition promotes autophagy and sensitizes PTEN-null tumors
to lysosomotropic agents. J Cell Biol. 183:101–116. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Puyal J, Ginet V, Grishchuk Y, Truttmann
AC and Clarke PG: Neuronal autophagy as a mediator of life and
death: contrasting roles in chronic neurodegenerative and acute
neural disorders. Neuroscientist. 2011. View Article : Google Scholar
|