Introduction
Birc5 is a member of the inhibitors of apoptosis
(IAPs) family. Unlike other IAPs, Birc5 is a bifunctional protein
that functions as a key regulator of mitosis and inhibitor of
programmed cell death. Its marked expression in cancer as opposed
to normal tissue (1) and the fact
that its upregulated expression correlates with tumor resistance to
chemotherapy (2,3), increased tumor grade, recurrence risk
and decreased cancer patient survival rate, have made Birc5 a
promising target for anticancer interventions (4–6).
Certain in vitro and in vivo studies have indicated
that the downregulation of Birc5 can sensitize human tumor cells to
conventional chemotherapeutic drugs, including Taxol, doxorubicin,
etoposide and cisplatin (7,8). We
previously reported that Birc5 ribozyme and siRNA resulted
in the inhibition of both the transcription and expression of Birc5
in tumor cells, triggering cell apoptosis (9,10).
However, Birc5 antagonists, at least those tested so far in
clinical and preclinical studies, have not shown ideal outcomes in
inducing cancer cell apoptosis. The details of the multiple
pathways emanating from the Birc5 networks are yet to be fully
elucidated.
It has been shown that the association of Birc5 with
the heat shock protein (Hsp) is required for its stability and
function. For example, Hsp90 binds to the mature form of Birc5 to
stabilize it in cells (11,12).
The targeted antibody-mediated disruption of the Birc5-Hsp90
complex in cancer cells could result in proteasomal degradation of
Birc5, mitochondrial dependent apoptosis and mitotic arrest
(13). Various studies have
investigated the possibility of targeting Birc5 using Hsp90
inhibitors. However, Cheung et al (14) showed that targeting Hsp90 with
small molecule inhibitors induces the overexpression of Birc5 in
certain cancer cell lines and subsequently enhances the ability of
cell survival in drug-treated situations. This suggests that the
dual inhibition of Hsp90 and Birc5 may be warranted for cancer
therapy.
The glucose-regulated protein, Hspa5, belongs to the
Hsp70 protein family, which is involved in a number of cellular
processes that maintain the homeostasis of cells. It is strongly
induced in tumors and plays critical roles in the stress of
oncogenesis (15–18). We previously showed that the
overexpression of Hsp70-2 protected cancer cells against apoptosis
induced by hypoxia and that the upregulated expression of Hspa5
protected NIT-1 cells from death induced by streptozotocin,
cytokines or cytotoxic T lymphocytes (19–21).
Hspa5 may be an important survival factor for the tumor to escape
immune surveillance and allow tumor cells proliferate.
We therefore hypothesized that the silencing the
Birc5 and Hspa5 simultaneously could induce a
stronger apoptotic effect than the silencing of Birc5 or
Hspa5 alone. Therefore, the purpose of this study was to
determine whether the co-silencing of Birc5 and Hspa5
is more effective in inducing apoptosis in hepatoma cells than
single gene interference.
Materials and methods
Plasmid vectors
Plasmid-based siRNA against Birc5 and/or
Hspa5 was designed using the Ambion Target finder
(http://www.ambion.com/techlib/misc/sirna_finder.html).
A BLAST search was used for selecting two target non-cross matching
sequences among the siRNA candidates generated. The 21-nucleotide
target sequences of exon 1 (5′-GGACCACCGCATCTCTACATT-3′) and exon 3
(5′-AGCATTCGTCCGGTTGCGCTT-3′) of the human Birc5 mRNA
(Genbank accession no. NM_001168.1) were selected and named as
pgsiRNA-Bir1 and pgsiRNABir2, respectively. Both sequences were
designed against all Birc5 mRNA splicing variants. The
19-nucleotide target sequence (5′-AGTGTTGGAAGATTCTGAT-3′) of the
human Hspa5 mRNA (Genbank accession no. NM_005347) was
selected and named pgsiRNA-Hsp. The co-silencing plasmid,
pgsiRNA-Bir+Hsp, was designed specifically against the exon 3
sequence of Birc5 and the above-mentioned Hspa5
sequence. The control siRNA plasmid, pgsiRNA-C, containing a
sequence (5′-CGTACGCGGAATACTTCGATT-3′) with no significant homology
to any known human mRNA, was used throughout the study. All
pGenesil plasmids (Genesil, Wuhan, China) contain genes for
enhanced green fluorescent protein (EGFP) as a fluorescence
marker.
Cell culture and transfection
The HepG2 human hepatocellular liver carcinoma cells
were cultured in DMEM complete growth medium (Invitrogen, Carlsbad,
CA, USA) supplemented with 10% fetal bovine serum (FBS;
Invitrogen). Cells were maintained in a humidified 37°C incubator
with 5% CO2. Plasmids were transfected into HepG2 cells
using Lipofectamine 2000 (Invitrogen) according to the
manufacturer’s instructions.
Twenty-four hours before transfection, cells
(2×105/well) were plated into 6-well plates. Twelve
hours before transfection, the medium was replaced by
antibiotics-free medium. When cells reached 80–90% confluence, the
medium was replaced with OPTI-MEM® I Reduced Serum
Medium (Invitrogen). Plasmids (4 μg) complexed with 10
μl of Lipofectamine™ Plus were added to the wells and
incubated for 5 h before being replaced by fresh medium containing
10% (v/v) FBS. All the following experiments were performed at 48 h
post-transfection.
Immunofluorescence assay
HepG2 cells were fixed in 4% paraformaldehyde for 1
h to detect the expression of Birc5 or Hspa5 on the membrane of
HepG2 cells or were permeabilized by 0.1% saponins in PBS for 30
min after fixation to detect the expression of Birc5 or Hspa5 in
the cytoplasm of HepG2 cells. Fixed cells were incubated with
rabbit anti-human Birc5 (Santa Cruz Biotechnology, Santa Cruz, CA,
USA) at 1:200 dilution, or goat anti-human Hspa5 (Santa Cruz
Biotechnology) at 1:200 dilution. The antigen-antibody complexes
were detected using FITC-conjugated goat anti-rabbit IgG (1:200
dilution; GE Healthcare UK Ltd., Buckinghamshire, UK), or donkey
anti-goat IgG (1:200 dilution; GE Healthcare UK Ltd.) and the mean
fluorescence intensities were determined by flow cytometry
(FACSCalibur, BD Biosciences, San Jose, CA, USA) and analyzed with
CellQuest software (BD Biosciences).
Immunohistochemistry
Tumor and adjacent non-tumor tissues from the
primary liver cancer patients were surgically collected and
immediately frozen at −80°C until use. For immunohistochemical
analysis, 31 liver tissues with stage I to IV hepatocellular
carcinoma (HCC) (obtained from Professor Yuan Li, Department of
Pathology, Tongji hospital of Huazhong University of Science and
Technology, Wuhan, China) were used. Permission for the use of all
of these tissues for research purposes was obtained from the
Institutional Review Board of Huazhong University of Science and
Technology.
Paraffin-embedded tissue sections were dewaxed by
xylene and rehydrated orderly in sequential alcohols. After 3
washes, slides were immersed in 0.6% hydrogen peroxide. The
sections were then subjected to incubation with rabbit anti-human
Birc5 (1:200 diluation) or goat anti-human Hspa5 (1:200 dilution)
for 30 min. Then, the tissue sections were incubated with HRP
conjugated goat anti-rabbit IgG (1:200 dilution; Santa Cruz
Biotechnology) or HRP conjugated donkey anti-goat IgG (1:200
dilution; Santa Cruz Biotechnology, USA) for 30 min at room
temperature and developed with diaminobenzidine (DAB) as the
choromogenic agent. Counterstaining was performed with hematoxylin.
The tissue sections were counterstained with hematoxylin for 30
sec, dehydrated reversely using sequential alcohols and xylene, and
then mounted using Histo-mount. Images of stained tissue sections
were captured using an Olympus DP70 CCD camera with an Olympus BX51
microscope. The immunohistochemical expression of Birc5 and Hspa5
was evaluated by creating a system of 10% steps (range, 0–100%).
All immunopositive tumor cells in HCC and hepatocytes in paratumor
tissues were counted in at least 10 consecutive high-power fields
(×400), and the scoring guideline was based on the percentage of
positive cells: −, negative staining (−10%); +, positive weak
staining (10–50%); ++, positive strong staining (50–100%).
Real-time PCR
Forty-eight hours after transfection, RNA was
purified from HepG2 cells using TRIzol reagent (Invitrogen)
according to the manufacturer’s instructions. mRNA (1 μg)
was reverse-transcribed into cDNA using RevertAid (Fermentas,
Vilnius, Lithuania) in a 20-μl system. cDNA (2 μl)
was amplified with the SYBR-Green Master Mix (Applied Biosystems,
Carlsbad, CA, USA) using the ABI 7900 Sequence Detection System
(Applied Biosystems) using the following primers: Actb
(β-actin), 5′-CCTAGAAGCATTTGCGGTGG-3′ (forward) and
5′-GAGCTACGAGCTGCCTG ACG-3′ (reverse); Birc5,
5′-GCCCAGTGTTTCTTCTGCTTCA-3′ (forward) and 5′-GC
ACTTTCTCCGCAGTTTCCTC-3′ (reverse); and Hspa5, 5′-GG
AGGACAAGAAGGAGGACG-3′ (forward) and 5′-CAGGAG TGAAGGCGACATAGG-3′
(reverse). PCR was performed by using the following parameters:
50°C for 2 min, 95°C for 2 min, and 40 cycles of 95°C for 15 sec
and 60°C for 30 sec. The values are presented as the means ±
SD.
Western blot analysis
Whole cell lysates at 48 h after transfection were
loaded on 10% sodium dodecyl sulfate (SDS) polyacrylamide gels. The
transferred membrane was then incubated with 1:200 diluted rabbit
anti-human Birc5, or goat anti-human Hspa5. The antigen-antibody
complexes were detected using HRP-conjugated anti-rabbit IgG (1:500
dilution, GE Healthcare UK Ltd.), or anti-goat IgG (1:500 dilution,
GE Healthcare UK Ltd.), and visualized using ECL system (GE
Healthcare UK Ltd.). ACTB (β-actin) (rabbit polyclonal antibody at
1:200 dilution, Santa Cruz Biotechnology) signals were used to
normalize the Hspa5 and Birc5 signals. Relative amounts of protein
were quantified using Multi Gauge Ver2.2 image analyzing software
(Fuji Photo Film Co. Ltd., Tokyo, Japan).
Proliferation assay
Effects of transfection on cellular proliferation
were determined using the MTT staining method. Cells in the
logarithmic growth phase were plated in 96-well plates and
incubated for 24 h in fresh medium. The cells were then transfected
with siRNA plasmid for 24, 48 and 72 h, respectively. At 4 h prior
to the end of incubation, 20 μl MTT was added to each well,
followed by 150 μl DMSO to terminate the reaction. The
optical density (OD) of each well was measured using a microculture
plate reader at wave length of 492 nm. The cell proliferation rate
(PR) was calculated using the following equation: PR = OD value in
the treated samples/OD value in the control samples × 100%.
Apoptosis analysis
Forty-eight hours after transfection, cells were
collected, washed twice with PBS and resuspended in APC-labeled
Annexin V (BD Biosciences) and PI according to the manufacturer’s
instructions. Cells were then incubated for 20 min in the dark.
Cell-associated fluorescence was analyzed using WinMDI2.8
software.
Experiments in vivo
Transfected (3×106) HepG2 cells
(dissolved in 0.2 ml PBS) were injected subcutaneously into
5-week-old male BALB/c nude mice (n=6 in each group). The mice were
kept in a pathogen-free environment at a constant temperature
(23±2°C) and humidity (50–70%) with a 12-h light-dark cycle. Tumor
sizes were measured every 3 days and calculated by the following
formula: volume (mm3) = ½(width)2 × length.
The growth curve of the tumor was shown.
Statistical analysis
All experiments were performed in triplicate and
data were expressed as the means ± SD. Statistical analyses were
conducted with the Student’s t-test and performed with SPSS 17.0
(SPSS Inc., Chicago, IL, USA). p-values <0.05 were considered to
indicate statistically significant differences.
Results
siRNA-mediated silencing of Birc5 and
upregulation of Hspa5
In an attempt to downregulate the expression of the
antiapoptotic protein, Birc5, HepG2 cells were transfected with
plasmids encoding siRNA against Birc5 exon 1 (pgsiRNA-Bir1)
and exon 3 (pgsiRNA-Bir2), as well as the control plasmid,
pgsiRNA-C. The expression levels of Birc5 were evaluated at
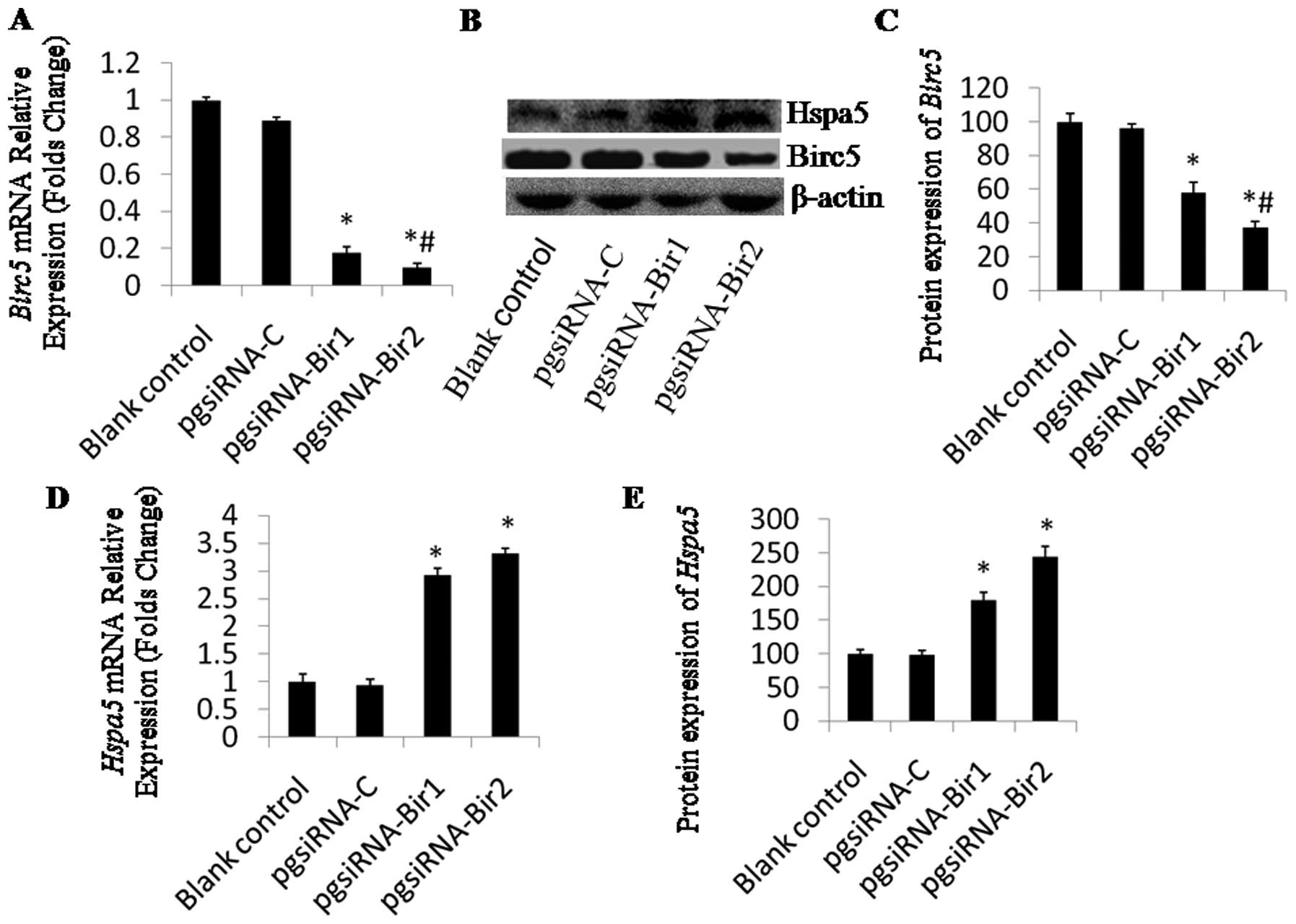
48 h post-transfection by real-time PCR and western blot analysis.
The mRNA level of Birc5 in the cells transfected with
pgsiRNA-Bir1 or pgsiRNA-Bir2 was decreased by 18 or 10% compared to
the control cells (Fig. 1A). The
densitometric analysis of the western blots revealed that the
protein level of Birc5 in cells transfected with pgsiRNA-Bir1 or
pgsiRNA-Bir2 was reduced by 58 or 37% compared to the control
levels, respectively, which indicated that pgsiRNA-Bir2 was more
efficient than pgsiRNA-Bir1 (Fig.
1C).
The expression of Hspa5 was also evaluated at
48 h post-transfection by real-time PCR (Fig. 1D) and western blot analysis
(Fig. 1B). The expression of
Hspa5 was significantly upregulated in HepG2 cells
transfected with pgsiRNA-Bir compared to the controls. For samples
transfected with pgsiRNA-Bir1 or pgsiRNA-Bir2, the mRNA level of
Hspa5 increased 2.9-fold and 3.3-fold compared to that of
the non-transfected cells (Fig.
1D), respectively. Densitometric analysis revealed that the
levels of Hspa5 in the cells transfected with pgsiRNA-Bir1 and
pgsiRNA-Bir2 were increased by 1.6- and 2.5-fold, respectively
(Fig. 1E). Transfection of
pgsiRNA-C had no significant effect on HSPA5 expression.
High expression of Birc5 and Hspa5 in
human liver tumors
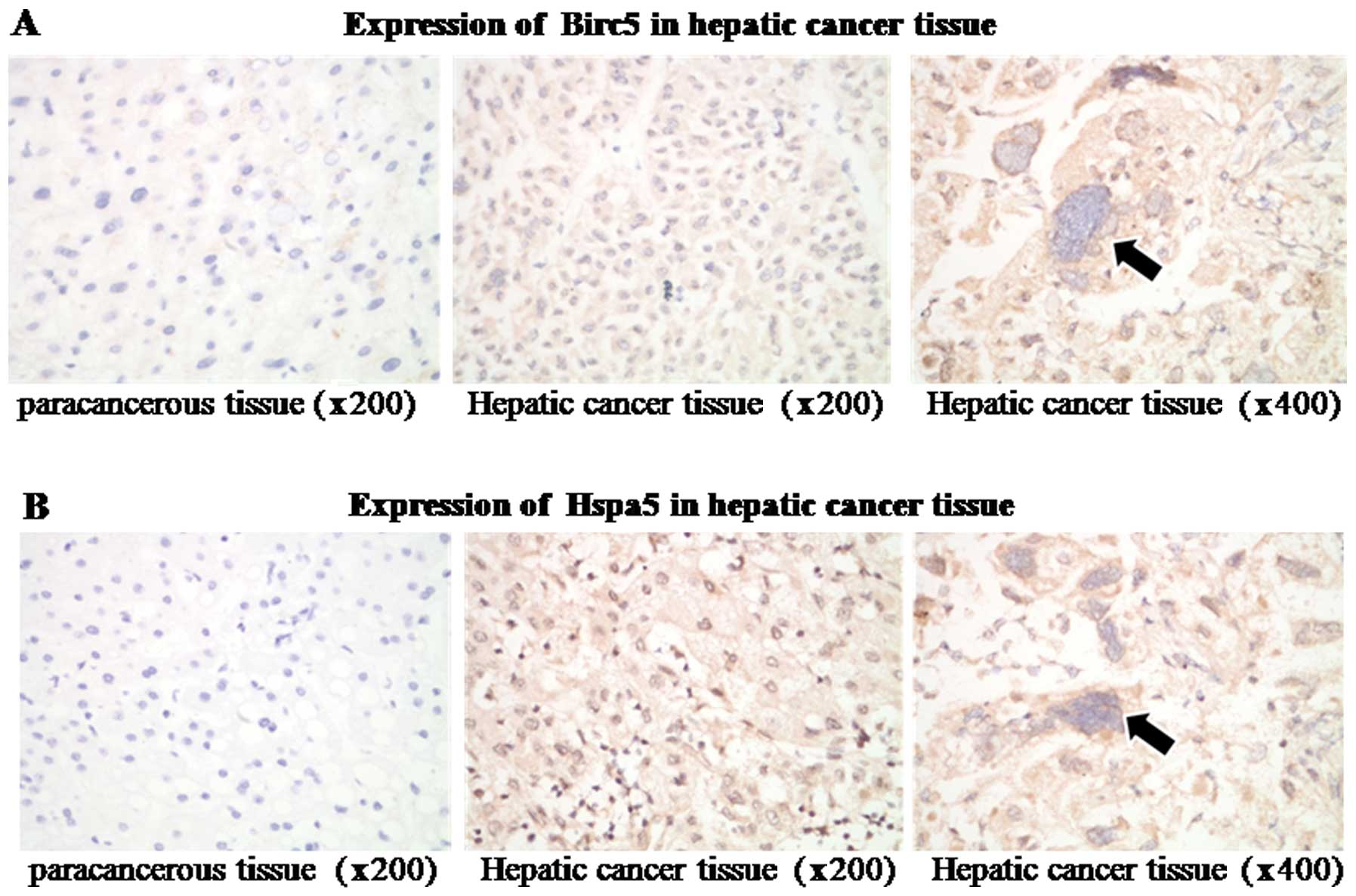
Immunofluorescence staining showed that Birc5 and
Hspa5 were highly expressed in HCC tissues, whereas were weak or
undetectable in paracancerous regions (Fig. 2). Out of 31 cases, 28 (90.3%)
showed a higher expression of Birc5 and Hspa5 in the tumor tissue
than in paracancerous areas (Table
I). Only 3 (array nos. 7, 12 and 23) exhibited similar
expression levels in both tumor and paracancerous regions.
 | Table IAnalysis of relative Birc5 and Hspa5
expression levels between paracancerous and tumor regions by
immunohistochemistry. |
Table I
Analysis of relative Birc5 and Hspa5
expression levels between paracancerous and tumor regions by
immunohistochemistry.
Expression and cellular localization of
Birc5 and Hspa5 in HepG2 cells
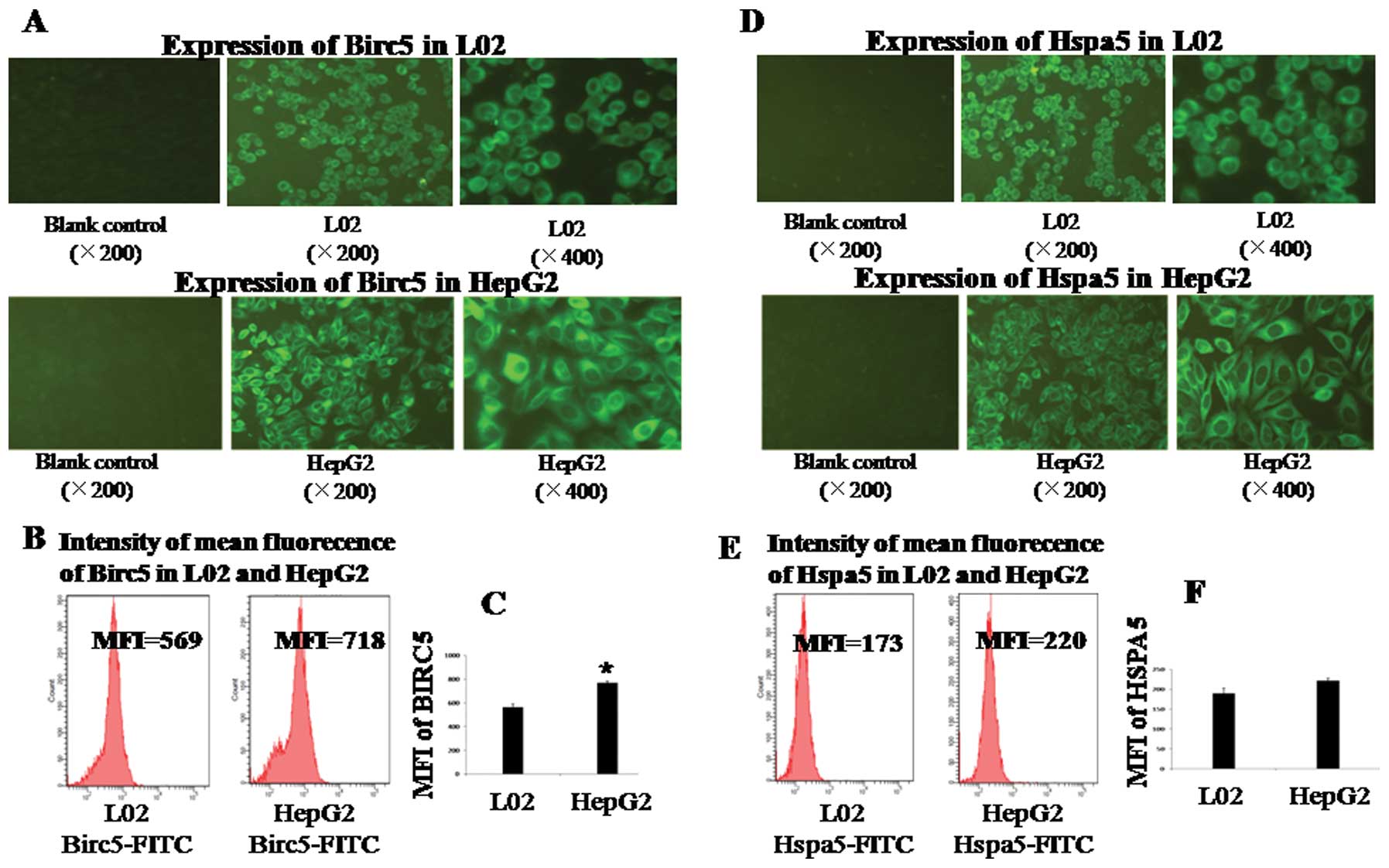
Immunofluorescence assay showed that both Birc5 and
Hspa5 were expressed in the cytoplasm of HepG2 cells (Fig. 3A and D). Mean fluorecence
intensities (MFIs) in HepG2 cells were monitored by flow cytometry,
which confirmed the observation of immunofluorescence staining
(Fig. 3B and E). The MFI of Birc5
in HepG2 cells showed a significant difference (p<0.05 vs. L02)
(Fig. 3A and B). However, the MFI
of Hspa5 in HepG2 cells showed no significant difference (p>0.05
vs. L02). The Hspa5 protein was quantitatively overexpressed in HCC
tumor tissues (Fig. 2B) but not in
the cell lines (Fig. 3D and
E).
Knockdown of Birc5 and Hspa5 by dual
interference siRNA
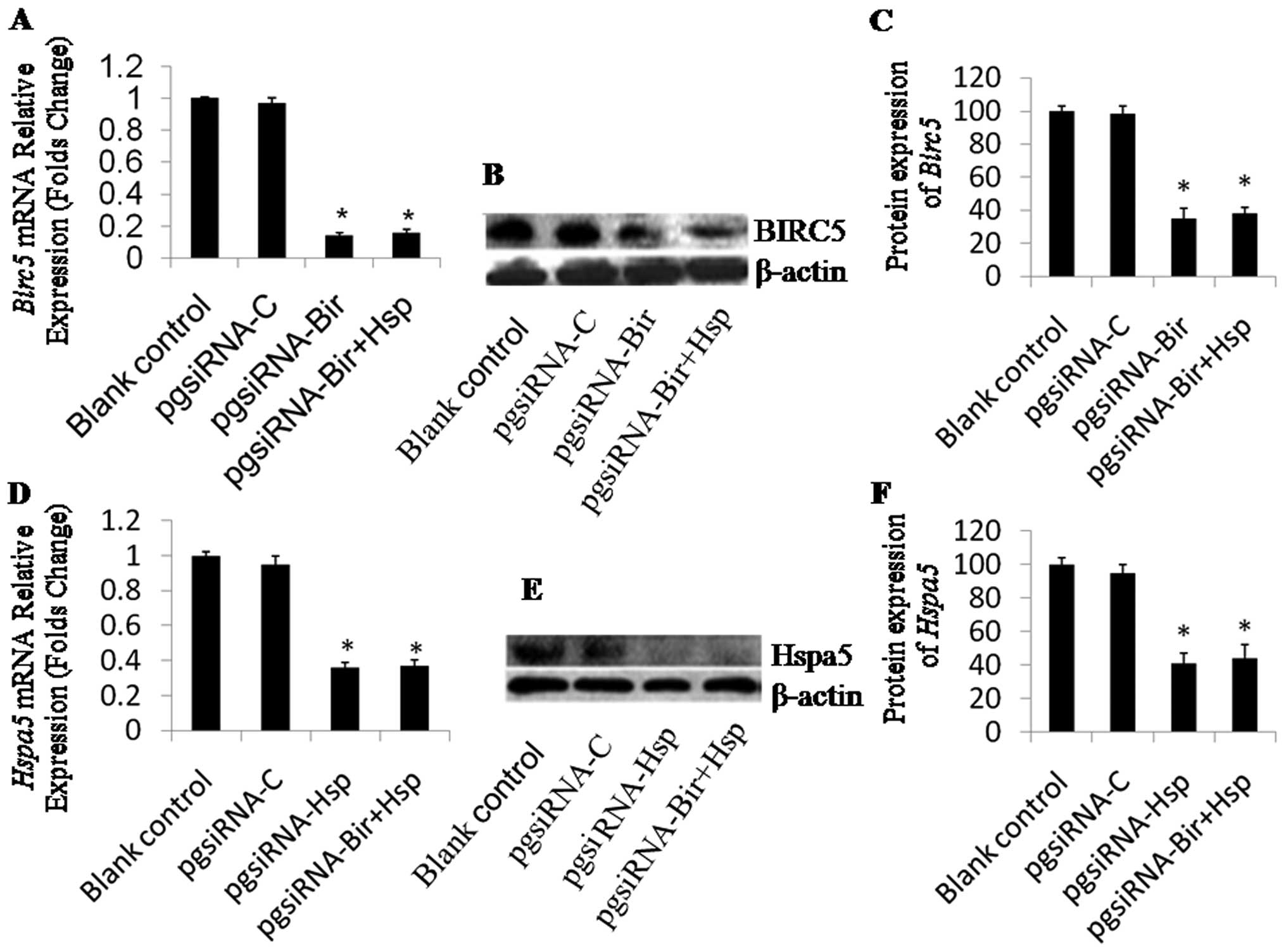
The expression of Birc5 was significantly
downregulated in HepG2 cells transfected with pasiRNA-Bir2 and
pgsiRNA-Bir+Hsp compared to the controls. pgsiRNA-Bir2 or
pgsiRNA-Bir+Hsp transfection resulted in the mRNA level of
Birc5 in HepG2 cells decreasing by 14 and 15% to that of the
non-transfected cells (Fig. 4A),
and the protein level decreasing by 35 and 38% to that of the
non-transfected cells (Fig. 4B and
C). The expression of Hspa5 was also significantly
downregulated in HepG2 cells transfected with pgsiRNA-Hsp and
pgsiRNA-Bir+Hsp compared with the controls. After transfection by
pgsiRNA-Hsp or pgsiRNA-Bir+Hsp, the HepG2 cells had a decreased
mRNA expression of Hspa5 of 36 and 37% compared to that of
the control cells (Fig. 4D), and a
decreased protein expression of 41 and 44% compared to that of the
control cells (Fig. 4E and F),
indicating that pgsiRNA-Hsp and pgsiRNA-Bir+Hsp were efficient in
silencing Hspa5 gene expression (Fig. 4D). These results indicate that the
designed siRNAs efficiently knocked down Birc5 and
Hspa5 expression in HepG2 cells.
Effect on proliferation of HepG2 cells by
different siRNA plasmids
To confirm whether transfection affects the
proliferation of HepG2 cells,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT)
assay was used to evaluate the cell proliferation. The PRs of HepG2
cells transfected with pgsiRNA-C, pgsiRNA-Hsp, pgsiRNA-Bir or
pgsiRNA-Bir+Hsp were 93.8±2.5, 78.1±3.1, 68.3±2.3 and 44.2±3.4%,
respectively at the 48-h time-point. These data confirmed that
pgsiRNA-Bir, pgsiRNA-Hsp and pgsiRNA-Bir+Hsp decreased the PR of
HepG2 cells significantly compared with the controls (p<0.05)
(Fig. 5A and B). Among these,
pgsiRNA-Bir+Hsp showed the most significant anti-proliferative
effect.
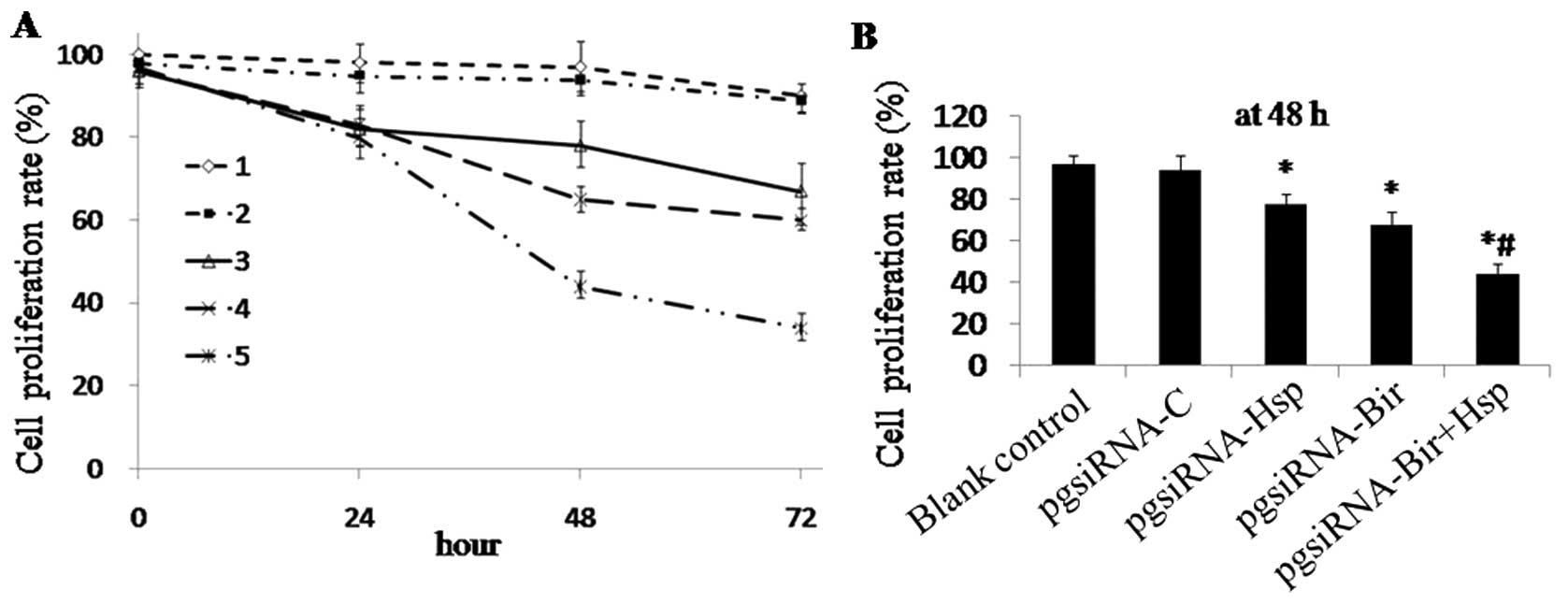 | Figure 5The effect of siRNA plasmids on
proliferation of HepG2 cells. (A) HepG2 cells were treated for
24-72 h with pgsiRNA-C, pgsiRNA-Bir, pgsiRNA-Hsp or siRNA-Bir+Hsp.
Proliferation assay was used to determine the growth rate relative
to the control cells. 1, Blank control group; 2, pgsiRNA-C group;
3, pgsiRNA-Hsp group; 4, pgsiRNA-Bir group; 5, pgsiRNA-Bir+Hsp
group; *p<0.05 compared with the blank control group,
#p<0.05. compared with the pgsiRNA-Hsp and
pgsiRNA-Bir groups. (B) The proliferation rates of HepG2 cells
transfected with pgsiRNA-C, pgsiRNA-Hsp, pgsiRNA-Bir or
pgsiRNA-Bir+Hsp at the 48-h time-point. *p<0.05 vs.
blank control, #p<0.05 vs. pgsiRNA-Hsp and
pgsiRNA-Bir. |
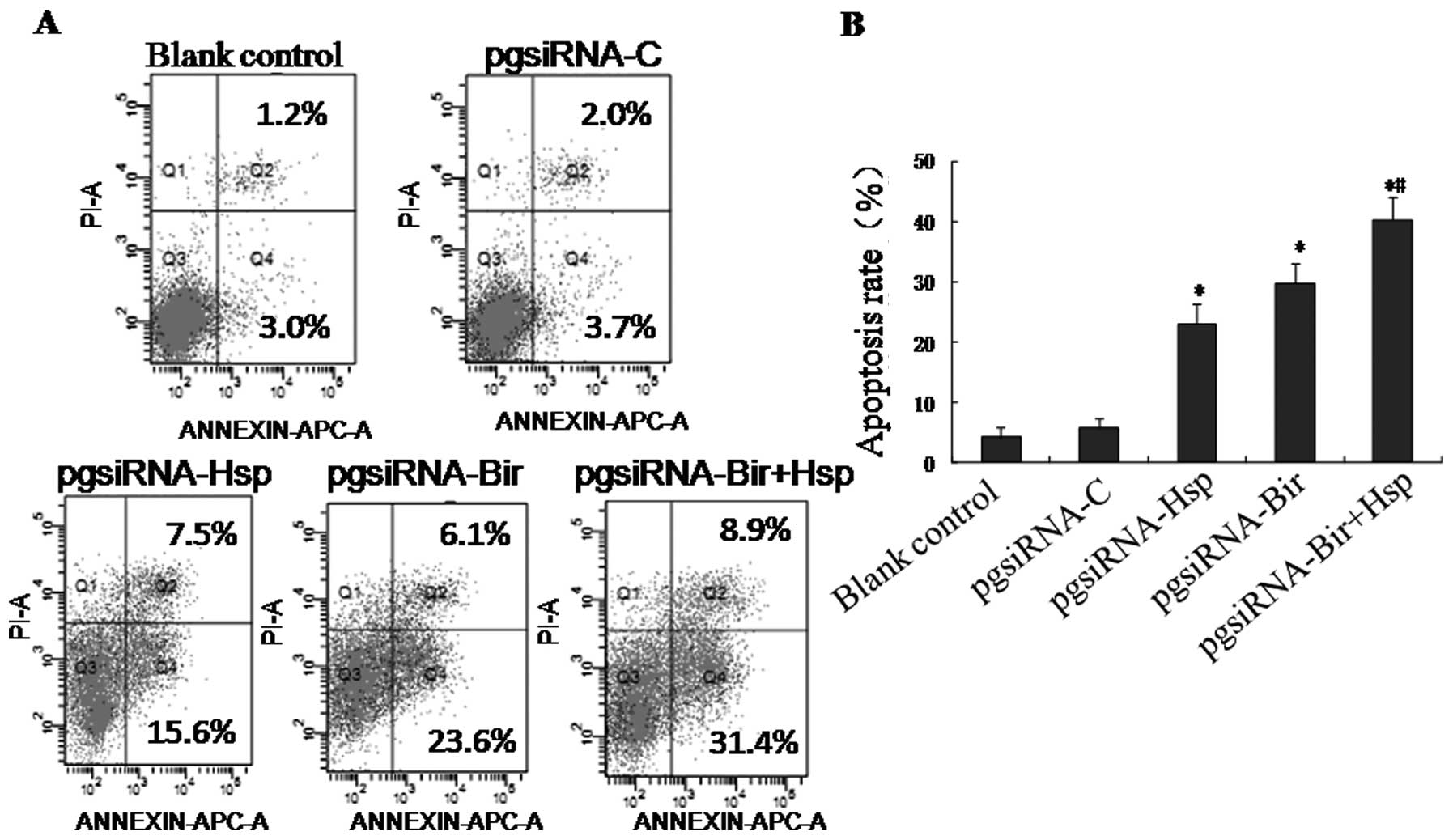
Co-silencing of Birc5 and Hspa5 results
in increased HepG2 cell apoptosis
Flow cytometry analysis revealed that the apoptotic
rates of HepG2 cells transfected with pgsiRNA-C, pgsiRNA-Hsp,
pgsiRNA-Bir or pgsiRNA-Bir+Hsp were 6.3±1.5, 23.1±2.1, 29.8±3.3 and
40.2±3.7%, respectively, whereas only 3.0±1.5% of non-transfected
cells underwent apoptosis at the 48-h time-point (Fig. 6). pgsiRNA-Hsp, pgsiRNA-Bir and
pgsiRNA-Bir+Hsp significantly induced apoptosis in HepG2 cells
(p<0.05 vs. blank control) and pgsiRNA-Bir+Hsp was the most
effective in inducing apoptosis (p<0.05 vs. pgsiRNA-Hsp or
pgsiRNA-Bir) (Fig. 6A and B).
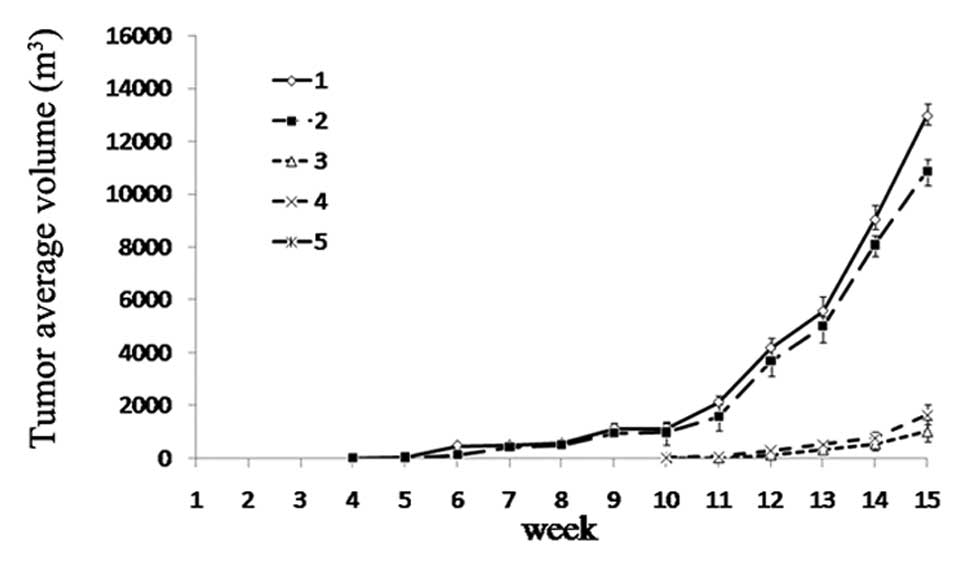
Inhibitory effect of co-silencing of
Birc5 and Hspa5 in vivo
With the above findings of the effects of
pgsiRNA-Bir+Hsp in vitro, we next examined whether
pgsiRNA-Bir+Hsp plays a critical role in tumor formation in
vivo, and whether it can be used in clinical gene therapy.
Equal numbers of HepG2 cells transfected with pgsiRNA-C,
pgsiRNA-Bir, pgsiRNA-Hsp and pgsiRNA-Bir+Hsp, and untreated cells
were injected subcutaneously into nude mice. In mice inoculated
with untreated cells or pgsiRNA-C-transfected cells, tumors formed
4 weeks later. However, in those mice inoculated with cells
transfected with pgsiRNA-Bir, pgsiRNA-Hsp, tumors formed 10 weeks
later (Fig. 7). Furthermore, in
the inoculated co-silenced cells, tumor formation in nude mice was
not observed. These results indicate that the co-silencing of
Birc5 and Hspa5 exerts a stronger growth suppressive
effect on liver cancer in vivo.
Discussion
Birc5 is considered one of the most promising cancer
targets.Certain Birc5 antagonists, such as antisense
oligonucleotides LY2181308 (22),
the locked nucleic acid EZN-3042 (23) and the promoter inhibitor YM155
(24,25), have been used in Phase I or II
clinical trials for treating HCCs, acute myeloid leukemias,
melanomas and several other advanced solid tumors. siRNA is
evolving as a promising strategy for cancer therapy due to its high
efficiency and specificity in blocking target mRNA expression
(26).
In this study, plasmid-based siRNAs specific for
Birc5 were designed and transfected into HepG2 cells. It was
found that siRNA targeting Birc5 exon 3 was more effective
than that targeting exon 1 in inducing cell apoptosis.
Nevertheless, a small portion of cells (29.7±3.3%) underwent
apoptosis. This could be due to the low transfection efficiency.
However, in our previous studies, the folate receptor-mediated
specific delivery of Birc5 siRNA caused approximately 35% of
HeLa or MCF-7 cells to become apoptotic (10). Tirrò et al (27) also reported that the viability of
WRO thyroid cancer cells transfected with siRNAs for Birc5
showed no difference with that of the scramble control. When
Birc5-specific siRNA was transfected into US 8–93 human
sarcoma cells, only a slight increase (2–4%) in the percentage of
cells undergoing apoptosis was observed, as well as in other 4
sarcoma cell lines. Even after an apoptotic stimulus was applied,
only a small percentage of cells underwent apoptosis (28). Certain studies have shown that the
use of anti-Birc5 oligonucleotides led to the same problem
(29,30). The reason could be that the
Birc5 knockdown resulted in the upregulation of certain
molecules functionally involved in survival, anti-apoptosis in the
tumor, which could counteract the effect of the Birc5
antagonists. Hendruschk et al showed that the RNAi-mediated
Birc5 knockdown in U373-MG cells induced the expression of
hypoxia inducible factor-1a (HIF-1a) (31). Similar to HIF-1a, Hspa5 is highly
induced in the presence of low oxygen levels and plays important
roles in tumor angiogenesis, apoptosis resistance, tumor cell
survival (17,32–34),
and furthermore, in the stress of oncogenesis (15–18).
Therefore, we wished to determine whether the Birc5
knockdown can induce the upregulation of Hspa5.
The results from our presen study demonstrate a
marked increase in the Hspa5 mRNA and protein levels upon
the depletion of Birc5 in HepG2 cells and a high expression
of Birc5 and Hspa5 in the HCC tissues. This suggests that the
RNAi-mediated Birc5 knockdown in HepG2 cells induced the
upregulation of Hspa5. However, the mechanism by which the
Birc5 depletion regulates the expression of Hspa5 is
still under investigation.
Taking all these results into account, we
hypothesized that the co-silencing of Birc5 and Hspa5
would be more effective in apoptosis induction than single gene
interference. The data confirmed that Birc5 and Hspa5
co-silencing induced an evident increase in the percentage of cells
undergoing apoptosis and an evident decrease in the percentage of
cell proliferation compared with the Birc5- or
Hspa5-silenced cells. Tumor formation was also not observed
in the nude mice inoculated with Birc5 and Hspa5
co-silenced cells (p<0.05). These results suggest that the
upregulation of Hspa5 which can suppress the activities of ER
stress sensors for maintaining tumor growth could be a defense
mechanism in response to Birc5-specific siRNA-mediated
apoptosis in HepG2 cells. An inhibitor of Hspa5 may be of clinical
benefit by targeting the Hspa5-protected cells after Birc5
knockdown. The dual inhibition of Birc5 and Hspa5 may be warranted
for cancer therapy.
The MFI of cytoplasmic expression of Hspa5 showed no
significant difference between the HepG2 and L02 cells (p>0.05),
which disaccorded with the expression of Hspa5 in human HCC
samples. One likely explanation is that cancer cells exhibit
elevated glucose metabolism with increased glycolytic activity, and
solid tumors often grow faster than their blood supply. The latter
creates a tumor microenvironment characterized by glucose
deprivation, acidosis and severe hypoxia (34). Solid tumor cells suffer more from
these microenvironmental stresses, which leads to the activation of
the unfolded protein response (UPR) in cancer cells. The UPR occurs
through the transcriptional and translational regulatory mechanisms
that improve the capacity of the endoplasmic reticulum (ER) to fold
and traffic proteins and allows the cell to survive under stress
conditions (35). The widely used
sentinel marker for ER stress and UPR activation is Hspa5 (36). Therefore, solid tumor cells in
these major physiological conditions are more likely to overexpress
Hspa5 than tumor cell lines.
To increase the transfection efficiency, a
transferrin receptor (TfR)-targeted gene delivery system is now
under investigation with the aim to shed light on the potential
therapeutic utilities of Birc5 and Hspa5
dual-targeting interfering siRNA.
Acknowledgements
This study was supported by: the
Natural Science Foundation of China (No. 81102219, 30972734), State
Project on Major Infectious Diseases Prevention (No.
2012ZX10002006-003), the New Faculty Member Program of the Chinese
Department of Education (No. 20070487103), the Fundamental Research
Funds for the Central Universities (No. 2010JC022, 2011TS070) and
the Natural Science Foundation of Hubei Province of China (No.
2010CDB03503).
References
|
1
|
Altieri DC: Survivin, versatile modulation
of cell division and apoptosis in cancer. Oncogene. 22:8581–8589.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chawla-Sarkar M, Bae SI, Reu FJ, et al:
Downregulation of Bcl-2, FLIP or IAPs (XIAP and survivin) by siRNAs
sensitizes resistant melanoma cells to Apo2L/TRAIL-induced
apoptosis. Cell Death Differ. 11:915–923. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nomura T, Yamasaki M, Nomura Y and Mimata
H: Expression of the inhibitors of apoptosis proteins in
cisplatin-resistant prostate cancer cells. Oncol Rep. 14:993–997.
2005.PubMed/NCBI
|
|
4
|
Li F, Yang J, Ramnath N, Javle MM and Tan
D: Nuclear or cytoplasmic expression of survivin: what is the
significance? Int J Cancer. 114:509–512. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hinnis AR, Luckett JC and Walker RA:
Survivin is an independent predictor of short-term survival in poor
prognostic breast cancer patients. Br J Cancer. 96:639–645. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Altieri DC: Validating survivin as a
cancer therapeutic target. Nat Rev Cancer. 3:46–54. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Olie RA, Simoes-Wust AP, Baumann B, et al:
A novel antisense oligonucleotide targeting survivin expression
induces apoptosis and sensitizes lung cancer cells to chemotherapy.
Cancer Res. 60:2805–2809. 2000.
|
|
8
|
Pennati M, Binda M, Colella G, et al:
Ribozyme-mediated inhibition of survivin expression increases
spontaneous and drug-induced apoptosis and decreases the
tumorigenic potential of human prostate cancer cells. Oncogene.
23:386–394. 2004. View Article : Google Scholar
|
|
9
|
Liu H, Guo S, Roll R, et al: Phi29 pRNA
vector for efficient escort of hammerhead ribozyme targeting
survivin in multiple cancer cells. Cancer Biol Ther. 6:697–704.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li L, Liu J, Diao Z, et al: Evaluation of
specific delivery of chimeric phi29 pRNA/siRNA nanoparticles to
multiple tumor cells. Mol Biosyst. 5:1361–1368. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pennati M, Folini M and Zaffaroni N:
Targeting survivin in cancer therapy: fulfilled promises and open
questions. Carcinogenesis. 28:1133–1139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gyurkocza B, Plescia J, Raskett CM, et al:
Antileukemic activity of shepherdin and molecular diversity of
hsp90 inhibitors. J Natl Cancer Inst. 98:1068–1077. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fortugno P, Beltrami E, Plescia J, et al:
Regulation of survivin function by Hsp90. Proc Natl Acad Sci USA.
100:13791–13796. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cheung CH, Chen HH, Cheng LT, et al:
Targeting Hsp90 with small molecule inhibitors induces the
over-expression of the anti-apoptotic molecule, survivin, in human
A549, HONE-1 and HT-29 cancer cells. Mol Cancer. 9:772010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tanimoto R, Sakaguchi M, Abarzua F, et al:
Down-regulation of BiP/GRP78 sensitizes resistant prostate cancer
cells to gene-therapeutic overexpression of REIC/Dkk3. Int J
Cancer. 126:1562–1569. 2010.PubMed/NCBI
|
|
16
|
Schardt JA, Weber D, Eyholzer M, Mueller
BU and Pabst T: Activation of the unfolded protein response is
associated with favorable prognosis in acute myeloid leukemia. Clin
Cancer Res. 15:3834–3841. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Q, He Z, Zhang J, et al:
Overexpression of endoplasmic reticulum molecular chaperone GRP94
and GRP78 in human lung cancer tissues and its significance. Cancer
Detect Prev. 29:544–551. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Arnaudeau S, Arboit P, Bischof P, et al:
Glucose-regulated protein 78: a new partner of p53 in trophoblast.
Proteomics. 9:5316–5327. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang WJ, Xia LM, Zhu F, et al:
Transcriptional upregulation of HSP70-2 by HIF-1 in cancer cells in
response to hypoxia. Int J Cancer. 124:298–305. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang M, Zhao XR, Wang P, et al: Glucose
regulated proteins 78 protects insulinoma cells (NIT-1) from death
induced by streptozotocin, cytokines or cytotoxic T lymphocytes.
Int J Biochem Cell Biol. 39:2076–2082. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang M, Wang P, Peng JL, et al: The
altered expression of glucose-regulated proteins 78 in different
phase of streptozotocin-affected pancreatic beta-cells. Cell Stress
Chaperones. 14:43–48. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carrasco RA, Stamm NB, Marcusson EG, et
al: Antisense inhibition of survivin expression as a cancer
therapeutic. Mol Cancer Ther. 10:221–232. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sapra P, Wang M, Bandaru R, et al:
Down-modulation of survivin expression and inhibition of tumor
growth in vivo by EZN-3042, a locked nucleic acid antisense
oligonucleotide. Nucleosides Nucleotides Nucleic Acids. 29:97–112.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakahara T, Kita A, Yamanaka K, et al:
Broad spectrum and potent antitumor activities of YM155, a novel
small-molecule surviving suppressant, in a wide variety of human
cancer cell lines and xenograft models. Cancer Sci. 102:614–621.
2011. View Article : Google Scholar
|
|
25
|
Nakahara T, Takeuchi M, Kinoyama I, et al:
YM155, a novel small-molecule survivin suppressant, induces
regression of established human hormone-refractory prostate tumor
xenografts. Cancer Res. 67:8014–8021. 2007. View Article : Google Scholar
|
|
26
|
Ashihara E, Kawata E and Maekawa T: Future
prospect of RNA interference for cancer therapies. Curr Drug
Targets. 11:345–360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tirrò E, Consoli ML, Massimino M, et al:
Altered expression of c-IAP1, survivin, and Smac contributes to
chemotherapy resistance in thyroid cancer cells. Cancer Res.
66:4263–4272. 2006.PubMed/NCBI
|
|
28
|
Kappler M, Bache M, Bartel F, et al:
Knockdown of survivin expression by small interfering RNA reduces
the clonogenic survival of human sarcoma cell lines independently
of p53. Cancer Gene Ther. 11:186–193. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu YF, Liang XJ, Liu YY, et al: Antisense
oligonucleotide targeting survivin inhibits growth by inducing
apoptosis in human osteosarcoma cells MG-63. Neoplasma. 57:501–506.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xia C, Xu Z, Yuan X, et al: Induction of
apoptosis in mesothelioma cells by antisurvivin oligonucleotides.
Mol Cancer Ther. 1:687–694. 2002.PubMed/NCBI
|
|
31
|
Hendruschk S, Wiedemuth R, Aigner A, et
al: RNA interference targeting survivin exerts antitumoral effects
in vitro and in established glioma xenografts in vivo. Neuro Oncol.
13:1074–1089. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kern J, Untergasser G, Zenzmaier C, et al:
GRP-78 secreted by tumor cells blocks the antiangiogenic activity
of bortezomib. Blood. 114:3960–3967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reddy RK, Mao C, Baumeister P, et al:
Endoplasmic reticulum chaperone protein GRP78 protects cells from
apoptosis induced by topoisomerase inhibitors: role of ATP binding
site in suppression of caspase-7 activation. J Biol Chem.
278:20915–20924. 2003. View Article : Google Scholar
|
|
34
|
Li J and Lee AS: Stress induction of
GRP78/BiP and its role in cancer. Curr Mol Med. 6:45–54. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Saito S and Tomida A: Use of chemical
genomics in assessment of the UPR. Methods Enzymol. 491:327–341.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ni M, Zhou H, Wey S, et al: Regulation of
PERK signaling and leukemic cell survival by a novel cytosolic
isoform of the UPR regulator GRP78/BiP. PLoS One. 4:e68682009.
View Article : Google Scholar : PubMed/NCBI
|