Introduction
As an integral part of the innate immune system,
natural killer (NK) cells respond rapidly to infectious diseases
and malignant transformation to restrict the dissemination of
diseases and to elicit the long-lasting, antigen-specific adaptive
immune reactions (1). Numerous
studies have illustrated that NK cells can effectively control
tumor growth through perforin (2)
and cytokines (3). The critical
role of NK cells in anti-tumor immunity has been further
demonstrated by clinical studies showing that patients with
dysfunctional NK cells have an increased risk of developing
leukemia (4).
Normal NK cells are usually not found in the cell
cycle; they are found only in response to infection from certain
pathogens or to the stimulation with high doses of appropriate
cytokines [type I interferons (IFNs), interleukin (IL)-2, IL-12 or
IL-15] (5). The early pioneering
study on lymphokine-activated killer cell-based immunotherapy by
Rosenberg was based on this protocol (6). Current techniques for activating NK
cells are all based on cytokines, usually with high doses which are
not well-tolerated by the host. IL-2 is known as the growth factor
for both T lymphocytes and NK cells, and potentially enhances the
cytotoxicity of these cells as well. Resting NK cells from humans
and mice respond readily to recombinant IL-2 (rIL-2) stimulation,
without the help of accessory cells and co-factors (7). It has been suggested that the in
vivo survival, tumor localization, and consequently, the
antitumor effect of the activated NK (A-NK) cells are strongly
dependent on the continuous support of relatively high doses of
exogenous IL-2 (8). It has been
shown that optimal tumor localization and antitumor activity of the
adoptively transferred NK cells are difficult to achieve even when
IL-2 was systemically administered every 4 h for 3 consecutive days
due to the short in vivo half-life of IL-2 (∼6.9 min)
(9). When long-lived (0.5 to 4 h)
polyethylene glycol-conjugated IL-2 (PEG-IL-2) was used, tumor
localization and antitumor activity were substantially improved
(10). Other cytokines, such as
IL-12 (11), IL-15 and IL-21,
which strongly augment NK cell proliferation and effector
functions, have also been used to substitute for IL-2. However, all
the cytokines systemically used in vivo have been associated
with dose-related toxicity (12).
It is clear that an alternative has to be found to circumvent the
demand of A-NK cells for exogenous IL-2 to sustain their activities
before adoptive NK cell transfer can be clinically feasible.
The adenovirus transduction of A-NK cells with IL-12
not only can enhance their antitumor effectiveness but can also
reduce their reliance on systemically administered IL-2 (8). However, the expression of IL-12 in NK
cells may still have the potential to cause a systemic IL-12
effect. In a mouse B16 tumor model, the direct delivery of IL-12 to
the tumor site maintained its anticancer activity but avoided much
of the toxic side-effects (13).
The co-transfection of tumor cells with GPI-anchored IL-2 and IL-12
has been shown to synergistically enhance the anti-tumor effect and
to have less adverse effects (14). It has also been reported that
endoplasmic reticulum-restricted IL-2 offers autocrine growth
stimulation to NK-92 cells (15).
In this study, we designed a fusion gene between mouse sonic
hedgehog (Hh) C-terminal (Shh-C) auto-processing domain and IL-12
(IL-12/Shh-C) as a self-supporting autocrine system to reduce or
eliminate the dependency of NK cells on exogenous cytokines and
sustain their activated status.
Hh family proteins have been shown to play essential
roles in cell fate specification during development. As morphogens,
they are produced from localized sites and spread through the
extracellular matrix to form a gradient and determine cell fates
through multiple regulations in a concentration-dependent manner
(16–21). The Hh precursor protein undergoes
internal proteolysis between Gly-Cys residues to confer cholesterol
modification at its carboxy terminus and palmitoylation
modification at its amino terminus (22). The cholesterol moiety serves as a
membrane-anchorage to restrict long-range signaling and unregulated
spreading, thus increasing local Shh concentration (23). It has been demonstrated that Shh-C
can direct green fluorescent protein (GFP) to the cell membrane
with a cholesterol tail attached (24). The results from the present study
show that mouse NK cells transduced with lentiviruses encoding this
IL-12/Shh-C fusion gene have significantly reduced dependency on
IL-2 in vitro. Also, prolonged survival was observed when
ex vivo A-NK cells transduced with IL-12/Shh-C lentivirus
were adoptively transferred to B16 melanoma tumor-bearing mice.
Materials and methods
Fusion gene construction and lentivirus
production
Eleven-day mouse embryo total RNA was purchased from
Clontech. cDNA was generated by reverse transcription with a
RetroScript kit (Ambion). Mouse Shh-C was PCR-amplified and cloned
into the pCR2.1 vector (Invitrogen). Mouse mIL-12 fragment without
stop codon was amplified with PCR using the pORF-mIL-12 plasmid
(Invivogen) as the template. The enhanced GFP (EGFP) fragment
without stop codon was PCR-amplified with pIRES-EGFP (Clontech) as
the template. Both mIL-12 and EGFP fragments were subcloned into
pCDH-EF1-MCS (System Biosciences) to construct the plasmid
pCDH-EGFP and pCDH-fusion-IL-12. pCDH-ORF-mIL-12 was generated by
ligation of pCDH-EF1-MCS digested with SwaI and pORF-mIL-12
treated with SwaI and EcoRV.
The calcium phosphate transfection method was used
in lentivirus production. 293TN cells were seeded at the density of
7×106 cells per T-150 flask one day before transfection
without antibiotics. Different lentiviral expression vectors (40
μg) (pCDH-EGFP, pCDH-ORF-mIL-12 and pCDH-fusion IL-12) were
mixed with pMDLg/pRRE, pRSV-Rev and pMD2.G (Addgene) and used for
co-transfection of 293TN cells. Forty-eight hours
post-transfection, supernatant containing recombinant lentiviral
particles was clarified with 0.45 μm filter (Corning) and
stored at −80°C. Lentivirus was titered by CHO cell transduction.
Briefly, 1×105 CHO cells were plated into 24-well plate
and lentivirus was added with various dilution factors of
10−2 to 10−1 in the presence of 8
μg/ml of polybrene (Sigma-Aldrich). After 1-h centrifugation
at 1,200 × g, the cells were incubated for 5 h at 37°C and 5%
CO2. Four days after transduction, biological titer was
calculated by the following equation as previously described
(27): transduction unit (TU/ml) =
(% of GFP- or IL-12 positive cells × number of cells at time of
transduction) × dilution factor/(100× volume of lentivirus
added).
Mice and tumor cell lines
Female C57BL/6 mice and congenic B6.PL-Thy1a/CyJ
mice, purchased from Jackson Laboratory, at age 8–20 weeks were
used for the present study, and all experiments were carried out
with approval from the Institutional Animal Care and Use Committee.
The murine melonoma cell line, B16, was obtained from ATCC. B16
cells were maintained in RPMI-1640 (Hyclone) with 10%
heat-inactivated fetal bovine serum (FBS), 2 mM of glutamine and
antibiotics.
NK cell isolation and lentiviral
transduction
Congenic B6.PL-Thy1a/CyJ mice were sacrificed and
spleen tissues were homogenized by grinding the tissues with 2
frosted glass slides. After filtering through a cell strainer of 40
μm (BD Biosciences) to obtain a single cell suspension,
spleen cell were centrifuged at 300 × g for 15 min. Erythrocytes
were lysed by ACK lysing buffer (Lonza). The splenic lymphocytes
were then subjected to magnetic labeling and separation by using a
Natural Killer Cell Isolation kit (Miltenyi Biotec). The purity of
freshly isolated NK cells from the spleen was >90%
NK1.1+, <6% CD3+. NK cells were cultured
in RPMI-1640 medium (Hyclone) supplemented with 10%
heat-inactivated FBS (Hyclone), 50 μM β-mercaptoethanol, 2
mM glutamine, and 1X non-essential amino acids. For activation and
expansion purposes, 6,000 IU/ml of recombinant human IL-2 (rhIL-2)
(Peprotech) was added every other day. Seven-day rhIL-2-stimulated
NK cells were plated into 48-well plates at 2×105
cells/250 μl per well. NK cells were transduced at 1 MOI in
the presence of 5 μg/ml polybrene (Sigma-Aldrich) by
centrifugation at 1,200 × g for 1 h. After centrifugation,
transduced NK cells were incubated at 37°C and 5% CO2
for an additional 4 h then replaced with complete culture medim.
The following day, lentiviral transduction was repeated on the
transduced NK cells.
NK cell proliferation assay
NK cells were transduced with EGFP/Shh-C, IL-12 or
IL-12/Shh-C lentiviruses at an MOI of 1 on both days 7 and 8 after
isolation. Three days after double transduction, 15,000
NKEGFP/Shh-C, NKIL-12 and
NKIL-12/Shh-C cells were plated per well in 96-well
plates with 100 μl of culture medium, supported with 0, 60,
600 and 6,000 IU/ml of IL-2. IL-2 was added every other day.
Proliferation assays were performed 24, 48, 72 and 96 h after
plating. AlamarBlue (Invitrogen) reagent was added in an amount
equal to 10% of the culture volume. After being cultured for 4.5 h
at 37°C/5% CO2, the absorbance was measured at a
wavelength of 560 nm with a reference of 620 nm. The cell numbers
were determined by using a standard curve. All samples were run in
triplicate.
Analysis of NK cell markers by flow
cytometry
EGFP expression was directly detected by comparing
transduced versus non-transduced NK cells. Antibodies for surface
marker analysis of NK cells for anti-NK1.1, CD3, CD25 and CD11b
were all from BD Pharmingen. For intracellular staining, anti-IL-12
p40, perforin and ganzyme B were obtained from eBioscience. Prior
to staining with antibodies, mouse BD Fc Block (BD Biosciences) was
used to prevent non-specific binding. All samples were fixed with
BD Cytofix™ Fixation Buffer (BD Biosciences) and then stored in
phosphate-buffered saline supplemented with 2% FBS and 0.2% sodium
azide (Sigma). For intracellular staining, samples were fixed then
permeabilized with BD Cytoperm™ Permeabilization Buffer (BD
Biosciences) prior to antibody staining. A minimum of 15,000 cells
were used in all samples. Data acquisition was performed on a Guava
EasyCyte Mini system (Guava Technologies) and analyzed by FACS
Express 2 software. Data obtained from quadrant gating were
compared to the non-transduced control group.
Cytokine production analysis
Supernatants from overnight NK cell cultures 7 days
after lentiviral infection and serum from experimental mice were
used to determine IFN-γ, IL-12 and tumor necrosis factor-α (TNF-α)
levels. Mice serum from each group was collected from the tail vein
at 3, 7 and 14 days after the adoptive transfer of NK cells. The
mouse IFN-γ ELISA Ready-SET-Go kit, the mouse IL-12/IL-23 total p40
ELISA Ready-SET-Go kit and the mouse TNF-α ELISA Ready-SET-Go kit
(eBioscience) were used according to manufacturer’s
instructions.
NK cell cytotoxicity assay
Mouse lymphoma YAC-1 cells were used as the target
and stained with 0.5 μM Guava CFSE (Guava Technologies) per
2.5×106 cells. Triplicate wells of NK cells were mixed
with YAC-1 cells in 96-well round bottom plates to yield effector
cell to target cell (E:T) ratios of 2:1, 1:1, 0.5:1 and 0.2:1. The
plates were centrifuged at 50 × g for 2 min prior to 4-h incubation
at 37°C and 5% CO2. Subsequently, 50 μl of Guava
Express 7-AAD (Guava Technologies) per 1×105 cells were
added into each well. Five thousand events were acquired for each
sample. YAC-1 cells only were used as the control for spontaneous
cell death. The percentage of NK cell cytotoxicity was calculated
using the following equation: percentage of specific lysis = (%
dead targets − % spontaneous dead targets) × 100/(100 − %
spontaneous dead targets).
Adoptive transfer of NK cells
C57BL/6 mice received 3.75×105 B16
melanoma tumor cells intravenously (i.v.) to establish pulmonary
metastases. Three days later, 5×106
NKEGFP/Shh-C cells, NKIL-12 cells and
NKIL-12/Shh-C cells were injected via the lateral tail
vein without IL-2 administration.
Analysis of tumor infiltration by NK
cells in vivo
Seventy-two hours after adoptive transfer, lung
tissues obtained from mice were fresh frozen at −80°C and were then
sectioned into 8-μm tissue slides. After fixing in 4%
paraformaldehyde, tissues were stained with FITC-conjugated
anti-Thy 1.1 Ab (BD Pharmingen) at a 1:200 dilution for 30 min at
room temperature. FITC-conjugated IgG2a (BD Pharmingen) was used as
an isotype control. A fluorescent microscope was used to obtain
images.
Statistical analysis
Two-tailed, unpaired Student’s t-tests were used.
For survival experiments, data were analyzed by the Kaplan-Meier
method.
Results
Fusion gene construction and
expression
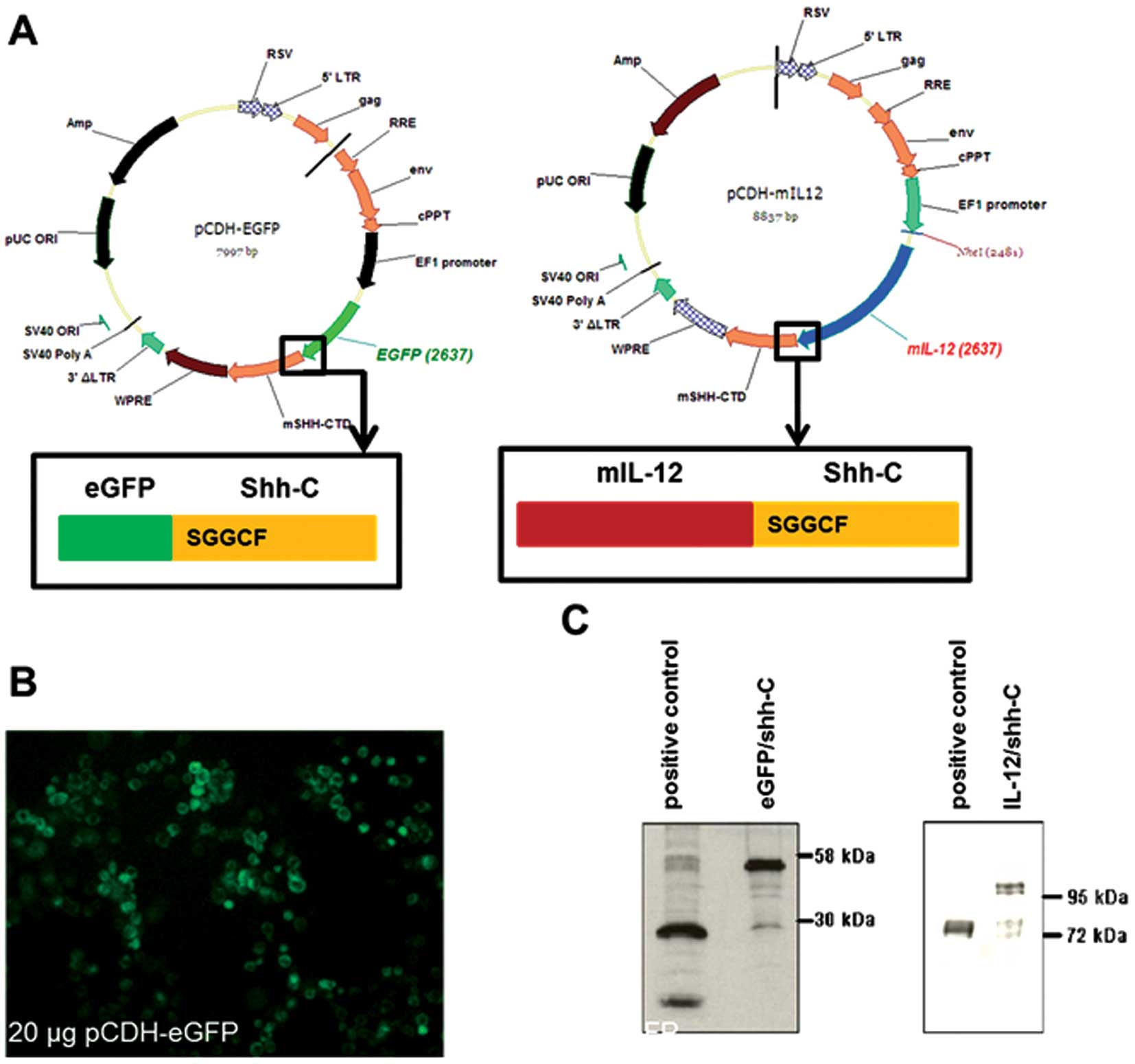
To construct the EGFP/Shh-C and murine IL-12/Shh-C
fusion genes, the N-terminal domain of the mouse sonic Hh gene was
replaced with EGFP or murine IL-12 (with signal peptides deleted),
leaving the conserved cleavage-processing motif, SGGCF, intact. The
fusion genes were then cloned into lentiviral expression vectors
(Fig. 1A). The fusion genes were
successfully expressed and processed in 293TN cells as expected. As
shown in Fig. 1B, EGFP was
concentrated around the periphery of cells forming fluorescent
holo-circles, indicating that the EGFP fusion protein was
successfully processed and tethered on the cell membrane. In
Fig. 1C, fusion EGFP (52 kDa),
processed EGFP (27 kDa), fusion IL-12 (95 kDa) and processed IL-12
(70 kDa) were all detected by western blot analysis.
NK cell transduction by fusion gene
recombinant lentiviruses
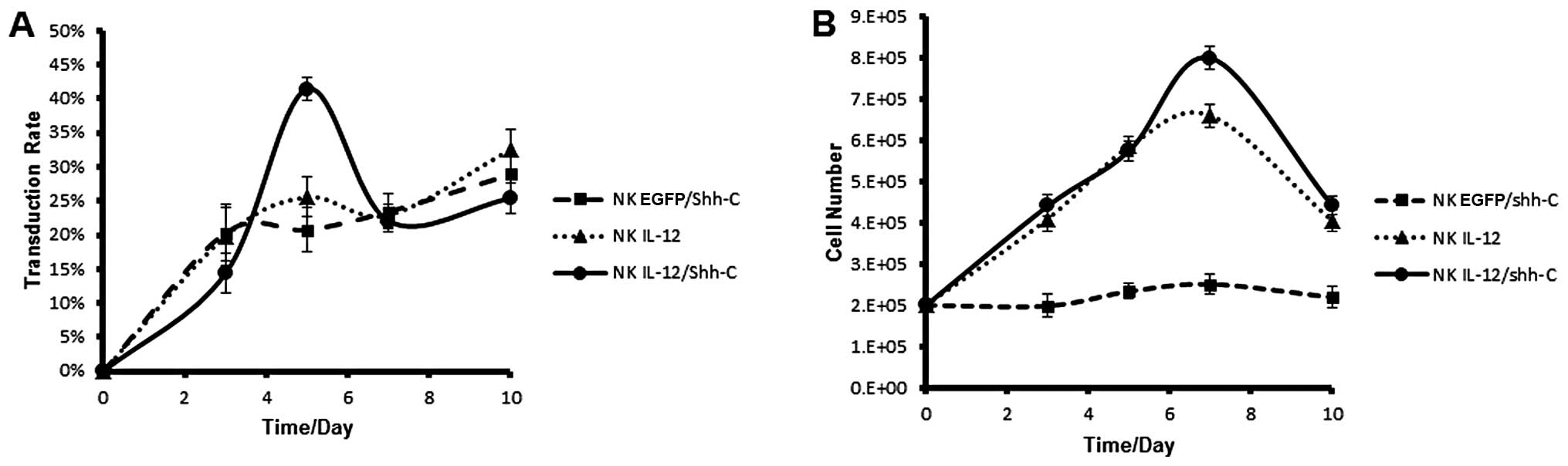
NK cells were doubly-transduced with EGFP/Shh-C,
IL-12 or IL-12/Shh-C recombinant lentiviruses at a MOI of 1 on both
days 7 and 8 after in vitro culture. On days 3, 5, 7 and 10
after the second transduction, the transduction rates were assayed
with FACS analysis. Cell numbers were determined by trypan blue
exclusion assay. All 3 recombinant lentiviruses had similar
transduction rates (∼30%) on day 10 after the second transduction
(Fig. 2A). However, NK cells
transduced with IL-12 and IL-12/Shh-C recombinant lentiviruses had
higher survival/proliferation rates resulting in significantly
higher cell numbers (Fig. 2B).
IL-12 and IL-12/Shh-C fusion proteins
enhance NK cell proliferation in vitro
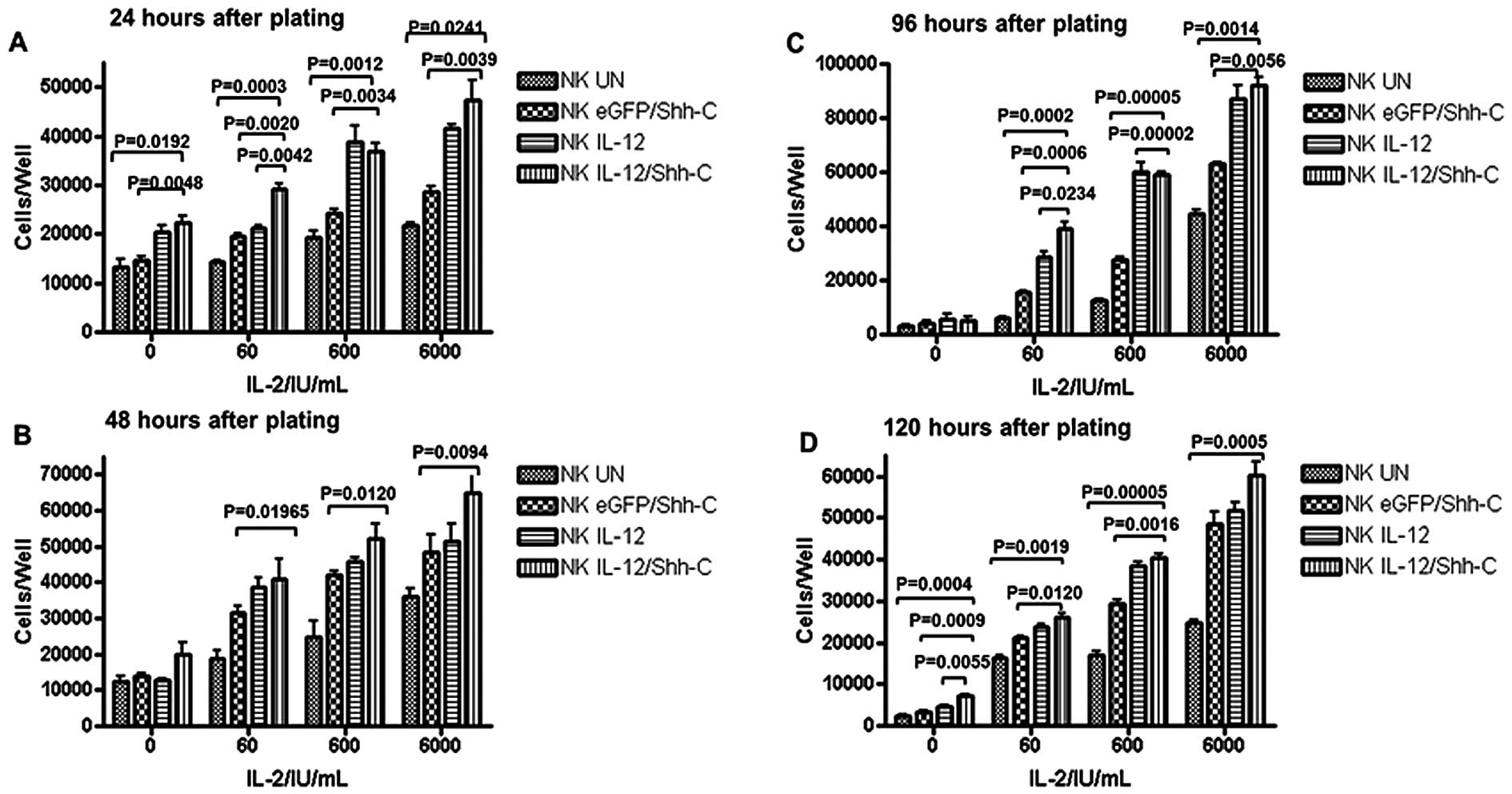
NK cells untransduced (NKUN) or
transduced with different recombinant lentiviruses were plated at a
concentration of 15,000 cells/well in a 96-well plate and cultured
with different concentrations of IL-2. NKIL-12/Shh-C
cells had a substantially faster growth rate compared to
NKUN cells and NKEGFP/Shh-C cells; even when
IL-2 was absent, NKIL-12/Shh-C cells showed an apparent
advantage in expansion compared to NKUN cells and
NKEGFP/Shh-C cells at 24 and 96 h after plating
(Fig. 3A–D). However, IL-12 cannot
be the sole driving force for NK cell survival in the long-term
cultures. The effect of IL-12 was most significant when IL-2 was
added at low or medium levels (Fig.
3A–D). NKIL-12/Shh-C cells, when given <10-fold
addition of IL-2, reached the same cell numbers as
NKEGFP/Shh-C cells in most cases.
Cytotoxicity of NK cells significantly
increased after lentivirus infection
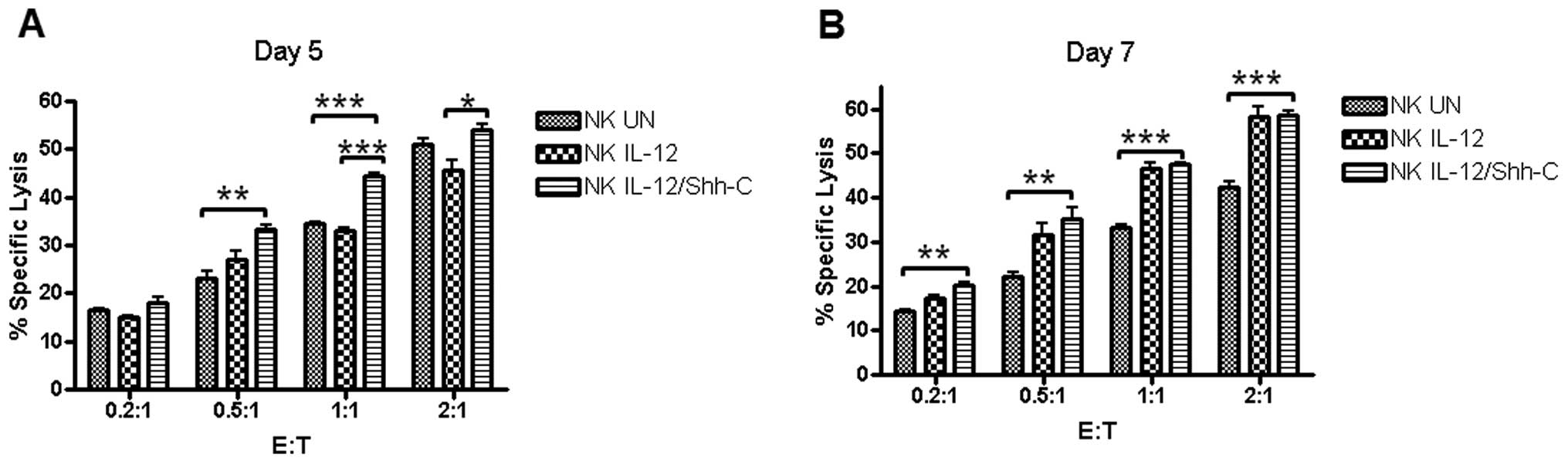
Mouse lymphoma YAC-1 cells were used as the target
and stained with 0.5 μM Guava CFSE (Guava Technologies) per
2.5×106 cells. NK cells were mixed with YAC-1 cells in
96-well round bottom plates to yield E:T ratios of 2:1, 1:1, 0.5:1
and 0.2:1. Five days after double transduction, statistically
significant differences between the cytotoxicities of
NKIL-12/Shh-C and NKIL-12 cells were observed
at E:T ratios of 1:1 (p=0.00017) and 2:1 (p=0.02152), as shown in
Fig. 4A. Additionally,
NKUN cells had a significantly lower cytotoxicity than
NKIL-12/Shh-C cells at E:T ratios of 0.5:1 (p=0.00413)
and 1:1 (p=0.00026). Furthermore, 7 days after double transduction,
significantly elevated cytotoxicities of NKIL-12/Shh-C
cells were observed compared to NKUN cells at all E:T
ratios, as shown in Fig. 4B.
NKIL-12 cells showed enhanced cytotoxicity at an E:T
ratio of 1:1 and 2:1 only, in comparison to the effect of
NKUN cells.
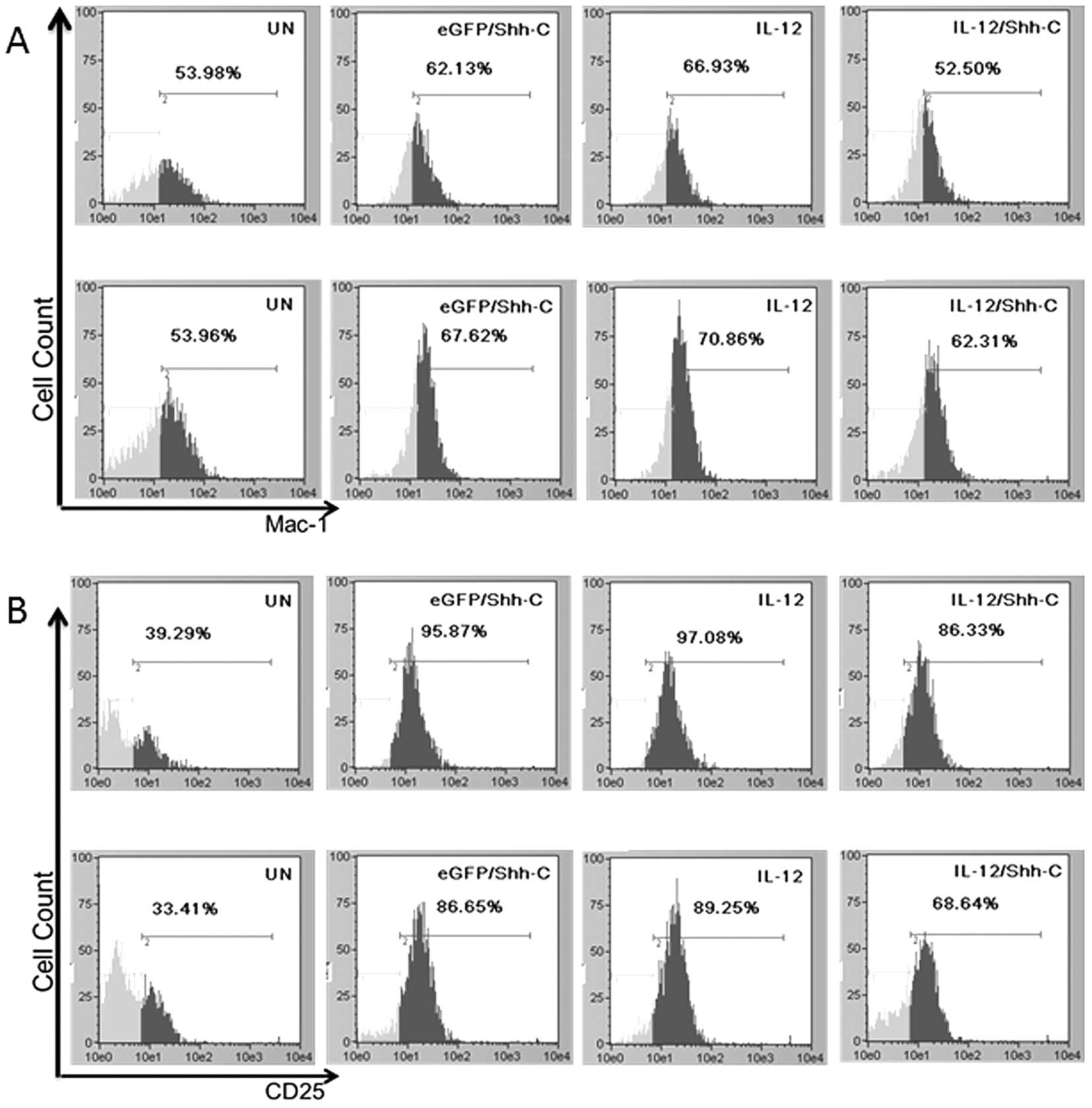
Phenotypic characterization of
recombinant lentivirus-infected NK cells
The percentage of the Mac-1-positive subpopulation
in lentivirus-transduced NK cells was significantly higher than
that of untransduced NK cells. Statistical analysis showed that
NKIL-12/Shh-C cells had insignificantly less
Mac-1hi population compared with NKUN cells
and NKEGFP/Shh-C cells at 7 days after lentivirus double
transduction. On day 9, NKIL-12/Shh-C cells had
considerably more Mac-1hi cells than NKUN
cells (p=0.00515) (Fig. 5A). For
CD25, all the recombinant lentivirus-transduced NK cells showed
significantly higher expression levels compared to the untransduced
NK cells on both days 7 and 9 after transduction.
NKIL-12/Shh-C cells had significantly lower CD25
expression compared to the NKIL-12 and
NKEGFP/Shh-C cells (Fig.
5B). Perforin and granzyme B levels were also significantly
higher in all lentivirus-transduced cells; however, granzyme B
expression in NKIL-12/Shh-C cells was significantly
lower than that in NKIL-12 cells (p=0.01349) or
NKEGFP/Shh-C cells (p=0.01816). Additionally,
significantly lower perforin production was observed in
NKIL-12/Shh-C cells when compared to NKIL-12
cells (p=0.04784) (Fig. 5C and
D).
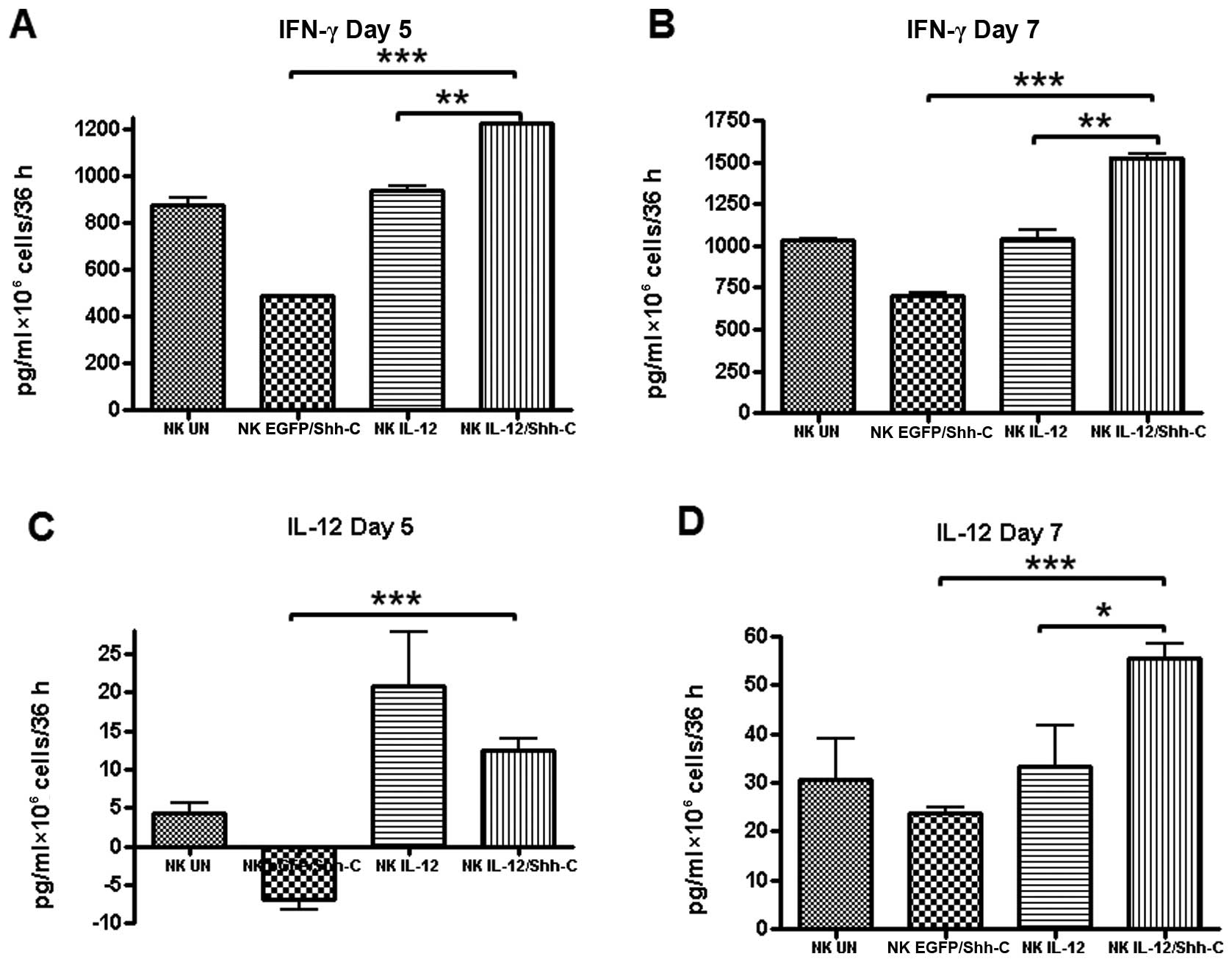
Enhanced IFN-γ and IL-12 production by
IL-12/Shh-C-infected NK cells
The cell culture supernatants were collected on the
5th and 7th day after double transduction and used for the
measurements of IFN-γ and IL-12 by ELISA. As shown in Fig. 6A and B, NKIL-12/Shh-C
cells secreted significantly higher amounts of IFN-γ on day 5
(p=0.00240) and even higher on day 7 (p=0.00121) than those
secreted by NKIL-12 cells. In addition, the IFN-γ yield
of NKIL-12/Shh-C cells was also considerably higher than
that of NKEGFP/Shh-C cells on both days (p<0.001).
The production of IL-12 was substantially higher by
NKIL-12/Shh-C cells compared to that by
NKIL-12 cells at 7 days after double transduction
(p=0.01186). NKEGFP/Shh-C cells released significantly
less IL-12 than NKIL-12/Shh-C on both days
(p<0.001).
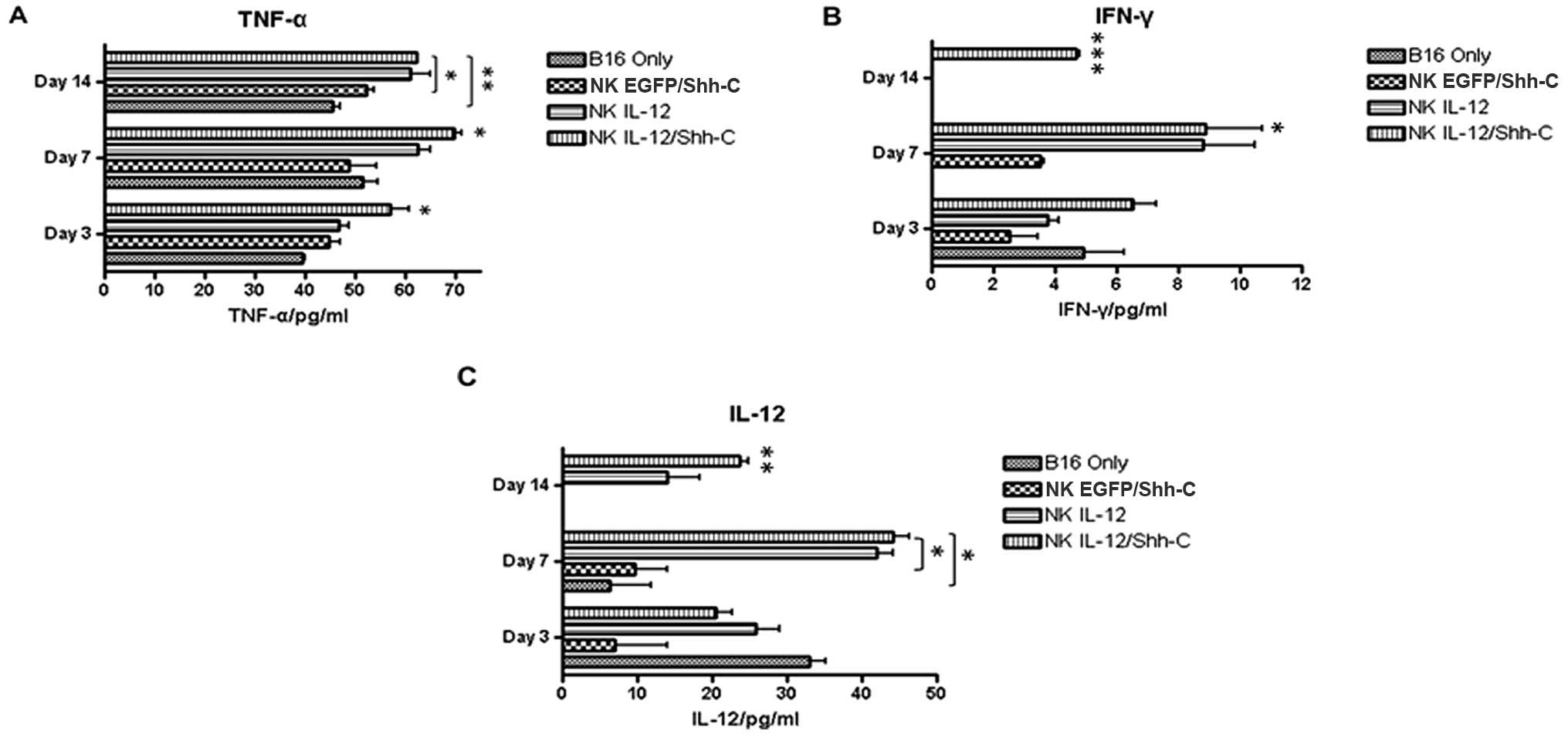
Elevated plasma cytokine secretion in
mice receiving adoptively transferred A-NK cells
Five million transduced or non-transduced NK cells
from B6.PL-Thy1a/CyJ mice were adoptively transferred into C57BL/6
mice i.v. challenged with B16 melanoma tumor cells 3 days earlier.
Serum samples from mouse tail veins were collected on days 3, 7 and
14 after adoptive transfer. Mice which had received
NKIL-12/Shh-C cells showed significantly higher levels
of TNF-α on days 3 (p=0.04410), 7 (p=0.03010) and 14 (p=0.00880)
compared to the B16 only group, as shown in Fig. 7A. At 14 days after adoptive
transfer, only mice which had received NKIL-12/Shh-C
cells showed a detectable level of IFN-γ (Fig. 7B). The IL-12 level was
significantly higher on day 7 (p=0.01898) in the mice which had
received NKIL-12/Shh-C cells compared with the mice
which had received NKEGFP/Shh-C cells (Fig. 7B and C).
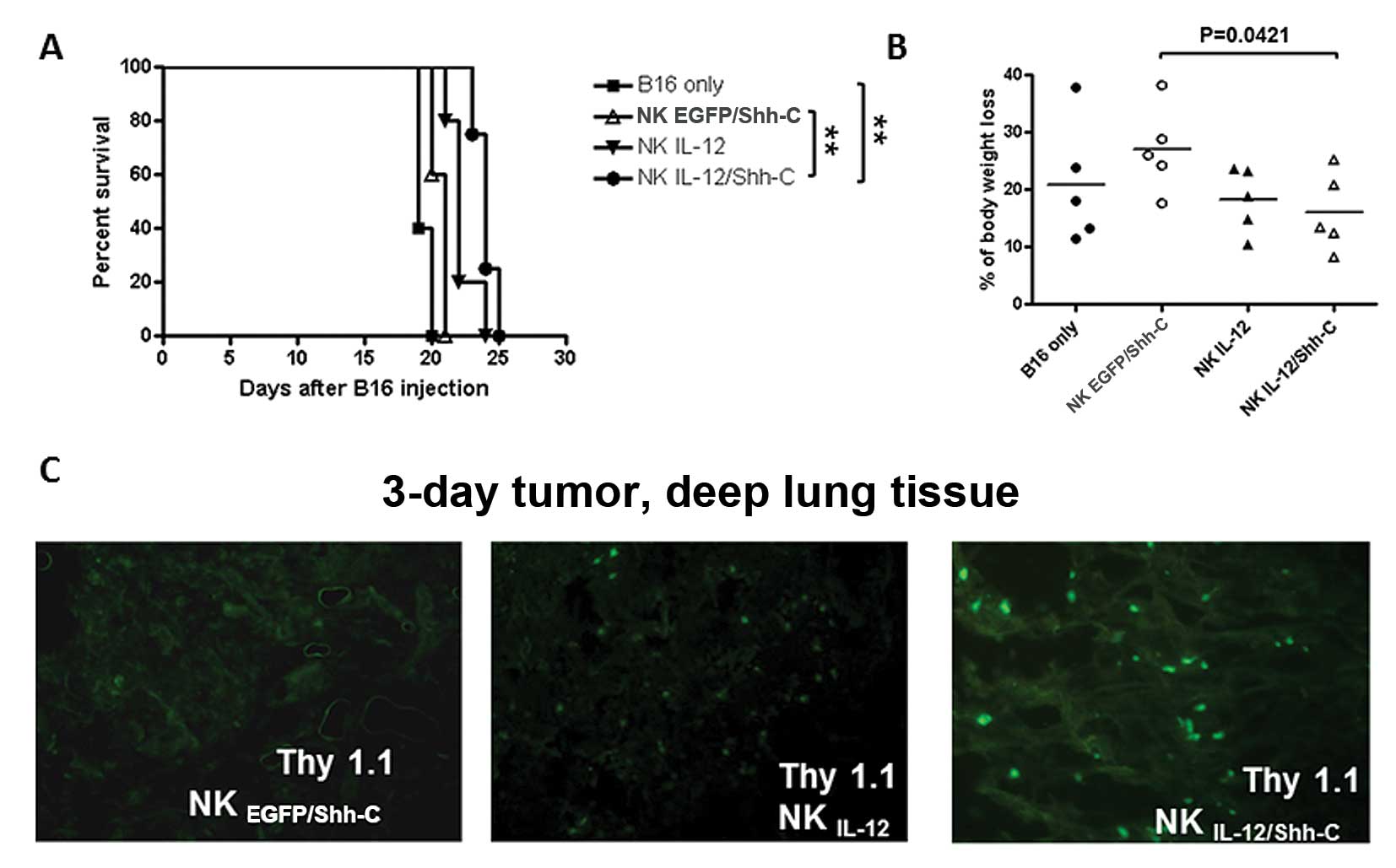
Adoptively transferred NK cells
infiltrate into lung tissue and prolong the survival time of
tumor-bearing mice
Five million transduced or non-transduced NK cells
from B6.PL-Thy1a/CyJ mice were adoptively transferred into C57BL/6
mice i.v. challenged with B16 melanoma tumor cells 3 or 10 days
earlier without IL-2 systemic administration. Tumor-bearing mice
which had received NKEGFP/Shh-C, NKIL-12 and
NKIL-12/Shh-C cells all resulted in significantly higher
overall survival rates than those of the B16 only mice (p=0.0179,
p=0.0023 and p=0.0052, respectively; Fig. 8A). In addition, mice which had
received NKIL-12 and NKIL-12/Shh-C cells
showed significantly prolonged survival times than mice which had
received NKEGFP/Shh-C cells (p=0.0116 and p=0.0067,
respectively).
The localization of adoptively transferred NK cells
in lung tissues with 3-day metastasized B16 tumor cells was
examined at 72 h after adoptive transfer. Though no exogenous IL-2
injection was given, all groups of donor NK cells were found to be
infiltrated into the deep lung tissues (Fig. 8B). More NKIL-12/Shh-C
cells were accumulated in the lung tissue indicated by the bigger
and brighter fluorescent dots. However, when the late tumor model
(10-day) was examined, donor NK cells were found to be difficult to
target in the deep lung tissues (Fig.
8C).
Discussion
The immunogenicity of tumor cells is frequently
suppressed by the down-regulation of major histocompatibility
complex (MHC)-I and -II expression (25). On the contrary, the NK
cell-mediated innate immune response does not require MHC for
antigen presentation (26).
Therefore, anti-tumor immune responses from NK cells would
potentially be a great alternative to T cell-mediated immunotherapy
for malignant tumors.
Two different approaches for the therapeutic use of
NK cells have been exploited, including endogenous NK cell
activation and adoptive transfer of in vitro activated
autologous NK cells (27). For
adoptive transfer, various animal studies have proven that
autologous NK cell transfer is effective in tumor therapy (28). However, clinical trials have failed
to meet the high expectations (29). One important contributing factor to
the poor outcome in a clinical setting is the severe side-effects
caused by IL-2 (30), which needs
to be administered in very high doses to patients to ensure the
survival and sustained activation of IL-2-A-NK cells. IL-12 has
been shown to enhance the antitumor activity of NK cells while
reducing their dependency on IL-2 (8). A fusion gene between mouse Shh-C
auto-processing domain and IL-12 (IL-12/Shh-C) was designed in the
present study as a self-supporting autocrine system aimed at
reducing or eliminating the dependency of NK cells on exogenous
IL-2 to sustain their activated status. The data from the present
study show that NKIL-12/Shh-C cells have a substantially
faster growth rate compared to the NKUN and
NKEGFP/Shh-C cells, even in the absence of IL-2.
However, IL-12 cannot be the sole driving force for NK cell
survival in long-term cultures. The effect of IL-12 was most
significant when IL-2 was added at low or medium levels. The
NKIL-12/Shh-C cells, when given <10-fold
supplementation of IL-2, reached the same growth rate as
NKEGFP/Shh-C cells. Also, the Mac-1hi cell
population in NKIL-12/Shh-C cells was not significantly
higher than that in NKUN cells 7 days after double
transduction, suggesting that NKIL-12/Shh-C cells are
less mature as a result of their significantly higher proliferation
rate. However, after an additional 2 days in culture, the newly
divided NK cells reached their maturation stage and the
Mac-1hi cell percentage was substantially higher than
the NKUN cells. Our results are in agreement with those
from previous reports that only DX5+Mac-1low
cells in the expansion stage have the potential to be proliferating
NK cells. In addition, NKIL-12/Shh-C cells had a
significantly lower level of granzyme B expression than that of
NKEGFP/Shh-C and NKIL-12 cells, further
suggesting that NKIL-12/Shh-C cells were less mature 7
days after double transduction.
When mouse lymphoma YAC-1 cells were used as a
target, the cytotoxicities of NKIL-12/Shh-C cells 5 days
after double transduction were significantly higher than those of
NKUN cells only at E:T ratios of 1:1 and 0.5:1. In
addition, 7 days after double transduction, significantly elevated
cytotoxicities of both NKIL-12/Shh-C cells were observed
compared to NKUN cells at all E:T ratios. Furthermore,
NKIL-12/Shh-C cells secreted significantly higher
amounts of IFN-γ on the 5th day and even higher on the 7th day than
those of NKIL-12 and NKEGFP/Shh-C cells. NK
cell cytotoxicity is mediated through 3 different pathways: granule
exocytosis pathway, death receptor pathway, and IFN-γ mediated
cytotoxicity (31). The present
study shows that the enhanced cytotoxicity of
NKIL-12/Shh-C cells is mainly due to their increased
secretion of cytotoxic cytokines, such as IFN-λ. Although perforin
and granzyme B expressions were significantly higher in all
lentivirus-transduced NK cells, the expression of these lytic
molecules in NKIL-12/Shh-C cells was significantly lower
than that in NKIL-12 and NKEGFP/Shh-C
cells.
The results from the current study showed that
adoptively transferred NK cells effectively localized to lung
tissues from mice which had received a tail vein injection of B16
tumor cells 3 days earlier. More NKIL-12/Shh-C cells
were accumulated in the lung tissues as indicated by the bigger and
brighter fluorescent dots, presumably due to the accumulation of
more NKIL-12/Shh-C cells or the increased survival of
the accumulated NK cells as a result of locally-accumulated IL-12.
Substantially prolonged mice survival and significantly less total
weight loss (data not shown) in mice were found following adoptive
transfer of NKIL-12/Shh-C cells when compared to mice
with NKEGFP/Shh-C cell transfer, even in the absence of
IL-2 systemic administration. Plasma cytokine analysis showed that
the levels of TNF-α and IFN-λ in the plasma of mice which had
received adoptive transfers of NKIL-12/Shh-C cells were
significantly higher than those in the plasma of mice which had
received NKEGFP/Shh-C or B16 tumor cells. Particularly
on day 14 after the adoptive transfers of NK cells, only mice which
had received NKIL-12/Shh-C cells showed a detectable
level of IFN-γ. It has been suggested that elevated IFN-γ and TNF-α
levels are able to promote nitric oxide production to recruit
macrophages to the tumor site. IFN-γ was also able to up-regulate
MHC class I expression to make target cells more susceptible to T
cell-mediated immune response (32). Both IFN-γ and TNF-α have been
reported to up-regulate tumor antigen expression, thus making
tumors more vulnerable to tumor-infiltrating lymphocyte killing.
However, when NK cells were adoptively transferred 10 days after
B16 tumor cell injection, very few adoptively transferred NK cells
were detected in the deep lung tissue, suggesting that it is very
difficult for the adoptively transferred NK cells to penetrate into
the tumor mass at this stage. B16 metastases have been documented
to be divided into loose and compact types morphologically
(8). The compact metastases have
several-fold less expression of chemoattractive proteins and they
also lack microvascular networks compared to the loose type. These
unique features of late tumor tissues may partially explain the
inability of NK cells to infiltrate into late tumor tissues.
Local or regional treatment eliminates the systemic
adverse reactions caused by soluble cytokines. Previous studies
have suggested that the efficiency of the delivery of cytokines
into the plasma membrane can be elevated through the reanchoring of
GPI-anchored proteins onto neighboring cells (14). With the current strategy, IL-12
with a cholesterol tail would not only anchor to the membrane, but
also diffuse into the near vicinity of the producing NK cells to
form a concentration gradient. Although the secreted IL-12 level is
low, as it can only be detected by ELISA, and not western blot
analysis, it should be enough to act on other NK cells or immune
cells coming into the gradient range, and would thus have more far
reaching effects than just the producing cell alone.
References
|
1
|
Moretta A, Bottino C, Mingari MC, Biassoni
R and Moretta L: What is a natural killer cell? Nat Immunol. 3:6–8.
2002. View
Article : Google Scholar
|
|
2
|
Smyth MJ, Thia KY, Cretney E, et al:
Perforin is a major contributor to NK cell control of tumor
metastasis. J Immunol. 162:6658–6662. 1999.PubMed/NCBI
|
|
3
|
Lauwerys BR, Garot N, Renauld JC and
Houssiau FA: Cytokine production and killer activity of NK/T-NK
cells derived with IL-2, IL-15, or the combination of IL-12 and
IL-18. J Immunol. 165:1847–1853. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Smith BR, Rosenthal DS and Ault KA:
Natural killer lymphocytes in hairy cell leukemia: presence of
phenotypically identifiable cells with defective functional
activity. Exp Hematol. 13:189–193. 1985.
|
|
5
|
Biron CA, Nguyen KB, Pien GC, Cousens LP
and Salazar-Mather TP: Natural killer cells in antiviral defense:
function and regulation by innate cytokines. Annu Rev Immunol.
17:189–220. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rosenberg S: Lymphokine-activated killer
cells: a new approach to immunotherapy of cancer. J Natl Cancer
Inst. 75:595–603. 1985.PubMed/NCBI
|
|
7
|
Trinchieri G, Matsumoto-Kobayashi M, Clark
SC, Seehra J, London L and Perussia B: Response of resting human
peripheral blood natural killer cells to interleukin 2. J Exp Med.
160:1147–1169. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goding SR, Yang Q, Knudsen KB, Potter DM
and Basse PH: Cytokine gene therapy using adenovirally transduced,
tumor-seeking activated natural killer cells. Hum Gene Ther.
18:701–711. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rosenberg SA, Lotze MT, Muul LM, et al:
Observations on the systemic administration of autologous
lymphokine-activated killer cells and recombinant interleukin-2 to
patients with metastatic cancer. N Engl J Med. 313:1485–1492. 1985.
View Article : Google Scholar
|
|
10
|
Yang Q, Hokland ME, Bryant JL, et al:
Tumor-localization by adoptively transferred,
interleukin-2-activated NK cells leads to destruction of
well-established lung metastases. Int J Cancer. 105:512–519. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Loza MJ and Perussia B: The IL-12
signature: NK cell terminal CD56+high stage and effector
functions. J Immunol. 172:88–96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maas RA, Dullens HF and Den Otter W:
Interleukin-2 in cancer treatment: disappointing or (still)
promising? A review. Cancer Immunol Immunother. 36:141–148. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Imboden M, Shi F, Pugh TD, et al: Safety
of interleukin-12 gene therapy against cancer: a murine
biodistribution and toxicity study. Hum Gene Ther. 14:1037–1048.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ji J, Li J, Holmes LM, et al:
Glycoinositol phospholipid-anchored interleukin 2 but not secreted
interleukin 2 inhibits melanoma yumor growth in mice. Mol Cancer
Ther. 1:1019–1024. 2002.PubMed/NCBI
|
|
15
|
Konstantinidis KV, Alici E, Aints A,
Christensson B, Ljunggren HG and Dilber MS: Targeting IL-2 to the
endoplasmic reticulum confines autocrine growth stimulation to
NK-92 cells. Exp Hematol. 33:159–164. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gurdon JB and Bourillot PY: Morphogen
gradient interpretation. Nature. 413:797–803. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cadigan KM: Regulating morphogen gradients
in the Drosophila wing. Semin Cell Dev Biol. 13:83–90. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lawrence PA and Struhl G: Morphogens,
compartments, and pattern: lessons from Drosophila? Cell.
85:951–961. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tabata T and Kornberg TB: Hedgehog is a
signaling protein with a key role in patterning Drosophila
imaginal discs. Cell. 76:89–102. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Teleman AA, Strigini M and Cohen SM:
Shaping morphogen gradients. Cell. 105:559–562. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vincent JP and Dubois L: Morphogen
transport along epithelia, an integrated trafficking problem. Dev
Cell. 3:615–623. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mann RK and Beachy PA: Novel lipid
modifications of secreted protein signals. Annu Rev Biochem.
73:891–923. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guerrero I and Chiang C: A conserved
mechanism of Hedgehog gradient formation by lipid modifications.
Trends Cell Biol. 17:1–5. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vincent S, Thomas A, Brasher B and Benson
JD: Targeting of proteins to membranes through hedgehog
auto-processing. Nat Biotechnol. 21:936–940. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sanda MG, Restifo NP, Walsh JC, et al:
Molecular characterization of defective antigen processing in human
prostate cancer. J Natl Cancer Inst. 87:280–285. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Robertson MJ and Ritz J: Biology and
clinical relevance of human natural killer cells. Blood.
76:2421–2438. 1990.PubMed/NCBI
|
|
27
|
Albertsson PA, Basse PH, Hokland M, et al:
NK cells and the tumour microenvironment: implications for NK-cell
function and anti-tumour activity. Trends Immunol. 24:603–609.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Basse PH, Whiteside TL and Herberman RB:
Cancer immunotherapy with interleukin-2-activated natural killer
cells. Mol Biotechnol. 21:161–170. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Law TM, Motzer RJ, Mazumdar M, et al:
Phase III randomized trial of interleukin-2 with or without
lymphokine-activated killer cells in the treatment of patients with
advanced renal cell carcinoma. Cancer. 76:824–832. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kammula US, White DE and Rosenberg SA:
Trends in the safety of high dose bolus interleukin-2
administration in patients with metastatic cancer. Cancer.
83:797–805. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Herberman RB, Reynolds CW and Ortaldo JR:
Mechanism of cytotoxicity by natural Killer (NK) cells. Annu Rev
Immunol. 4:651–680. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Skoskiewicz MJ, Colvin RB, Schneeberger EE
and Russell PS: Widespread and selective induction of major
histocompatibility complex-determined antigens in vivo by gamma
interferon. J Exp Med. 162:1645–1664. 1985. View Article : Google Scholar : PubMed/NCBI
|