Introduction
With approximately 738,000 deaths per year and an
overall five-year survival rate below 30%, gastric cancer is the
second most common cause of cancer mortality worldwide (1–3).
This high level of mortality is due to the fact that most gastric
cancer patients are diagnosed with locally advanced or metastatic
disease. Therefore, the treatment options are limited, and new
therapeutic approaches are urgently needed.
The dysregulation of the expression of the epidermal
growth factor receptor (EGFR) is considered an important step in
tumorigenesis. As EGFR is overexpressed in approximately 27 to 42%
of gastric tumors (4,5), it represents an interesting target
for therapeutic intervention. One therapeutic approach is the use
of monoclonal antibodies that target EGFR. Cetuximab is one such
monoclonal antibody and is approved for the treatment of recurrent
colorectal cancer (CRC) and squamous cell carcinoma of the head and
neck (SCCHN). Furthermore, a number of phase II studies on gastric
cancer patients treated with various combinations of cetuximab with
chemotherapy have shown promising results (6–9). A
multinational phase III trial of cetuximab plus chemotherapy is
on-going.
Experience with CRC and SCCHN therapies has shown
that only a subgroup of patients benefits from cetuximab-based
therapy. Well-known cetuximab resistance mechanisms include
activating mutations in the Kirsten-Ras gene (KRAS),
leading to a constantly activated EGFR pathway (10). Nevertheless, in many cases, the
underlying resistance mechanism remains unclear. For optimal
patient selection, it is therefore necessary to identify additional
markers that are predictive of the cetuximab response.
On the molecular level, cetuximab inhibits EGFR
activation by binding to the EGFR ligand binding site with a
significantly higher affinity than the members of the family of
EGFR-binding ligands (11). This
group of EGFR-binding ligands consists of the epidermal growth
factor (EGF), amphiregulin (AREG), epiregulin (EREG), epigen
(EPGN), transforming growth factor α (TGFα), betacellulin (BTC) and
heparin-binding EGF (HB-EGF) (12–17).
Ligand binding results in EGFR dimerization, the activation of its
tyrosine kinase domain and subsequent signal transduction via a
cascade of EGFR signaling pathway intermediates. Finally, processes
that play an important role in tumorigenesis are activated; these
processes include cell survival, proliferation and migration. In
contrast to ligand binding, cetuximab binding results in EGFR
internalization without further activation of the receptor and the
downstream signaling pathway (18).
Previously, it was shown that AREG expression and
EREG expression are positive predictive markers for the outcome of
CRC patients treated with cetuximab in combination with
chemotherapy (19,20). Among KRAS wild-type tumors,
patients with high expression levels of AREG and EREG are highly
likely to respond to these therapy regimens. In addition to
KRAS, AREG and EREG, a predictive role in EGFR-inhibitory
therapy has been discussed for EGFR, EGFR signaling intermediates,
such as PI3K, PTEN or BRAF, and other EGFR ligands (21). A number of studies have suggested a
negative predictive value for other receptor tyrosine kinases
(RTKs), such as the hepatocyte growth factor receptor, MET
(22–24).
In gastric cancer, the search for predictive markers
for the response to cetuximab-based therapies is on-going. As the
prevalence of KRAS mutations in gastric cancer is low
(25), a correlation between
KRAS mutations and therapy response is difficult to
establish. In contrast to CRC, no significant correlation between
AREG expression and the response rate was found in gastric cancer
patients treated with cetuximab in combination with oxaliplatin,
leucovorin and 5-fluorouracil in a recent clinical phase II trial
(26).
In the present study, we analyzed the cetuximab
responsiveness of several gastric cancer cell lines (AGS, AZ521,
Hs746T, LMSU and MKN1) and assessed the predictive value of EGFR
and its ligands AREG and EGF alone and in combination with
activating KRAS mutations and MET activation. We introduced
a hierarchy between these markers and established a model that
facilitates the correct classification of all five gastric cancer
cell lines as cetuximab-responsive or -non-responsive. The model
was validated with three other human gastric cancer cell lines,
KATOIII, MKN28 and MKN45.
Materials and methods
Cell lines and cultivation
conditions
The human gastric cancer cell lines AGS, KATOIII,
MKN1, MKN28 and MKN45 were cultured in RPMI-1640 medium
(Invitrogen/Gibco, Darmstadt, Germany) supplemented with 10% fetal
bovine serum Sera Plus (PAN Biotech, Aidenbach, Germany), 2 mM
l-glutamine (Invitrogen/Gibco) and penicillin-streptomycin (PAA
Laboratories, Pasching, Austria; 100 international units (IU)/ml,
100 μg/ml) as reported previously (24). LMSU cells were grown in Nutrient
Mixture F-10 Ham medium (Sigma-Aldrich Chemie GmbH, Steinheim,
Germany), AZ521 cells were cultured in Minimum Essential Medium
Eagle (MEM, Sigma-Aldrich Chemie GmbH), and Hs746T cells were grown
in Dulbecco’s modified Eagle’s medium (DMEM) with GlutaMAX™ I
(Invitrogen/Gibco), 4500 mg/l d-glucose and sodium pyruvate; all
three media were supplemented with 10% fetal bovine serum Sera Plus
and penicillin-streptomycin (100 IU/ml, 100 μg/ml). The
cells were grown at 37°C in a humidified 5% CO2
atmosphere. The absence of mycoplasma was ensured in the
conditioned medium after thawing frozen cells. Cell lines were used
until passage 30.
Cell line source and cell validation
testing
The AGS and KATOIII cells were obtained from the
European Collection of Cell Cultures (ECACC), a Health Protection
Agency Culture Collection Supplier of authenticated and quality
controlled cell lines and nucleic acids (Porton Down, Salisbury,
UK; http://www.hpacultures.org.uk/collections/ecacc.jsp).
MKN1, AZ521 and LMSU cells were supplied by the Cell Bank RIKEN
BioResource Center (Tsukuba, Japan). MKN28 cells were kindly
provided by Dr V. Wachek (Medical University of Vienna, Vienna,
Austria). MKN45 cells were obtained from Professor M. Ebert
(Technische Universität München, Klinikum rechts der Isar, Munich,
Germany). Hs746T cells were obtained from the ATCC Cell Biology
Collection (LGC Standards GmbH, Wesel, Germany). The cell
validation testing was performed as described previously (24). In addition, authentication of the
Hs746T cell line was performed by short tandem repeat profiling
using the Cell ID™ system (Promega, Mannheim, Germany).
XTT cell proliferation assay
The XTT cell proliferation kit II (Roche
Diagnostics, Mannheim, Germany) was used according to the
manufacturer’s instructions to assess growth inhibitory effects, as
described previously (24). A
modification of the US National Cancer Institute (NCI) protocol for
in vitro cancer screens was used to determine cellular
sensitivity to cetuximab (27,28).
According to the individual doubling times of the different cell
lines, cells were plated at densities between 1×103 and
4×103 cells per well in 80 μl of culture
medium.
After 24 h of incubation at 37°C and 5%
CO2, cetuximab (Merck, Darmstadt, Germany) was added at
concentrations between 0 and 200 μg/ml in 20 μl of
culture medium. A cetuximab concentration of 100 μg/ml is
comparable to the active drug concentrations achieved in cancer
patients (29–31). After 48 h of incubation, 50
μl of XTT labeling mixture was added per well, and after 2 h
at 37°C and 5% CO2, the absorbance of the samples was
measured using a microplate reader (Asys Expert Plus, Biochrom,
Berlin, Germany).
DNA isolation and sequencing
DNA isolation was performed using a standard
protocol. Cells (1×106) were harvested, washed twice
with phosphate-buffered saline (PBS) [centrifugation at 1,000
rounds per minute (rpm), 3 min] and transferred to 1.5 ml reaction
tubes. The cell sediment was resuspended in 200 μl of 50 mM
Tris-HCl pH 8.5, 1 mM EDTA, 0.5 % Tween-20 and 0.2 mg/ml proteinase
K. After a 3-h incubation at 55°C, proteinase K was inactivated by
boiling for 10 min, and aliquots were used for the mutation
analysis.
The sequences of the primers used for the polymerase
chain reaction (PCR) were as follows (from 5′ to 3′): BRAF forward
(F), ACAGTAAAAATAGGTGATTTTGGTCTAGCTACAGA; BRAF reverse (R),
CTATGAAAATACTATAGTTGAGACCTTCAATGACTTTC; EGFR exon 18 F,
AGGGCTGAGGTGACCCTTGT; EGFR exon 18 R, TCCCCACCAGACCATGAGAG; EGFR
exon 19 F, GCACCATCTCACAATTGCCAGTTA; EGFR exon 19 R,
AAAAGGTGGGCCTGAGGTTCA; EGFR exon 21 F, CCTCACAGCAGGGTCTTCTCTGT;
EGFR exon 21 R, TCAGGAAAATGCTGGCTGACCTA; KRAS exon 2 F,
GGTGGAGTATTTGATAGTGTATTAACC; KRAS exon 2 R,
CCTCTATTGTTGGATCATATTCG; PIK3CA exon 9 F,
TTGCTTTTTCTGTAAATCATCTGTG; PIK3CA exon 9 R,
CTGCTTTATTTATTCCAATAGGTATG; PIK3CA exon 20 F, TGACATTTGAGCAAA
GACCTG; PIK3CA exon 20 R, CCTATGCAATCGGTCTTTGC.
The BRAF hotspot mutation, V600E, was
analyzed using allele-specific PCR following an established
protocol (32). Positive and
negative controls that had been verified by direct sequencing were
included in this analysis. The primers for the EGFR mutation
analysis were described previously (33,34).
The KRAS mutation analysis was performed as reported
previously (7). The EGFR,
KRAS and PIK3CA mutation analysis was performed using
PCR amplification followed by direct sequencing.
DNA sequencing analysis was performed using the
BigDye Terminator v1.1 Cycle Sequencing kit (Applied Biosystems,
Foster City, CA, USA), and the products were separated using an
automated sequencing system (3130 Genetic Analyzer, Applied
Biosystems).
Enzyme-linked immunosorbent assay
(ELISA)
For the ELISA-based detection of AREG and EGF,
DuoSet ELISA kits (R&D Systems, Minneapolis, MN, USA) were used
according to the manufacturer’s instructions. The ligand
concentrations were measured in the conditioned medium and in the
cellular extract.
For the determination of the AREG and EGF levels in
the conditioned medium of the cell cultures, 1×106 cells
were seeded into cell culture plates (10 cm in diameter) and
cultured in 10 ml of medium. EGF and cetuximab were added 2 h after
the cultures were seeded. At the indicated time-points, the
conditioned medium was harvested and centrifuged to remove the cell
debris (13,000 rpm, 4°C, 10 min). The supernatant was used for
subsequent analysis.
For the determination of AREG and EGF levels in the
cellular extract, 5×105 cells (AGS, AZ521),
8×105 cells (MKN28) or 1×106 cells (Hs746T,
KATOIII, LMSU, MKN1 and MKN45) were seeded as described above and
cultured for 48 h. Subsequently, the cells were lysed in 350
μl of lysis buffer (7X lysis buffer: 20 mM Tris-HCl, pH 7.5;
1 mM EDTA; Complete mini protease inhibitor cocktail tablets, Roche
Applied Science, concentration according to the manufacturer’s
instructions) and sonicated (25 sec, amplitude 70%). The cell
debris was removed by centrifugation as described above. A volume
of 100 μl of total protein (100 μg/ml) was used for
each sample.
Western blot analysis
The expression levels of EGFR, phosphorylated EGFR
(Y1068) and phosphorylated MET (Y1234/1235) were determined using a
standard protocol. For the western blot analysis, cells were seeded
at a density of 1×106 cells per 10-cm tissue culture
dish. After 48 h, the cells were washed with ice-cold PBS and lysed
on ice with 150 μl of L-CAM lysis buffer as described
previously (35). Between 10 and
30 μg of total protein were separated by SDS-polyacrylamide
gel electrophoresis and transferred to PVDF membranes (GE
Healthcare, Munich, Germany, no. RPN303F).
After 1 h of blocking, the membranes were probed
with appropriate primary antibodies overnight at 4°C: anti-EGFR
rabbit polyclonal antibody (no. 2232, Cell Signaling Technology,
distributed by New England Biolabs, Frankfurt, Germany; dilution
1:2,000), anti-phosphorylated EGFR (pEGFR) rabbit polyclonal
antibody directed against tyrosine residue 1068 (no. 44788G,
Invitrogen, Karlsruhe, Germany; dilution 1:2,000),
anti-phosphorylated MET (pMET) rabbit monoclonal antibody directed
against tyrosine residues 1234 and 1235 (no. 3077, Cell Signaling
Technology, dilution 1:1,000), anti-α-tubulin mouse monoclonal
antibody (no. T6199, Sigma-Aldrich Chemie GmbH; dilution 1:10,000)
and anti-β-actin mouse monoclonal antibody (no. A1978,
Sigma-Aldrich Chemie GmbH; dilution 1:5,000).
Detection was performed using horseradish
peroxidase-conjugated secondary antibodies by enhanced
chemiluminescence (Amersham, Braunschweig, Germany). For signal
quantification, blots were scanned and densitometrically analyzed
using Image J software 1.42q (National Institute of Health, MD,
USA).
Flow cytometry analysis
Cells were seeded at densities of 3×105
to 1×106 cells per 10-cm tissue dish and cultured for 72
h. After washing with PBS (without
Ca2+/Mg2+), the cells were detached using 1
ml of Versene (Invitrogen/Gibco) for 10–15 min at 37°C. Detached
cells were resuspended in ice-cold FACS buffer [1% (w/v) bovine
serum albumin (BSA; Sigma-Aldrich Chemie GmbH) in PBS (without
Ca2+/Mg2+)]. After two additional washing
steps [each comprising i) resuspension of the cells in FACS buffer,
ii) sedimentation at 300 g (4°C) for 3 min and iii) discarding the
supernatant], the cells (1×105) were incubated with
monoclonal antibody directed against EGFR (Ab-1, clone 528, Thermo
Fisher Scientific, Ulm, Germany; 2.5 μg/ml) in 50 μl
of FACS buffer in 96-well microtiter plates. Additionally, an
isotype control antibody [IgG2a, clone PPV-04, Exbio, Prague, Czech
Republic, distributed by Biozol (Eching, Germany; 2.5
μg/ml)] was routinely applied. After incubation for 1 h at
4°C in the dark, the cells were washed twice as described above and
incubated with 50 μl of secondary antibody solution [Alexa
Fluor 647 F(ab)2-fragment (H+L), Invitrogen, 5 μg/ml] for 1
h (4°C) in the dark. Two washing steps were performed to remove
unbound secondary antibodies. The cells were then resuspended in
300 μl of FACS buffer and subsequently analyzed using a
FACSCanto flow cytometer (BD Bioscience, Heidelberg, Germany). For
live-dead discrimination, propidium iodide was added to each sample
at a final concentration of 1 μg/ml directly before
measurement.
Statistical analysis
The data are presented as the means ± standard
deviation (SD). Pairwise comparisons of samples were performed by
two-sided Welch’s t-tests. In the XTT cell proliferation assay, one
sample t-tests were used to test the activity ratio of treated
samples to untreated samples against a reference value of 100%
which indicates equality of activity. All analyses were performed
on an explorative significance level of 0.05 using the statistical
software package R (The R Foundation for Statistical Computing,
Vienna, Austria). P-values at a significance level of <0.05 are
indicated by (*) and <0.01 by (**). A summary of all statistical
data is available from the authors upon request.
Results
Cetuximab responsiveness of gastric
cancer cell lines
The EGFR signaling pathway is involved in the
regulation of tumor cell proliferation. To determine the cetuximab
responsiveness of a panel of five human gastric cancer cell lines
(AGS, AZ521, Hs746T, LMSU and MKN1), cells were treated with
varying concentrations of the therapeutic antibody (0–200
μg/ml cetuximab). The metabolic activity of the cell lines
as a surrogate marker for cell viability was analyzed with the XTT
cell viability assay.
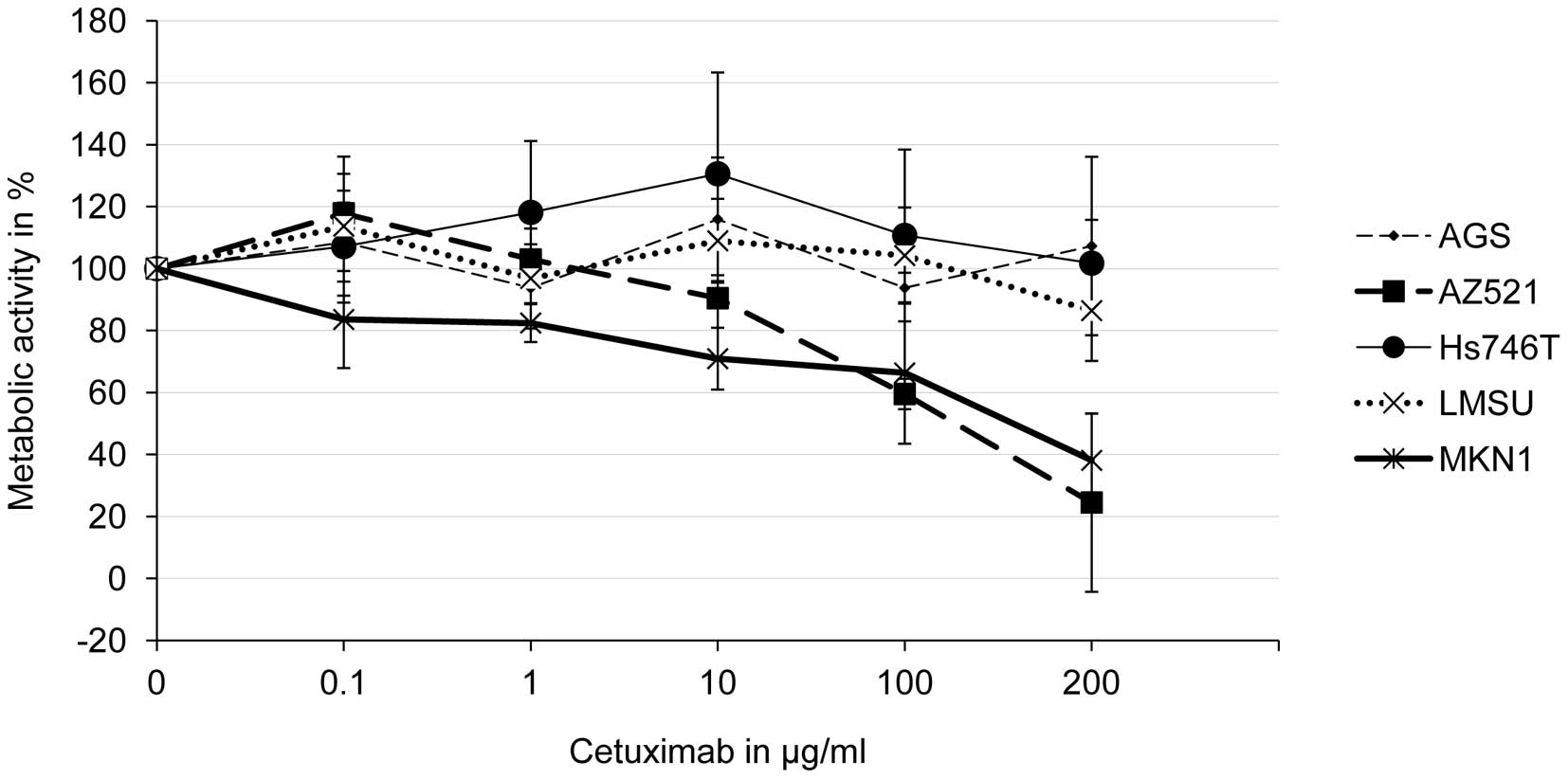
The concentration-response curves indicate that the
metabolic activities of AZ521 and MKN1 cells were significantly
reduced by cetuximab in a concentration-dependent manner (Fig. 1). Significant effects on MKN1 cells
were observed at a cetuximab concentration of 1 μg/ml and on
AZ521 cells at a cetuximab concentration of 100 μg/ml. The
cell lines, AGS, LMSU and Hs746T, were not
cetuximab-responsive.
Based on these results, the gastric cancer cell
lines, AZ521 and MKN1, were classified as cetuximab-responsive,
whereas the other considered cell lines (AGS, Hs746T and LMSU) were
regarded as cetuximab-resistant.
Association of cetuximab responsiveness
with the AREG and EGF levels
Previously, a correlation between the levels of EGFR
ligands and the response to cetuximab-based therapy was
demonstrated. The ligand, AREG, was shown to have a high predictive
value for the response to EGFR inhibitory therapy in CRC patients
(19), whereas a low EGF level was
associated with a higher probability to respond to cetuximab plus
chemotherapy in gastric cancer patients (26). To clarify the predictive role of
EGFR ligands in our gastric cancer model, we determined the amounts
of AREG and EGF secreted into the conditioned medium and in the
cellular extract by ELISA. Using this approach, it was possible to
distinguish between proteolytically released soluble ligands and
membrane-bound ligands.
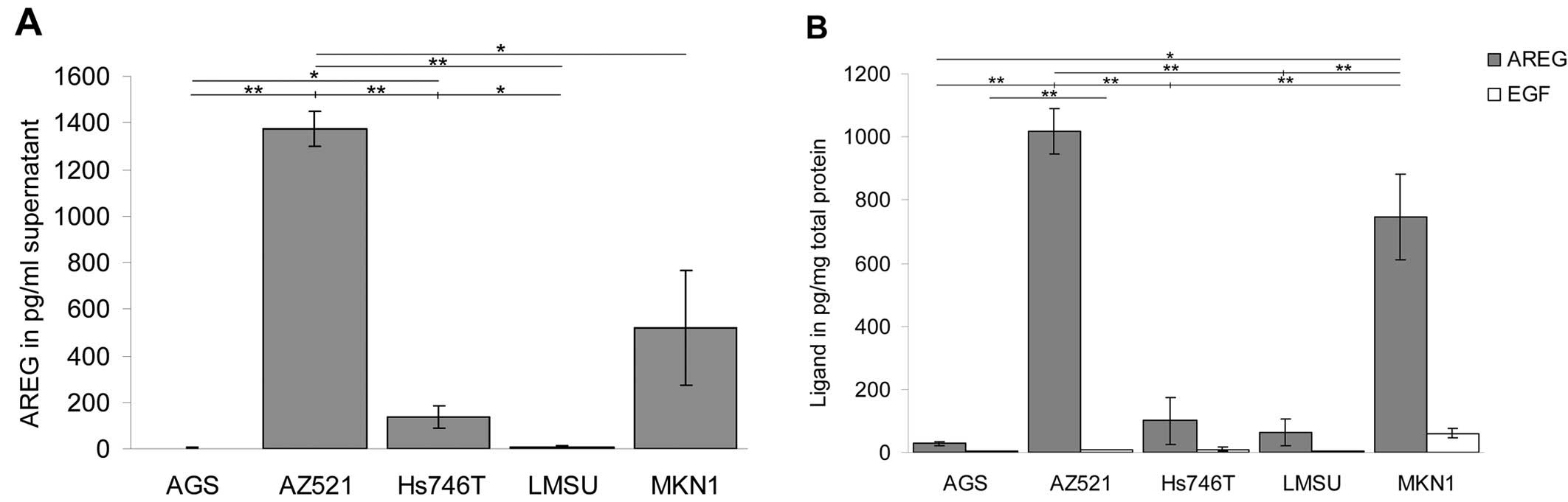
As shown in Fig. 2,
AREG was expressed and secreted by several cell lines. AZ521 cells
displayed the highest amount of AREG, with a mean concentration of
1,375 pg/ml in the conditioned medium and 1,017 pg/mg in the
cellular extract. A high level of AREG was also detected in the
MKN1 cells (519 pg/ml in the conditioned medium and 746 pg/mg in
the cellular extract). In comparison, only low amounts of AREG were
observed for the cell line Hs746T. In these cells, the AREG
concentrations were 135 pg/ml in the conditioned medium and 100
pg/mg in the cellular extract, levels 10-fold lower than those in
the AZ521 cells. It was not possible to detect significant
concentrations of AREG in the conditioned media of the AGS and LMSU
cell cultures. Furthermore, analysis of the cellular extract
revealed only marginal amounts of the ligand in these two cell
lines.
EGF was not found in significant concentrations in
the cell extracts or in the conditioned medium of most of the cell
lines. Only in MKN1 cells was a low amount of EGF detected in the
cellular extract; this EGF level was very close to the detection
limit of the assay.
These findings indicate that AREG is expressed in
the cetuximab-sensitive cell lines, AZ521 and MKN1, suggesting a
positive predictive value of the ligand in gastric cancer cells. By
contrast, due to the low detected EGF levels, no correlation could
be established between the presence of EGF and the cetuximab
responsiveness of the cell lines that were studied.
Effect of cetuximab treatment on AREG
levels in AZ521 and MKN1 cells
To examine the role of the EGFR signaling pathway in
the regulation of AREG, the levels of secreted AREG were determined
after the treatment of cells with cetuximab or combinations of EGF
and cetuximab. The AREG concentrations were determined in the
conditioned medium of the cetuximab-sensitive cell lines, AZ521 and
MKN1, using ELISA.
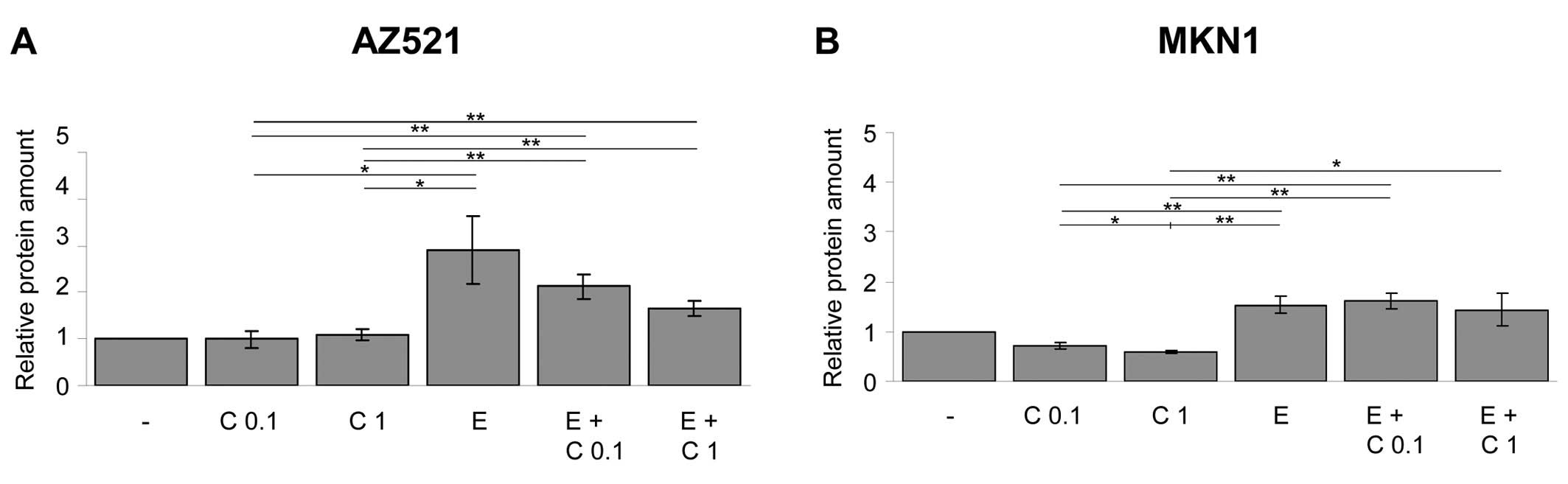
As shown in Fig. 3,
cetuximab treatment caused a significant decrease in the level of
secreted AREG in the MKN1 cells, but no such decrease was observed
in the AZ521 cells. In both cell lines, the addition of exogenous
EGF to the cells resulted in an increase in AREG secretion. This
EGF-induced AREG secretion was blocked by simultaneous treatment of
the cells with increasing concentrations of cetuximab to some
extent in the MKN1 cells and almost completely in the AZ521 cells.
By contrast, Hs746T cells did not respond to EGF or cetuximab
treatment (data not shown).
These results demonstrate that the EGFR signaling
pathway itself is involved in the regulation of the ligand AREG.
This important finding suggests that there exists an autoregulatory
mechanism in gastric cancer cell lines.
Analysis of EGFR expression, localization
and activation
Ligand binding results in EGFR dimerization,
stimulation of its tyrosine kinase activity and activation of
downstream signaling cascades. The total and activated levels of
EGFR were determined in the five gastric cancer cell lines using
immunoblot and flow cytometry analyses.
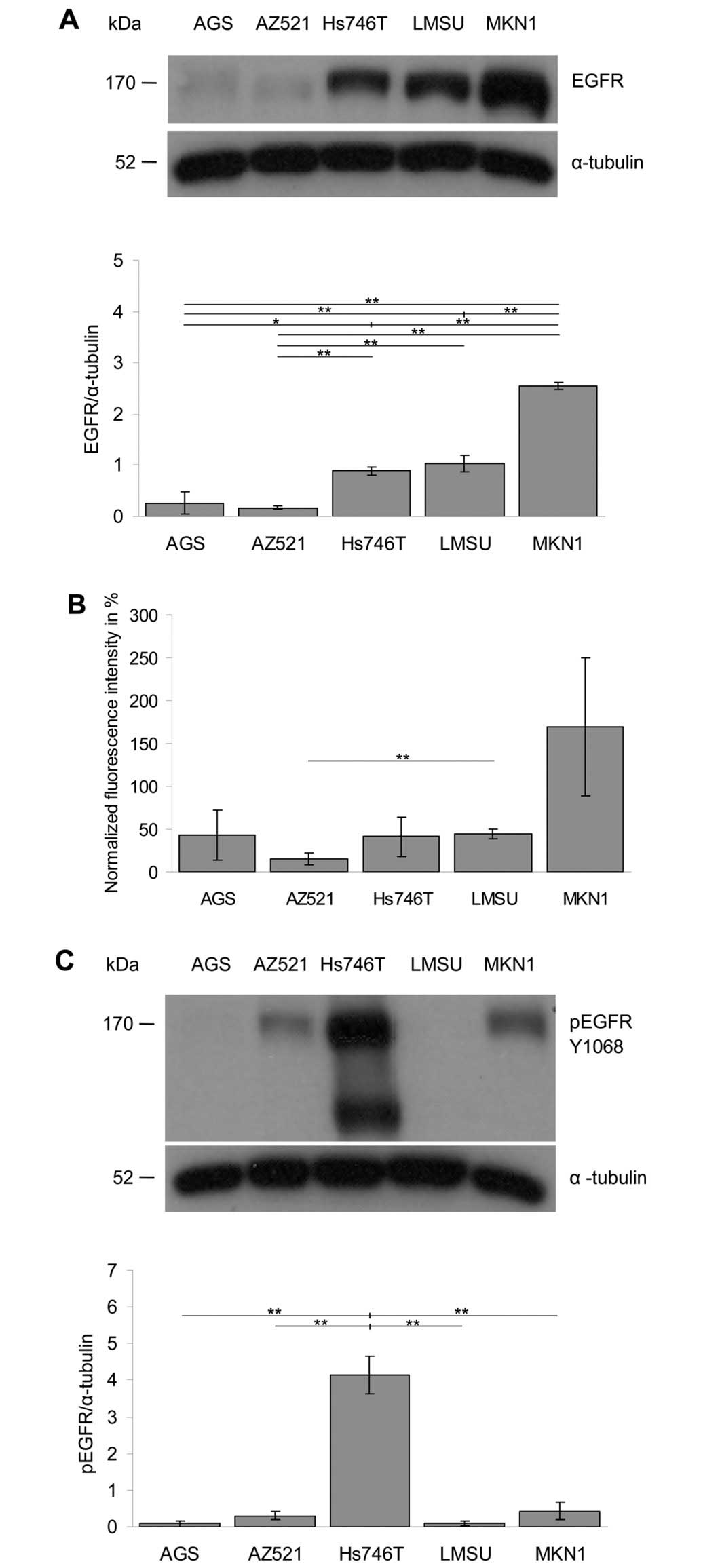
The analysis of the expression level of EGFR in the
different cell lines by western blotting revealed the following
order: MKN1 > LMSU = Hs746T > AGS = AZ521 (Fig. 4A). Essentially the same order was
obtained when the surface localization of EGFR was analyzed with
flow cytometry (Fig. 4B).
The activation level of EGFR was determined by
western blot analysis of the level of EGFR phosphorylation on
tyrosine residue Y1068 (Fig. 4C).
The following order of the EGFR activation levels was obtained:
Hs746T >> AZ521 = MKN1 > AGS = LMSU. Together, these
findings demonstrate that the studied cell lines express EGFR at
considerably different levels and that the expression and
activation levels of EGFR are not correlated.
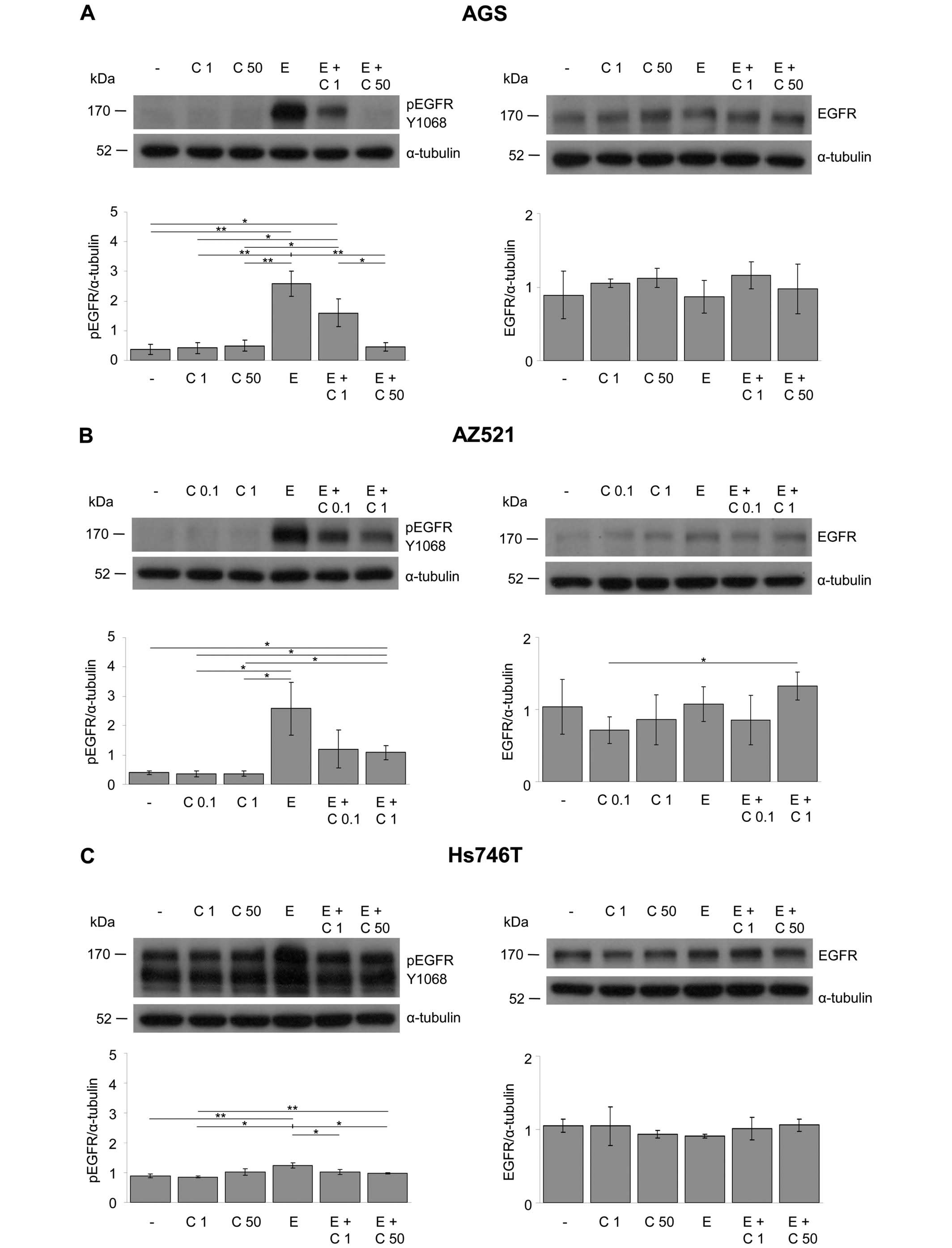
Effect of EGF and cetuximab treatment on
the phosphorylation of EGFR
The effects of EGF and cetuximab on the expression
and activation of EGFR were determined using western blot
analysis.
The detection of EGFR phosphorylated on tyrosine
residue Y1068 revealed that EGFR was activated by EGF in the AGS,
AZ521, LMSU and MKN1 cell lines and that this EGF-induced
activation of EGFR was blocked by cetuximab in a
concentration-dependent manner (Fig.
5A, B, D and E). By contrast, Hs746T cells were EGF- and
cetuximab-non-responsive (Fig.
5C). In all cell lines, the EGFR expression levels remained
essentially unchanged during all treatment conditions.
Together, EGF and/or cetuximab had only minor
effects on the degree of EGFR phosphorylation in the Hs746T cell
line, whereas EGFR activation was modulated by the treatment in the
four other cell lines (AGS, AZ521, LMSU and MKN1).
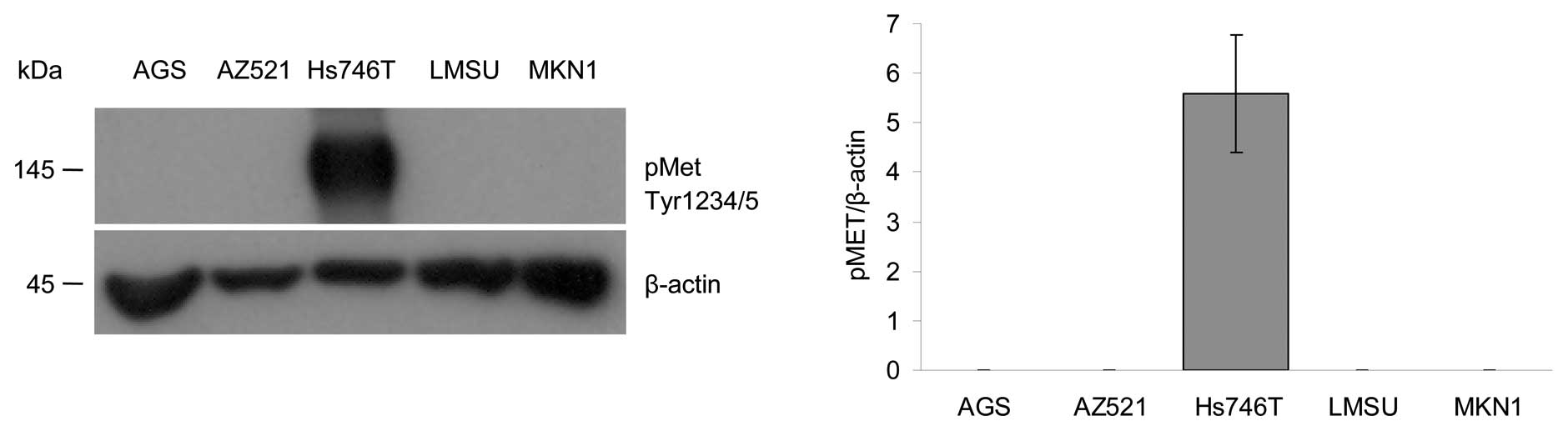
Analysis of the MET activation
A number of studies have suggested a predictive role
for MET in EGFR inhibitory therapy (36–38).
To determine the role of MET activation in our gastric cancer
model, the phosphorylation status of the MET receptor was
determined using western blot analysis. Detection of MET
phosphorylated on tyrosine residue Y1234/1235 revealed that the
activation of MET was very strong in Hs746T cells, whereas no
signals were detected in the AGS, AZ521, LMSU and MKN1 cell lines
(Fig. 6).
Mutation analysis
EGFR, KRAS, BRAF and PI3K are key components of the
EGFR-signaling pathway, and oncogenic alterations in these genes
are related to the response to EGFR-targeting therapeutics.
Therefore, hotspot mutation regions in these genes were analyzed in
the panel of gastric cancer cell lines.
In detail, for BRAF, the cell lines were
screened for the activating mutation V600E. Exons 18, 19 and 21 of
EGFR, exon 2 of KRAS and exons 9 and 20 of
PIK3CA were sequenced.
The presence of the KRAS mutation G12D in AGS
cells has been reported previously, and we confirmed this finding.
We also confirmed the presence of the recently described
PIK3CA mutation, E545K, in MKN1 cells. No further genetic
alterations were identified (Table
I).
 | Table IGenetic alterations in hotspot
mutation regions of key components of the EGFR signaling pathway in
gastric cancer cell lines. |
Table I
Genetic alterations in hotspot
mutation regions of key components of the EGFR signaling pathway in
gastric cancer cell lines.
| Mutation | AGS | AZ521 | Hs746T | LMSU | MKN1 |
|---|
| EGFRa | WT | WT | WT | WT | WT |
| KRASb | G12Dc | WT | WT | WT | WT |
|
PIK3CAd | WT | WT | WT | WT |
E545Ke |
| BRAFf | WT | WT | WT | WT | WT |
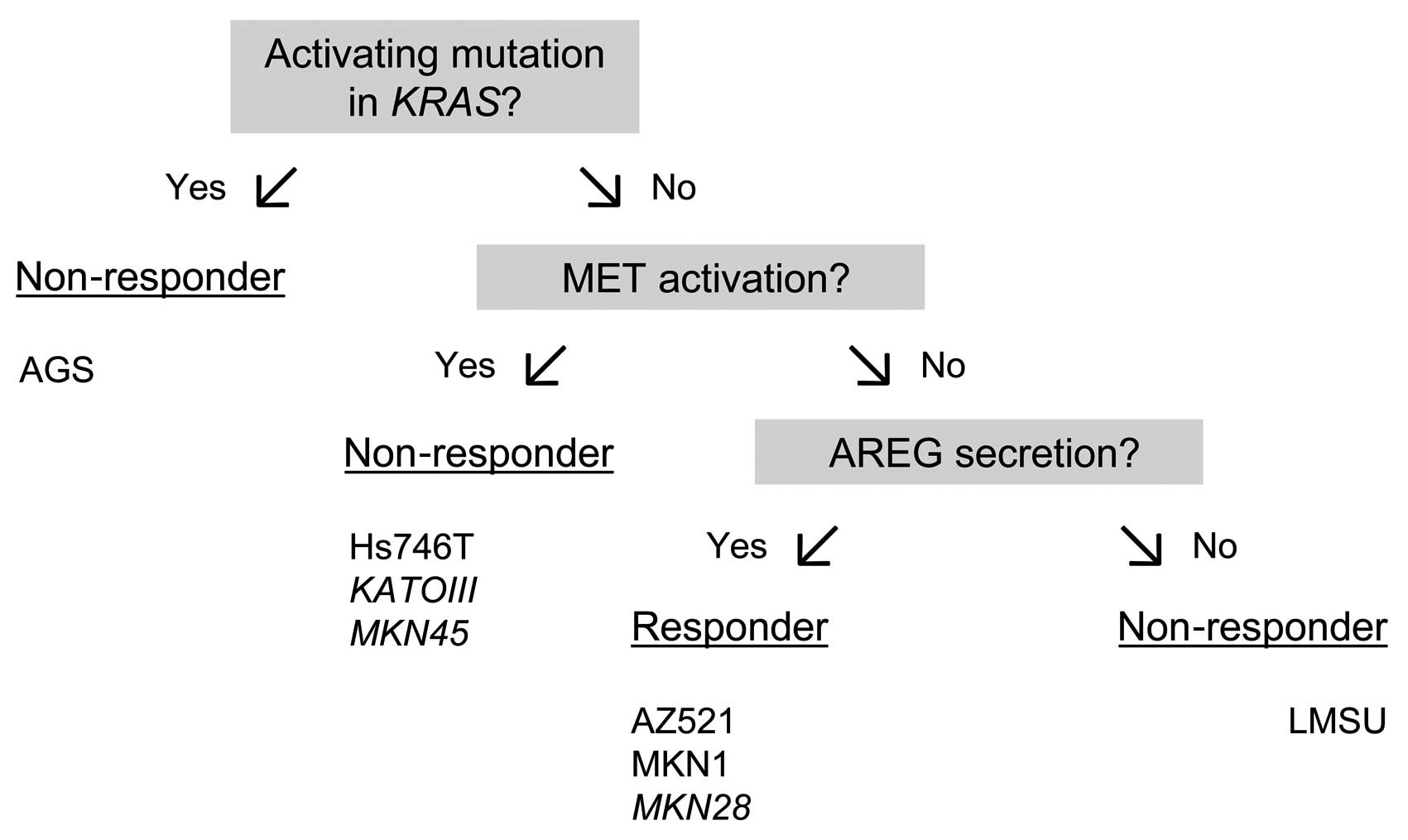
Model for predicting cetuximab
sensitivity in gastric cancer cell lines
In our study, we found that the gastric cancer cell
lines, AZ521 and MKN1, were cetuximab-sensitive, whereas the AGS,
Hs746T and LMSU cell lines were found to be cetuximab-resistant
based on the results of the XTT cell viability assay.
The cetuximab responsiveness of the AZ521 and MKN1
cells was associated with AREG expression and secretion. The
cetuximab resistance of the AGS cells is most likely due to the
KRAS mutation, whereas the cetuximab insensitivity of Hs746T
cells may be explained by MET activation. Together, we were able to
explain the cetuximab response of four of the five investigated
cell lines.
We propose the model presented in Fig. 7 to predict the cetuximab response
of gastric cancer cell lines. To validate this model, we measured
the amount of secreted AREG in three additional human gastric
cancer cell lines, KATOIII (523 pg/ml in the conditioned medium and
927 pg/mg in the cellular extract), MKN28 (918 pg/ml in the
conditioned medium and 106 pg/mg in the cellular extract) and MKN45
(7 pg/ml in the conditioned medium and 168 pg/mg in the cellular
extract) (Table II). Among these
three cell lines, there was one cetuximab-responsive cell line
(MKN28) and two cetuximab-non-responsive cell lines (KATOIII,
MKN45) (24). These cell lines
have previously been characterized with respect to the KRAS
mutation and MET activation status (24). MET has been shown to be activated
in the cetuximab-resistant cell lines, KATOIII and MKN45, but not
in the MKN28 cell line (24). All
three cell lines were correctly classified by the model.
| Table IIMolecular characteristics of the
gastric cancer cell lines. |
Table II
Molecular characteristics of the
gastric cancer cell lines.
Discussion
Responsiveness of gastric cancer cell
lines to cetuximab treatment
In this study, the predictive value of several
molecular markers for the cetuximab responsiveness of the human
gastric cancer cell lines, AGS, AZ521, Hs746T, LMSU and MKN1, was
analyzed. The analysis of cell viability after cetuximab treatment
identified two cetuximab-responsive cell lines, AZ521 and MKN1,
whose metabolic activity was significantly reduced by cetuximab in
a dose-dependent manner. The other three cell lines, AGS, Hs746T
and LMSU, were not responsive to cetuximab. For the MKN1 and AGS
cells, these results are in agreement with the results reported
previously by our group (24). All
considered cell lines expressed EGFR at different levels.
Model for the prediction of cetuximab
responsiveness
In the present study, we performed a detailed
molecular analysis of different putative predictive markers for
cetuximab responsiveness, including the expression and secretion of
the ligands, AREG and EGF, activation of the RTK, MET, and the
presence of activating mutations in KRAS.
The cetuximab responsiveness of AZ521 and MKN1 cells
was associated with AREG expression and secretion. By contrast, the
cetuximab resistance of AGS cells was most likely due to the
KRAS mutation (24). As a
number of studies have suggested an association between MET
activation and resistance to cetuximab (22–24),
one possible explanation for the cetuximab insensitivity of Hs746T
cells is the high level of MET tyrosine kinase activity. Notably,
the Hs746T cell line harbors a splice site mutation in cMET,
resulting in the deletion of the juxtamembrane domain (39).
We established a model to facilitate the correct
classification of gastric cancer cell lines as cetuximab-responsive
and -non-responsive cell lines. In an attempt to establish a
hierarchy of predictive molecular markers, the highest priority was
allocated to activating KRAS mutations. This decision was
based on the experience that patients with colorectal cancers
lacking oncogenic activation of the EGFR downstream effectors,
KRAS, BRAF, PIK3CA and PTEN, are the most likely to benefit from
anti-EGFR therapies (21). The
second place in the hierarchy was assigned to MET activation.
Finally, gastric cancer cell lines lacking activated KRAS
mutations and MET activation were classified according to their
level of secreted AREG. Using this approach, it was possible to
correctly classify the cetuximab responsiveness of all five cell
lines included in this study (AGS, AZ521, Hs746T, LMSU and MKN1).
The reason for the cetuximab resistance of LMSU cells is presently
unknown.
The model was validated with three other human
gastric cancer cell lines, KATOIII, MKN28 and MKN45, among which
one was cetuximab-responsive (MKN28) and two were
cetuximab-non-responsive (KATOIII and MKN45) (24). None of these three cell lines
harbors a known activating KRAS mutation (24). We have previously shown that MET is
activated in the cetuximab-resistant cell lines, KATOIII and MKN45,
but not in the MKN28 cell line (24). In the present study, we detected
high levels of secreted AREG in the MKN28 cells and used the model
to classify this cell line as cetuximab-responsive. AREG secretion
was not detectable in the cetuximab-resistant cell line MKN45,
which falls into the non-responder category due to the elevated MET
activation status. Despite the high levels of secreted AREG, the
KATOIII cell line was classified as non-responsive due to the high
level of activated MET.
In this study, the advantage of the model as a tool
to determine the cetuximab sensitivity of gastric cancer cell lines
becomes evident. However, it also becomes clear that it is
difficult to predict the therapy response by evaluation of a single
marker. In order to ensure a correct classification, we suggest
defining a panel of predictive markers and establishing a hierarchy
among them. Of course, we make no claim of completeness as regards
this study.
Positive predictive role of AREG
secretion for cetuximab responsiveness
In the present study, we found a positive predictive
value of AREG secretion for cetuximab responsiveness when the
KRAS mutation status and MET activation were taken into
account.
This finding is in agreement with the positive
predictive role that was attributed to AREG and EREG as markers for
the outcome of CRC patients treated with cetuximab and chemotherapy
who have no KRAS mutations in their tumors (19,20).
In contrast to the situation in CRC, in 38 advanced gastric cancer
patients treated with cetuximab in combination with oxaliplatin,
leucovorin and 5-fluorouracil in a recent clinical phase II trial,
no significant correlation between AREG expression and the response
rate was found (26). Notably, the
level of activated MET was not determined in that study.
One main technical difference between the CRC study
and the gastric cancer study is that the AREG mRNA expression level
was examined in the CRC tumors, whereas the serum levels of AREG
were determined in the gastric cancer patients. When the
correlation between tumor mRNA expression and the level of protein
in the blood (measured by ELISA) was assessed, only a modest
correlation between the systemic protein and tumor mRNA levels of
AREG was found (19).
Cetuximab is approved for the treatment of recurrent
SCCHN in addition to CRC. For these tumors, the findings regarding
the predictive value for AREG are contradictory. In one study, high
EGFRvIII and AREG expression levels identified SCCHN patients who
were less likely to benefit from combination treatment with
cetuximab and docetaxel (40). In
a different study that included SCCHN tumors and cell lines,
autocrine production of AREG was found to predict sensitivity to
both gefitinib and cetuximab in EGFR wild-type cancers (41).
A number of studies have suggested that AREG
produced by tumor cells may be involved in the pathogenesis and/or
progression of human gastric carcinoma (42,43).
Notably, 20 out of 32 tumors (62.5%) expressed AREG mRNA at higher
levels than their corresponding normal mucosas. By contrast, no
obvious correlation was observed between the AREG mRNA levels and
the histological types or tumor staging of gastric carcinoma
(43). Additionally, an
association between AREG expression and the development of
peritoneal carcinomatosis in gastric cancer patients was reported
(42).
Recently, it was shown that low levels of the EGFR
ligands, EGF and TGFα, in combination with EGFR expression
positively correlated with the response rates of gastric cancer
patients to a cetuximab/modified FOLFOX6 regimen (26). Considering the lack of EGF
expression in both cetuximab-sensitive and cetuximab-resistant
gastric cancer cell lines, we conclude that EGF expression is not
of predictive value in our study. This lack of soluble EGF suggests
that although EGF is expressed in all cell lines to a certain
extent, it is not proteolytically released. Previously, it was
proposed that the proteolytic release of EGF is essential for its
activity as it cannot act in a juxtacrine manner (44). Due to the absence of soluble EGF,
the EGF-based induction of the EGFR signaling pathway most likely
plays no role or only a minor role in our gastric cancer cell
lines.
Association of MET activation and
cetuximab resistance
As mentioned above, a number of studies have
suggested a predictive role for MET in EGFR inhibitory therapy
(36–38,44,45).
As reported previously, the activation of MET was accompanied with
the activation of EGFR in the KATOIII and MKN45 cell lines
(24). A similar observation was
made in the present study: Hs746T cells showed high levels of MET
and EGFR activation. Notably, cetuximab had only minor effects on
the degree of EGFR phosphorylation in this cell line, whereas EGFR
activation was modulated by the treatment in the four other
considered cell lines (AGS, AZ521, LMSU and MKN1).
As discussed previously (24), there is a close correlation between
MET and EGFR, including physical interaction and ligand-dependent
or -independent transactivation, and the co-activation of multiple
RTKs in cancer cells results in resistance to single-agent therapy
(37,46). Consequently, the co-activation of
MET would explain the failure of anti-EGFR therapy in the
non-responsive cell lines, Hs746T, KATOIII and MKN45.
In conclusion, in this study, to our knowledge, we
present the first model that allows the response of gastric cancer
cell lines to cetuximab treatment to be predicted. The considered
markers were the status of KRAS mutation, MET activation and
AREG secretion. Even if cell culture models are too simple to
explain the complex in vivo situation in patients, we
believe that the inclusion of a reasonable number of cell lines and
accurate statistical analysis will allow for the generation of
hypotheses that can be tested in clinical studies.
Abbreviations:
|
AREG
|
amphiregulin
|
|
CRC
|
colorectal cancer
|
|
DMEM
|
Dulbecco’s modified Eagle’s medium
|
|
ECACC
|
European Collection of Cell
Cultures
|
|
EGF
|
epidermal growth factor
|
|
EGFR
|
epidermal growth factor receptor
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
EREG
|
epiregulin
|
|
IU
|
international units
|
|
KRAS
|
Kirsten-Ras gene
|
|
MEM
|
Minimum Essential Medium Eagle
|
|
PBS
|
phosphate-buffered saline
|
|
PCR
|
polymerase chain reaction
|
|
rpm
|
rounds per minute
|
|
RTK
|
receptor tyrosine kinase
|
|
SCCHN
|
squamous cell carcinoma of the head
and neck
|
|
SD
|
standard deviation
|
Acknowledgements
This study was supported by the German
Federal Ministry of Education and Research and the Austrian Federal
Ministry for Science and Research as part of the program
‘Medizinische Systembiologie-MedSys’ (CANCERMOTISYS project,
www.cancermotisys.eu). We thank Birgit Geist and
Susanne Plaschke for their excellent technical assistance.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang SJ, Emery R, Fuller CD, Kim JS,
Sittig DF and Thomas CR: Conditional survival in gastric cancer: a
SEER database analysis. Gastric Cancer. 10:153–158. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Verdecchia A, Santaquilani M and Sant M:
Survival for cancer patients in Europe. Ann Ist Super Sanita.
45:315–324. 2009.PubMed/NCBI
|
|
4
|
Kim MA, Lee HS, Lee HE, Jeon YK, Yang HK
and Kim WH: EGFR in gastric carcinomas: prognostic significance of
protein overexpression and high gene copy number. Histopathology.
52:738–746. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liang Z, Zeng X, Gao J, et al: Analysis of
EGFR, HER2, and TOP2A gene status and chromosomal polysomy in
gastric adenocarcinoma from Chinese patients. BMC Cancer.
8:3632008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pinto C, Di Fabio F, Barone C, et al:
Phase II study of cetuximab in combination with cisplatin and
docetaxel in patients with untreated advanced gastric or
gastro-oesophageal junction adenocarcinoma (DOCETUX study). Br J
Cancer. 101:1261–1268. 2009. View Article : Google Scholar
|
|
7
|
Lordick F, Luber B, Lorenzen S, et al:
Cetuximab plus oxaliplatin/leucovorin/5-fluorouracil in first-line
metastatic gastric cancer: a phase II study of the
Arbeitsgemeinschaft Internistische Onkologie (AIO). Br J Cancer.
102:500–505. 2010. View Article : Google Scholar
|
|
8
|
Kim C, Lee JL, Ryu MH, et al: A
prospective phase II study of cetuximab in combination with XELOX
(capecitabine and oxaliplatin) in patients with metastatic and/or
recurrent advanced gastric cancer. Invest New Drugs. 29:366–373.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luber B, Deplazes J, Keller G, et al:
Biomarker analysis of cetuximab plus
oxaliplatin/leucovorin/5-fluorouracil in first-line metastatic
gastric and oesophago-gastric junction cancer: results from a phase
II trial of the Arbeitsgemeinschaft Internistische Onkologie (AIO).
BMC Cancer. 11:5092011. View Article : Google Scholar
|
|
10
|
Lievre A, Bachet JB, Le Corre D, et al:
KRAS mutation status is predictive of response to cetuximab therapy
in colorectal cancer. Cancer Res. 66:3992–3995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goldstein NI, Prewett M, Zuklys K,
Rockwell P and Mendelsohn J: Biological efficacy of a chimeric
antibody to the epidermal growth factor receptor in a human tumor
xenograft model. Clin Cancer Res. 1:1311–1318. 1995.PubMed/NCBI
|
|
12
|
Harris RC, Chung E and Coffey RJ: EGF
receptor ligands. Exp Cell Res. 284:2–13. 2003. View Article : Google Scholar
|
|
13
|
Higashiyama S, Abraham JA, Miller J,
Fiddes JC and Klagsbrun M: A heparin-binding growth factor secreted
by macrophage-like cells that is related to EGF. Science.
251:936–939. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Komurasaki T, Toyoda H, Uchida D and
Morimoto S: Epiregulin binds to epidermal growth factor receptor
and ErbB-4 and induces tyrosine phosphorylation of epidermal growth
factor receptor, ErbB-2, ErbB-3 and ErbB-4. Oncogene. 15:2841–2848.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shoyab M, Plowman GD, McDonald VL, Bradley
JG and Todaro GJ: Structure and function of human amphiregulin: a
member of the epidermal growth factor family. Science.
243:1074–1076. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Strachan L, Murison JG, Prestidge RL,
Sleeman MA, Watson JD and Kumble KD: Cloning and biological
activity of epigen, a novel member of the epidermal growth factor
superfamily. J Biol Chem. 276:18265–18271. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Watanabe T, Shintani A, Nakata M, et al:
Recombinant human betacellulin. Molecular structure, biological
activities, and receptor interaction. J Biol Chem. 269:9966–9973.
1994.PubMed/NCBI
|
|
18
|
Prewett M, Rockwell P, Rockwell RF, et al:
The biologic effects of C225, a chimeric monoclonal antibody to the
EGFR, on human prostate carcinoma. J Immunother Emphasis Tumor
Immunol. 19:419–427. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Khambata-Ford S, Garrett CR, Meropol NJ,
et al: Expression of epiregulin and amphiregulin and K-ras mutation
status predict disease control in metastatic colorectal cancer
patients treated with cetuximab. J Clin Oncol. 25:3230–3237. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jacobs B, De Roock W, Piessevaux H, et al:
Amphiregulin and epiregulin mRNA expression in primary tumors
predicts outcome in metastatic colorectal cancer treated with
cetuximab. J Clin Oncol. 27:5068–5074. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bardelli A and Siena S: Molecular
mechanisms of resistance to cetuximab and panitumumab in colorectal
cancer. J Clin Oncol. 28:1254–1261. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Krumbach R, Schuler J, Hofmann M,
Giesemann T, Fiebig HH and Beckers T: Primary resistance to
cetuximab in a panel of patient-derived tumour xenograft models:
activation of MET as one mechanism for drug resistance. Eur J
Cancer. 47:1231–1243. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liska D, Chen CT, Bachleitner-Hofmann T,
Christensen JG and Weiser MR: HGF rescues colorectal cancer cells
from EGFR inhibition via MET activation. Clin Cancer Res.
17:472–482. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Heindl S, Eggenstein E, Keller S, et al:
Relevance of MET activation and genetic alterations of KRAS and
E-cadherin for cetuximab sensitivity of gastric cancer cell lines.
J Cancer Res Clin Oncol. Jan 31–2012.(Epub ahead of print).
|
|
25
|
Kim IJ, Park JH, Kang HC, et al:
Mutational analysis of BRAF and K-ras in gastric cancers: absence
of BRAF mutations in gastric cancers. Hum Genet. 114:118–120. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han SW, Oh DY, Im SA, et al: Phase II
study and biomarker analysis of cetuximab combined with modified
FOLFOX6 in advanced gastric cancer. Br J Cancer. 100:298–304. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Paull KD, Shoemaker RH, Hodes L, et al:
Display and analysis of patterns of differential activity of drugs
against human tumor cell lines: development of mean graph and
COMPARE algorithm. J Natl Cancer Inst. 81:1088–1092. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boyd MR and Paull KD: Some practical
considerations and applications of the National Cancer Institute in
vitro anticancer drug discovery screen. Drug Dev Res. 34:91–109.
1995. View Article : Google Scholar
|
|
29
|
Luo FR, Yang Z, Dong H, et al: Correlation
of pharmacokinetics with the antitumor activity of Cetuximab in
nude mice bearing the GEO human colon carcinoma xenograft. Cancer
Chemother Pharmacol. 56:455–464. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Robert F, Ezekiel MP, Spencer SA, et al:
Phase I study of anti-epidermal growth factor receptor antibody
cetuximab in combination with radiation therapy in patients with
advanced head and neck cancer. J Clin Oncol. 19:3234–3243.
2001.PubMed/NCBI
|
|
31
|
Baselga J, Pfister D, Cooper MR, et al:
Phase I studies of anti-epidermal growth factor receptor chimeric
antibody C225 alone and in combination with cisplatin. J Clin
Oncol. 18:904–914. 2000.PubMed/NCBI
|
|
32
|
Loughrey MB, Waring PM, Tan A, et al:
Incorporation of somatic BRAF mutation testing into an algorithm
for the investigation of hereditary non-polyposis colorectal
cancer. Fam Cancer. 6:301–310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pan Q, Pao W and Ladanyi M: Rapid
polymerase chain reaction-based detection of epidermal growth
factor receptor gene mutations in lung adenocarcinomas. J Mol
Diagn. 7:396–403. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Marchetti A, Martella C, Felicioni L, et
al: EGFR mutations in non-small-cell lung cancer: analysis of a
large series of cases and development of a rapid and sensitive
method for diagnostic screening with potential implications on
pharmacologic treatment. J Clin Oncol. 23:857–865. 2005. View Article : Google Scholar
|
|
35
|
Bremm A, Walch A, Fuchs M, et al: Enhanced
activation of epidermal growth factor receptor caused by
tumor-derived E-cadherin mutations. Cancer Res. 68:707–714. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Agarwal S, Zerillo C, Kolmakova J, et al:
Association of constitutively activated hepatocyte growth factor
receptor (Met) with resistance to a dual EGFR/Her2 inhibitor in
non-small-cell lung cancer cells. Br J Cancer. 100:941–949. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guo A, Villen J, Kornhauser J, et al:
Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl
Acad Sci USA. 105:692–697. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Asaoka Y, Tada M, Ikenoue T, et al:
Gastric cancer cell line Hs746T harbors a splice site mutation of
c-Met causing juxta-membrane domain deletion. Biochem Biophys Res
Commun. 394:1042–1046. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tinhofer I, Klinghammer K, Weichert W, et
al: Expression of amphiregulin and EGFRvIII affect outcome of
patients with squamous cell carcinoma of the head and neck
receiving cetuximab-docetaxel treatment. Clin Cancer Res.
17:5197–5204. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yonesaka K, Zejnullahu K, Lindeman N, et
al: Autocrine production of amphiregulin predicts sensitivity to
both gefitinib and cetuximab in EGFR wild-type cancers. Clin Cancer
Res. 14:6963–6973. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yasumoto K, Yamada T, Kawashima A, et al:
The EGFR ligands amphiregulin and heparin-binding egf-like growth
factor promote peritoneal carcinomatosis in CXCR4-expressing
gastric cancer. Clin Cancer Res. 17:3619–3630. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kitadai Y, Yasui W, Yokozaki H, et al:
Expression of amphiregulin, a novel gene of the epidermal growth
factor family, in human gastric carcinomas. Jpn J Cancer Res.
84:879–884. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dong J, Opresko LK, Chrisler W, et al: The
membrane-anchoring domain of epidermal growth factor receptor
ligands dictates their ability to operate in juxtacrine mode. Mol
Biol Cell. 16:2984–2998. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Inno A, Salvatore MD, Cenci T, et al: Is
there a role for IGF1R and c-MET pathways in resistance to
cetuximab in metastatic colorectal cancer? Clin Colorectal Cancer.
10:325–332. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Stommel JM, Kimmelman AC, Ying H, et al:
Coactivation of receptor tyrosine kinases affects the response of
tumor cells to targeted therapies. Science. 318:287–290. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kim IJ, Park JH, Kang HC, et al:
Mutational analysis of BRAF and K-ras in gastric cancers: absence
of BRAF mutations in gastric cancers. Hum Genet. 114:118–120. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mita H, Toyota M, Aoki F, et al: A novel
method, digital genome scanning detects KRAS gene amplification in
gastric cancers: involvement of overexpressed wild-type KRAS in
downstream signaling and cancer cell growth. BMC Cancer. 9:1982009.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kuniyasu H, Yasui W, Kitadai Y, Yokozaki
H, Ito H and Tahara E: Frequent amplification of the c-met gene in
scirrhous type stomach cancer. Biochem Biophys Res Commun.
189:227–232. 1992. View Article : Google Scholar : PubMed/NCBI
|