Introduction
Apoptin, a chicken anemia virus-encoded protein,
consists of 121 amino acids (1).
An attractive feature of apoptin is that it induces apoptosis in a
large number of human tumor or transformed cells but not in their
normal, healthy counterparts (2,3).
This is correlated with the subcellular localization of the
protein. In normal cells, apoptin is found in the cytoplasm whereas
in tumor cells it is found in the nucleus (4–6).
This suggests that apoptin may be used as an agent that selectively
eliminates malignant cells, if it can be delivered into target
cells in vivo in sufficient amounts. One straightforward
strategy to reach this goal would be the application of the apoptin
gene, rather than the protein itself.
The transacting activator of transcription (TAT)
protein transduction domain (PTD) from human immunodeficiency virus
type 1 (aa 47–57; YGRKKRRQRRR) is one of the most commonly used
protein transduction systems (7,8). In
some studies, this TAT-PTD transduction domain has been
successfully used to deliver apoptin into cancer cells in
vitro. The data from these studies show that TAT-apoptin
efficiently kills human tumor, but not normal cells (9–11).
However, one disadvantage of using the TAT sequence in in
vivo studies is that physical delivery of TAT fusion gene such
as direct injection into the tumor bed is inefficient, as the
produced recombinant protein may not reach all the cells within the
tumor mass.
To enable the efficient transduction of tumor cells
with apoptin, we have developed a novel mammalian expression system
for the in vitro secretion of proteins (12,13).
We have previously shown the efficient and tumor-specific killing
of cells by TAT-apoptin fused to a signal peptide (SP) whose amino
acid sequence and corresponding cDNA sequences were generated by an
SP hidden Markov model (SP-HMM) (14). This SP directs strong protein
secretion and expression and the TAT-apoptin fusion protein
secreted from transfected cells can re-enter the adjacent
untransfected cells. It should be noted that in normal cells
resistant to apoptin-mediated cell death, fusion protein is
localized in the cytoplasm, whereas in sensitive cells it is in the
nucleus. Nuclear localization was shown to be crucial for the
cell-killing ability of apoptin (12). We have demonstrated that
SP-TAT-apoptin induces apoptosis only in malignant cells, and its
secretory property might greatly increase its potency, once it is
delivered in vivo for cancer therapy. We recently generated
a lentivirus-based vector to express SP-TAT-apoptin and apoptosis
induced by apoptin was observed in human hepatocellular carcinoma
(HCC) cells (13). We thus
hypothesize that lentivirus-based SP-TAT-apoptin kills tumor cells;
in addition, normal cells infected with the lentivirus-based
SP-TAT-apoptin secrete TAT-apoptin fusion protein persistently,
which leads to more extensive tumor cell death.
In this study, we investigated if Lentivirus-based
delivery of SP-TAT-apoptin could serve as a therapeutic agent for
HCC treatment in a mouse xenograft model. Our findings showed that
systemic delivery of the Lentivirus-based SP-TAT-apoptin viruses
eradicated xenograft tumors.
Materials and methods
Cell lines
Human immortalized liver cell line (HL-7702; catalog
no. GNHu 6), and human HCC cell lines (HepG2; catalog no. TCHu 72
and Bel-7402; catalog no. TCHu 10) were obtained from the Shanghai
Institutes for Biological Sciences (SIBS). Human Umbilical Vein
Endothelia (HUVEC; catalog no. CRL-1730™) cell line was obtained
from American Type Culture Collection. Cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (FBS), 50 μg/ml streptomycin and 100
μg/ml penicillin.
Constructs
GFP, apoptin, TAT-GFP, TAT-apoptin, SP-TAT-apoptin,
and SP-TAT-GFP constructs contain the cDNA for GFP, apoptin,
TAT-GFP, TAT-apoptin, SP-TAT-apoptin, and SP-TAT-GFP, respectively
cloned into the pLenti6/V5-DTOPO® vector (catalog no.
K4950-00, Invitrogen) (13).
Lentivirus (LV) production
LV-SP-TAT-GFP and LV-SP-TAT-apoptin viruses were
produced by Biomics Biotechnologies (Nantong, China) at a titer of
1.0×1011 viral particle per ml, as described in our
previous publication (13).
Transient transfection
HUVEC cells (1.0×106) per well were
seeded in 6-well slides. The following day, cells were transfected
with 600 ng DNA per well of the different constructs using
Lipofectamine™ 2000 (catalog no. 11668-027, Invitrogen) according
to the supplier's instructions. At 24 or 48 h after transfection,
cells were fixed, permeabilized and apoptin/GFP expression was
examined by fluorescence microscopy. To confirm TAT fusion protein
secretion by HUVEC cells transfected with these constructs,
supernatant from the wells was centrifuged for 5 min at 200 g and
three volumes of supernatant were mixed with one volume of 3X
concentrated Laemmli sample buffer (3X LSB).
Stable transfection
HUVEC cells (7×105) were seeded in 10-cm
culture dishes and transfected the following day with 20 μg
of GFP, apoptin, TAT-GFP, TAT-apoptin, SP-TAT-apoptin, and
SP-TAT-GFP constructs using Lipofectamine 2000 (catalog no.
11668-027, Invitrogen) according to the instructions. After 3 days,
cells were put under selection with 8 μg/ml Blasticidin
(catalog no. R21001, Invitrogen) after transfection with GFP,
apoptin, TAT-GFP, TAT-apoptin, SP-TAT-apoptin, and SP-TAT-GFP
constructs. Selection medium was changed every 3–4 days and clones
were picked after 3 weeks. To examine TAT-apoptin/GFP and
SP-TAT-apoptin/GFP expression in each clone, 5×105 cells
were seeded per well in 6-well slides. In order to confirm the
expression of recombinant GFP, cells were fixed, permeabilized,
counter stained with 4′,6-diamino-2-phenylindole (DAPI, catalog no.
D9542, Sigma) containing mounting solution and examined by
fluorescence microscopy (Olympus). The expression of recombinant
apoptin was confirmed by immunoblot analysis using anti-V5 antibody
(catalog no. R960-25, Invitrogen). After immunolabeling, cells were
washed, stained with DAPI (catalog no. D9542, Sigma), and then
viewed with fluorescent microscopy (Olympus). To confirm TAT fusion
protein secretion, supernatant from HUVEC/TAT-GFP,
HUVEC/SP-TAT-GFP, HUVEC/TAT-apoptin, and HUVEC/SP-TAT-apoptin
clones was centrifuged for 5 min at 200 g and three volumes were
mixed with one volume of 3X LSB. To check SP-TAT-apoptin secretion
by HUVEC/SP-TAT-apoptin, supernatant was precipitated in 20%
trichloroacetic acid for 10 min at 4°C, centrifuged for 5 min at
13,000 rpm and then the pellet was washed twice with acetone and
dried at 95°C for 5–10 min. Finally, the pellet was lysed in 3X LSB
and boiled for 5 min.
Fluorescence microscopy
Cells were fixed in 4% (wt/vol) paraformaldehyde in
phosphate-buffered saline (PBS) for 3 h, washed three times in PBS,
permeabilized in 0.2% (vol/vol) Triton X-100 in PBS for 15 min and
then washed three times in PBS. For apoptin/GFP detection, nuclei
were counterstained in medium containing DAPI. Cells were examined
under a fluorescent microscope (Olympus).
Confocal microscopy
Cells were prepared as described for fluorescence
microscopy and fluorescence images were recorded on an Olympus AX
70 (Olympus) confocal imaging system.
Flow cytometry assay
The sensitivity of cells to apoptosis was examined
by fluorescence activated cell sorting (FACS) analysis using
propidium iodide (PI) and fluorescein isothiocyanate (FITC)
conjugated anti-Annexin V antibody according to the manufacturer's
instructions (catalog no. 11858777001, Roche). Briefly, after
incubating the cells for 48 h under normal glucose conditions or
glucose-deprived conditions, the cells were stained with PI and
FITC conjugated anti-Annexin V and then analyzed with a FACS
Calibur (Coulter).
Western blot analysis
Cells were lysed in LSB (62.5 mmol/l Tris-Cl pH 6.7,
100 mmol/l β-mercaptoethanol, 2% sodium dodecyl sulfate, 1 mg/ml
aprotinin, 100 mg/ml phenylmethylsulphoxide). Equal amounts of
proteins were subjected to SDS-PAGE and then western blotting for
assessing GFP, TAT-GFP, SP-TAT-GFP, apoptin, TAT-apoptin, and
SP-TAT-apoptin expression. Primary antibodies for GFP (catalog no.
sc-9996) were purchased from Santa Cruz Biotechnology. β-actin blot
served as protein loading control.
Concentration of supernatant
Two milliliters of supernatant from HUVEC,
HUVEC/TAT-GFP, HUVEC/SP-TAT-GFP, HUVEC/TAT-apoptin, and
HUVEC/SP-TAT-apoptin cells prepared for transduction were
transferred to a vivaspin 2 column (catalog no. VS0291, Sartorius)
which was centrifuged for 25 min at 2,000 g at 4°C resulting in a
20-fold concentration. Target cells were treated for 30 min with
100 μl concentrated supernatant per well, fixed,
permeabilized, and examined for apoptin/GFP expression by
fluorescence microscopy. To assess those protein levels
concentrated supernatant was mixed with 3X LSB and examined by
western blot analysis.
RT-PCR
Total RNA was prepared using TRIzol®
reagent (catalog no. 15596-018, Invitrogen) from xenograft tissues.
To assess mRNA expression, RT-PCR was carried out using a
PrimeScript RT-PCR Kit (catalog no. DRR014A, Takara). The primers
for the apoptin gene and β-actin were described in our previous
report (13). The amplification
profile was as follows: 95°C for 45 sec, 56°C for 45 sec, and 72°C
for 1 min running in 30 cycles. After 30 amplification cycles, the
expected PCR products were size fractionated onto a 1% agarose gel
and stained with ethidium bromide.
Mouse xenografts and lentivirus
injection
A total of 20 BALB/C null mice and 16 male ICR mice
at 5 weeks of age were used for the experiment. The mice were
subcutaneously injected in the upper portion of the hind limb with
107 HepG2 cells. Three days after tumor cell engrafting,
the mice were randomly divided into four groups of five mice each.
When tumors were palpable (∼50 mm3 in 4–6 weeks),
animals were treated once by one of the two routes: via the tail
vein or intra-tumoral injection, with LV-SP-TAT-GFP or
LV-SP-TAT-apoptin at a dose indicated in the figure legends. Tumor
growth was monitored by measuring the length (L) and the width (W),
and the volume was calculated by the formula of
V=LxW2/2. The wet weight of dissected xenograft tumors
was recorded at the end of the experiment. The protocols used in
the study were approved by the Hospital's Protection of Human
Subjects Committee.
Immunohistochemistry and TUNEL assay
For protein analysis, xenograft tumors were
snap-frozen and stored at −80°C before processing. For
immunostaining analysis, specimens were fixed in 4%
paraformadehyde, paraffin-embedded and 4-micron tissue sections
were cut. Tumor sections were stained with hematoxylin and eosin
(H&E) to evaluate tumor structure. Primary antibody for V5
(catalog no. R960-25) was purchased from Invitrogen. Tissue
sections were immunostained with primary antibody against V5
followed by exposure to FITC labeled secondary antibodies and by
DAPI for nuclei (Sigma). Apoptotic cell death was determined by
in situ TUNEL analysis with the In Situ Cell Death Detection
Kit, POD (catalog no. 11684817910, Roche) as described in the
specifications.
Results
Signal-peptide-linked TAT-apoptin is
secreted, but not TAT-apoptin
To enable the efficient transduction of tumor cells
with apoptin, we have developed a novel mammalian expression system
for the in vivo secretion of proteins. Previously we
designed a secretory TAT-apoptin fusion protein by adding a
secretory signal SP to the N-terminus of the recombinant SP
sequence (12). In this study,
HUVEC cells were stably transfected with either TAT-apoptin/GFP or
SP-TAT-apoptin/GFP vectors, and the clones expressing
TAT-apoptin/GFP or SP-TAT-apoptin/GFP were selected using
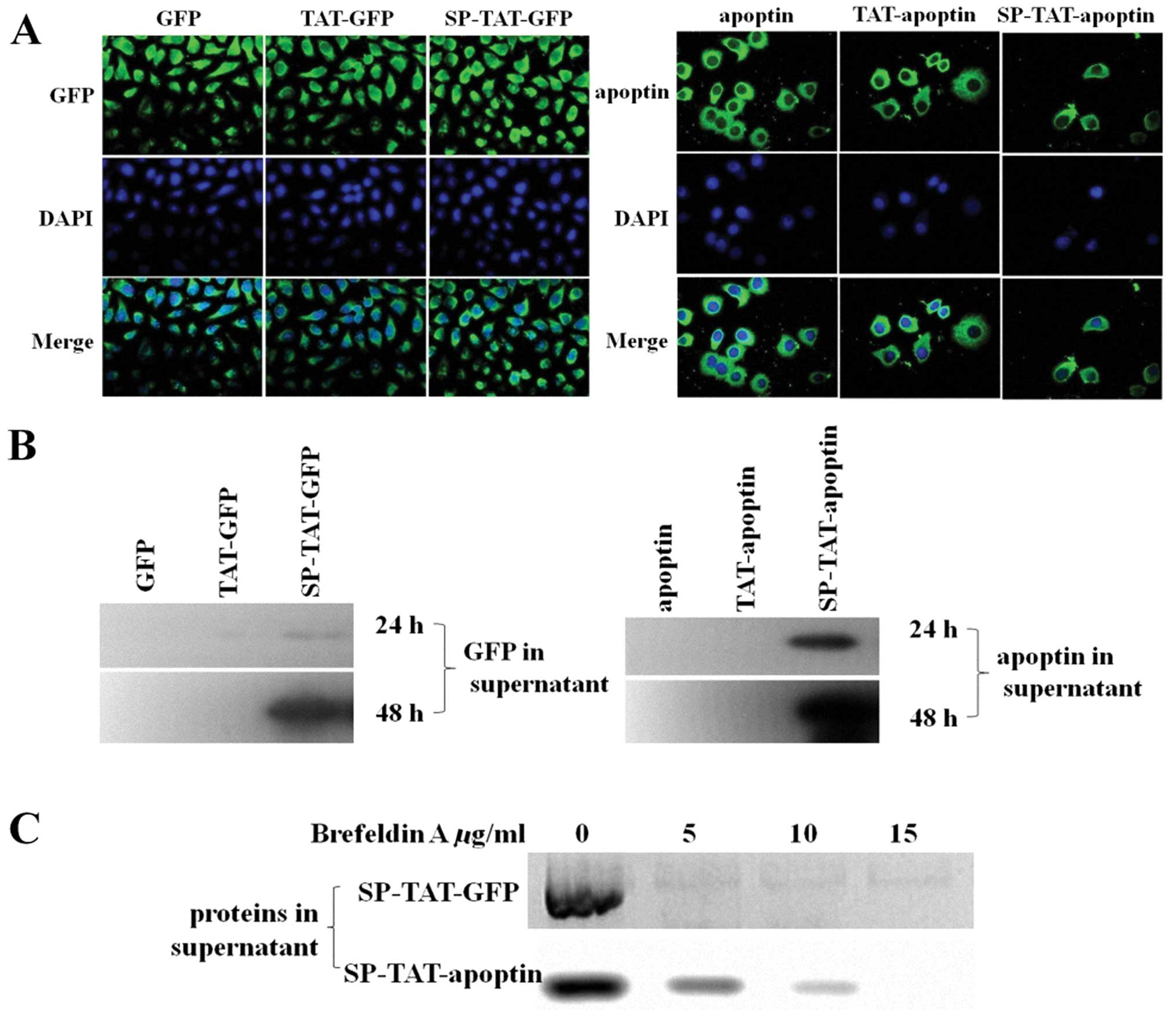
Blasticidin (8 μg/ml). Fluorescence microscopy detected no
difference with the different apoptin/GFP modifications (Fig. 1A). However, western blot analysis
showed the SP-TAT-apoptin/GFP HUVEC stable clones secreted high
levels of SP-TAT-apoptin/GFP in the culture medium while the
secretion from the TAT-apoptin/GFP HUVEC stable clones of
TAT-apoptin/GFP was very low (Fig.
1B). Brefeldin A (BFA) is a fungal metabolite that inhibits
protein transport out of the Golgi apparatus, causes disassembly of
this organelle and eventually blocks the secretion of proteins from
cells (15,16). HUVEC cells expressing
SP-TAT-apoptin/GFP were treated with different concentrations of
BFA. As shown in Fig. 1C,
secretion of SP-TAT-apoptin/GFP by HUVEC was significantly reduced
with BFA treatment.
Lentivirus-based secretable TAT-apoptin
is efficiently secreted and kills HCC cells in vitro
To further assess the secretory ability of
SP-TAT-apoptin/GFP, lentiviruses LV-SPTAT-apoptin/GFP were
generated as described in our previous publication (13). LV-SP-TAT-apoptin/GFP-infected HUVEC
cells were examined for the secretion of TAT-apoptin/GFP in the
supernatant by western blot analysis. High levels of recombinant
protein were detected in the supernatant (Fig. 2A). Furthermore, apoptosis,
estimated by flow cytometry assay (Annexin V and PI), was detected
in HepG2 Bel-7402 cells treated with the concentrated supernatant
of LV-SP-TAT-apoptin-infected HUVEC cells (41.22 and 50.31%,
respectively), in contrast to no induction of cell death in HepG2
and Bel-7402 cells treated with the concentrated supernatant of
LV-SP-TAT-GFP-infected HUVEC cells. Moreover, there was no
induction of cell death in HL-7702 cells treated with the
concentrated supernatant of LV-SPTAT-apoptin/GFP-infected HUVEC
cells (Fig. 2B). Using
fluorescence microscopy, HepG2, Bel-7402 and HL-7702 cells treated
with the concentrated supernatant of HUVEC cells infected with
LV-SP-TAT-GFP for 30 min showed positive GFP expression in target
cells. However, apoptin nuclear localization was detected only in
HepG2 and Bel-7402 cells treated with the concentrated supernatant
of HUVEC cells infected with LV-SP-TAT-apoptin for 30 min, while
apoptin nuclear localization was not detected in HL-77002 cells
treated with the concentrated supernatant of HUVEC cells infected
with LV-SP-TAT-apoptin for 30 min (Fig. 2C).
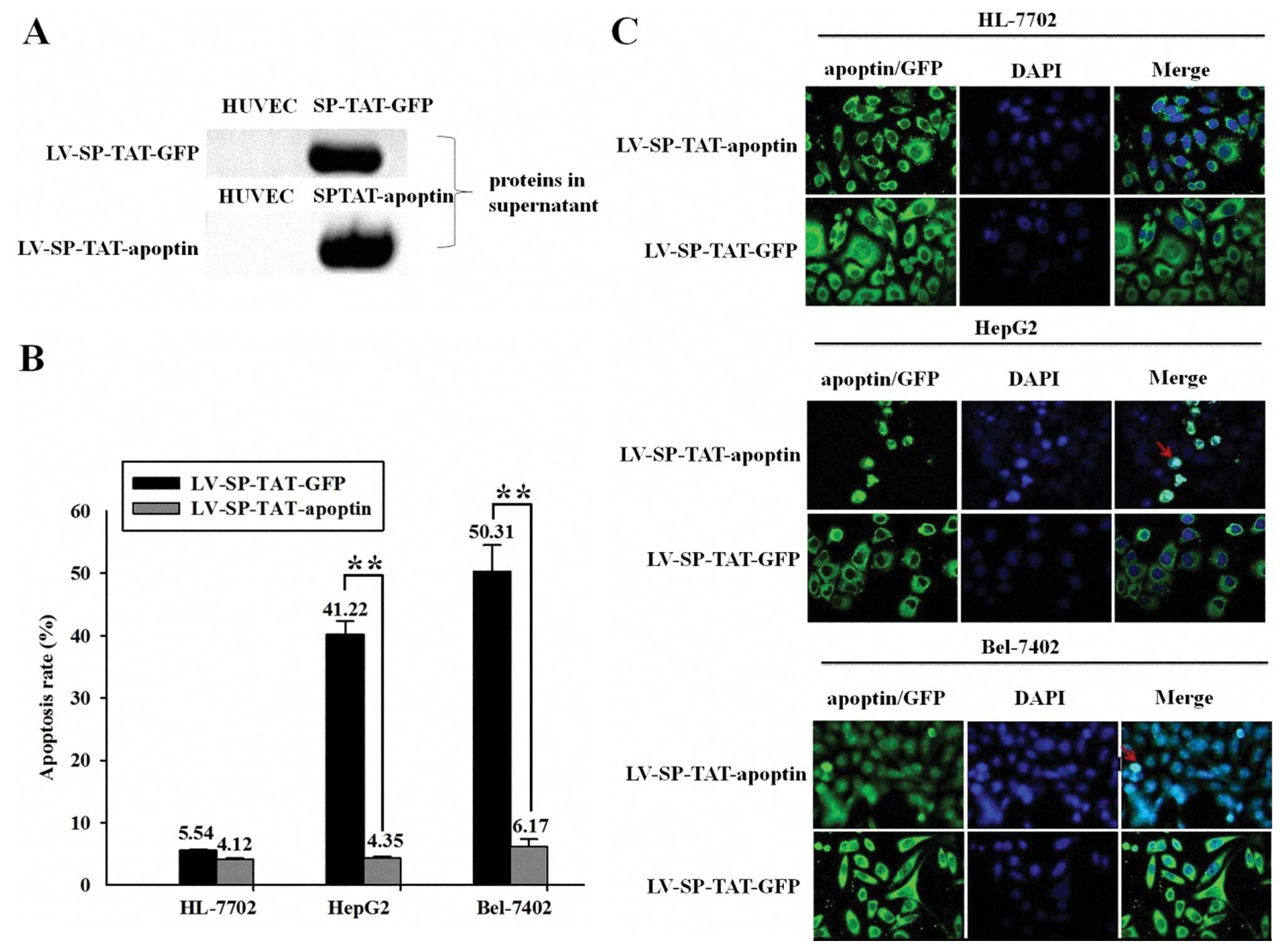 | Figure 2Lentivirus-based secretable
TAT-apoptin is efficiently secreted and kills HCC cells. (A)
LV-SP-TAT-apoptin/GFP-infected HUVEC cells expressing secretable
TAT-apoptin/GFP were examined for the secretion of TAT-apoptin/GFP
in the supernatant by western blot analysis. High levels of
recombinant protein were detected in the supernatant. (B)
Apoptosis, estimated by flow cytometry assay (Annexin V and PI) was
detected in HepG2 Bel-7402 cells treated with the concentrated
supernatant of LV-SP-TAT-apoptin-infected HUVEC cells (41.22 and
50.31%, respectively), while there was no induction of cell death
in HepG2 and Bel-7402 cells treated with the concentrated
supernatant of LV-SP-TAT-GFP-infected HUVEC cells. Moreover, there
was no induction of cell death in HL-7702 cells treated with the
concentrated supernatant of LV-SP-TAT-apoptin/GFP-infected HUVEC
cells. (Student's t-test, **P<0.05). (C) Using
fluorescence microscopy, HepG2, Bel-7402 and HL-7702 cells treated
with the concentrated supernatant of HUVEC cells infected with
LV-SP-TAT-GFP for 30 min showed positive GFP expression in target
cells. However, apoptin nuclear localization was detected only in
HepG2 and Bel-7402 cells treated with the concentrated supernatant
of HUVEC cells infected with LV-SP-TAT-apoptin for 30 min, while
apoptin nuclear localization was not detected in HL-77002 cells
treated with the concentrated supernatant of HUVEC cells infected
with LV-SP-TAT-apoptin for 30 min. Magnification, ×600. |
Lentivirus-based secretable TAT-apoptin
eradicates HCC xenograft tumors in vivo after systemic
delivery
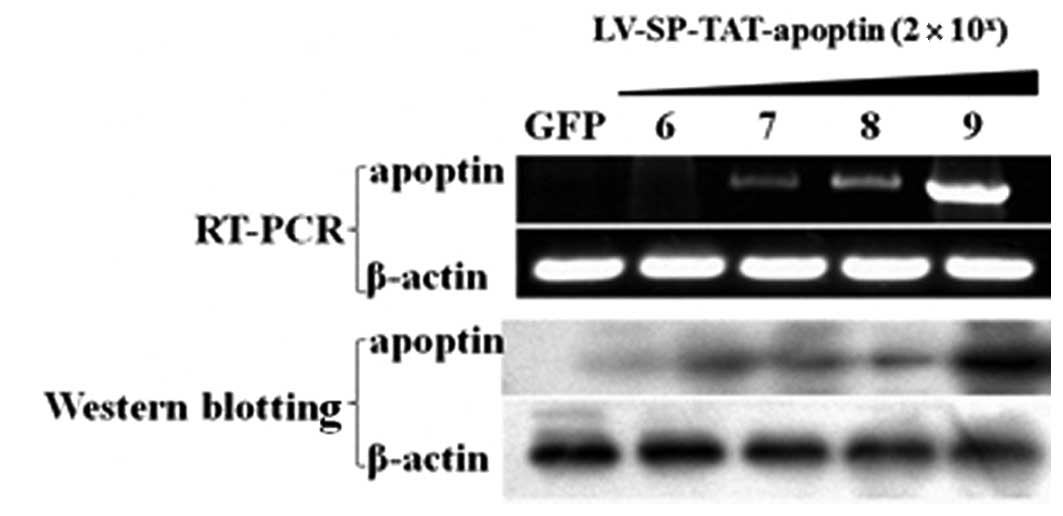
A pilot experiment was initially carried out to
determine a proper dose for efficient SP-TAT-apoptin expression in
xenograft tumors. Xenograft tumors were generated in nude mice
using the HCC cell line HepG2. When tumors were palpable (around 50
mm3 in size), 4 different doses (log-dilution,
2×106 – 2×109 viral particles in 10 μl
total volume) of the recombinant LV-SPTAT-apoptin viruses were
injected into the tumor (multiple sites per tumor). In addition,
two other animals received the virus LV-SP-TAT-GFP
(2×109 viral particles in 10 μl) as the negative
controls. One week later, xenograft tumors were harvested for
further analysis. RT-PCR and western blotting results showed a
gradually increasing pattern in the apoptin mRNA and protein levels
(Fig. 3), reflecting a
dose-dependent expression of viral genes. The peak effect was seen
at the dose of 2×109 particles per 50
mm3.
Subsequently, we tested if the secretable
TAT-apoptin could tackle the critical obstacle, HCC. HepG2 cells
were injected into the upper portion of the hind limb of BALB/C
null mice, and allowed to grow. Once HepG2 xenograft tumors were
palpable (∼50 mm3), animals were randomly divided into
two groups to receive an intra-tumoral injection of either
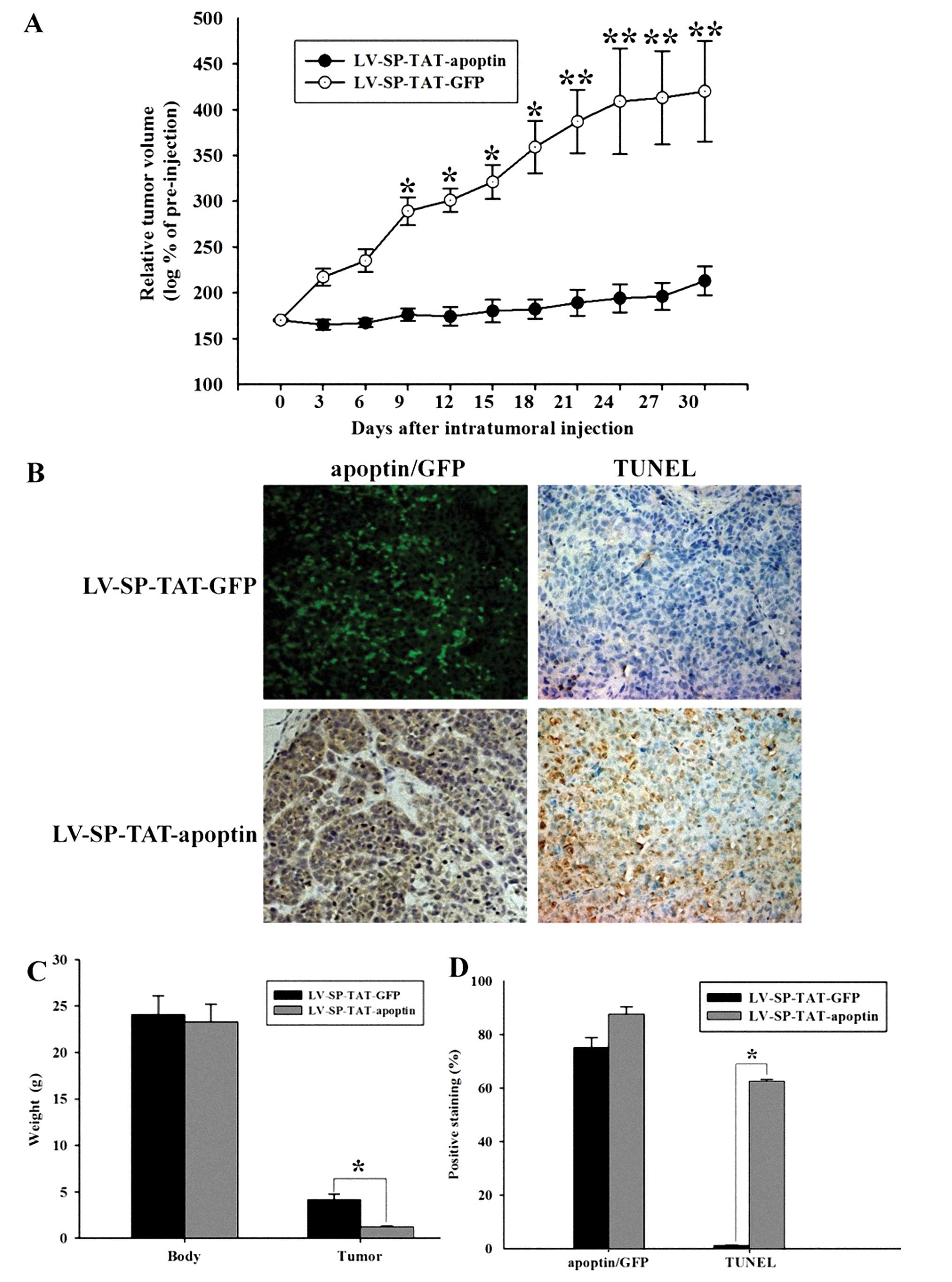
LV-SP-TAT-GFP or LV-SP-TAT-apoptin viruses. Tumor growth was
monitored for another 30 days. As shown in Fig. 4A and C, following intra-tumoral
injection, LV-SPTAT-GFP virus-injected xenograft tumors grew
constantly. By contrast, LV-SP-TAT-apoptin virus-injected xenograft
tumors were completely suppressed. These data indicate that
LV-SP-TAT-apoptin eliminated progression of HCC. At the end of the
experiments, xenograft tumors were harvested for further analysis,
including virus distribution (apoptin/GFP expression) and apoptotic
cell death (TUNEL assay). As shown in Fig. 4B and D, a diffused pattern of
apoptin/GFP expression was observed in virus-injected xenografts,
although some spots displayed higher levels of apoptin/GFP
expression, perhaps due to uneven distribution of injected viruses.
In accordance with the tumor growth pattern, higher apoptotic index
(TUNEL assay) was found in the LV-SP-TAT-apoptin virus-treated
xenografts compared to the LV-SP-TAT-GFP virus-treated xenografts,
which is consistent with our previous finding that
LV-SP-TAT-apoptin virus leads to apoptotic cell death in HCC
cells.
We showed that intra-tumoral injection of
LV-SP-TAT-apoptin virus abrogated tumor growth but did not
eradicate xenograft tumors. A plausible explanation might be the
uneven distribution of the locally injected viruses, which affected
the number of tumor cells (SP-TAT-apoptin producer cells) killed by
TAT-apoptin. In order to achieve a better virus distribution and to
induce a profound tumor recession, we went on to inject the viruses
via the tail vein for systemic delivery of the viruses. Once HepG2
cell-based xenograft tumors were palpable (∼50 mm3) in
nude mice, LV-SP-TAT-apoptin or LV-SP-TAT-GFP viruses were injected
via the tail vein at a dose of 2.0×109 viral particles
per injection. Tumor growth and animal condition were monitored for
one month. No animal presented any obvious complication and no
visible side-effect was noticed due to the virus injection other
than xenograft-related stress. As shown in Fig. 5A, LV-SP-TAT-GFP virus-injected
tumor showed a rapid and continuous growth. However, in a group of
eight animals that received LV-SP-TAT-apoptin injection, five
tumors disappeared within 10 days and two other tumors disappeared
within 2 weeks. The last tumor remained a steady size for a long
period of time. Following dissection, H&E staining revealed
that it was a fibrous scar with a few HCC cells (Fig. 5B). These data strongly indicate
that the LV-SP-TAT-apoptin virus injection eradicated xenograft
tumors. Lentivirus-based secretable TAT-apoptin eradicates HCC
xenograft tumors in vivo after systemic delivery.
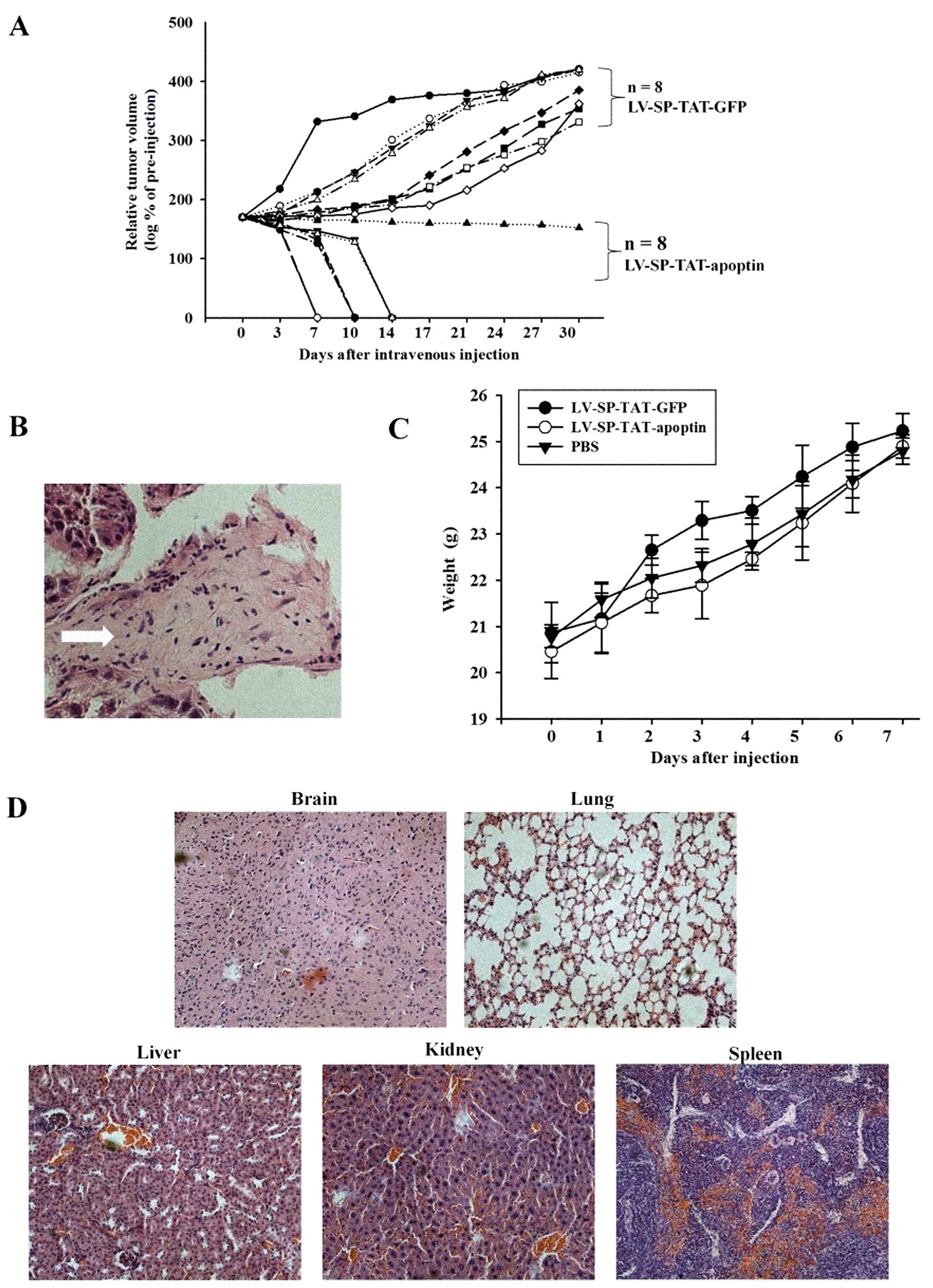 | Figure 5Systemic injection of
LV-SP-TAT-apoptin virus eradicates HepG2 xenograft tumors in nude
mice and the toxicity of LV-SP-TAT-apoptin in mice. (A) Data
presented are the LV-SP-TAT-apoptin virus-injected relative tumor
volume compared to the initial value from individual xenografts. Of
note, 7 tumors disappeared within 2 weeks and the last one
displayed as a steady node, whose image is shown in panels B, C
following dissection. However, LV-SP-TAT-GFP virus-injected tumor
showed continuous growth. Quantitative data (mean ± SEM) are
presented in panel A. The asterisk indicates a significant
difference compared to the control (Student's t-test, P<0.05).
(B) H&E staining revealed that LV-SP-TAT-apoptin-injected tumor
was a fibrous scar with a few HCC cells. Magnification, ×200.
Arrow, the fibrous scar. (C) Following virus injection, all groups
gained weight similar to control animals (range within 10%).
Quantitative data (mean ± SEM) are presented in panel A. (D) No
overt pathology was noted in the lung, spleen, kidney, and liver
tissues. Magnification, ×200. |
Toxicity of lentivirus-based secretable
TAT-apoptin
To evaluate the possible toxic effects of
LV-SP-TAT-apoptin, we injected 2.0×109 viral particles
(LV-SP-TAT-apoptin, LV-SP-TAT-GFP or saline) in healthy ICR mice
via the tail vein. Since body weight is generally an accurate
indicator of health status within age-matched groups, we used this
parameter as a criterion of well-being. It can be seen in Fig. 5C that all groups gained weight
similar to control animals (ranging within 10%) after injection.
After 1 week, all animals were sacrificed. Brain, lung, spleen,
kidney, and liver samples were isolated for macroscopic evaluation
and histological examination. No overt pathology was noted in these
tissues (Fig. 5D). The general
parameter measured (the weights of total body) were similar to
those of the control groups at the end of the experiment (Fig. 5C). Based on our data, we conclude
that significant amounts of the lentivirus-based secretable
TAT-apoptin can be administered without acute fatal toxicity.
Discussion
This report describes the generation of recombinant
lentivirus expressing secretable TAT-apoptin and its effects on
xenograft tumors in vivo. Our previous in vitro
studies (12,13), have shown that SP-TAT-apoptin
induces apoptosis in HepG2 cells, but not in HUVEC cells. We
expected, therefore, that the generation and production of
recombinant lentivirus expressing the secretable SP-TAT-apoptin
gene might greatly increase its potency once it is delivered in
vivo for cancer therapy.
We report here that the resultant SP-TAT-apoptin/GFP
was capable of being secreted from the producer cells and was taken
up by several target cell lines. The efficient delivery of the
secreted TAT-apoptin/GFP was demonstrated using both harvested
culture supernatant as well as mixing experiments of the culture
supernatant from producer cells and target cells. The
TAT-apoptin/GFP was unable to enter the target cells probably due
to the fact that TAT-apoptin/GFP fusion protein without SP can be
secreted into the culture supernatant. BFA, a drug that blocks
secretion, prevents the secretion of SP-TAT-apoptin/GFP via the
constitutive pathway. These results suggest that SP
linked-TAT-apoptin/GFP is secreted through the constitutive
pathway, but not TAT-apoptin/GFP.
To further assess the secretory ability of
SP-TAT-apoptin/GFP, lentivirus LV-SP-TAT-apoptin/GFP was generated
as described in our previous publication (13). Our data showed the successful
secretion of high levels of TAT-apoptin/GFP into the culture medium
from HUVEC cells infected by LV-SP-TAT-apoptin/GFP. Significantly,
the secreted TAT-apoptin displaced to nucleus and induced cell
death in target cancer cells. The next objective would be to test
the efficacy of LV-SP-TAT-apoptin infected cells to secrete this
fusion protein and kill cancer cells within the tumor environment
in vivo. There are two possible ways for the in vivo
delivery of LV-SP-TAT-apoptin viruses: i) through intra-tumoral
injection and ii) by injection via the tail vein. To mimic the
clinical situation of cancer treatment, in our study, we first
injected the LV-SP-TAT-apoptin viruses directly into existing
xenograft tumors. Similar to other reports, this approach resulted
only in the suppression of the xenograft tumor growth, and not in
tumor elimination as we had expected, due to the uneven
distribution of the locally injected viruses. After careful
evaluation of the tumor section, we realized that the injected
LV-SP-TAT-apoptin viruses were not evenly distributed inside the
tumor mass. To achieve a more profound effect, we injected the
LV-SP-TAT-apoptin viruses via the tail vein. Almost all the
xenograft tumors disappeared following the treatment while the
xenografts that received the control LV-SP-TAT-GFP viruses
continued to grow, indicating a superior tumor-targeting efficiency
through the systemic delivery.
Moreover, the animal studies presented in this paper
demonstrate a low toxicity of SP-TAT-apoptin in vivo,
confirming and extending the results of the in vitro
studies. Healthy ICR mice were injected via the tail vein with
LV-SP-TAT-apoptin, LV-SP-TAT-GFP, and phosphate buffered saline
(PBS), respectively. After 1 week, all animals were sacrificed.
Lung, spleen, kidney, and liver samples were isolated for
macroscopic evaluation and histological examination. No overt
pathology was noted in these tissues. The general parameters
measured (the weights of total body), were similar to those of the
control groups at the end of the experiment. From these data, we
conclude that significant amounts of the lentivirus-based
secretable TAT-apoptin can be administered without acute fatal
toxicity.
In this study, we demonstrated that systemic
delivery of the lentivirus-based SP-TAT-apoptin viruses eradicated
xenograft tumors without acute fatal toxicity. Our results indicate
that secretable TAT-apoptin might be a potential strategy in
hepatocellular carcinoma. However, as a clinical therapy for HCC,
there are still a large number of issues to be resolved. Although
lentivirus has been shown to be very efficient for delivering genes
into cells, there are restrictions for its therapeutic use, such as
risk of insertional mutagenesis and possible immunogenicity. Thus,
side-effects from lentivirus-based SP-TAT-apoptin therapy may limit
its application. Indeed, one of our future directions is to develop
a novel delivery system combined with the secretable proteins to
provide a safer and more effective therapeutic strategy. In
summary, our data strongly suggest that systemic delivery of
lentivirus-based secretable TAT-apoptin is feasible to eradicate
liver cancer in vivo.
Abbreviations:
|
TAT
|
transacting activator of
transcription
|
|
PTD
|
protein transduction domain
|
|
SP
|
signal peptide
|
|
HCC
|
hepatocellular carcinoma
|
|
BFA
|
Brefeldin A
|
|
TUNEL
|
terminal deoxynucleotidyl transferase
dUTP nick end labeling
|
|
H&E
|
hematoxylin and eosin
|
|
PI
|
propidium iodide
|
|
PBS
|
phosphate buffered saline
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
FBS
|
fetal bovine serum
|
|
LSB
|
Laemmli sample buffer
|
Acknowledgements
This study was supported by the
National Natural Scientific Foundation of China (no. 81071692). We
thank Professor Pei-jun Liu, Dr Lei Li, and Dr Lei Zhao for their
helpful insight in the course of these studies and Professor Huang
Chen for his technical advice.
References
|
1.
|
Noteborn MH and van der Eb AJ:
Apoptin-induced apoptosis: potential for antitumor therapy. Drug
Resist Updat. 1:99–103. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Los M, Panigrahi S, Rashedi I, Mandal S,
Stetefeld J, Essmann F, et al: Apoptin, a tumor-selective killer.
Biochim Biophys Acta. 1793:1335–1342. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Russo A, Terrasi M, Agnese V, Santini D
and Bazan V: Apoptosis: a relevant tool for anticancer therapy. Ann
Oncol. 17:vii115–23. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Alvisi G, Poon IK and Jans DA:
Tumor-specific nuclear targeting: promises for anti-cancer therapy?
Drug Resist Updat. 9:40–50. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Maddika S, Mendoza FJ, Hauff K, Zamzow CR,
Paranjothy T and Los M: Cancer-selective therapy of the future:
apoptin and its mechanism of action. Cancer Biol Ther. 5:10–19.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Poon IK, Oro C, Dias MM, Zhang JP and Jans
DA: A tumor cell-specific nuclear targeting signal within chicken
anemia virus VP3/apoptin. J Virol. 79:1339–1341. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Chauhan A, Tikoo A, Kapur AK and Singh M:
The taming of the cell penetrating domain of the HIV Tat: myths and
realities. J Control Release. 117:148–162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Dietz GP and Bahr M: Delivery of bioactive
molecules into the cell: the Trojan horse approach. Mol Cell
Neurosci. 27:85–131. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Wang H, Zhong CY, Wu JF, Huang YB and Liu
CB: Enhancement of TAT cell membrane penetration efficiency by
dimethyl sulphoxide. J Control Release. 143:64–70. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Flinterman M, Farzaneh F, Habib N, Malik
F, Gaken J and Tavassoli M: Delivery of therapeutic proteins as
secretable TAT fusion products. Mol Ther. 17:334–342. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Guelen L, Paterson H, Gaken J, Meyers M,
Farzaneh F and Tavassoli M: TAT-apoptin is efficiently delivered
and induces apoptosis in cancer cells. Oncogene. 23:1153–1165.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Han SX, Ma JL, Lv Y, Huang C, Liang HH and
Duan KM: Secretory Transactivating Transcription-apoptin fusion
protein induces apoptosis in hepatocellular carcinoma HepG2 cells.
World J Gastroenterol. 14:3642–3649. 2008. View Article : Google Scholar
|
|
13.
|
Han SX, Zhao J, Ma JL, Huang C, Lu Y, Ou
W, et al: The effect of the fused gene of SP-TAT-Apoptin
transfected by lentivirus on HepG2 cells. Xi Bao Yu Fen Zi Mian Yi
Xue Za Zhi. 26:310–312. 2010.(In Chinese).
|
|
14.
|
Barash S, Wang W and Shi Y: Human
secretory signal peptide description by hidden Markov model and
generation of a strong artificial signal peptide for secreted
protein expression. Biochem Biophys Res Commun. 294:835–842. 2002.
View Article : Google Scholar
|
|
15.
|
Lippincott-Schwartz J, Yuan LC, Bonifacino
JS and Klausner RD: Rapid redistribution of Golgi proteins into the
ER in cells treated with brefeldin A: evidence for membrane cycling
from Golgi to ER. Cell. 56:801–813. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Pelletier L, Jokitalo E and Warren G: The
effect of Golgi depletion on exocytic transport. Nat Cell Biol.
2:840–846. 2000. View
Article : Google Scholar : PubMed/NCBI
|