Introduction
Glioblastoma (GB, WHO grade IV glioma) and
anaplastic gliomas (WHO grade III gliomas) are lethal brain tumors
for which only limited therapeutic options exist. The standard
treatment for high-grade gliomas (WHO grade III and IV) consists of
a maximal safe surgical resection followed by adjuvant
radiotherapy. While concomitant temozolomide during the radiation
therapy followed by six cycles of adjuvant chemotherapy improves
the survival of patients with GB, the role of chemotherapy in the
initial treatment of anaplastic glioma remains to be defined
(1).
Clinically relevant molecular subtypes of GB have
recently been identified (2). A
small proportion of GB are characterized by mutation of the
isocitrate dehydrogenase 1 and −2 (IDH1 and −2) genes, a genotype
that is most frequently found in low-grade and anaplastic gliomas
(3). Genomic alterations of the
epidermal growth factor receptor (EGFR) gene play a crucial role in
pathogenesis of a subgroup of GB (4). The most common gain-of-function
alterations for EGFR in GB are mutation, amplification, and
overexpression (5,6). Expression of a constitutively
phosphorylated EGFR-mutant, EGFR variant III (EGFRvIII), is found
in approximately 20%–30% of GB patients. EGFRvIII has an in-frame
deletion of exons 2–7 resulting in the loss of the amino acid
residues that contribute to the ligand binding area. Consequently,
EGFRvIII causes ligand-independent constitutive activation of
downstream signaling pathways such as the Mitogen-activated protein
kinase (MAPK) pathway that contributes to the malignant phenotype
(7–9). In vitro studies have
demonstrated that expression of EGFRvIII leads to a growth and
survival advantage in several types of cancer cells (10–13).
Another frequent EGFR mutant in GB is the EGFR variant IV
(EGFRvIV), which has a genomic truncation of the carboxyl-terminus
domain (CTD, exon 25, 26 and 27 in EGFRvIVa or exon 25 and 26 in
EGFRvIVb) (14,15). Both EGFRvIV variants have a
transforming capacity in vitro and in animals (16). Furthermore, a novel deletion
mutation with the transforming capacity was found also in EGFR CTD
which is the deletion of exon 27 (17).
In high-grade gliomas, the EGFRvIII and EGFRvIV
mutations have been found exclusively in association with EGFR gene
amplification (18,19). Expression of EGFRvIII identifies a
particular subtype of anaplastic astrocytoma with a poor prognosis.
In the global GB-population this correlation with survival is less
distinct (20–24). However, within the subpopulation of
GB patients with EGFR amplification, a strong correlation was
reported between EGFRvIII expression and a poor overall survival
(4). No study has yet reported a
correlation between EGFRvIV expression and survival of GB
patients.
The importance of EGFRvIII expression as a
predictive marker for anti-EGFR therapies has been a matter of
debate. Co-expression of EGFRvIII and a phosphatase and tensin
homologue on chromosome 10 (PTEN) was reported to be a significant
predictor of responsiveness to small molecule EGFR kinase
inhibitors in retrospectively identified GB patients benefitting
from such therapy (25).
Furthermore, concomitant EGFRvIII expression and loss of PTEN were
shown to synergistically induce genomic instability in vitro
and result in enhanced tumor formation (26). In additional studies it was
reported that EGFR amplification and protein kinase B/akt
activation to predict the response to treatment with a small
molecule EGFR kinase inhibitor in GB patients (27–28).
These findings were however not confirmed in a prospective
randomized study with erlotinib (29).
In vitro data indicate that cetuximab is able
to bind to and down regulate EGFRvIII. This may however not be
sufficient to inhibit the proliferation of the glioma cells
(30). Other observations have
indicated that cetuximab not only reduced the phosphorylated
EGFRvIII but also inhibited significantly the proliferation of
EGFRvIII expressing cells (31).
Moreover, cetuximab proved to be active in animal models with EGFR
amplified GB (32). In addition,
radiation and chemotherapeutic agents augmented the effect of
cetuximab on GB cells in vitro and in vivo (33). Recently, the anti-tumor activity of
cetuximab against EGFR CTD-mutant GB in both cells and animals was
reported (17).
We have previously reported the results of a
stratified 2-arm prospective phase II clinical trial of recurrent
high-grade glioma patients treated with the EGFR monoclonal
antibody cetuximab. A numerically superior outcome, not reaching
statistical significance, was found in patients with EGFR gene
amplification (34). In this work,
we have further investigated the correlation between EGFR
amplification, EGFRvIII and EGFRvIV expression, PTEN expression,
and IDH1 mutation status and the survival of patients treated with
cetuximab in this clinical trial.
Materials and methods
Patients and tumor material
Tumor material was obtained from 35 patients with
recurrent high-grade glioma who participated in the prospective
phase II trial (34). Tumor
tissues were obtained during therapeutic resections performed
before the time of study participation. Tumor sections with a
thickness of 4 μm were used for H&E staining and centralized
review of tumor histopathology and grading according to WHO 2007
criteria (35).
EGFR and IDH1 molecular analysis
The IDH1 mutation status was assessed with a
PCR-DGGE-Sequencing system as reported previously (36). The amplification status of EGFR was
assessed by FISH, as described in the report on the clinical trial
with cetuximab (34).
For the molecular analysis of EGFRvIII,
formalin-fixed paraffin-embedded (FFPE) tissue blocks were
sectioned at a thickness of 10 μm (3 sections for RNA isolation).
As a positive control an EGFRvIII transfected U87 cell line,
obtained from Professor Webster Cavenee (Ludwig Institute for
Cancer Research, University of California, San Diego, CA, USA), was
used (10). Tissues were dewaxed
by xylene and ethanol. The total RNA was isolated from tumor
sections using the RNeasy FFPE kit (Qiagen, Venlo, Netherlands)
according to the manufacturer’s instructions with modifications by
changing the incubation time after mixing with proteinase K for 36
h at 55°C, meanwhile, adding proteinase K every 12 h. Depending on
the size of the tumor sample, the RNA ranged from 0.02 to 2 μg
(Nanodrop2000, Thermo Scientific, Wilmington, DE, USA). The RNA
isolation of the positive control cell line (U87δ) was performed
with the Absolute RNA Wash Solution (Applied Biosystems, Carlsbad,
CA, USA). The reverse transcription was accomplished using the
SuperScript® II Reverse Transcriptase (Invitrogen)
according to the manufacturer’s protocol. The entire cDNA sample
was treated with Ribonuclease H (Invitrogen, Ghent, Belgium) to
eliminate the residual RNA after reverse transcription.
Two pairs of primers for EGFRvIII were derived from
the previous reference to perform a hemi-nested PCR (19) with some modifications. For the
first step PCR, a forward primer GAGCT CTTCGGGGAGCAG and a reverse
primer TCCTCCATCTCA TAGCTGTCG were used to generate a 178 bp
fragment that span the junctions of the deletion c.158_1136del. A
standard PCR Master mix (25 μl in total) is composed of cDNA (1
μl), 1X PCR buffer, 1 μg/μl bovine serum albumin (BSA), 0.8 mM
dNTPs, 0.025 U/μl Taq DNA polymerase (Qiagen, 5 U/μl) and 2 ng/μl
of each primer. The reaction was run for 35 cycles denaturation at
94°C for 1 min, annealing at 64°C for 1 min and extension at 72°C
for 1 min. During the second step, a 131 bp fragment was amplified
25 cycles using 1 μl of product from the first step PCR with the
forwards primer GAGCTCTTCGGGG AGCAG and the reverse primer
GTGATCTGTCACCACATA ATTACCTTTCT. All other PCR conditions were the
same as step 1. The final PCR product was visualized on a 2%
agarose gel and confirmed by sequencing (ABI 310 Genetic Analyzer,
Applied Biosystems, Foster city, CA, USA) after purification with
the High Pure PCR Product Purification kit (Roche, Penzberg,
Germany). The analysis of each sample was performed in triplicate.
EGFRvIV mutation analysis was performed with the same method as
EGFRvIII.
Immunohistochemistry of PTEN
PTEN expression was evaluated by
immunohistochemistry on 4 μm FFPE tissue sections by using a rabbit
IgG monoclonal antibody (1:100 dilution, PTEN, 138G6, Cell
Signaling Technology Inc., Beverly, MA, USA) and an anti-rabbit IgG
secondary antibody (ultraView Universal DAB Detection Kit, Ventana
Medical Systems Inc., Tucson, AZ, USA) according to the
manufacturer’s protocol. Immunoreactivity was visualized with a DAB
solution provided in the kit (ultraView Universal DAB Detection
Kit, Ventana Medical Systems Inc.). Positive controls were
visualized on the same sections with epithelial cells and neurons.
PTEN staining was both nuclear and cytoplasmic and was evaluated
with a two-score system including intensity and percentage of
positive cells as described (37).
Clinical data and survival analysis
Clinical information was collected from each patient
to investigate possible correlation between EGFRvIII mutation
status and clinical parameters including glioma WHO grade, age,
date of birth, date of initial diagnosis and of recurrence, date of
progression and date of death or last contact. Kaplan-Meier
survival analysis was used to examine the correlation between
molecular baseline factors and survival data. We also analyzed the
association between EGFRvIII, EGFRvIV, PTEN expression, EGFR
amplification and IDH1 mutation.
Results
Analysis of EGFR gene amplification,
EGFRvIII-, EGFRvIV-RNA expression, and PTEN-protein expression and
IDH1 mutation status
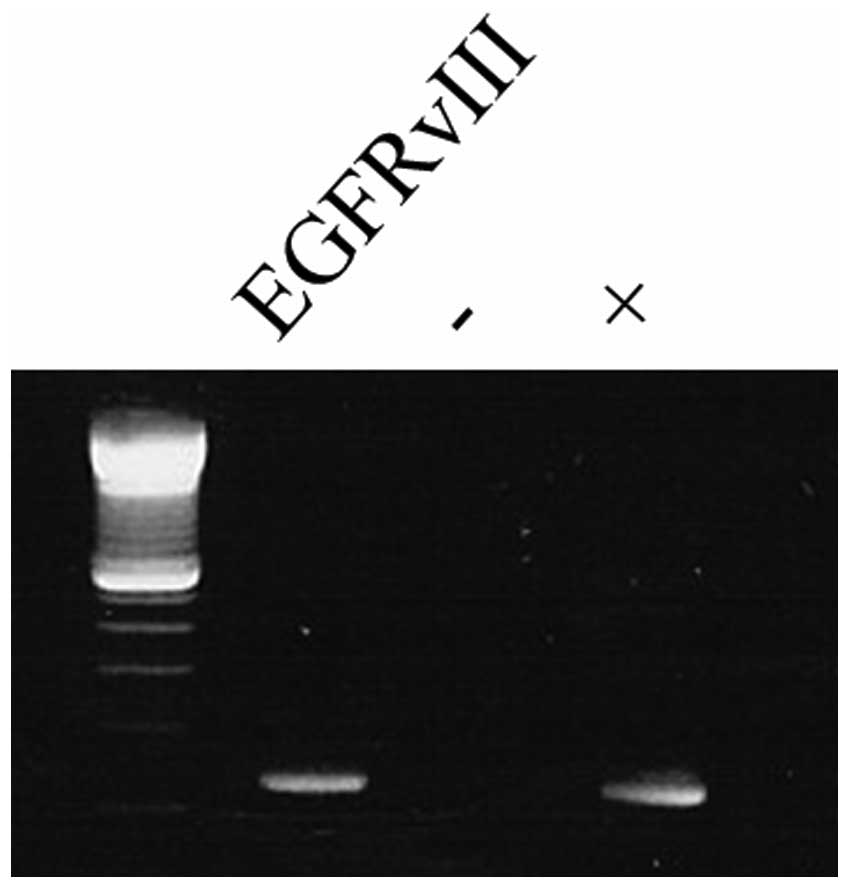
EGFR amplification, EGFRvIII-, and EGFRvIV
expression were detected in, respectively 19/35 (54%), 11/35 (31%)
and 7/35 (20%, EGFRvIVa in 5/7 and EGFRvIVb in 2/7) of the gliomas
(Fig. 1, Table I). EGFRvIII and EGFRvIV expression
were exclusively found in patients with an EGFR amplification
(11/19, 58%; 7/19, 37%). EGFRvIII was most frequently found in
de novo GBs (9/26, 35%), and less frequently in secondary
GBs and grade II or III glioma (2/9, 22%). Likewise, EGFRvIV was
detected in 6/26 (23%) de novo GB and 1/9 (11%) of the
anaplastic astrocytoma.
 | Table ISummary of clinical and molecular
characteristics. |
Table I
Summary of clinical and molecular
characteristics.
| Characteristic
(N=35) | N |
| Sex, M/F | 22/13 |
| Median age,
range | 54, 33–73 |
| Histology at first
diagnosis | |
| WHO grade II and
III glioma | 9 |
| WHO grade IV, de
novo GB | 26 |
| Histology at
recurrence | |
| WHO grade II and
III glioma | 3 |
| WHO grade IV,
secondary GB | 6 |
| WHO grade IV,
de novo GB | 26 |
| KPS at
recurrence | |
| 100 | 3 |
| 90–80 | 10 |
| 70–60 | 22 |
| EGFR amplification
status | |
|
Amplification | 19 |
| Wild-type | 16 |
| EGFRvIII
status | |
| Mutation | 11 |
| Wild-type | 24 |
| EGFRvIV status | |
| Mutation | 7 |
| Wild-type | 28 |
| IDH1 status | |
| Mutation | 6 |
| Wild-type | 29 |
| PTEN status
(N=31) | |
| Positive | 6 |
| Negative | 25 |
IDH1 mutation was found in 6/35 (17%) patients
(Table I) and correlated with a
younger age at first diagnosis, and a histological diagnosis of
low-grade or secondary GB. IDH1 mutation was only rarely found in
association with an EGFR amplification or expression of EGFRvIII
and EGFRvIV (2/19 patients with EGFR amplification, 1/11 patients
with EGFRvIII and 0/7 patients with EGFRvIV were found with a
concomitant IDH1 mutation, Table
II).
| Table IICorrelation between molecular
markers. |
Table II
Correlation between molecular
markers.
| EGFRvIII |
|---|
| + | − |
|---|
|
|
|---|
| IDH1 mutation | IDH1 mutation |
|---|
| + | − | + | − |
|---|
| N=31 EGFR
amplification |
| + | PTEN | + | 0 | 2 | 0 | 2 |
| | − | 1 | 8 | 1 | 5 |
| − | PTEN | + | 0 | 0 | 0 | 2 |
| | − | 0 | 0 | 2 | 8 |
| N=35 EGFR
amplification |
| + | EGFRvIV | + | 0 | 5 | 0 | 2 |
| | − | 1 | 5 | 1 | 5 |
| − | EGFRvIV | + | 0 | 0 | 0 | 0 |
| | − | 0 | 0 | 4 | 12 |
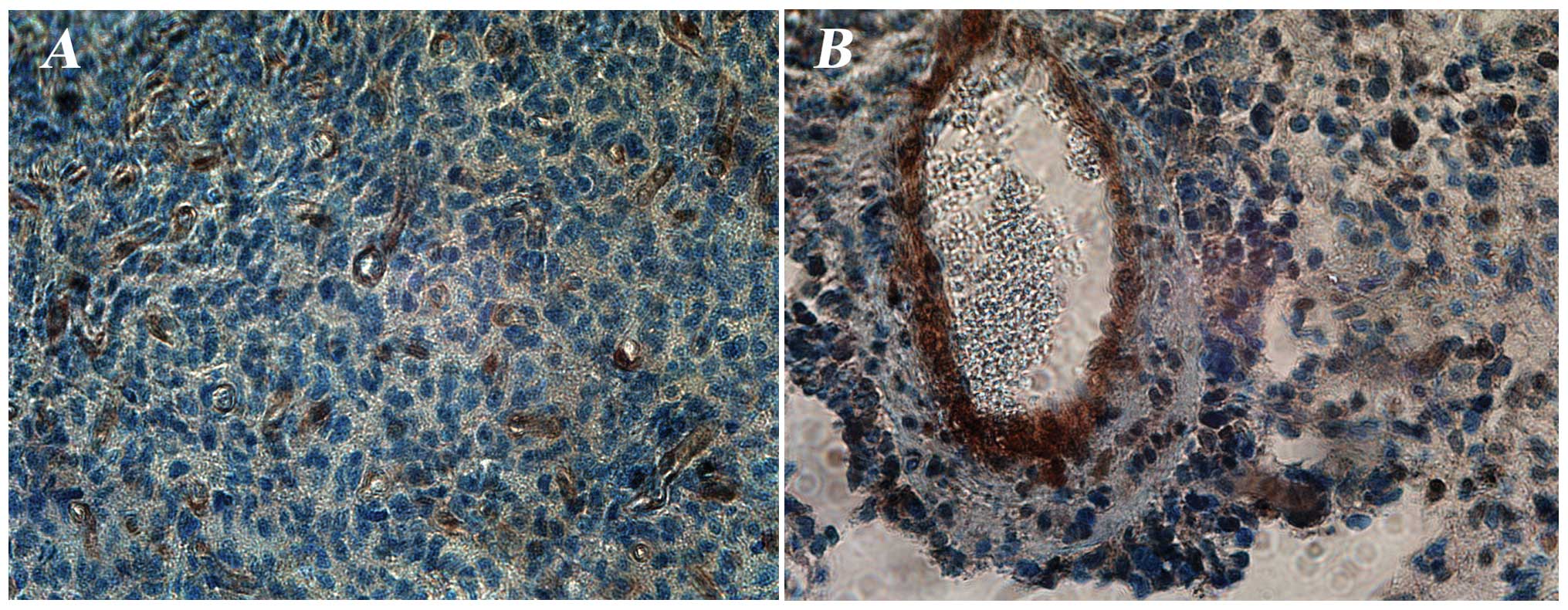
Strong positive IHC staining for PTEN expression was
observed in 6 out of the 31 tested gliomas (Table I; Fig.
2) and found only in WHO grade IV gliomas (5/6 de novo
GB and 1/6 secondary GB). Of these 6 patients with a PTEN positive
de novo GB, 4 had an EGFR amplification, two of which also
expressed EGFRvIII, none of them had EGFRvIV and all 6 patients had
an IDH1 wild-type status (Table
II). No significant correlation was found between PTEN
expression and clinical baseline characteristics such as age,
gender and performance status.
Correlation between molecular markers and
the survival of patients treated with cetuximab
At the time of this analysis, 34 out of 35 patients
died, all because of tumor progression; one patient was lost to
follow-up. The median overall survival (OS) and median progression
free survival (PFS) from the time of recruitment to the clinical
trial were respectively 4.8 months (95% CI: 3.9–5.6) and 1.8 months
(95% CI: 1.5–2.0) (Table II).
In our previous report on this phase II trial, EGFR
amplification was found to correlate with a numerical superior PFS
and OS in patients treated with cetuximab (34). In this updated subgroup analysis,
the correlation between EGFR amplification and superior survival of
patients was still present for PFS [median PFS 2.10 in patients
with EGFR amplification vs. 1.63 in patients with wild-type EGFR
(p=0.09)] (Fig. 3A), however, was
no longer found for OS (median OS 4.73 in patients with EGFR
amplification vs. 4.80 in patients with wild-type EGFR, p=0.69). A
numerically worse survival (PFS and OS) was observed for patients
with expression of EGFRvIII but this difference did not reach the
level of statistical significance [median PFS of 1.63 vs. 1.93
months (p=0.21) and median OS of 3.27 vs. 4.93 months (p=0.21)]
(Fig. 3B and C). A better PFS and
OS were found for patients with EGFR amplification lacking EGFRvIII
expression [median PFS of 3.03 vs. 1.63 months (p=0.006); median OS
of 5.57 vs. 3.97 months (p=0.12)] (Fig. 3D and E). When analyzed within the
cohort of patients with EGFR amplification (n=19), expression of
EGFRvIII correlated significantly with a worse outcome in survival
[median PFS 1.63 vs. 3.03 months (p=0.01); median OS 3.27 vs. 5.57
months (p=0.08)] (Fig. 3F and
G).
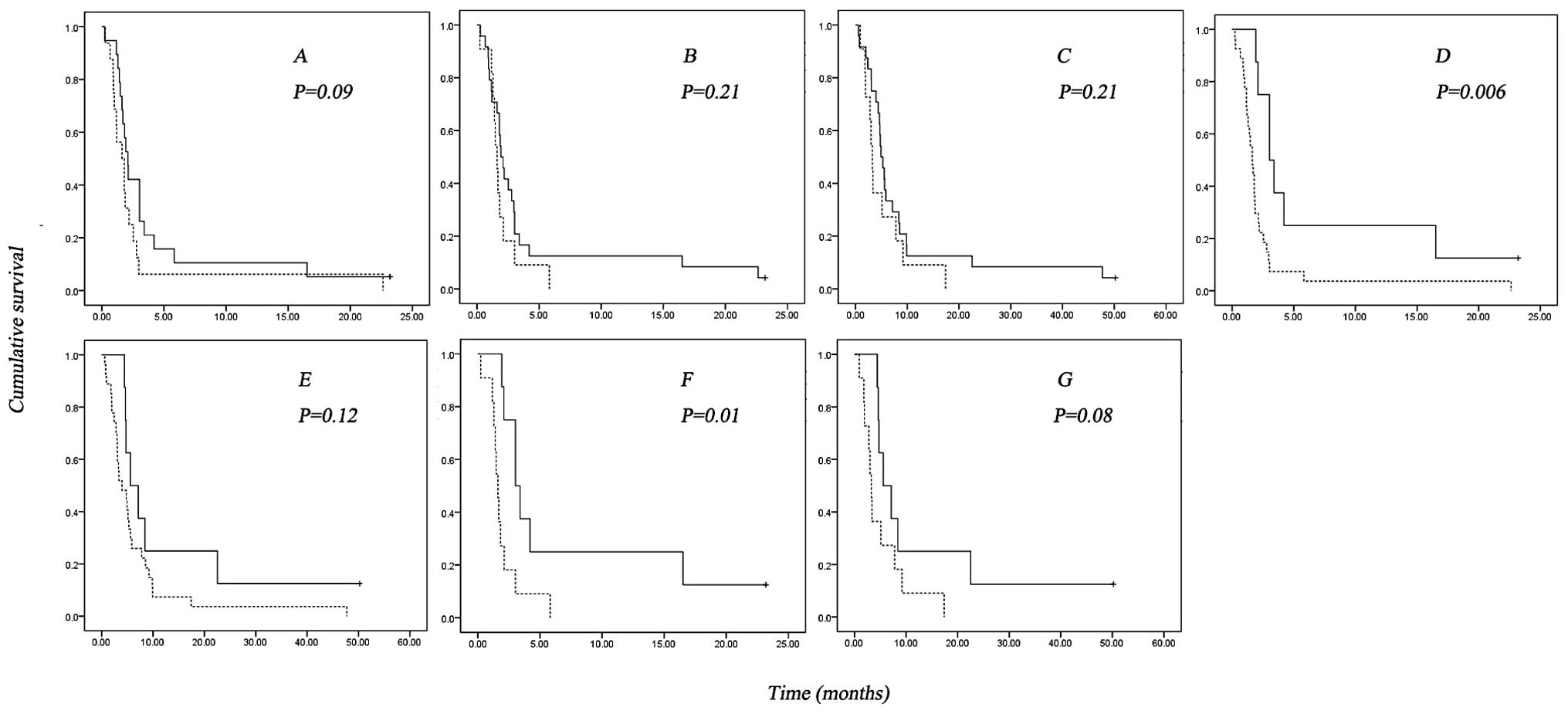 | Figure 3Kaplan-Meier survival estimates
(p-value according to the Log-rank test). (A) Progression free
survival (PFS) from the time of cetuximab treatment on EGFR
amplification status in the global study population (p=0.09), solid
line, EGFR amplification; dash line, EGFR wild-type; +, censored.
(B and C) PFS and OS from the time of treatment of cetuximab in
global population EGFRvIII status (p=0.21 and 0.21), solid line,
EGFRvIII negative; dash line, EGFRvIII positive; +, censored. (D
and E) PFS and OS from the time of treatment of cetuximab in global
population based on EGFR amplification/EGFRvIII status (p=0.006 and
0.12), solid line, EGFR amplification without EGFRvIII; dash line,
others; +, censored. (F and G) PFS and OS from the time of
treatment of cetuximab in cohort of patients with EGFR
amplification based on EGFRvIII status (p=0.01 and 0.08), solid
line, EGFRvIII negative; dash line, EGFRvIII positive; +,
censored. |
In univariate analysis, EGFRvIV mutation and PTEN
expression were not correlated with the survival of the total
population or within the subgroup of EGFR-amplified GBs.
Discussion
In this study of 35 patients with recurrent
high-grade gliomas treated with the EGFR monoclonal antibody
cetuximab, we find that EGFRvIII/vIV expression is restricted to
patients with EGFR amplification, confirming prior reports in the
literature. EGFRvIII and EGFRvIV expression were rarely found in
patients with an IDH1 mutation (1/6 and 0/6), again confirming the
previous reports on the association of IDH1 mutation with WHO grade
II and III glioma and secondary GB (3) and EGFR amplification/mutation with
de novo GB (38).
Overall treatment with cetuximab has low activity
against recurrent GB but shows a trend towards a higher activity in
patients with EGFR-amplified GB, as was reported in our initial
clinical study report (34). In
this molecular sub-study we investigated the potential influence of
specific EGFR-mutants and found that the expression of EGFRvIII
correlated with a significantly worse survival in the cohort of
patients with an EGFR amplification in our study. Similar
observations have been reported previously (4). Since we observed that, the PFS of
patients treated with cetuximab was significantly superior in
patients with EGFR gene amplification but without EGFRvIII
expression, we hypothesize that EGFR amplification without EGFRvIII
may identify a subpopulation with a higher sensitivity for
cetuximab. Given the single-arm study design, we however cannot
exclude a naturally worse prognosis of EGFRvIII mutant GB among
patients with EGFR-amplified tumors.
Although it was reported that cetuximab showed
effective inhibition to EGFR CTD deletion mutation in glioblastoma
cells and animal models (17), we
did not find a correlation between EGFRvIV deletion and survival of
patients in our study in total population or any sub-cohort. We can
however not exclude that this is related to the small sample size
of patients with an EGFRvIV mutated GB in our study.
In the literature, PTEN expression detected by IHC
has been reported in about 62% of primary glioma (39) and in 17% of recurrent GB (40). PTEN expression as detected by IHC
is also more frequently found in low-grade glioma as compared to
high-grade glioma. We detected a positive PTEN expression in 21% of
high-grade glioma (in 19% of the whole population), which is
comparable with the data in the literature. It was previously
reported that coexpression of EGFRvIII and PTEN was a predictor for
response to EGFR inhibitors in patients with recurrent GB (25). In this study only two patients were
found with combined EGFRvIII and PTEN expression making it
impossible to assess this correlation. A study with larger sample
size would be needed to assess the role of EGFRvIII and PTEN in
survival of glioma patients treated with cetuximab at
recurrence.
In summary, within the recurrent glioma patients who
were treated with cetuximab we have confirmed that EGFR mutation is
exclusively present in patients with EGFR amplification. In
addition, we have found a particularly favorable survival for the
sub-group of patients with EGFR amplification, but without EGFRvIII
expression. Our study supports further documentation of both the
glioma EGFRvIII expression and amplification status in studies with
cetuximab.
Acknowledgements
This study was supported by the Free
University Brussels (VUB)/the Chinese Scholarship Council (CSC) PhD
program. We gratefully acknowledge the supply of U87/EGFRvIII cell
from Professor Webster Cavenee (Ludwig Institute for Cancer
Research, University of California, San Diego, CA, USA). We thank
Mr. Kurt De Neef (Laboratory of Molecular Oncology, Free University
Brussels, Belgium), Mr. Pascal Verhavert, Mr. Tim Walravens and
Miss Sara Laceur (Pathology Department, University Hospital
Brussels, Belgium) for their technical help. We also thank Miss
Katrien Van den Bossche (data manager, University Hospital
Brussels, Belgium) for her assistance and help. Bart Neyns has
received research funding from Merck.
References
|
1.
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ,
Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M,
Lacombe D, Cairncross JG and Mirimanoff RO; European Organisation
for Research and Treatment of Cancer Brain Tumour and Radiation
Oncology Groups and National Cancer Institute of Canada Clinical
Trials Group: Effects of radiotherapy with concomitant and adjuvant
temozolomide versus radiotherapy alone on survival in glioblastoma
in a randomised phase III study: 5-year analysis of the EORTC-NCIC
trial. Lancet Oncol. 10:459–466. 2009.
|
|
2.
|
Verhaak RG, Hoadley KA, Purdom E, Wang V,
Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe
G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W,
Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN,
Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW,
Meyerson M, Getz G, Perou CM and Hayes DN; Cancer Genome Atlas
Research Network: Integrated genomic analysis identifies clinically
relevant subtypes of glioblastoma characterized by abnormalities in
PDGFRA, IDH1, EGFR and NF1. Cancer Cell. 17:98–110. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Yan H, Parsons DW, Jin G, McLendon R,
Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ,
Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW,
Velculescu VE, Vogelstein B and Bigner DD: IDH1 and IDH2 mutations
in gliomas. N Engl J Med. 360:765–773. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Shinojima N, Tada K, Shiraishi S, Kamiryo
T, Kochi M, Nakamura H, Makino K, Saya H, Hirano H, Kuratsu J, Oka
K, Ishimaru Y and Ushio Y: Prognostic value of epidermal growth
factor receptor in patients with glioblastoma multiforme. Cancer
Res. 63:6962–6970. 2003.PubMed/NCBI
|
|
5.
|
Ekstrand AJ, James CD, Cavenee WK, Seliger
B, Pettersson RF and Collins VP: Genes for epidermal growth factor
receptor, transforming growth factor alpha, and epidermal growth
factor and their expression in human gliomas in vivo. Cancer Res.
51:2164–2172. 1991.PubMed/NCBI
|
|
6.
|
Wikstrand CJ, McLendon RE, Friedman AH and
Bigner DD: Cell surface localization and density of the
tumor-associated variant of the epidermal growth factor receptor,
EGFRvIII. Cancer Res. 57:4130–4140. 1997.PubMed/NCBI
|
|
7.
|
Ekstrand AJ, Longo N, Hamid ML, Olson JJ,
Liu L, Collins VP and James CD: Functional characterization of an
EGF receptor with a truncated extracellular domain expressed in
glioblastomas with EGFR gene amplification. Oncogene. 9:2313–2320.
1994.PubMed/NCBI
|
|
8.
|
Huang PH, Xu AM and White FM: Oncogenic
EGFR signaling networks in glioma. Sci Signal. 2:re62009.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Wheeler SE, Suzuki S, Thomas SM, Sen M,
Leeman-Neill RJ, Chiosea SI, Kuan CT, Bigner DD, Gooding WE, Lai SY
and Grandis JR: Epidermal growth factor receptor variant III
mediates head and neck cancer cell invasion via STAT3 activation.
Oncogene. 29:5135–5145. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Nishikawa R, Ji XD, Harmon RC, Lazar CS,
Gill GN, Cavenee WK and Huang HJ: A mutant epidermal growth factor
receptor common in human glioma confers enhanced tumorigenicity.
Proc Natl Acad Sci USA. 91:7727–7731. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Tang CK, Gong XQ, Moscatello DK, Wong AJ
and Lippman ME: Epidermal growth factor receptor vIII enhances
tumorigenicity in human breast cancer. Cancer Res. 60:3081–3087.
2000.PubMed/NCBI
|
|
12.
|
Theys J, Jutten B, Dubois L, Rouschop KM,
Chiu RK, Li Y, Paesmans K, Lambin P, Lammering G and Wouters BG:
The deletion mutant EGFRvIII significantly contributes to stress
resistance typical for the tumour microenvironment. Radiother
Oncol. 92:399–404. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Sok JC, Coppelli FM, Thomas SM, Lango MN,
Xi S, Hunt JL, Freilino ML, Graner MW, Wikstrand CJ, Bigner DD,
Gooding WE, Furnari FB and Grandis JR: Mutant epidermal growth
factor receptor (EGFRvIII) contributes to head and neck cancer
growth and resistance to EGFR targeting. Clin Cancer Res.
12:5064–5073. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Ekstrand AJ, Sugawa N, James CD and
Collins VP: Amplified and rearranged epidermal growth factor
receptor genes in human glioblastomas reveal deletions of sequences
encoding portions of the N- and/or C-terminal tails. Proc Natl Acad
Sci USA. 89:4309–4313. 1992. View Article : Google Scholar
|
|
15.
|
Frederick L, Eley G, Wang XY and James CD:
Analysis of genomic rearrangements associated with EGRFvIII
expression suggests involvement of Alu repeat elements. Neuro
Oncol. 2:159–163. 2000.PubMed/NCBI
|
|
16.
|
Pines G, Huang PH, Zwang Y, White FM and
Yarden Y: EGFRvIV: a previously uncharacterized oncogenic mutant
reveals a kinase autoinhibitory mechanism. Oncogene. 29:5850–5860.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Cho J, Pastorino S, Zeng Q, Xu X, Johnson
W, Vandenberg S, Verhaak R, Cherniack AD, Watanabe H, Dutt A, Kwon
J, Chao YS, Onofrio RC, Chiang D, Yuza Y, Kesari S and Meyerson M:
Glioblastoma-derived epidermal growth factor receptor
carboxyl-terminal deletion mutants are transforming and are
sensitive to EGFR-directed therapies. Cancer Res. 71:7587–7596.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Frederick L, Wang XY, Eley G and James CD:
Diversity and frequency of epidermal growth factor receptor
mutations in human glioblastomas. Cancer Res. 60:1383–1387.
2000.PubMed/NCBI
|
|
19.
|
Yoshimoto K, Dang J, Zhu S, Nathanson D,
Huang T, Dumont R, Seligson DB, Yong WH, Xiong Z, Rao N, Winther H,
Chakravarti A, Bigner DD, Mellinghoff IK, Horvath S, Cavenee WK,
Cloughesy TF and Mischel PS: Development of a real-time RT-PCR
assay for detecting EGFRvIII in glioblastoma samples. Clin Cancer
Res. 14:488–493. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Newcomb EW, Cohen H, Lee SR, Bhalla SK,
Bloom J, Hayes RL and Miller DC: Survival of patients with
glioblastoma multi-forme is not influenced by altered expression of
p16, p53, EGFR, MDM2 or Bcl-2 genes. Brain Pathol. 8:655–667. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Sonnweber B, Dlaska M, Skvortsov S,
Dirnhofer S, Schmid T and Hilbe W: High predictive value of
epidermal growth factor receptor phosphorylation but not of
EGFRvIII mutation in resected stage I non-small cell lung cancer
(NSCLC). J Clin Pathol. 59:255–259. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Nieto Y, Nawaz F, Jones RB, Shpall EJ and
Nawaz S: Prognostic significance of overexpression and
phosphorylation of epidermal growth factor receptor (EGFR) and the
presence of truncated EGFRvIII in locoregionally advanced breast
cancer. J Clin Oncol. 25:4405–4413. 2007. View Article : Google Scholar
|
|
23.
|
Heimberger AB, Hlatky R, Suki D, Yang D,
Weinberg J, Gilbert M, Sawaya R and Aldape K: Prognostic effect of
epidermal growth factor receptor and EGFRvIII in glioblastoma
multiforme patients. Clin Cancer Res. 11:1462–1466. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Aldape KD, Ballman K, Furth A, Buckner JC,
Giannini C, Burger PC, Scheithauer BW, Jenkins RB and James CD:
Immunohistochemical detection of EGFRvIII in high malignancy grade
astrocytomas and evaluation of prognostic significance. J
Neuropathol Exp Neurol. 63:700–707. 2004.PubMed/NCBI
|
|
25.
|
Mellinghoff IK, Wang MY, Vivanco I,
Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute
DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R,
Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF,
Sawyers CL and Mischel PS: Molecular determinants of the response
of glioblastomas to EGFR kinase inhibitors. N Engl J Med.
353:2012–2024. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Li L, Dutra A, Pak E, Labrie JE 3rd,
Gerstein RM, Pandolfi PP, Recht LD and Ross AH: EGFRvIII expression
and PTEN loss synergistically induce chromosomal instability and
glial tumors. Neuro Oncol. 11:9–21. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Haas-Kogan DA, Prados MD, Lamborn KR,
Tihan T, Berger MS and Stokoe D: Biomarkers to predict response to
epidermal growth factor receptor inhibitors. Cell Cycle.
4:1369–1372. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Haas-Kogan DA, Prados MD, Tihan T,
Eberhard DA, Jelluma N, Arvold ND, Baumber R, Lamborn KR, Kapadia
A, Malec M, Berger MS and Stokoe D: Epidermal growth factor
receptor, protein kinase B/Akt, and glioma response to erlotinib. J
Natl Cancer Inst. 97:880–887. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
van den Bent MJ, Brandes AA, Rampling R,
Kouwenhoven MC, Kros JM, Carpentier AF, Clement PM, Frenay M,
Campone M, Baurain JF, Armand JP, Taphoorn MJ, Tosoni A, Kletzl H,
Klughammer B, Lacombe D and Gorlia T: Randomized phase II trial of
erlotinib versus temozolomide or carmustine in recurrent
glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol.
27:1268–1274. 2009.PubMed/NCBI
|
|
30.
|
Jeuken J, Sijben A, Alenda C, Rijntjes J,
Dekkers M, Boots-Sprenger S, McLendon R and Wesseling P: Robust
detection of EGFR copy number changes and EGFR variant III:
technical aspects and relevance for glioma diagnostics. Brain
Pathol. 19:661–671. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Patel D, Lahiji A, Patel S, Franklin M,
Jimenez X, Hicklin DJ and Kang X: Monoclonal antibody cetuximab
binds to and down-regulates constitutively activated epidermal
growth factor receptor vIII on the cell surface. Anticancer Res.
27:3355–3366. 2007.
|
|
32.
|
Eller JL, Longo SL, Hicklin DJ and Canute
GW: Activity of anti-epidermal growth factor receptor monoclonal
antibody C225 against glioblastoma multiforme. Neurosurgery.
51:1005–1013. 2002.PubMed/NCBI
|
|
33.
|
Eller JL, Longo SL, Kyle MM, Bassano D,
Hicklin DJ and Canute GW: Anti-epidermal growth factor receptor
monoclonal antibody cetuximab augments radiation effects in
glioblastoma multiforme in vitro and in vivo. Neurosurgery.
56:155–162. 2005.
|
|
34.
|
Neyns B, Sadones J, Joosens E, Bouttens F,
Verbeke L, Baurain JF, D’Hondt L, Strauven T, Chaskis C, In’t Veld
P, Michotte A and De Greve J: Stratified phase II trial of
cetuximab in patients with recurrent high-grade glioma. Ann Oncol.
20:1596–1603. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Lv S, Teugels E, Sadones J, Quartier E,
Huylebrouck M, Du Four S, Le Mercier M, De Witte O, Salmon I,
Michotte A, De Greve J and Neyns B: Correlation between IDH1 gene
mutation status and survival of patients treated for recurrent
glioma. Anticancer Res. 31:4457–4463. 2011.PubMed/NCBI
|
|
37.
|
Loupakis F, Pollina L, Stasi I, Ruzzo A,
Scartozzi M, Santini D, Masi G, Graziano F, Cremolini C, Rulli E,
Canestrari E, Funel N, Schiavon G, Petrini I, Magnani M, Tonini G,
Campani D, Floriani I, Cascinu S and Falcone A: PTEN expression and
KRAS mutations on primary tumors and metastases in the prediction
of benefit from cetuximab plus irinotecan for patients with
metastatic colorectal cancer. J Clin Oncol. 27:2622–2629. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Mason WP and Cairncross JG: Invited
article: the expanding impact of molecular biology on the diagnosis
and treatment of gliomas. Neurology. 71:365–373. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Fults D and Pedone C: Immunocytochemical
mapping of the phosphatase and tensin homolog (PTEN/MMAC1) tumor
suppressor protein in human gliomas. Neuro Oncol. 2:71–79.
2000.PubMed/NCBI
|
|
40.
|
de Groot JF, Gilbert MR, Aldape K, Hess
KR, Hanna TA, Ictech S, Groves MD, Conrad C, Colman H, Puduvalli
VK, Levin V and Yung WK: Phase II study of carboplatin and
erlotinib (Tarceva, OSI-774) in patients with recurrent
glioblastoma. J Neurooncol. 90:89–97. 2008.PubMed/NCBI
|