Introduction
Leukemia, a malignant hematopoietic tumor, is a
cancer of the blood or bone marrow characterized by the abnormal
proliferation of white blood cells; it ranks sixth in the
prevalence of human tumors worldwide (1). Leukemias are classified into two
subtypes: acute lymphocytic leukemia originating from lymphocytes
in the bone marrow and myelogenous leukemia originating from
granulocytes or monocytes (2,3).
Treatment of leukemia is difficult, and the probability of
recurrence is high due to chemoresistance. To overcome these
defects, the development of novel therapeutic strategies is needed
for more effective treatment of this serious disease.
Apoptosis (programmed cell death), plays a
fundamental role in the normal development and differentiation of
multicellular organisms. Apoptosis also occurs as a reaction to
protect cells damaged by diseases or noxious agents. The two types
of apoptosis include an extrinsic pathway that involves
transmembrane death, receptor-mediated interactions and an
intrinsic pathway that involves mitochondria-mediated stimuli
(4,5). The death receptor pathway begins with
the ligation of cell surface death receptors and the activation of
caspase-8, which activates the downstream effector caspases (−3,
−6, and/or −7). The mitochondrion-mediated pathway begins with the
disruption of the mitochondrial membrane potential (MMP,
ΔΨm) and release of apoptogenic proteins such as cytochrome
c into the cytosol. Once in the cytosol, cytochrome c
can activate caspase-9, which in turn cleaves and activates the
executioner caspases. Following activation of effector caspases
such as caspase-3, cleavage of several specific substrates occurs,
including poly(ADP-ribose) polymerase (PARP), eventually leading to
apoptosis (6,7). In some cells, caspase-8 also mediates
the intrinsic pathway via cleavage of the pro-apoptotic Bid, a
BH3-only protein (8,9).
The caspase-cascade signaling system is also
regulated by a number of different molecules, such as proteins from
the Bcl-2 and the inhibitor of apoptosis protein (IAP) families.
The Bcl-2 family, which has both anti-apoptotic (Bcl-2 and Bcl-xL)
and pro-apoptotic (Bax, Bak, and Bid) members, acts on the
mitochondrion to prevent or to facilitate the release of
apoptogenic factors (10,11). The IAP family proteins, on the
other hand, are all endogenous inhibitors of apoptosis, and are
able to bind and inhibit caspases (12,13).
Reactive oxygen species (ROS) include highly
reactive hydroxyl radicals (OH·), superoxide anions
(O2−), singlet oxygen
(1O2), and hydrogen peroxide
(H2O2), which form as natural byproducts of
the normal cellular metabolism of oxygen (14). Although ROS have important roles as
intracellular messengers in cell signaling, extended high levels of
ROS can cause severe damage to DNA, RNA, and proteins, eventually
leading to cell death via either apoptotic or necrotic mechanisms
(15,16). ROS induced by a diverse range of
stimuli are also produced inside organelles such as mitochondria
(17,18). During oxidative stress-induced cell
death, ROS can target the mitochondrial membrane potential
(16,18).
DNA methylation is critically involved in embryonic
development, chromatin structure, genomic imprinting, and
chromosome inactivation and stability (19,20).
Aberrant hypermethylation represses transcription by way of CpG
islands in the promoter region and is associated with gene
inactivation. Decitabine (5-aza-2′-deoxycytidine) is a synthesized
cytosine analogue that incorporates itself into the DNA strands of
proliferating cells (21). This
compound effectively inhibits DNA methylation and increases
re-expression-silenced genes by covalently binding to the promoter
regions of DNA methyltransferase (22,23).
Recently, the therapeutic activity of decitabine in acute leukemias
has been tested in phase clinical trials for its excellent
capability to reactivate the expression of several methylated genes
(24–26). Furthermore, there are also reports
on the effectiveness of decitabine for the treatment of solid
tumors (27,28), the connection between decitabine’s
clinical activity and hypomethylation activity remains unclear. In
addition, other mechanisms that are independent of hypomethylation
are also important for its anticancer activity, inducing cell cycle
arrest, differentiation, and apoptosis, and inhibiting invasion.
For example, at low concentrations, decitabine acts as an
S-phase-specific inducer, causing de novo DNA
hypermethylation and silencing of transcription process in some
cancer cells (29–31). However, DNA damage caused by a high
concentration of decitabine induces apoptosis in a
caspase-dependent or independent manner (30,32–35).
While well known as a therapeutic agent due to its unique
properties, the functional mechanisms by which decitabine induces
anti-leukemic activity remain to be investigated.
In the present study, we demonstrated that
decitabine induced several characteristic apoptotic symptoms in
human leukemia cell lines U937 and HL60, including DNA
fragmentation, chromatin condensation, activation of caspases,
cleavage of PARP, and mitochondrial depolarization. We also showed
that ROS are potential causes of the caspase activation that leads
to decitabine-induced apoptosis.
Materials and methods
Cell culture and MTT assay
Human leukemia U937 and HL60 cells were purchased
from the American Type Culture Collection (Rockville, MD) and
cultured in RPMI-1640 medium (Invitrogen Corp., Carlsbad, CA)
supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco BRL,
Grand Island, NY), 1 mM L-glutamine, 100 U/ml penicillin, and 100
mg/ml streptomycin at 37°C in a humidified atmosphere of 95% air
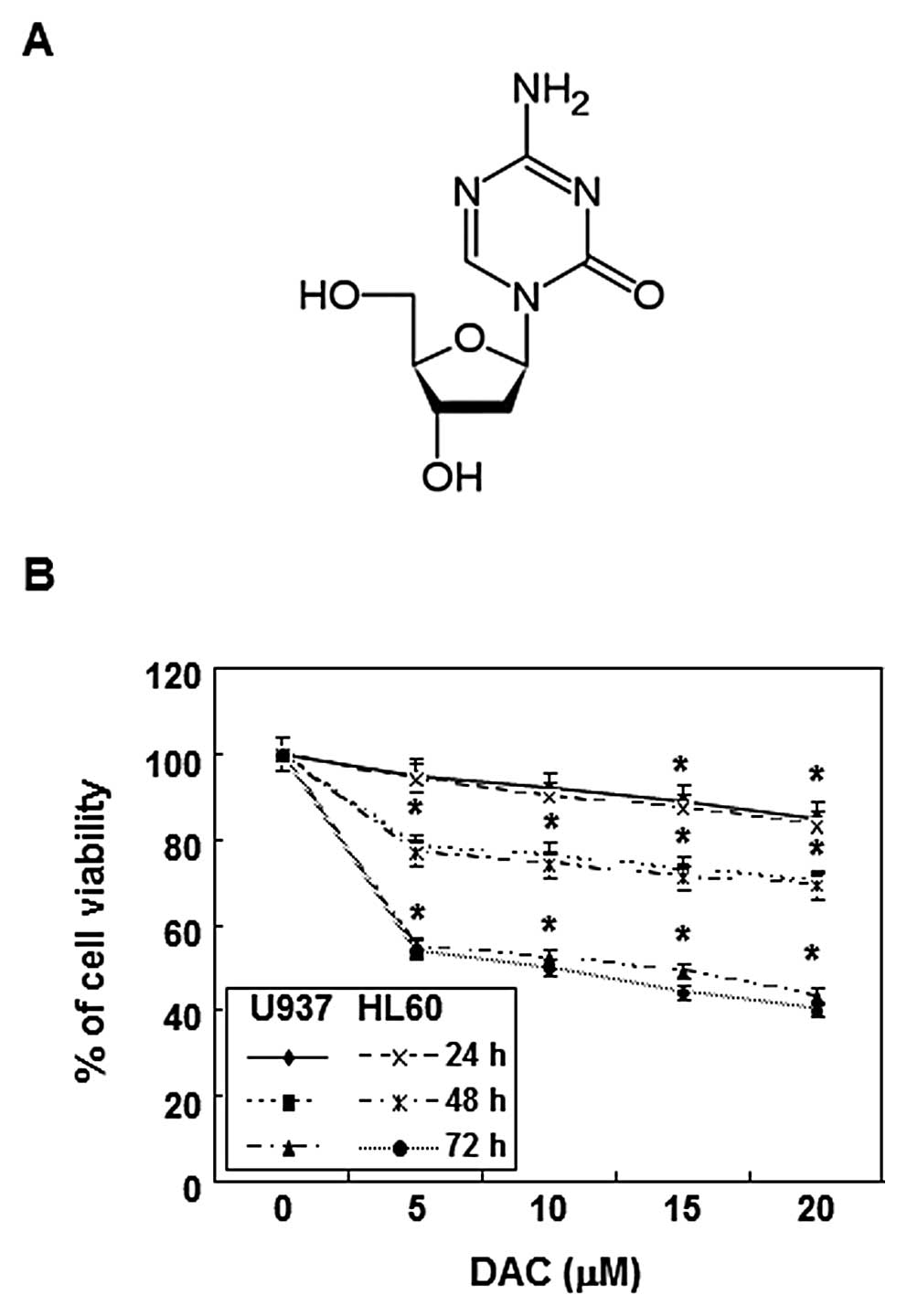
and 5% CO2. Decitabine (Fig. 1A) was purchased from Sigma-Aldrich
Chemical Co. (St. Louis, MO), dissolved in 100% dimethyl sulfoxide
(DMSO) to a stock concentration 10 mM, and stored at −80°C. For the
cell viability study, the cells were seeded onto 6-well plates at a
concentration of 5×104 cells/well, grown to 70%
confluence, and then treated with various concentrations of
decitabine for the indicated times. Following treatment, cell
viability was determined using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT,
Sigma-Aldrich) assay, which is based on the conversion of MTT to
MTT-formazan by mitochondrial enzymes.
Nuclear staining with DAPI
For nuclear staining, cells were washed with
phosphate-buffered saline (PBS) and fixed with 3.7%
paraformaldehyde (Sigma-Aldrich) in PBS for 10 min at room
temperature. Fixed cells were washed with PBS and stained with 2.5
μg/ml 4,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich)
solution for 10 min at room temperature. Cells were then washed
twice with PBS and analyzed using a fluorescence microscope (Carl
Zeiss, Germany).
Agarose gel electrophoresis for DNA
fragmentation assay
Following decitabine treatment, cells were lysed in
a buffer containing 10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA,
and 0.5% Triton X-100 for 1 h at room temperature. Lysates were
vortexed and cleared by centrifugation at 19,000 g for 30 min at
4°C. A 25:24:1 (v/v/v) equal volume of neutral
phenol:chloroform:isoamyl alcohol (Sigma-Aldrich) was used for
extraction of DNA in the supernatant, followed by electrophoretic
analysis on 1.2% agarose gels containing 0.1 μg/ml ethidium
bromide (EtBr, Sigma-Aldrich).
Flow cytometry analysis for measurement
of sub-G1 phase
The cells were harvested and washed once with PBS,
fixed in ice-cold 70% ethanol, and stored at 4°C. Prior to
analysis, the cells were washed once again with PBS, suspended in 1
ml of a cold propidium iodide (PI, Sigma-Aldrich) solution
containing 100 μg/ml RNase A, 50 μg/ml PI, 0.1% (w/v)
sodium citrate, and 0.1% (v/v) NP-40, and further incubated on ice
for 30 min in the dark. Flow cytometric analyses were carried out
using a flow cytometer (FACSCalibur, Becton-Dickinson, San Jose,
CA). CellQuest software was used to determine the relative DNA
content, based on the presence of a red fluorescence.
Measurement of intracellular ROS and MMP
(ΔΨm)
ROS production was monitored using the stable
non-polar dye 2,7 dichlorofluorescein diacetate (DCF-DA, Molecular
Probes, Leiden, The Netherlands), which readily diffuses into cells
(36). The cells were seeded in
24-well plates and incubated in the absence or presence of
decitabine for different periods of time, after which they were
incubated with 10 μM DCF-DA for 30 min. ROS production in
the cells was monitored by flow cytometer, using CellQuest
software. To measure MMP, ΔΨm, the dual-emission
potential-sensitive probe 5,5 V, 6,6 V-tetrachloro-1,1 V,3,3
V-tetraethyl-imidacarbocyanine iodide (JC-1, Sigma-Aldrich), was
used. After treatment with decitabine, 5×105 cells were
collected, stained with 2 mg/l JC-1 at 37°C for 20 min, and then
analyzed with a flow cytometer (37).
Protein extraction and western
blotting
The cells were harvested and lysed. The protein
concentrations were measured using a Bio-Rad protein assay (Bio-Rad
Laboratories, Hercules, CA), according to the manufacturer’s
instructions. For western blot analysis, an equal amount of protein
was subjected to electrophoresis on SDS-polyacrylamide gel and
transferred by electroblotting to a nitrocellulose membrane
(Schleicher & Schuell, Keene, NH). The blots were probed with
the desired antibodies for 1 h, incubated with the diluted
enzyme-linked secondary antibody, and visualized by enhanced
chemiluminescence (ECL), according to the recommended procedure
(Amersham Corp., Arlington Heights, IL) (38). The primary antibodies were
purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA) and
Cell Signaling Technology, Inc. (Boston, MA). The
peroxidase-labeled donkey anti-rabbit immunoglobulin and
peroxidase-labeled sheep anti-mouse immunoglobulin were purchased
from Amersham Corp.
In vitro caspase activity assay
Activities of caspases were determined by use of
colorimetric assay kits, which utilize synthetic tetrapeptides
(Asp-Glu-Val-Asp (DEAD) for caspase-3; Ile-Glu-Thr-Asp (IETD) for
caspase-8; Leu-Glu-His-Asp (LEHD) for caspase-9, respectively)
labeled with p-nitroaniline (pNA). Briefly, decitabine-treated and
untreated cells were lysed in the supplied lysis buffer.
Supernatants were collected and incubated with the supplied
reaction buffer containing DTT and DEAD-pNA, IETD-pNA, or LEHD-pNA
as substrates at 37°C. The reactions were measured by changes in
absorbance at 405 nm using the VERSAmax tunable microplate
reader.
Statistical analysis
Unless otherwise indicated, each result is expressed
as the mean ± SD of data obtained from triplicate experiments.
Statistical analysis was performed using a paired Student’s t-test.
Differences at p<0.05 were considered statistically
significant.
Results
Inhibition of cell viability by
decitabine in human leukemia cells
To evaluate the effects of decitabine on cell
viability, U937 and HL60 cells were stimulated with various
concentrations of decitabine for the indicated times, and an MTT
assay was performed. As shown in Fig.
1B, decitabine induced a decrease in cell viability in a
concentration- and time-dependent manner. For example, treatment
with 20 μM decitabine for 72 h resulted in 58 and 60%
inhibition in U937 and HL60 cells, respectively, which was
associated with many morphological changes (data not shown).
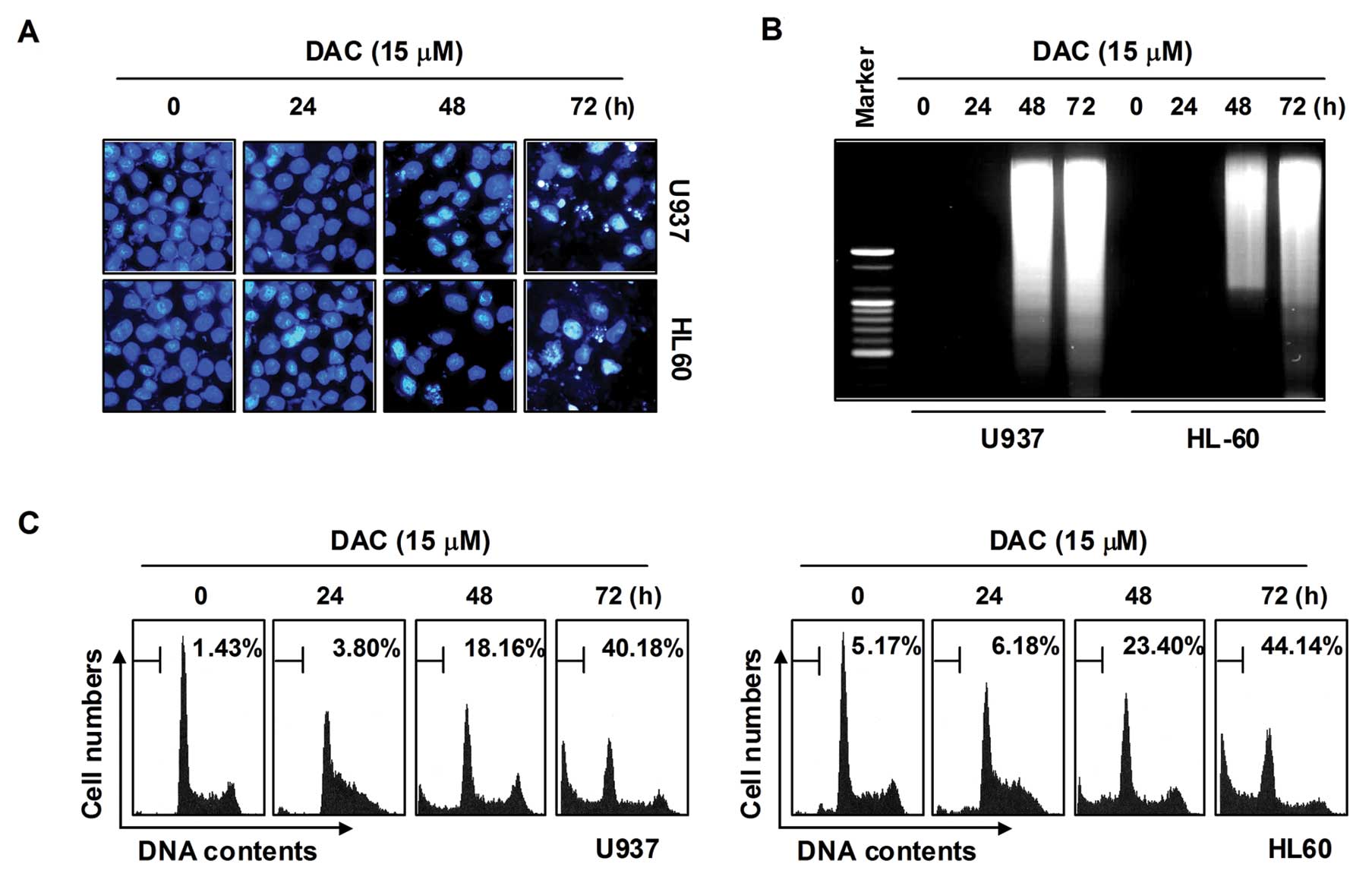
Induction of apoptosis by decitabine in
human leukemia cells
In order to determine whether the decrease in cell
viability in U937 and HL60 cells by decitabine treatment was due to
the induction of apoptosis, three established criteria were
subsequently used for the assessment of apoptosis. First,
morphological changes of cells were determined using DAPI staining;
as shown in Fig. 2A, treatment
with decitabine resulted in observation of a significant number of
cells with chromatin condensation, loss of nuclear construction,
and formation of apoptotic bodies in a time-dependent fashion,
whereas these features were not observed in control cells. Second,
we analyzed DNA fragmentation, which is another hallmark of
apoptosis. Following agarose gel electrophoresis of DNAs from cells
treated with decitabine, a typical ladder pattern of
internucleosomal fragmentation was observed. In contrast, DNA
fragmentation was barely detected in control cells (Fig. 2B). In addition, the degree of
apoptosis in cells treated with decitabine was determined using
flow cytometric analysis for detection of hypodiploid cell
populations. As shown in Fig. 2C,
the addition of decitabine to cells resulted in increased
accumulation of cells in the sub-G1 phase in a manner similar to
that observed with decitabine-induced viability inhibition, the
formation of apoptotic bodies, and accumulation of extranuclear
fragmented DNA. This finding suggests that U937 and HL60 cells may
undergo apoptosis after exposure to decitabine, and there is a good
correlation between the extent of apoptosis and inhibition of
growth.
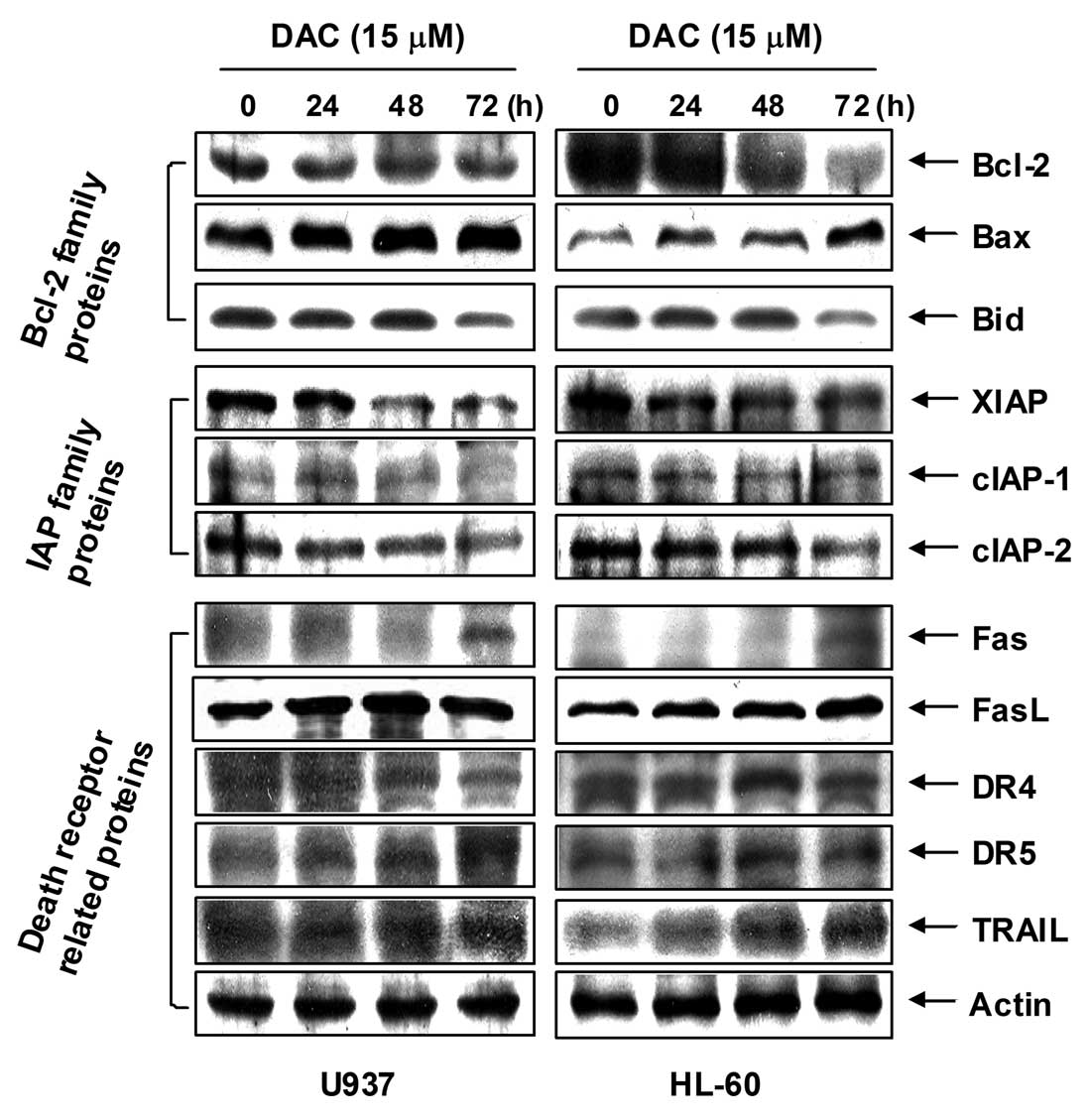
Effects of decitabine on the expression
of apoptosis-related proteins in human leukemia cells
The role of Bcl-2 and the IAP family, as well as
death receptor-related members in decitabine-mediated apoptosis,
was determined by western blotting for measurement of expression of
their proteins. As shown in Fig.
3, the levels of anti-apoptotic Bcl-2 proteins were decreased
in response to decitabine treatment; however, the levels of
pro-apoptotic Bax were increased in a concentration-dependent
manner. Under these conditions, the levels of total pro-apoptotic
protein Bid, a BH3-only pro-apoptotic member of the Bcl-2 family,
concentration-dependently decreased in response to decitabine
treatment. In addition, the levels of anti-apoptotic IAP family
proteins such as XIAP, cIAP-1, and cIAP-2 were markedly inhibited
by decitabine treatment in a concentration-dependent manner.
Furthermore, the results showed that decitabine treatment resulted
in a concentration-dependent increase in the level of death
receptor-related proteins including Fas, Fas ligand (FasL), death
receptor (DR) 4, DR5, and tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL).
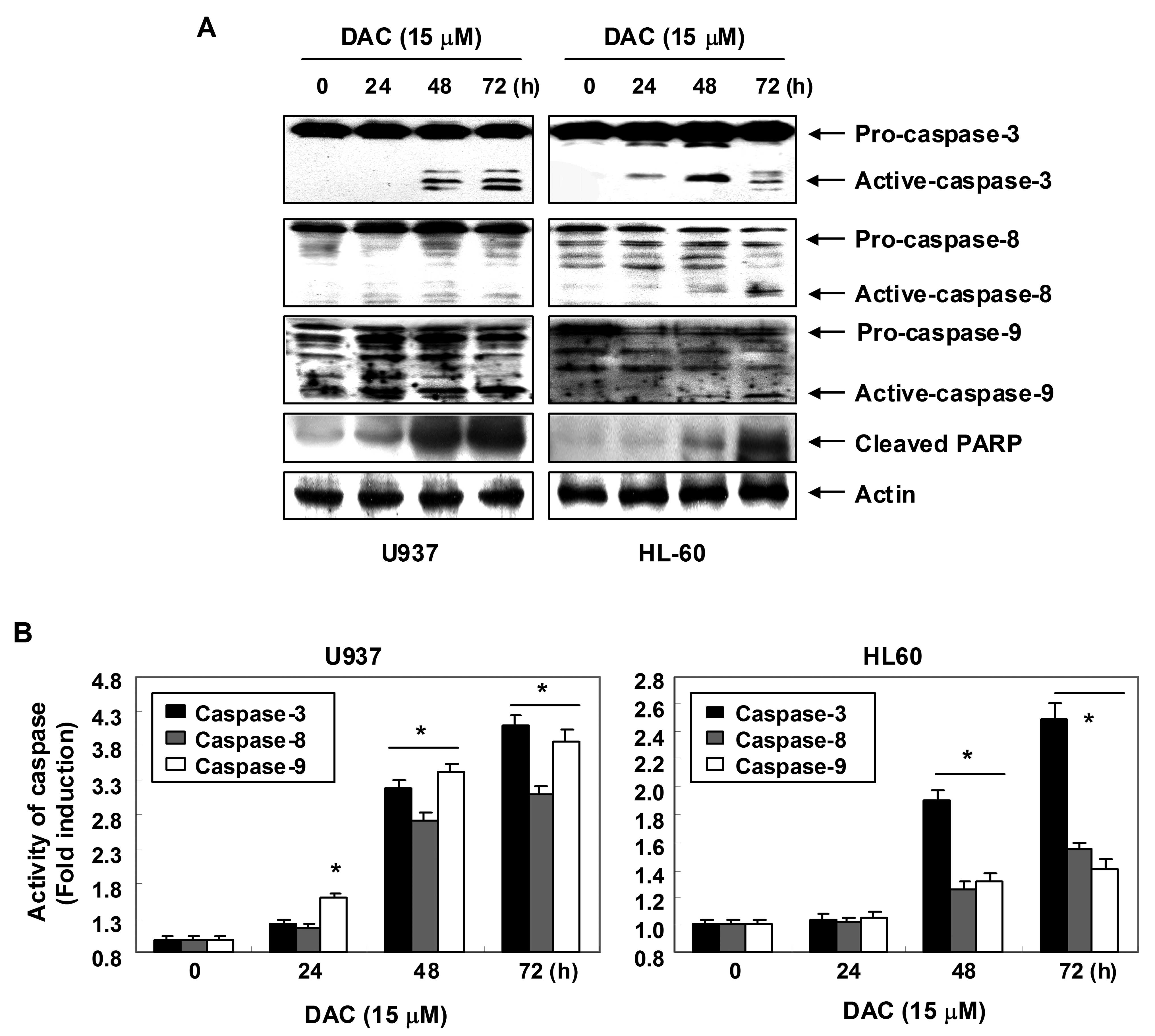
Activation of caspases by decitabine in
human leukemia cells
Expression levels and activities of caspase-3, -8
and -9 in U937 and HL60 cells exposed to decitabine were measured
in order to determine whether decitabine-induced apoptosis is
associated with the activation of caspases. As shown in Fig. 4A, immunoblotting results showed
that decitabine treatment induced a concentration-dependent
increase in levels of active-caspase-3, -8 and -9 proteins. For
further quantification of the proteolytic activation of caspases,
protein in the lysates of cells treated with decitabine was
normalized and then assayed for in vitro activities using
fluorogenic substrates. As shown in Fig. 4B, treatment with decitabine
resulted in a significant concentration-dependent increase of the
activities of caspase-3, -8 and -9 compared with control cells. In
addition, decitabine treatment led to progressive proteolytic
cleavage of PARP, a well-known substrate protein of activated
caspase-3.
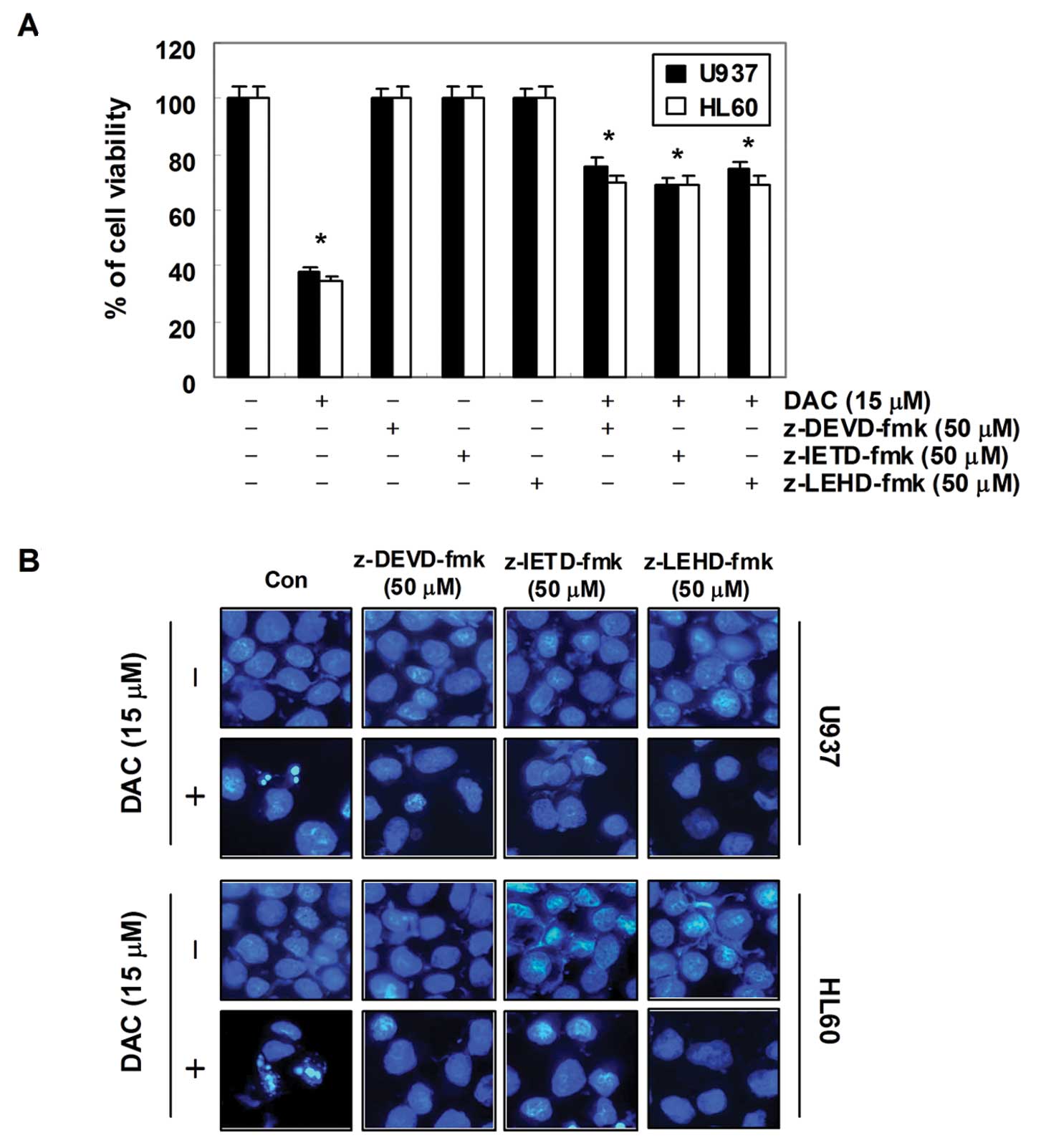
In order to demonstrate that the activation of
caspases is a key step in the apoptotic pathway induced by
decitabine, U937 and HL60 cells were pretreated with potential
caspase-specific inhibitors (z-DEVD-fmk, z-IETD-fmk, and z-LEHD-fmk
for the inactivation of caspase-3, -8 and -9, respectively) for 1
h, followed by treatment with decitabine for 72 h. As shown in
Fig. 5, pretreatment with caspase
inhibitors significantly attenuated chromatin condensation and the
formation of apoptotic bodies, and restored the decreased
viability. These results indicate that decitabine treatment induces
apoptosis in U937 and HL60 cells through a caspase-dependent
pathway.
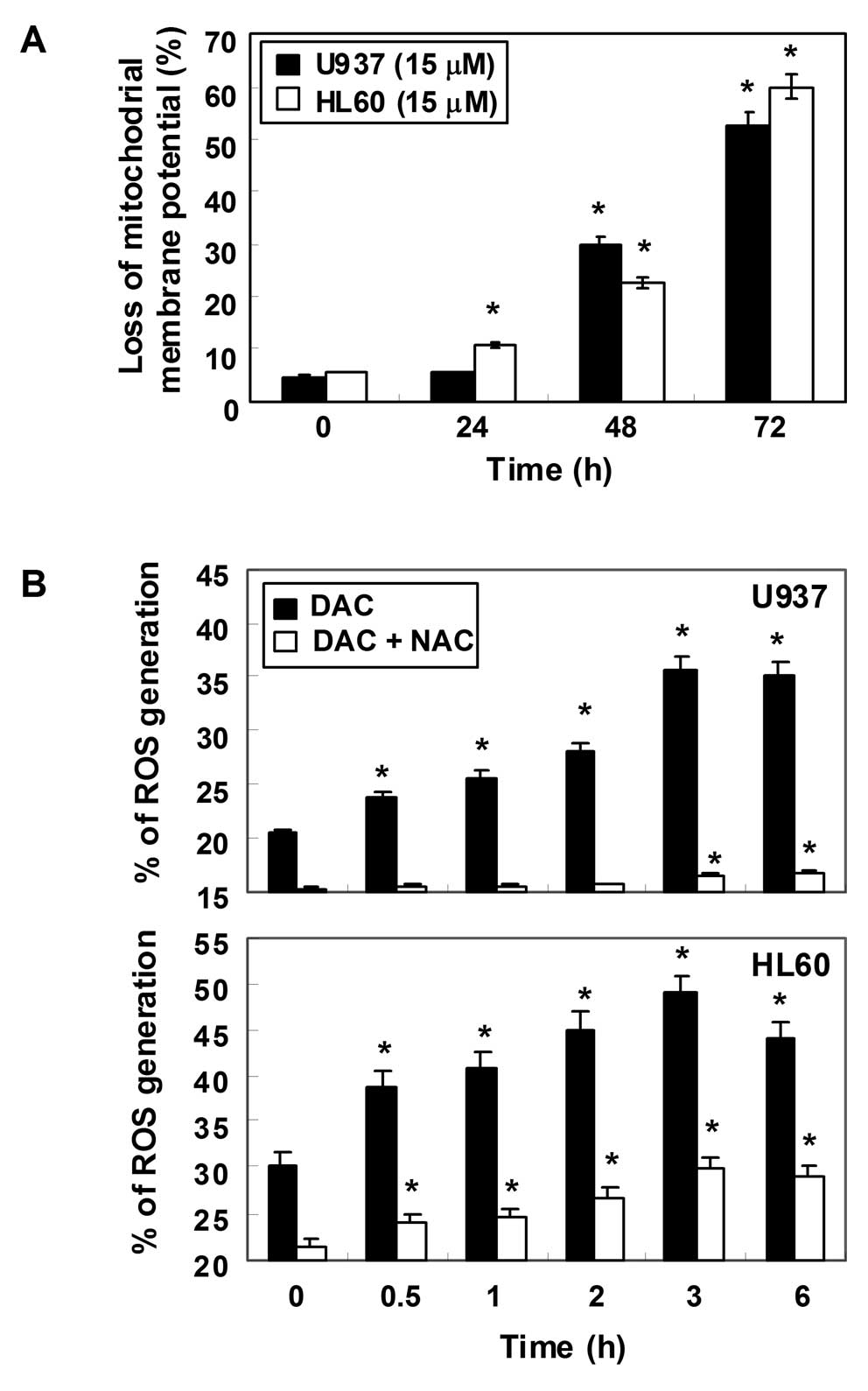
Loss of MMP values and increase of ROS
generation by decitabine in human leukemia cells
To examine the role of mitochondria in apoptosis
induced by decitabine, we attempted to characterize the
relationship between changes in the MMP and ROS production. For
this study, the effects of decitabine on the levels of MMP were
monitored via a flow cytometer using the mitochondrial-specific
probe, JC-1. As shown in Fig. 6A,
the loss of MMP values showed a time-dependent increase by
decitabine treatment, indicating that decitabine induced
mitochondrial membrane hyperpolarization by depolarization. Next,
ROS production was measured using a cell-permeant,
oxidation-sensitive dye, DCF-DA. In U937 cells exposed to
decitabine for the indicated time periods, generation of ROS was
observed at 0.5 h and the levels continued to increase at 3 h
(Fig. 6B). A similar trend in ROS
generation was also observed in HL60 cells in response to
decitabine treatment. As expected, the ROS scavenger
N-acetyl-L-cysteine (NAC), a commonly used reactive oxygen
intermediate scavenger, markedly blocked the levels of ROS from the
decitabine-treated U937 and HL60 cells at 10 mM.
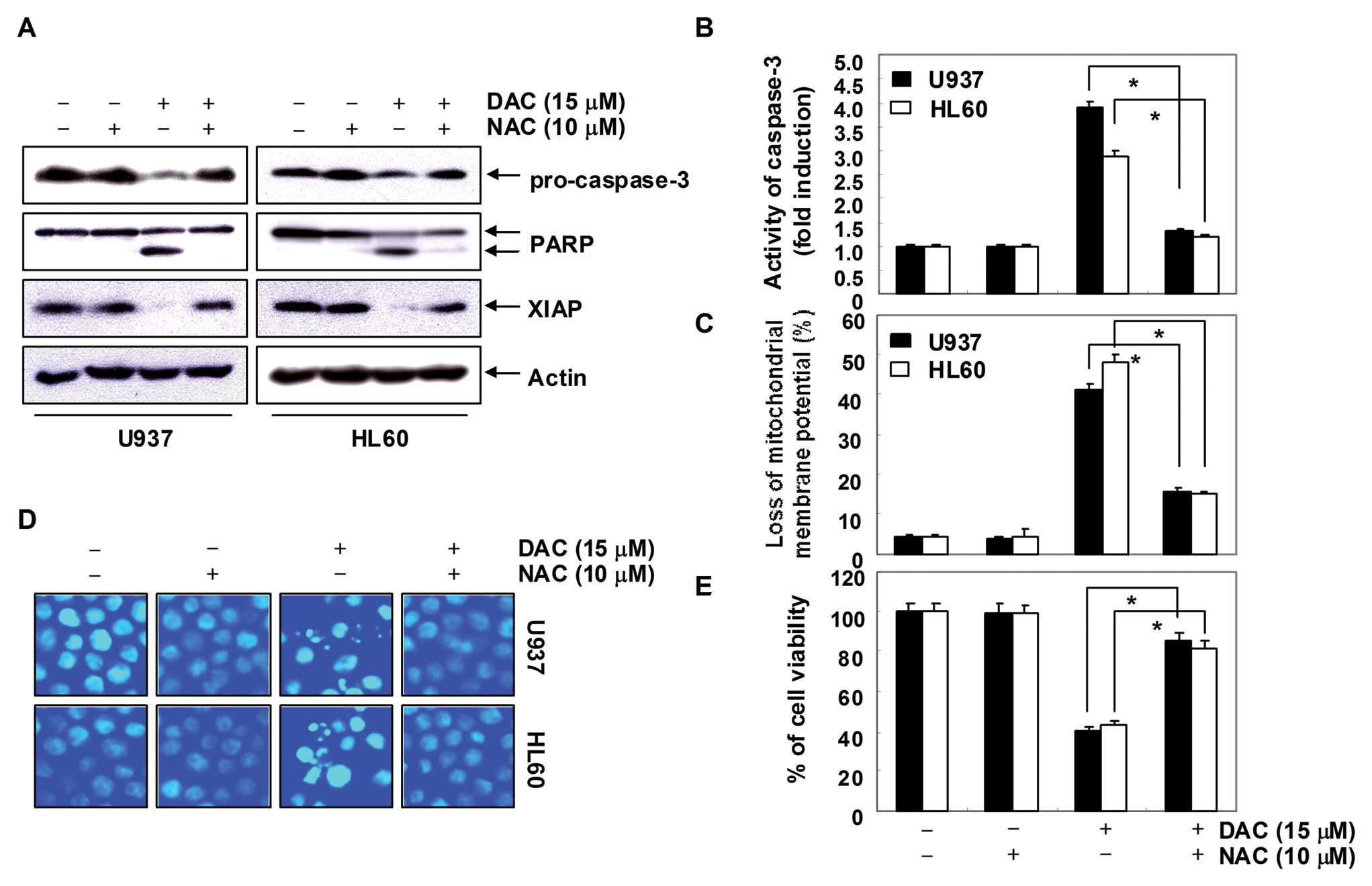
Decitabine-induced apoptosis is
associated with the generation of ROS in human leukemia cells
In order to show that generation of ROS is a key
step in the decitabine-induced apoptotic pathway, cells were
pretreated with 10 mM NAC for 1 h, followed by treatment with
decitabine for 72 h. As shown in Fig.
7A–C, blocking of ROS generation by pretreatment of cells with
NAC effectively prevented decitabine-induced downregulation of
pro-caspase-3 and XIAP expression, cleavage of PARP activation of
caspase-3, and loss of MMP. In addition, pretreatment of cells with
NAC also prevented decitabine-induced chromatin condensation
(Fig. 7D), which was associated
with recovered cell viability (Fig.
7E). Collectively, these findings suggest that an increase in
ROS generation is required for the occurrence of decitabine-induced
apoptosis in U937 and HL60 cells.
Discussion
Although an increasing amount of data indicate that
decitabine, a well-known demethylating agent in cancer therapy, can
suppress the growth of cultured cancer cells by causing cell cycle
arrest and apoptosis induction (29–35),
the signaling pathways involved in apoptosis induction by this
compound are poorly defined. In the present study, we demonstrated
that decitabine-induced anti-proliferative effects in human
leukemia U937 and HL60 cells were related to the induction of
apoptosis, as confirmed by measurement of chromatin condensation of
nuclei, DNA fragmentation, and the induction of the sub-G1 phase.
Our data also demonstrated that decitabine induced apoptosis of
U937 and HL60 cells through generation of ROS and mitochondrial
dysfunction, suggesting that ROS act as upstream signaling
molecules for the initiation of cell death.
Apoptosis plays an important role in the normal
development and differentiation of multicellular organisms, which
are characterized by morphological and biological changes such as
cytoplasmic shrinkage, chromatin condensation, and DNA degradation.
Apoptosis also serves as a critical protective mechanism against
carcinogenesis caused by mutations of genetic materials of normal
cells or various carcinogens (39). A variety of stimuli can trigger it,
including death receptor-mediated signaling (extrinsic pathway) or
intracellular stresses (intrinsic pathway) (4,5). Our
data indicated that decitabine-induced apoptosis of U937 and HL60
cells was associated with increased enzymatic activity of both the
extrinsic and intrinsic caspase cascades, such as caspase-8 and -9
(Fig. 4). Although the truncated
Bid form was not detected, the levels of intact Bid proteins were
gradually downregulated in a concentration-dependent manner by
decitabine in both cell lines (Fig.
3). Decitabine treatment also decreased the levels of IAP
family proteins, such as XIAP, cIAP-1, and cIAP-2, and the
anti-apoptotic Bcl-2, whereas the levels of pro-apoptotic Bax and
death receptor-related proteins, including Fas, FasL, DR4, DR5, and
TRAIL, were markedly increased in response to decitabine treatment
(Fig. 3). Additionally, decitabine
caused a significant reduction in the MMP values, which was
connected with the activation of caspase-3 and the concomitant
degradation of PARP. Thus, the results indicated that caspase-8
activation by decitabine triggered mitochondrial apoptotic events
by inducing conformational changes in apoptotic proteins.
Furthermore, treatment with decitabine in the presence of caspase
inhibitors was found to significantly prevent apoptosis (Fig. 5). Therefore, the data suggest that
decitabine-induced apoptosis in both the U937 and HL60 cell lines
is caspase-dependent, and the apoptotic effects of decitabine
appear to involve activation of both the intrinsic and extrinsic
pathways.
In addition to apoptosis, ROS are known to mediate
other intracellular signaling cascades. Oxidative stress is
generally considered to be an important regulator of apoptosis
(15,17). Many studies have suggested that a
disproportionate production of ROS leads to oxidative stress,
dysfunction of cell organelles like the mitochondria, and
eventually apoptosis or necrosis (16). Recently, ROS-mediated caspase
activation and mitochondrial dysfunction have been suggested as
critical for decitabine-induced apoptosis in several cancer cell
lines (35,40,41);
however, the current role of mitochondrial functional changes
associated with ROS generation in the response of human leukemia
cells to decitabine has not yet been well explored. Therefore, we
next investigated the question of whether apoptosis by decitabine
in U937 and HL60 cells was associated with the generation of
ROS.
We found a significant early overproduction of ROS
in decitabine-treated U937 and HL60 cells; furthermore, co-culture
with NAC, a commonly used ROS scavenger, effectively blocked this
ROS generation (Fig. 6). The
present results revealed that the activation of caspase-3 and
degradation of PARP proteins in decitabine-treated cells were
ROS-dependent, and that co-culture with NAC effectively blocked
decitabine-induced growth inhibition and apoptosis in U937 as well
as HL60 cells. The findings from the present study also indicated
that the loss of MMP in decitabine-treated cells is ROS-dependent,
which suggests that ROS may act upstream of caspase activation. In
addition, blocking of ROS generation prevented decitabine-induced
downregulation of XIAP. Because IAP family proteins are also
substrates of activated caspase-3 (42,43),
the observed decrease in XIAP expression may be due to
caspase-3-mediated processing following decitabine treatment. Since
ROS have the potential to induce the collapse of the MMP, and
consequently trigger the series of events leading to the
mitochondria-associated apoptotic pathway (15,44),
our findings suggest the involvement of ROS production and
mitochondrial dysfunction in decitabine-induced caspase-mediated
apoptosis in U937 and HL60 cells.
In conclusion, our study demonstrated that human
leukemia cells undergo apoptosis in response to treatment with
decitabine, and that this occurs through a mitochondria-mediated
pathway that requires ROS generation upstream for disruption of the
MMP, which leads to subsequent activation of caspases. The present
data emphasize the key role of ROS in apoptosis induced by
decitabine in human leukemia cells, and indicate that a positive
correlation exists between ROS and mitochondrial events leading to
apoptosis, and may aid in understanding of the mechanisms for the
anticancer activity of human leukemia. Taken together, although
further investigation of its activity in vivo is necessary
to elaborate and exploit this promise, these results suggest that
human leukemia may be a potential chemotherapeutic agent for the
treatment of leukemia patients.
Acknowledgements
This study was supported by Basic
Science Research Program through the National Research Foundation
of Korea (NRF) funded by the Ministry of Education, Science, and
Technology (2010-0008843 and 2010-001730).
References
|
1.
|
Chang H, Lin H, Yi L, Zhu J, Zhou Y, Mi M
and Zhang Q: 3,6-Dihydroxyflavone induces apoptosis in leukemia
HL-60 cells via reactive oxygen species-mediated p38 MAPK/JNK
pathway. Eur J Pharmacol. 648:31–38. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Abramson N and Melton B: Leukocytosis:
basics of clinical assessment. Am Fam Physician. 62:2053–2060.
2000.PubMed/NCBI
|
|
3.
|
Gilliland DG, Jordan CT and Felix CA: The
molecular basis of leukemia. Hematology Am Soc Hematol Educ
Program. 80–97. 2004. View Article : Google Scholar
|
|
4.
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Siegelin MD, Habel A and Gaiser T: 17-AAG
sensitized malignant glioma cells to death-receptor mediated
apoptosis. Neurobiol Dis. 33:243–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Lazebnik YA, Kaufmann SH, Desnoyers S,
Poirier GG and Earnshaw WC: Cleavage of poly(ADP-ribose) polymerase
by a proteinase with properties like ICE. Nature. 371:346–347.
1994. View
Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Jin Z and El-Deiry WS: Overview of cell
death signaling pathways. Cancer Biol Ther. 4:139–163.
2005.PubMed/NCBI
|
|
8.
|
Li H, Zhu H, Xu CJ and Yuan J: Cleavage of
BID by caspase 8 mediates the mitochondrial damage in the Fas
pathway of apoptosis. Cell. 94:491–501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Luo X, Budihardjo I, Zou H, Slaughter C
and Wang X: Bid, a bcl-2 interacting protein, mediates cytochrome c
release from mitochondria in response to activation of cell surface
death receptors. Cell. 94:481–490. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Dlugosz PJ, Billen LP, Annis MG, Zhu W,
Zhang Z, Lin J, Leber B and Andrews DW: Bcl-2 changes conformation
to inhibit Bax oligomerization. EMBO J. 25:2287–2296. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Park C, Jin CY, Kwon HJ, Hwang HJ, Kim GY,
Choi IW, Kwon TK, Kim BW, Kim WJ and Choi YH: Induction of
apoptosis by esculetin in human leukemia U937 cells: roles of Bcl-2
and extracellular-regulated kinase signaling. Toxicol In Vitro.
24:486–494. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
De Laurenzi V and Melino G: Apoptosis. The
little devil of death. Nature. 406:135–136. 2000.PubMed/NCBI
|
|
13.
|
Gao Z, Tian Y, Wang J, Yin Q, Wu H, Li YM
and Jiang X: A dimeric Smac/diablo peptide directly relieves
caspase-3 inhibition by XIAP. Dynamic and cooperative regulation of
XIAP by Smac/Diablo. J Biol Chem. 282:30718–30727. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Crack PJ and Taylor JM: Reactive oxygen
species and the modulation of stroke. Free Radic Boil Med.
38:1433–1444. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Fiers W, Beyaert R, Declercq W and
Vandenabeele P: More than one way to die: apoptosis, necrosis and
reactive oxygen damage. Oncogene. 18:7719–7730. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Skulachev VP: Bioenergetic aspects of
apoptosis, necrosis and mitoptosis. Apoptosis. 11:473–485. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Chakraborti T, Das S, Mondal M,
Roychoudhury S and Chakraborti S: Oxidant, mitochondria and
calcium: an overview. Cell Signal. 11:77–85. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Choi WY, Kim GY, Lee WH and Choi YH:
Sanguinarine, a benzophenanthridine alkaloid, induces apoptosis in
MDA-MB-231 human breast carcinoma cells through a reactive oxygen
species-mediated mitochondrial pathway. Chemotherapy. 54:279–287.
2008. View Article : Google Scholar
|
|
19.
|
Robertson KD: DNA methylation and human
disease. Nat Rev Genet. 6:597–610. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Watanabe Y and Maekawa M: Methylation of
DNA in cancer. Adv Clin Chem. 52:145–167. 2010. View Article : Google Scholar
|
|
21.
|
Jones PA and Taylor SM: Cellular
differentiation, cytidine analogs and DNA methylation. Cell.
20:85–93. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Bender CM, Pao MM and Jones PA: Inhibition
of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth
of human tumor cell lines. Cancer Res. 58:95–101. 1998.
|
|
23.
|
Karpf AR and Jones DA: Reactivating the
expression of methylation silenced genes in human cancer. Oncogene.
21:5496–5503. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Kihslinger JE and Godley LA: The use of
hypomethylating agents in the treatment of hematologic
malignancies. Leuk Lymphoma. 48:1676–1695. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Santos FP, Kantarjian H, Garcia-Manero G,
Issa JP and Ravandi F: Decitabine in the treatment of
myelodysplastic syndromes. Expert Rev Anticancer Ther. 10:9–22.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Robak T: New nucleoside analogs for
patients with hematological malignancies. Expert Opin Investig
Drugs. 20:343–359. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Issa JP and Kantarjian HM: Targeting DNA
methylation. Clin Cancer Res. 15:3938–3946. 2009. View Article : Google Scholar
|
|
28.
|
Boumber Y and Issa JP: Epigenetics in
cancer: what’s the future? Oncology. 25:220–206. 2011.
|
|
29.
|
Hurtubise A and Momparler RL: Effect of
histone deacetylase inhibitor LAQ824 on antineoplastic action of
5-Aza-2′-deoxycytidine (decitabine) on human breast carcinoma
cells. Cancer Chemother Pharmacol. 58:618–625. 2006.PubMed/NCBI
|
|
30.
|
Valdez BC, Li Y, Murray D, Corn P,
Champlin RE and Andersson BS: 5-Aza-2′-deoxycytidine sensitizes
busulfan-resistant myeloid leukemia cells by regulating expression
of genes involved in cell cycle checkpoint and apoptosis. Leuk Res.
34:364–372. 2010.
|
|
31.
|
Ng KP, Ebrahem Q, Negrotto S, Mahfouz RZ,
Link KA, Hu Z, Gu X, Advani A, Kalaycio M, Sobecks R, et al: p53
independent epigenetic-differentiation treatment in xenotransplant
models of acute myeloid leukemia. Leukemia. 25:1739–1750. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Festuccia C, Gravina GL, D’Alessandro AM,
Millimaggi D, Di Rocco C, Dolo V, Ricevuto E, Vicentini C and
Bologna M: Downmodulation of dimethyl transferase activity enhances
tumor necrosis factor-related apoptosis-inducing ligand-induced
apoptosis in prostate cancer cells. Int J Oncol. 33:381–388.
2008.
|
|
33.
|
Deng T and Zhang Y: 5-Aza-2′-deoxycytidine
reactivates expression of RUNX3 by deletion of DNA
methyltransferases leading to caspase independent apoptosis in
colorectal cancer Lovo cells. Biomed Pharmacother. 63:492–500.
2009.
|
|
34.
|
Capper D, Gaiser T, Hartmann C, Habel A,
Mueller W, Herold-Mende C, von Deimling A and Siegelin MD:
Stem-cell-like glioma cells are resistant to TRAIL/Apo2L and
exhibit down-regulation of caspase-8 by promoter methylation. Acta
Neuropathol. 117:445–456. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Ruiz-Magaña MJ, Rodríguez-Vargas JM,
Morales JC, Saldivia MA, Schulze-Osthoff K and Ruiz-Ruiz C: The DNA
methyltransferase inhibitors zebularine and decitabine induce
mitochondria-mediated apoptosis and DNA damage in p53 mutant
leukemic T cells. Int J Cancer. 130:1195–1207. 2012.PubMed/NCBI
|
|
36.
|
Kim SY, Kang HT, Choi HR and Park SC:
Biliverdin reductase A in the prevention of cellular senescence
against oxidative stress. Exp Mol Med. 43:15–23. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Choi JH, Choi AY, Yoon H, Choe W, Yoon KS,
Ha J, Yeo EJ and Kang I: Baicalein protects HT22 murine hippocampal
neuronal cells against endoplasmic reticulum stress-induced
apoptosis through inhibition of reactive oxygen species production
and CHOP induction. Exp Mol Med. 42:811–822. 2010. View Article : Google Scholar
|
|
38.
|
Jeong JH, Ryu DS, Suk DH and Lee DS:
Anti-inflammatory effects of ethanol extract from Orostachys
japonicus on modulation of signal pathways in LPS-stimulated RAW
264.7 cells. BMB Rep. 44:399–404. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Lin CT, Lin WH, Lee KD and Tzeng PY: DNA
mismatch repair as an effector for promoting phorbol ester-induced
apoptotic DNA damage and cell killing: implications in tumor
promotion. Int J Cancer. 119:1776–1784. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Brodská B and Holoubek A: Generation of
reactive oxygen species during apoptosis induced by DNA-damaging
agents and/or histone deacetylase inhibitors. Oxid Med Cell Longev.
2011:2535292011.PubMed/NCBI
|
|
42.
|
Hell K, Saleh M, Crescenzo GD,
O’Connor-McCourt MD and Nicholson DW: Substrate cleavage by
caspases generates protein fragments with Smac/Diablo-like
activities. Cell Death Differ. 10:1234–1239. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Choi YE, Butterworth M, Malladi S, Duckett
CS, Cohen GM and Bratton SB: The E3 ubiquitin ligase cIAP1 binds
and ubiquitinates caspase-3 and -7 via unique mechanisms at
distinct steps in their processing. J Biol Chem. 284:12772–12782.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Fleury C, Mignotte B and Vayssière JL:
Mitochondrial reactive oxygen species in cell death signaling.
Biochimie. 84:131–141. 2002. View Article : Google Scholar : PubMed/NCBI
|