Introduction
Most breast and ovarian cancers are sporadic and
only about 5–10% of breast and 10% of ovarian cancers are thought
to be hereditary, causing the hereditary breast and ovarian cancer
(HBOC) syndrome (1,2). Majority of HBOC cases have underlying
cause in germline mutations in the BRCA1 and BRCA2
susceptibility genes (3,4). Carriers of known deleterious
mutations in the BRCA genes have a lifetime risk of
approximately 60 to 80% for development of breast cancer (BC) and a
15 to 40% lifetime risk for ovarian cancer (OC) and are also at a
heightened risk for some other cancer types (4–6). So
far, genome-wide association studies have not identified other
highly penetrant susceptibility genes linked with HBOC, as reviewed
in Mavaddat et al(7).
Genetic screening of BRCA1 and BRCA2 therefore
remains the only verified strategy for identification of
individuals at high risk for hereditary BC and/or OC. To reduce
cancer risk, healthy carriers of deleterious BRCA mutations
are presented with various preventive options, such as regular
intensive screenings, prophylactic mastectomy with breast
reconstruction and/or oophorectomy or chemoprevention in the
setting of a clinical trial (8,9).
Additionally, genetic counseling and BRCA screening can be
offered to first degree relatives of the carrier.
The present report continues the previous report of
our group from 2011 where pathogenic mutations in the BRCA1
and BRCA2 genes in the Slovene population were described
(10). We describe novel
pathogenic mutations that have not yet been described in the
literature or BRCA mutational databases, such as Breast
Cancer Information Core Database (BIC), Human Gene Mutation
Database (HGMD-Professional), Universal Mutation Database (UMD) and
Leiden Open Variation Database (LOVD). We also report pathogenic
mutations for which records already exist but were detected for the
first time in the Slovene HBOC families tested between January 2009
and December 2011. The possible effects of novel and pathogenic
BRCA1 and BRCA2 mutations which have been detected in
Slovene HBOC families for the first time are discussed.
Patients and methods
Tested individuals
In the period from January 2009 to December 2011,
559 new individuals from 379 Slovene HBOC families were submitted
through mutational screening of the BRCA1 and/or the
BRCA2 genes at the Institute of Oncology Ljubljana, which is
the only public institution performing BRCA screenings in
Slovenia. Probands were chosen after genetic counseling according
to the ASCO guidelines for genetic and genomic testing for cancer
susceptibility (11). The family
history data were verified in the Slovenian state cancer registry
established in 1950. All tested individuals provided written
informed consent and attended genetic counseling sessions before
and after testing.
Mutation screening
In 362 probands admitted for complete screening of
all BRCA1/2 exons, methods for variations searching
consisted of multiplex ligation-dependent probe amplification
analysis (MLPA; MRC Holland, Amsterdam, Netherlands) for detection
of large genomic deletions and insertions and screening for small
mutations of all BRCA1 and BRCA2 exons with
high-resolution melting (HRM), denaturing gradient gel
electrophoresis (DGGE) and direct sequencing methods (10). Probands (197) from cancer-affected
families with already confirmed pathogenic BRCA mutation
were tested only for the familial pathogenic mutation. The
nomenclature of this study follows the Nomenclature for Description
of Genetic Variations approved by the Human Genome Variation
Society (HGVS).
Results
Since the screening for BRCA mutations began
in Slovenia in the year 1999, altogether 45 distinct pathogenic
BRCA mutations have been detected in the tested Slovene
families - 22 in the BRCA1 and 23 in the BRCA2
(Table I). The overall mutation
detection rates for the period between January 1999 to December
2008 and from January 2009 to December 2011 were 29.8 and 21.2%,
respectively (Table II). The
majority of detected pathogenic mutations were nonsense mutations
creating premature stop codons or missense mutations and small
deletions and/or insertions that cause frameshifting and also lead
to premature termination of translation. Of all detected
BRCA1 mutations four were large deletions, all of more than
one exon. No large deletions or insertions were detected in the
BRCA2 gene so far. In the period of the last three years
(January 2009 to December 2011) 559 probands were tested either for
the known familial mutation or were submitted through the complete
screening of all BRCA exons (Table II). Of the tested probands 115 were
positive for BRCA1 pathogenic mutation and 41 for
BRCA2 pathogenic mutation. In the stated period, three novel
mutations were found which have not yet been described, one in the
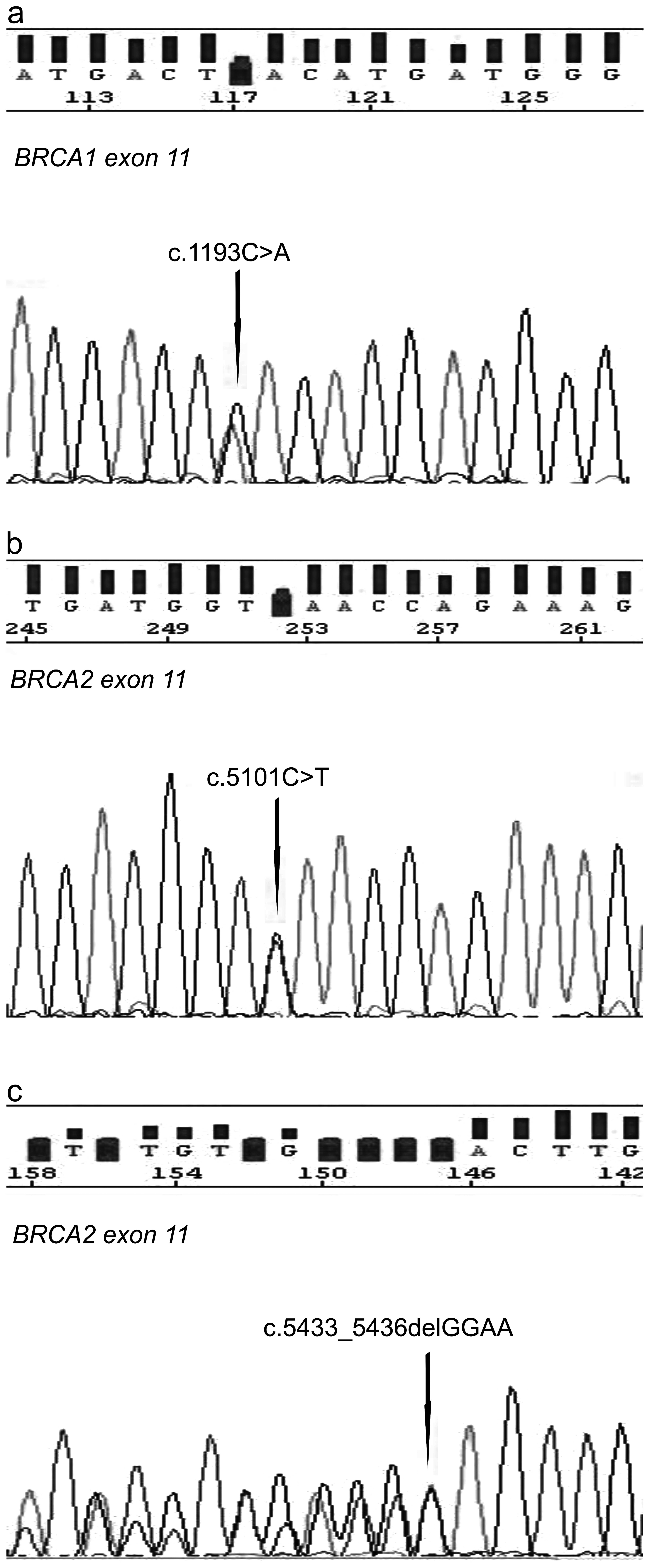
BRCA1 and two in the BRCA2 gene (Table III). The novel BRCA1
pathogenic mutation was detected in a healthy female from a HBOC
family (Table III, Fig. 1). All novel BRCA2 mutations
were detected in female BC patients (Table III, Fig. 1).
 | Table I.All pathogenic mutations in
BRCA1 and BRCA2 detected in Slovene HBOC
families. |
Table I.
All pathogenic mutations in
BRCA1 and BRCA2 detected in Slovene HBOC
families.
| Mutationa | Amino acid
changeb | Type of
mutation | No. of positive
families |
|---|
| BRCA1 | c.66_67delAG |
p.Glu23Valfs*17 | Frameshift | 1 |
| c.116G>A | p.Cys39Tyr | Missense | 8 |
| c.181T>G | p.Cys61Gly | Missense | 31 |
| c.181T>A | p.Cys61Ser | Missense | 5 |
| c.191G>A | p.Cys64Tyr | Missense | 3 |
| c.457_458delAG |
p.Ser153Cysfs*5 | Frameshift | 1 |
|
c.844_850dupTCATTAC |
p.Gln284Leufs*5 | Frameshift | 14 |
|
c.843_846delCTCA |
p.Ser282Tyrfs*15 | Frameshift | 2 |
| c.1193C>A |
p.Ser398* | Nonsense | 1 |
| c.1687C>T |
p.Gln563* | Nonsense | 23 |
|
c.2269_2270delG |
p.Val757Phefs*8 | Frameshift | 1 |
|
c.3018_3021delTTCA |
p.His1006Glnfs*17 | Frameshift | 3 |
|
c.3436_3439delTGTT |
p.Cys1146Leufs*8 | Frameshift | 1 |
| c.3718C>T |
p.Gln1240* | Nonsense | 2 |
|
c.5177_5180delGAAA |
p.Arg1726Lysfs*3 | Frameshift | 2 |
| c.5251C>T |
p.Arg1751* | Nonsense | 2 |
| c.5266dupC |
p.Gln1756Profs*74 | Frameshift | 9 |
| c.5377A>T |
p.Lys1793* | Nonsense | 3 |
| Exon 1–2del | | Large deletion | 2 |
| Exon 5–10del | | Large deletion | 4 |
| Exon 5–8del | | Large deletion | 1 |
| Exon 5–7del | | Large deletion | 3 |
| BRCA2 | c.262_263delCT |
p.Leu88Alafs*12 | Frameshift | 1 |
| c.658_659delGT |
p.Val220Ilefs*4 | Frameshift | 1 |
| c.775A>T |
p.Arg259* | Nonsense | 1 |
| c.1528G>T |
p.Glu510* | Nonsense | 1 |
|
c.1773_1776delTTAT |
p.Ile591Metfs*22 | Frameshift | 1 |
| c.1813insA |
p.Ile605Asnfs*11 | Frameshift | 1 |
| c.3265C>T |
p.Gln1089* | Nonsense | 2 |
|
c.3975_3978dupTGCT |
p.Ser1328Cysfs*3 | Frameshift | 5 |
|
c.4936_4939delGAAA |
p.Glu1646Glnfs*23 | Frameshift | 1 |
| c.5101C>T |
p.Gln1701* | Nonsense | 2 |
|
c.5213_5216delCTTA |
p.Thr1738Ilefs*2 | Frameshift | 1 |
| c.5291C>G |
p.Ser1764* | Nonsense | 5 |
| c.5351insA |
p.Asn1784Lysfs*3 | Frameshift | 1 |
|
c.5433_5436delGGAA |
p.Glu1811Aspfs*3 | Frameshift | 1 |
|
c.5609_5610delTCinsAG |
p.Phe1870* | Nonsense | 2 |
|
c.6491_6494delAGTT |
p.Gln2164Argfs*3 | Frameshift | 1 |
| c.6641insC |
p.Thr2214Asnfs*10 | Frameshift | 1 |
| c.6814delA |
p.Arg2272Glufs*8 | Frameshift | 1 |
| c.7303C>T |
p.Gln2435* | Nonsense | 1 |
| c.7806-2A>G | aberrant
splicing | Splicing | 13 |
| c.8175G>A |
p.Trp2725* | Nonsense | 2 |
| c.9117G>A | p.Pro3039Pro | Splicing | 1 |
| c.9286C>T |
p.Glu3096* | Nonsense | 1 |
| Table II.Screening for mutations in
BRCA genes in probands from HBOC families in Slovenia. |
Table II.
Screening for mutations in
BRCA genes in probands from HBOC families in Slovenia.
| Period | No. of tested
probands | No. of new
families | No. of new
BRCA1 positive families | No. of new
BRCA2 positive families |
|---|
| January 1999 –
December 2008a | 521 | 322 | 68 | 28 |
| January 2009 –
December 2011 | 559 | 349 | 54 | 20 |
| Total | 1080 | 671 | 122 | 48 |
| Table III.Novel pathogenic mutations in
BRCA1 and BRCA2 genes. |
Table III.
Novel pathogenic mutations in
BRCA1 and BRCA2 genes.
| Gene | HGVS
nomenclaturea | BIC
nomenclatureb | Amino acid
changec | No. of
families | Proband
characteristics (age at onset) | Other confirmed
carriers in the family | Family history of
the BC and/or OC (age at onset) |
|---|
| BRCA1 | c.1193C>A | 1312C>A |
p.Ser398* | 1 | Healthy, age
34 | / | Mother - BC
(53) |
| Maternal aunt - BC
(43) and OC (54) |
| BRCA2 | c.5101C>T | 5329C>T |
p.Gln1701* | 2 | BC (39) | Healthy daughter,
age 34 | / |
| OC (49) and BC
(51) | Healthy daughter,
age 31 | Mother - OC
(73) |
| Healthy sister, age
51 | |
| Healthy sister’s
daughter, age 30 | |
|
c.5433_5436delGGAA |
5661_5664delGGAA |
p.Glu1811Aspfs*3 | 1 | BC (53) | / | Maternal
grandmother - BC (54) |
| Maternal aunt - BC
(57) |
| Maternal cousin -
bilateral BC (38, 62) |
Besides three novel mutations, eleven known
pathogenic BRCA mutations were discovered for the first time
in the Slovene HBOC families, three BRCA1 and eight
BRCA2 (Tables IV and
V). All these newly detected
pathogenic mutations were detected in female BC and/or OC patients
(Tables IV and V). All novel and newly detected
pathogenic mutations in Slovenia were small mutations dictating
premature stop codon formation and subsequent truncation of BRCA1
or BRCA2 proteins.
| Table IV.Known BRCA1 pathogenic
mutations that have been detected for the first time in Slovene
HBOC families |
Table IV.
Known BRCA1 pathogenic
mutations that have been detected for the first time in Slovene
HBOC families
| HGVS
nomenclaturea | BIC
nomenclatureb | Amino acid
changec | No. of
families | Proband
characteristics (age at onset) | Other confirmed
carriers in the family | Family history of
BC and OC (age at onset) | Other cancers in
the family (age at onset) |
|---|
| c.66_68delAG | 185_186delAG |
p.Glu23Valfs*17 | 1 | BC (39), OC
(42) | / | Mother - BC
(45) | Maternal aunt - UC
(73) |
|
c.3436_3439delTGTT |
3555_3558delTGTT |
p.Cys1146Leufs*8 | 1 | OC (55) | Healthy daughter,
age 31 | Sister - OC
(53) | / |
| Sister - BC
(66) | |
| Mother - BC
(75) | |
| c.3718C>T | 3837C>T |
p.Gln1240* | 2 | BC (60) | / | Sister - bilateral
BC (46, 49) | Maternal uncle - LC
(57) |
| Mother - OC
(69) | Maternal aunt - FTC
(56) |
| Maternal aunt - OC
(65) | |
| Maternal aunt - BC
(70) | |
| OC (38) | Paternal aunt - OC
(41) | Father - PC
(74) |
| Paternal aunt - BC
(42) and OC (56) | Sister - CC
(32) |
| Paternal aunt - CRC
(51) |
| Paternal aunt - OC
(71) | |
| Table V.Known BRCA2 pathogenic
mutations that have been detected for the first time in Slovene
HBOC families |
Table V.
Known BRCA2 pathogenic
mutations that have been detected for the first time in Slovene
HBOC families
| HGVS
nomenclaturea | BIC
nomenclatureb | Amino acid
changec | No. of
families | Proband
characteristics (age at onset) | Other confirmed
carriers in the family (age at onset) | Family history of
BC and OC (age at onset) | Other cancers in
the family (age at onset) |
|---|
| c.262_263delCT | 490_491delCT |
p.Leu88Alafs*12 | 1 | BC (46) | / | Sister - BC
(55) | Mother - EC
(44) |
| Father - BRC
(56) |
| c.658_659delGT | 886_887delGT | p.
Val220Ilefs*4 | 1 | BC (72) and OC
(74) | / | Sister - OC
(59) | / |
| Sister - OC
(64) |
| Sister - OC
(78) |
|
c.1773_1776delTTAT |
2001_2004delTTAT |
p.Ile591Metfs*22 | 1 | BC (54) | Daughter - BC (38)
Healthy daughter, age 37 | Mother - BC
(68) | / |
|
c.5213_5216delCTTA |
5441_5444delCTTA |
p.Thr1738Ilefs*2 | 1 | OC (54) | Healthy daughter,
age 35 | Mother - BC
(35) | / |
| c.6641insC | 6869insC |
p.Thr2214Asnfs*10 | 1 | BC (47) | / | Sister - BC
(36) | / |
| Brother - BC
(44) |
| c.6814delA | 7042delA |
p.Arg2272Glufs*8 | 1 | BC (32) | / | Mother - bilateral
BC (58) | Maternal
grandmother - GC (70) |
| Maternal aunt - BC
(59) | Maternal aunt - BRC
(63) |
| c.8175G>A | 8403G>A |
p.Trp2725* | 2 | BC (43) | Healthy brother,
age 43 | Mother - BC
(46) | / |
| Paternal
grandmother - BC (72) |
| Paternal aunt - BC
(31) |
| Paternal aunt - BC
(41) |
| BC (26) | / | Mother - BC
(45) | / |
| Maternal
grandmother - BC (65) |
| c.9117G>A | 9345G>A | p.Pro3039Pro | 1 | BC (49) | Daughter - OC (24)
Healthy daughter, age 24 | Daughter - OC
(24) | Mother - CHC
(53) |
Discussion
Several recent studies have associated specific
BRCA mutations with specific cancer risks and phenotypes
(12,13). Many HBOC studies therefore have
focused on predicting effects of specific BRCA mutations and
reveal possible underlying molecular mechanisms (7). In this context, we discuss here the
predicted effects of the individual novel and newly detected
Slovene BRCA1 and BRCA2 pathogenic mutations.
Novel mutations
All three novel mutations described here
-c.1193C>A (p.Ser398*) in the BRCA1 and c.5101C>T
(p.Gln1701*) and c.5433_5436delGGAA (p.Glu1811Aspfs*3)
in the BRCA2 gene are located in exon 11 of BRCA1 or
BRCA2, which is the largest exon in both genes and also
carry the majority of pathogenic mutations described so far. As
BRCA mutations causing truncation of the BRCA proteins are
regarded as pathogenic, with some exceptions of truncating
mutations in the last 27th exon of the BRCA2, we predict
that all three novel mutations have deleterious effects (14,15).
More detailed descriptions are given below.
BRCA1
Mutation c.1193C>A (p.Ser398*) in exon
11 causes stop codon formation at codon 398. In the BIC database a
similar mutation discovered in Asian population, which leads to
formation of stop codon 398 (c.1193C>G), is described as a
clinically significant variant, but no references are given. Codon
398 lies in one of five conserved regions located at the 5′ end of
exon 11 (codons 282–554), which include putative interacting sites
for several proteins thought to be involved in transcription
(16). Codon 398 also forms a part
of interacting site (codons 341–748) for DNA repair protein RAD50
which participates in DNA repair by homologous recombination and by
non-homologous end joining (16,17).
Accordingly, we predict c.1193C>A mutation to severely impair
BRCA1-mediated DNA repair.
BRCA2
Mutation c.5101C>T (p.Gln1701*) is a nonsense
mutation causing formation of a stop codon at position 1701 which
is located in exon 11 in the ovarian cancer cluster region (OCCR)
spanning nucleotides 3035 to 6629. Several studies have shown that
truncating mutations in the OCCR region confer a higher ratio of
ovarian cancer relative to breast cancer (18–20).
Also, higher risk of prostate cancer was recently detected in males
with mutations in the BRCA2 OCCR region (21). Consistently with these studies, one
of the two Slovene BRCA2 c.5101C>T families exhibits a
high incidence of OC, besides BC (Table III).
Frameshift mutation c.5433_5436delGGAA (p.Glu1811
Aspfs*3) results in translation termination at amino
acid position 1813, which lies within the BRC repeat region in exon
11, between BRC5 (amino acids 1649–1735) and BRC6 (amino acids
1822–1914). Jara et al described a similar mutation,
c.5439delT (p.Leu1813fs*1), that might be
disease-causing (22). This
mutation dictates formation of a stop codon at amino acid position
1814 in the BRC repeat region (22).
The BRC repeat region is a region of eight highly
conserved internal BRC repeats separated by conserved nucleotide
stretches (23,24). The eight BRC repeats bind the RAD51
recombinase and control its activity in homologous DNA
recombination (23,24). Truncating mutation within the
BRCA2 BRC repeat domain, such as the novel
c.5433_5436delGGAA, are therefore predicted to seriously impair the
cell’s ability to repair DNA double-strand breaks (23–25).
Known BRCA pathogenic mutations that have
been detected for the first time in Slovene HBOC families
BRCA1
From January 2009 to December 2011 three known
pathogenic mutations in the BRCA1 and eight in the
BRCA2 gene were detected for the first time in the Slovene
HBOC families. Except one, all cause premature formation of stop
codons leading to truncation of the respective BRCA1 or BRCA2
proteins.
The mutation c.66_67delAG
(p.Glu23Valfs*17) is the most common BRCA1
mutation worldwide which occurs at a frequency of 1.1% in the
Ashkenazi Jews (26). Despite
being so widespread, this is the first recording of c.66_67delAG in
Slovenia, which to note has only very small Jewish population
(estimated 500–1,000 people). The c.66_67delAG dictates formation
of stop codon in the BRCA1 exon 2 thus forming a truncated
BRCA1 protein, BRAt, which lacks all known BRCA1 functional domains
(26). Studies have shown that
besides being non-functional the truncated BRCA proteins can also
impair the function of wild-type BRCA proteins (26,27).
It was further suggested that the BRAt mutant protein increases
transcription of the protein maspin (mammary serine protease
inhibitor), which has been implicated in inhibition of growth,
invasion, and metastatic potential of cancer cells (26,28).
Jiang et al also demonstrated that maspin sensitizes BRCA
deficient breast carcinoma cells to staurosporine-induced apoptosis
thus leading to an increased chemosensitivity (29).
The other two newly detected BRCA1 mutations
are located in exon 11. Mutation c.3718C>T
(p.Gln1240*) is reported few times in the BRCA
mutational databases but is published only once by Kwong et
al, who detected it in an endometrial cancer patient of
European origin (30). We detected
the c.3718C>T in two Slovene families who are, interestingly,
both affected by various cancer types (Table IV). As this mutation was first
detected in endometrial cancer, this could imply that the
c.3718C>T predisposes to other cancer types besides BC/OC.
Further studies are needed to corroborate this observation and
uncover possible underlying molecular mechanisms.
Mutation c.3436_3439delTGTT
(p.Cys1146Leufs*8) in the 11th exon of BRCA1 was
before only found once in the Slovene neighboring country Austria
(31). It is predicted to cause
termination of protein translation at codon 1153. It can be
compared to a similar mutation c.3481_3491del11
(p.Glu1161Phefs*3) that creates stop codon at 1163
(32). The c.3481_3491del11 is a
widespread French founder mutation that is frequently detected in
hereditary OC (33,34). Comparably, the Slovene
c.3436_3439delTGTT family is characterized by higher incidence of
OC relative to BC. Future studies are needed to determine whether
increased incidence of OC is associated with specific exon 11
truncating BRCA1 mutations.
BRCA2
The eight newly detected BRCA2 mutations are
all rather rare with only few existing records or publications.
Mutation c.262_263delCT (p.Leu88Alafs*12) is located in
exon 3. It was first described in one Polish HBOC family (described
as 488_489delCT) and was recently detected in a Spanish BC patient
(35,36). Salgado et al suggested that
abrogation of the amino-terminal exon 3 transcription activation
domain in the BRCA2 protein affects BRCA2 role in transcriptional
regulation and DNA repair processes through replication protein A
(RPA) (36). They further
suggested that abrogation of most (3320 amino acids) of the 3418
BRCA2 amino acids has more severe biological consequences besides
disrupted transcriptional regulation (36).
Mutation, c.658_659delGT
(p.Val220Ilefs*4), is located in exon 8 and is predicted
to truncate the protein before the eight BRC repeats (37). Interestingly, the c.658_659delGT is
one of a few BRCA2 mutations found in BRCA2 biallelic
cases. These biallelic BRCA2 mutations are known to cause
the D1 subgroup of Fanconi anemia (FA-D1), a rare autosomal
recessive disorder characterized, among other defects, by
predisposition to several childhood cancers (38,39).
Studies have shown that FA-D1 patients are especially at a high
risk of developing brain tumors, in particular medulloblastomas,
compared to other subgroups which are caused by mutations in other
DNA-repair genes (40). To note,
BC and OC risk in biallelic BRCA2 patients is difficult to
determine as FA patients usually die at a young age, before BC or
OC would generally develop. Nevertheless, it could be useful to
follow whether carriers of monoallelic c.658_659delGT are also
burdened by an increased risk for medulloblastomas or other brain
tumors. The Slovene family which has monoallelic BRCA2
c.658_659delGT does not, however, exhibit any brain tumors and is
affected mostly by quite late onset of OC. No biallelic
BRCA2 mutations were detected in Slovenia so far.
Mutation c.1773_1776delTTAT
(p.Ile591Metfs*22) causes formation of a stop codon 612
in the exon 10 of BRCA2. It has been described for Western
European and Chinese population (41–43).
A similar truncating deletion c.1787_1799del13 forming stop codon
near at 609 was recently discovered in a prostate cancer patient
with family history of stomach cancer but no BC or OC (44). No functional characterizations have
yet been published for c.1773_1776delTTAT, however, the Slovene
family having c.1773_1776delTTAT is to date affected only by BC
(Table V).
Three BRCA2 mutations were detected in the
exon 11, c.5213_5216delCTTA, c.6641insC and c.6814delA. Mutation
c.5213_5216delCTTA (p.Thr1738Ilefs*2) in exon 11 has been already
found in several HBOC families, mainly in the USA, the Netherlands
and in Belgium (45–49). It causes formation of termination
signal at codon 1739 located between BRC5 and BRC6 in the BRC
repeat region, similarly to the novel mutation c.5433_5436delGGAA
discussed above. According to the literature no other cancers
besides BC and OC are associated with this mutation. This also
applies to the Slovene c.5213_5216delCTTA family.
Mutation c.6641insC (p.Thr2214Asnfs*10)
in exon 11 is a frameshift mutation reported only once in BIC
database. Mutation is predicted to form a stop codon at position
2223 located at the 3′ end of exon 11. The mutation is causing the
truncated BRCA2 protein for the subsequent exons 12 to 27. Mutation
c.6641insC was identified in Slovene BC patient diagnosed at age
47, with a history of two BC cases in her family, diagnosed at ages
36 and 44. Interestingly, one was male BC (Table V). Similar mutation c.6641dupC
(p.Lys2215Tyrfs*10) was detected in nearby Croatia in
two unrelated families (50).
Mutation c.6814delA (p.Arg2272Glufs*8) in
exon 11 was detected in Slovene BC patient diagnosed at 32 years of
age, whose mother had bilateral BC. It is described only once in
the UMD database, without references, and is predicted to form stop
codon at position 2279 near the 3′ end of exon 11, therefore
abrogating exons 12 to 27.
Mutation c.8175G>A (p.Trp2725*) was
first reported just recently by Levanat et al(50). Mutation c.8175G>A was identified
in two unaffected siblings (with a family history of two BC cases)
from Croatia (50). Mutation
c.8175G>A lies in the frequently mutated exon 18 of BRCA2
leading to the truncation of the BRCA2 oligonucleotide binding
domain (OB1) in the DNA-binding domain (DBD) (32). The BRCA2 DBD region is needed for
binding of single-stranded DNA (ssDNA) that results from DNA damage
or replication errors (51).
Through this binding of ssDNA the BRCA2 protein mediates delivery
of RAD51 to the sites of exposed single-stranded DNA thus enabling
the RAD51 to catalyze homologous pairing and DNA strand exchange
(51). Through affecting this
recruitment of RAD51 to the ssDNA, mutations in the BRCA2 DBD are
predicted to affect the homologous recombination needed for
maintaining the integrity of the genome. Besides binding ssDNA, OB1
also binds the 70-amino acid DSS1 which is needed for BRCA2
stability and is also crucial for the BRCA2 functioning in one of
the homologous recombination pathways (52,53).
Mutation c.9117G>A (p.Pro3039Pro) is located in
exon 23 of BRCA2. This splicing mutation was shown to be
truncating (54). By this mutation
the OB2 functional domain of BRCA2 protein is affected most
probably causing impaired repair of double-strand DNA breaks
(51,55). Mutation c.9117G>A was identified
in three tested members from one Slovene family. Proband (mother)
was diagnosed with BC at the age of 49. Her two daughters were both
identified as carriers; one diagnosed with OC at the age 24 and one
still unaffected. Mutation c.9117G>A has been already found in
several HBOC families of Western/Central/East European origin
(56).
The present report describes three novel BRCA
pathogenic mutations that have been detected in Slovene HBOC
families thereby contributing to the ever-expanding spectrum of the
world-wide pathogenic BRCA mutations. Eleven previously
known pathogenic mutations that have been discovered for the first
time in Slovenia are also presented. For the probands bearing novel
or pathogenic BRCA1 and BRCA2 mutations which have
been detected in Slovene population for the first time, relevant
clinical data and family history are given. Recent literature is
reviewed to provide new data, which should help to create specific
plans for preventive and/or therapeutic strategies for individual
carriers according to their specific mutation.
Acknowledgements
The authors gratefully thank the
genetic counselors and nurses as well as laboratory technicians who
helped to recruit patients, collect their clinical data and
performed the analyses - Nikola Bešič, Alenka Vrečar and Simona
Traven.
References
|
1.
|
J FerlayDM ParkinE
Steliarova-FoucherEstimates of cancer incidence and mortality in
Europe in 2008Eur J
Cancer46765781201010.1016/j.ejca.2009.12.01420116997
|
|
2.
|
CR JamesJE QuinnPB MullanPG JohnstonDP
HarkinBRCA1, a potential predictive biomarker in the treatment of
breast
cancerOncologist12142150200710.1634/theoncologist.12-2-14217296808
|
|
3.
|
J XuB WangY ZhangR LiY WangS ZhangClinical
implications for BRCA gene mutation in breast cancerMol Biol
Rep3930973102201210.1007/s11033-011-1073-y21691706
|
|
4.
|
HT LynchE SilvaC SnyderJF LynchHereditary
breast cancer: part I. Diagnosing hereditary breast cancer
syndromesBreast
J14313200810.1111/j.1524-4741.2007.00515.x18086272
|
|
5.
|
D ThompsonDF EastonBreast Cancer Linkage
ConsortiumCancer incidence in BRCA1 mutation carriersJ Natl Cancer
Inst9413581365200210.1093/jnci/94.18.135812237281
|
|
6.
|
L KadouriD BercovichA ElimelechA novel
BRCA-1 mutation in Arab kindred from east Jerusalem with breast and
ovarian cancerBMC Cancer714200710.1186/1471-2407-7-1417233897
|
|
7.
|
N MavaddatAC AntoniouDF EastonM
Garcia-ClosasGenetic susceptibility to breast cancerMol
Oncol4174191201010.1016/j.molonc.2010.04.011
|
|
8.
|
C KoumpisC DimitrakakisA
AntsaklisPrevalence of BRCA1 and BRCA2 mutations in unselected
breast cancer patients from GreeceHered Cancer Clin
Pract910201110.1186/1897-4287-9-1022085629
|
|
9.
|
KA MetcalfeJL SempleSA NarodTime to
reconsider subcutaneous mastectomy for breast-cancer
prevention?Lancet
Oncol6431434200510.1016/S1470-2045(05)70210-215925821
|
|
10.
|
V StegelM KrajcJ ZgajnarThe occurrence of
germline BRCA1 and BRCA2 sequence alterations in Slovenian
populationBMC Med Genet129201110.1186/1471-2350-12-921232165
|
|
11.
|
ME RobsonCD StormJ WeitzelDS WollinsK
OffitAmerican Society of Clinical OncologyAmerican Society of
Clinical Oncology policy statement update: genetic and genomic
testing for cancer susceptibilityJ Clin
Oncol28893901201010.1200/JCO.2009.27.066020065170
|
|
12.
|
R RoyJ ChunSN PowellBRCA1 and BRCA2:
different roles in a common pathway of genome protectionNat Rev
Cancer126878201210.1038/nrc318122193408
|
|
13.
|
N TungY WangLC CollinsEstrogen receptor
positive breast cancers in BRCA1 mutation carriers: clinical risk
factors and pathologic featuresBreast Cancer
Res12R12201010.1186/bcr247820149218
|
|
14.
|
P RadiceS De SummaL CalecaS
TommasiUnclassified variants in BRCA genes: guidelines for
interpretationAnn Oncol22Suppl
1i18i23201110.1093/annonc/mdq66121285146
|
|
15.
|
A BorgRW HaileKE MaloneCharacterization of
BRCA1 and BRCA2 deleterious mutations and variants of unknown
clinical significance in unilateral and bilateral breast cancer:
the WECARE studyHum
Mutat31E1200E1240201010.1002/humu.2120220104584
|
|
16.
|
MA FlemingJD PotterCJ RamirezGK
OstranderEA OstranderUnderstanding missense mutations in the BRCA1
gene: an evolutionary approachProc Natl Acad Sci
USA10011511156200310.1073/pnas.023728510012531920
|
|
17.
|
Q ZhongTG BoyerPL ChenWH LeeDeficient
nonhomologous end-joining activity in cell-free extracts from
Brca1-null fibroblastsCancer Res6239663970200212124328
|
|
18.
|
SA GaytherJ MangionP RussellVariation of
risks of breast and ovarian cancer associated with different
germline mutations of the BRCA2 geneNat
Genet15103105199710.1038/ng0197-1038988179
|
|
19.
|
D ThompsonD EastonVariation in cancer
risks, by mutation position, in BRCA2 mutation carriersAm J Hum
Genet68410419200110.1086/31818111170890
|
|
20.
|
U HamannX LiuS LangeHU UlmerA BennerRJ
ScottContribution of BRCA2 germline mutations to hereditary
breast/ovarian cancer in GermanyJ Med
Genet39E12200210.1136/jmg.39.3.e1211897832
|
|
21.
|
A MoranC O’HaraS KhanRisk of cancer other
than breast or ovarian in individuals with BRCA1 and BRCA2
mutationsFam
Cancer11235242201210.1007/s10689-011-9506-222187320
|
|
22.
|
L JaraS AmpueroE SantibanezBRCA1 and BRCA2
mutations in a South American populationCancer Genet
Cytogenet1663645200610.1016/j.cancergencyto.2005.08.01916616110
|
|
23.
|
E RajendraAR VenkitaramanTwo modules in
the BRC repeats of BRCA2 mediate structural and functional
interactions with the RAD51 recombinaseNucleic Acids
Res388296201010.1093/nar/gkp87319875419
|
|
24.
|
MK ShivjiSR MukundE RajendraThe BRC
repeats of human BRCA2 differentially regulate RAD51 binding on
single-versus double-stranded DNA to stimulate strand exchangeProc
Natl Acad Sci
USA1061325413259200910.1073/pnas.090620810619628690
|
|
25.
|
RB JensenA CarreiraSC
KowalczykowskiPurified human BRCA2 stimulates RAD51-mediated
recombinationNature467678683201010.1038/nature0939920729832
|
|
26.
|
JD O’DonnellRJ LingerPA KrukBRCA1 185delAG
mutant protein, BRAt, up-regulates maspin in ovarian epithelial
cellsGynecol Oncol1162622682010
|
|
27.
|
TA KingW LiE BrogiHeterogenic loss of the
wild-type BRCA allele in human breast tumorigenesisAnn Surg
Oncol1425102518200710.1245/s10434-007-9372-117597348
|
|
28.
|
CH StreuliMaspin is a tumour suppressor
that inhibits breast cancer tumour metastasis in vivoBreast Cancer
Res4137140200210.1186/bcr43712100737
|
|
29.
|
N JiangY MengS ZhangE Mensah-OsmanS
ShengMaspin sensitizes breast carcinoma cells to induced
apoptosisOncogene2140894098200210.1038/sj.onc.120550712037665
|
|
30.
|
A KwongEK NgEY TangA novel de novo BRCA1
mutation in a Chinese woman with early onset breast cancerFam
Cancer10233237201110.1007/s10689-011-9429-y21404118
|
|
31.
|
T WagnerD Stoppa-LyonnetE
FleischmannDenaturing high-performance liquid chromatography
detects reliably BRCA1 and BRCA2
mutationsGenomics62369376199910.1006/geno.1999.602610644434
|
|
32.
|
S CaputoL BenboudjemaO SinilnikovaE
RouleauC BeroudR LidereauDescription and analysis of genetic
variants in French hereditary breast and ovarian cancer families
recorded in the UMD-BRCA1/BRCA2 databasesNucleic Acids
Res40D992D1002201210.1093/nar/gkr116022144684
|
|
33.
|
SA JanezicA ZiogasLM KrumroyGermline BRCA1
alterations in a population-based series of ovarian cancer casesHum
Mol Genet8889897199910.1093/hmg/8.5.88910196379
|
|
34.
|
A MusolinoM MichiaraMA BellaMolecular
profile and clinical variables in BRCA1-positive breast cancers. A
population-based studyTumori91505512200516457150
|
|
35.
|
B GorskiA JakubowskaT HuzarskiA high
proportion of founder BRCA1 mutations in Polish breast cancer
familiesInt J Cancer110683686200410.1002/ijc.2016215146557
|
|
36.
|
J SalgadoC GutierrezC GilM RoblesJ
Garcia-FoncillasComparative disease pattern of a patient with a
novel BRCA2 truncation and knockout models for BRCA2Breast Cancer
Res Treat123291293201010.1007/s10549-010-0776-420151322
|
|
37.
|
TS FrankSA ManleyOI OlopadeSequence
analysis of BRCA1 and BRCA2: correlation of mutations with family
history and ovarian cancer riskJ Clin
Oncol162417242519989667259
|
|
38.
|
MA AdankH SegersSE van MilFanconi anemia
gene mutations are not involved in sporadic Wilms tumorPediatr
Blood Cancer55742744201010.1002/pbc.2258820589654
|
|
39.
|
NG HowlettT TaniguchiS OlsonBiallelic
inactivation of BRCA2 in Fanconi
anemiaScience297606609200210.1126/science.107383412065746
|
|
40.
|
BP AlterPS RosenbergLC BrodyClinical and
molecular features associated with biallelic mutations in
FANCD1/BRCA2J Med Genet4419200710.1136/jmg.2006.04325716825431
|
|
41.
|
ZL MaMZ CaoWF LiAnalysis of BRCA2 gene
mutations among familial and/or early-onset breast cancer patients
in eastern Shandong of ChinaZhonghua Yi Xue Yi Chuan Xue Za
Zhi251951982008(In Chinese).
|
|
42.
|
JJ van HarsselCE van RoozendaalY
DetischEfficiency of BRCAPRO and Myriad II mutation probability
thresholds versus cancer history criteria alone for BRCA1/2
mutation detectionFam Cancer9193201201019949876
|
|
43.
|
V Caux-MoncoutierL CasteraC TirapoEMMA, a
cost- and time-effective diagnostic method for simultaneous
detection of point mutations and large-scale genomic
rearrangements: application to BRCA1 and BRCA2 in 1,525 patientsHum
Mutat32325334201110.1002/humu.21414
|
|
44.
|
Z Kote-JaraiD LeongamornlertE
SaundersBRCA2 is a moderate penetrance gene contributing to
young-onset prostate cancer: implications for genetic testing in
prostate cancer patientsBr J
Cancer10512301234201110.1038/bjc.2011.38321952622
|
|
45.
|
CS SinclairC AdemA NaderiTBX2 is
preferentially amplified in BRCA1- and BRCA2-related breast
tumorsCancer Res6235873591200212097257
|
|
46.
|
C AdemC ReynoldsCL SoderbergPathologic
characteristics of breast parenchyma in patients with hereditary
breast carcinoma, including BRCA1 and BRCA2 mutation
carriersCancer97111200310.1002/cncr.1104812491499
|
|
47.
|
AH van der HoutAM van den OuwelandRB van
der LuijtA DGGE system for comprehensive mutation screening of
BRCA1 and BRCA2: application in a Dutch cancer clinic settingHum
Mutat27654666200616683254
|
|
48.
|
BB HermsenPJ van DiestJ BerkhofLow
prevalence of (pre) malignant lesions in the breast and high
prevalence in the ovary and Fallopian tube in women at hereditary
high risk of breast and ovarian cancerInt J
Cancer11914121418200610.1002/ijc.2198816615107
|
|
49.
|
A RomanoPJ LindseyDC FischerTwo
functionally relevant polymorphisms in the human progesterone
receptor gene (+331 G/A and progins) and the predisposition for
breast and/or ovarian cancerGynecol Oncol101287295200616360811
|
|
50.
|
S LevanatV MusaniML CvokThree novel
BRCA1/BRCA2 mutations in breast/ovarian cancer families in
CroatiaGene498169176201210.1016/j.gene.2012.02.01022366370
|
|
51.
|
WK HollomanUnraveling the mechanism of
BRCA2 in homologous recombinationNat Struct Mol
Biol18748754201110.1038/nsmb.209621731065
|
|
52.
|
H YangPD JeffreyJ MillerBRCA2 function in
DNA binding and recombination from a BRCA2-DSS1-ssDNA
structureScience29718371848200210.1126/science.297.5588.183712228710
|
|
53.
|
N SiaudMA BarberaA EgashiraPlasticity of
BRCA2 function in homologous recombination: genetic interactions of
the PALB2 and DNA binding domainsPLoS
Genet7e1002409201110.1371/journal.pgen.100240922194698
|
|
54.
|
PC FongTA YapDS BossPoly(ADP)-ribose
polymerase inhibition: frequent durable responses in BRCA carrier
ovarian cancer correlating with platinum-free intervalJ Clin
Oncol2825122519201010.1200/JCO.2009.26.9589
|
|
55.
|
AB SpurdleSR LakhaniS HealeyClinical
classification of BRCA1 and BRCA2 DNA sequence variants: the value
of cytokeratin profiles and evolutionary analysis - a report from
the kConFab InvestigatorsJ Clin
Oncol2616571663200810.1200/JCO.2007.13.277918375895
|
|
56.
|
HA RischJR McLaughlinDE ColePopulation
BRCA1 and BRCA2 mutation frequencies and cancer penetrances: a
kin-cohort study in Ontario, CanadaJ Natl Cancer
Inst9816941706200610.1093/jnci/djj46517148771
|