Introduction
It is widely accepted that normal somatic cells have
intrinsically limited division capacity and reach a
non-proliferative state called cellular senescence. Senescent cells
are characterized by irreversible growth arrest, a typical
gene-expression profile, hyporesponsiveness to external stimuli,
increased acidic β-galactosidase activity, and a flat and enlarged
morphology. Most tumors contain cell populations that have escaped
the senescence barriers to proliferative potential. This
capability, known as immortality, could trigger genomic instability
and tumorigenesis. Therefore, cellular senescence has been proposed
as a major tumor suppression mechanism (1), and therapy-induced senescence
represents a novel functional target that may improve cancer
therapy (2–5).
Gliomas are the most common primary brain tumor in
adults, and high-grade gliomas are almost universally fatal.
Despite recent advances in diagnosis and therapy, including
surgical resection followed by radiation and chemotherapy, the
prognosis for patients with malignant glioma remain unsatisfactory
due to the high recurrence rate and relative drug resistance of
high-grade glioma cells (6).
Conventional anticancer drugs are associated with relatively strong
toxic side-effects and drug resistance.
20(S)-ginsenoside Rg3 [20(S)-Rg3] is an effective
medicinal chemical compound extracted from Panax ginseng with a
C42H72O13 framework and 784.3 Da
molecular weight (7). 20(S)-Rg3
has been shown to be safe, and recent evidence from in vitro
experiments and animal models have demonstrated that 20(S)-Rg3
possesses a variety of anti-mutagenic and cancer-inhibitory
properties (8–12). However, the exact molecular
mechanism of its antitumor effects remains unclear.
Here, we show that chronic treatment with 20(S)-Rg3
at a sub-apoptotic concentration caused senescence-like growth
arrest in U87 glioma cells and that this was partially reliant on
20(S)-Rg3-induced ROS generation and induction of p53/ p21.
Moreover, we found that Akt plays a critical role in
20(S)-Rg3-induced ROS generation and senescence in glioma
cells.
Materials and methods
Materials
U87 glioma cell lines were purchased from American
Type Culture Collections (Manassas, VA, USA). These cell lines were
cultured in Dulbecco’s modified Eagle’s medium (HyClone, Logan, UT,
USA) supplemented with 10% fetal bovine serum and 25 U/ml
penicillin/streptomycin. Anti-phospho-p53, and phospho-Akt
polyclonal antibody were purchased from Cell Signaling Inc.
(Danvers, MA, USA). Antibodies for p21, p27, β-actin were purchased
from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Secondary
horseradish peroxidase-conjugated anti-rabbit and anti-mouse
antibodies were from Zymed Laboratories (San Francisco, CA, USA);
chemiluminescent detection systems were from Pierce (Rockford, IL,
USA); and Lipodin-Pro protein transfection reagent was from
Abbiotec (San Diego, CA, USA). Small interfering RNA duplexes
against Akt, p53, and p21 were purchased from Dharmacon Co.
Transfection reagent (Oligofectamine™ 2000) was
purchased from Invitrogen (Eugene, OR, USA). N-acetyl-l-cysteine
and doxorubicin were obtained from Calbiochem (San Diego, CA, USA),
and 2′, 7′-dichlorofluorescein diacetate and MitoSOX was purchased
from Invitrogen (Carlsbad, CA, USA). 20(S)-ginsenoside Rg3 were
obtained from International laboratory (USA). All other biochemical
reagents were from Sigma or Invitrogen.
SA β-galactosidase staining
Cellular senescence of cells was confirmed by a
senescence-associated β-galactosidase activity assay as described
by Dimri et al (13). After
being grown in a semi-confluent state, senescence-associated
β-galactosidase, pH 6.0, activity was examined. Cells were washed
with phosphate-buffered saline (PBS) and fixed with 2%
paraformaldehyde containing 0.2% glutaraldehyde in PBS for 5 min at
room temperature (RT). After washing with PBS, cells were incubated
with β-galactosidase reagent [1 mg/ml
5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal), 40 mm
citric acid/sodium phosphate buffer, pH 6.0, 5 mm potassium
ferrocyanide/potassium ferricyanide, 150 mm NaCl, 2 mm
MgCl2] at 37°C.
Western blot analysis
Akt, p16, p21, p27 and p53 band were determined by
western blotting. Briefly, cells were lysed with lysis buffer [50
mM Tris pH7.4, 150 mM NaCl, 1 mM EDTA pH 8.0, 1 mM protease
inhibitor (Roche), 1 mM PMSF, 1 mM NaF, 1 mM sodium orthovanadate],
and protein contents were determined using Bradford reagent. Equal
amounts of protein (40 μg) were then separated by SDS-PAGE
and transferred to nitrocellulose membranes (Schleicher &
Schuell BioScience Inc). After blocking with TBS containing
Tween-20 in the presence of 2.5% nonfat dry milk, the membranes
were incubated with the primary antibodies at 4°C for 16 h.
Secondary antibodies were added for 1 h at RT. The antibody-antigen
complexes were detected using the ECL detection system
(Pierce).
Analysis of apoptosis by Annexin V
staining
The amount of phosphatidylserine (PS) on cell
surfaces (a measure of apoptosis) was determined using an Annexin
V-FITC apoptosis detection kit (Abcam, USA), according to the
manufacturer’s instructions. Briefly, following
H2O2 or staurosporine treatment, cells were
harvested and washed twice with PBS. The cells were then
resuspended in 0.5 ml of cold 1X binding buffer (10 mM Hepes, pH
7.4, 150 mM NaCl, 2.5 mM CaCl2, 1 mM MgCl2,
4% BSA), and 2.5 μl of Annexin V-FITC (fluorescein
isothiocyanate) was added. Following incubation for 15 min, stained
cells were analyzed by flow cytometry (Becton-Dickinson FACSorter)
using CELLQuest software. Error bars represent standard deviations
of the means.
RNA interference and transfection
U87 cells were plated in a 100 mm dish, transfected
with 0.5 nmol of siRNA and Oligofectamine reagent in serum-free
medium and incubated for 4 h at 37°C in a CO2 incubator.
Following incubation, the cells were supplemented with growth
medium containing 10% fetal bovine seerum. Cells were harvested
after 72 h.
Measurement of intracellular ROS
level
Cells were stained with 50 μM of DCF-DA for
30 min and then harvested. For quantitation of mitochondrial ROS,
the cells were incubated with 0.2 μM MitoSOX for 30 min at
37°C, washed with PBS, trypsinized, collected in PBS, and analyzed
on a FACSCalibur. To examine the effect of N-acetylcysteine (NAC),
cells were treated with 20 mM of NAC for 2 days.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT)
assays
Cells were plated in 24-well tissue culture plates
and allowed to attach overnight. MTT was added to each well to a
final concentration of 200 μg/ml, and cells were incubated
for 4 h. After removing the medium completely, the formazan product
was solubilized with dimethylsulfoxide. Optical densities (OD) were
measured at 490 nm. Each experiment was performed three times.
Error bars represent standard deviations of the means.
Statistical analysis
Data are presented as the means ± S.D. and p-values
were calculated using a Student’s t-test. A value of p<0.05 was
considered to indicate a statistically significant difference. All
data presented are the representative of at least three separate
experiments.
Results
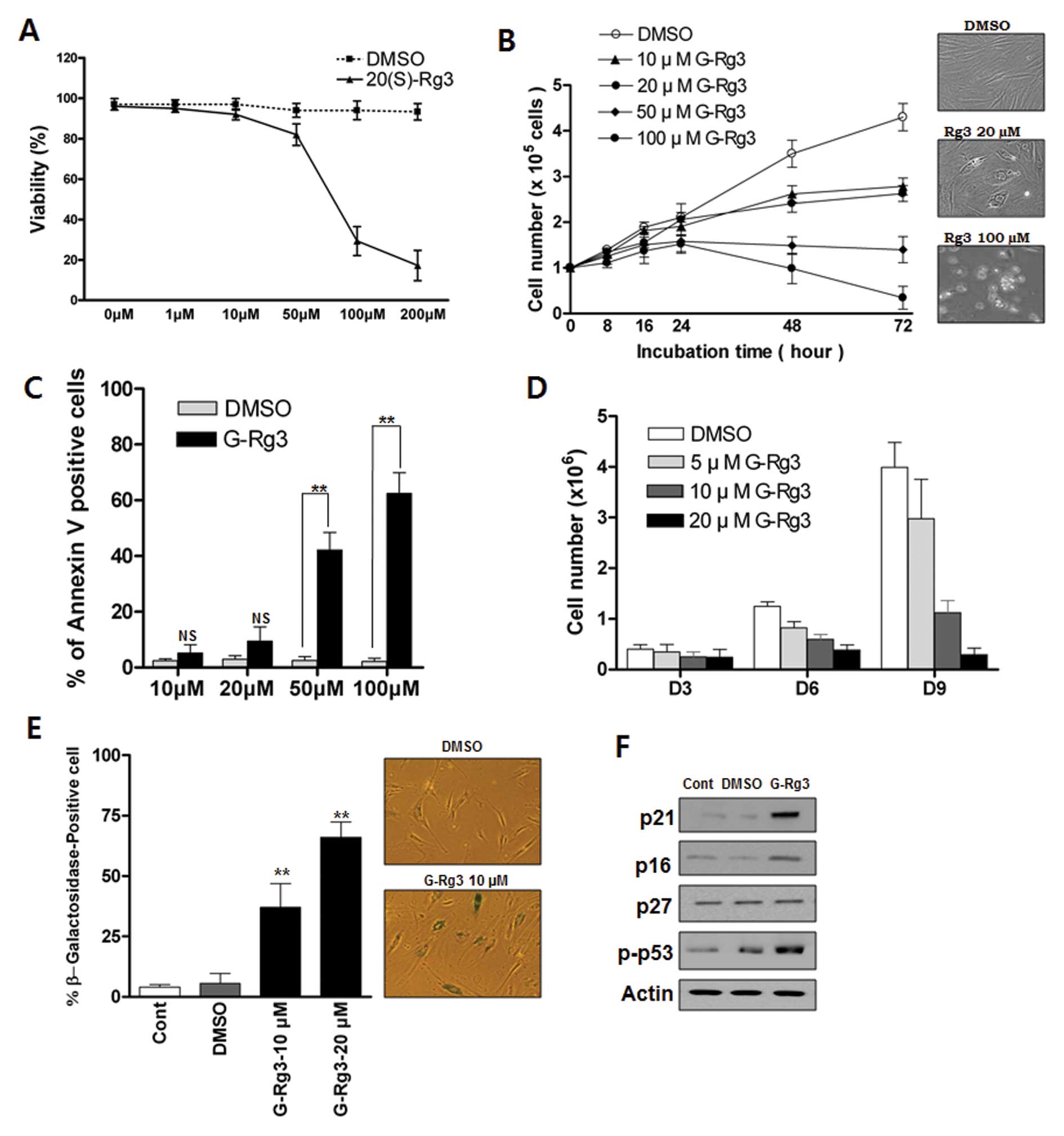
Low or high doses of 20(S)-Rg3 induces
either senescence or apoptosis, respectively, in glioma cells
To investigate the growth-regulatory activity of
20(S)-Rg3 in glioma cells, we treated glioma cells with various
concentrations of 20(S)-Rg3. Exposure of U87 cells to 20(S)-Rg3 for
3 days resulted in a dose-dependent inhibition of cell
proliferation (Fig. 1A and B).
Concentrations of ≥10 μM 20(S)-Rg3 suppress the growth of
U87 cells (Fig. 1B) whereas
concentrations <10 μM exhibit little effect (data not
shown). Next we examined the involvement of apoptosis in
20(S)-Rg3-induced growth arrest. Direct detection of apoptosis with
Annexin V staining showed that apoptotic cells were not
significantly increased in a sublethal dose of 20(S)-Rg3 (10
μM, 20 μM) compared with a high dose of 20(S)-Rg3 (50
μM, 100 μM) (Fig.
1C). We further examined the effects of chronic exposure of
sub-apoptotic 20(S)-Rg3 on cell proliferation. Direct cell count
assay indicated that the growth of U87 cells was gradually
inhibited by chronic exposure of a sublethal dose of 20(S)-Rg3,
which became more obvious on day 9 (Fig. 1D). 20(S)-Rg3 (20 μM)
completely suppressed cell proliferation for at least nine days,
while inducing only modest levels of cell death (data not shown).
To test the possibility that cell growth arrest in response to
20(S)-Rg3 was caused by the induction of cellular senescence, we
treated glioma cells with various sublethal concentrations of
20(S)-Rg3 and examined the expression of
senescence-associated-β-galactosidase (SA-β-gal), a commonly
accepted marker of senescence. As shown in Fig. 1E, ∼67% of U87 cells after chronic
20(S)-Rg3 treatment at a 20 μM concentration were stained
positively compared with only ∼5% cells with positive staining in
the DMSO control group (Fig. 1E).
Cells that were positive for SA-β-gal showed an enlarged and
flattened morphology that was consistent with cellular senescence
(Fig. 1E). These findings
indicated that 20(S)-Rg3 could induce senescence in addition to
apoptosis. An immunoblot analysis showed that the level of p53 and
cyclin-dependent kinase inhibitor p21CIP were markedly increased in
20(S)-Rg3-treated cells (Fig. 1F).
However, there was no increase in the expression of p27Kip1 and
only a moderate elevation of p16INK4 in 20(S)-Rg3-treated U87 cells
(Fig. 1F).
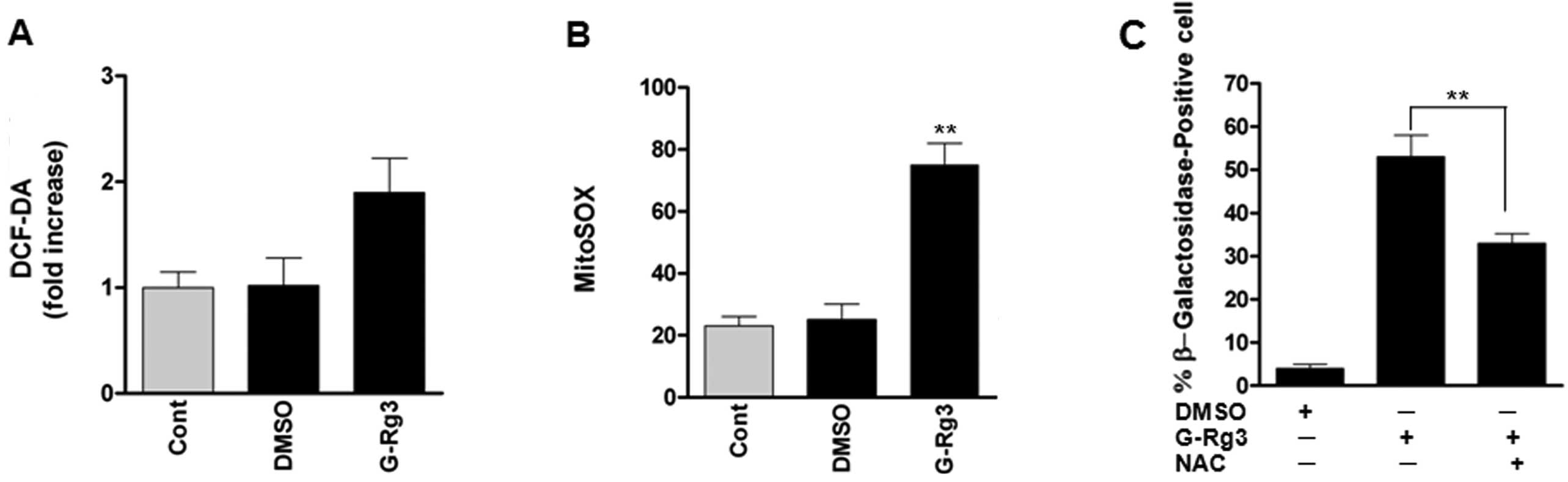
20(S)-Rg3 triggers senescence by an
increase of mitochondrial ROS level
As reactive oxygen species (ROS) are well-known
inducers of senescence, we evaluated whether treatment of glioma
cells with 20(S)-Rg3 increases oxidative stress. ROS levels, as
assessed by dichlorofluorescein (DCF), were increased after
20(S)-Rg3 treatment of U87 glioma cells (Fig. 2A). As mitochondrial ROS are the
major cellular source of ROS, we also examined the fluorescence of
MitoSOX Red as a mitochondrial superoxide indicator. Fluorescence
intensity of MitoSOX Red was significantly increased in U87 cells,
consistent with increased intracellular ROS levels (Fig. 2B). To address the role of ROS in
senescence induced by 20(S)-Rg3, we treated U87 cells with the ROS
scavenger N-acetyl-l-cysteine before 20(S)-Rg3 exposure. NAC
treatment reduced percentages of SA-β-gal-positive cells (Fig. 2C). These data suggest that elevated
ROS levels contribute to senescence induction by 20(S)-Rg3.
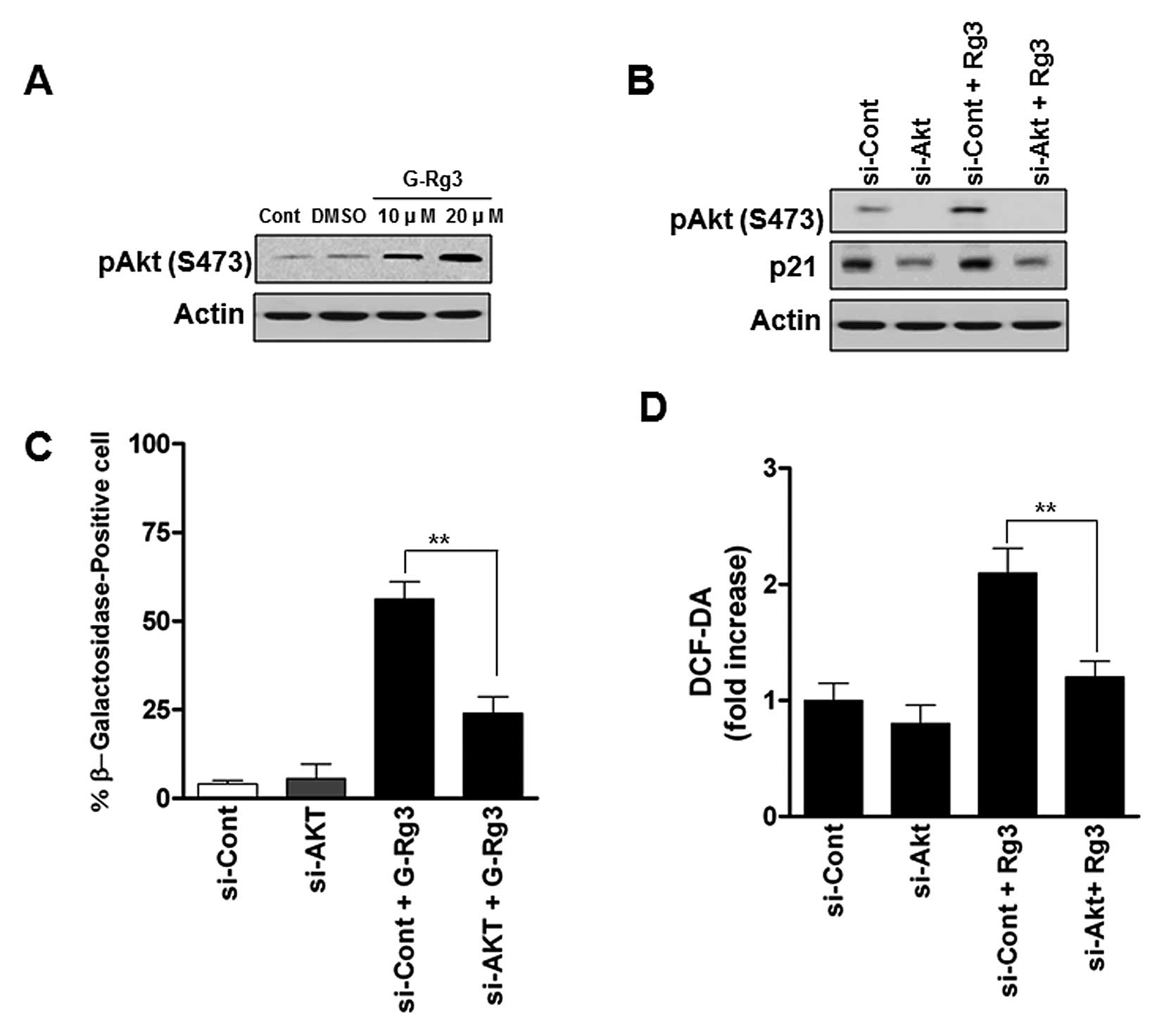
Activation of the Akt contributes to
senescence induction by 20(S)-Rg3 and is associated with elevation
of ROS
It has been reported that PI3K/Akt signaling
cascades are frequently deregulated in glioma and Akt activation is
involved in glioma cell senescence (14,15).
To examine whether Akt activity is involved in 20(S)-Rg3-induced
senescent glioma cells, we measured phospho-Akt levels. U87 cells
treated with 20 μM 20(S)-Rg3 were characterized by increases
in phospho-Akt (Fig. 3A). To
address whether the increase in levels of phospho-Akt is important
for the induction of senescence in 20(S)-Rg3-treated human glioma
cells, we employed si-RNA against Akt (Fig. 3B). As shown in Fig. 3C, depletion of Akt significantly
decreased the SA-β-gal activity compared with control
siRNA-infected cells. As ROS generation has been shown to be
involved in the Akt in glioma cells (16), we next examined whether the
increased ROS levels caused by 20(S)-Rg3 occurred as a result of
activation of Akt or was a secondary effect of this drug.
Transfection of Akt siRNA reduced ROS generation by approximately
32% (Fig. 3D). These data strongly
suggest that AKT may play an important role in regulating cellular
ROS status and senescence in glioma cells.
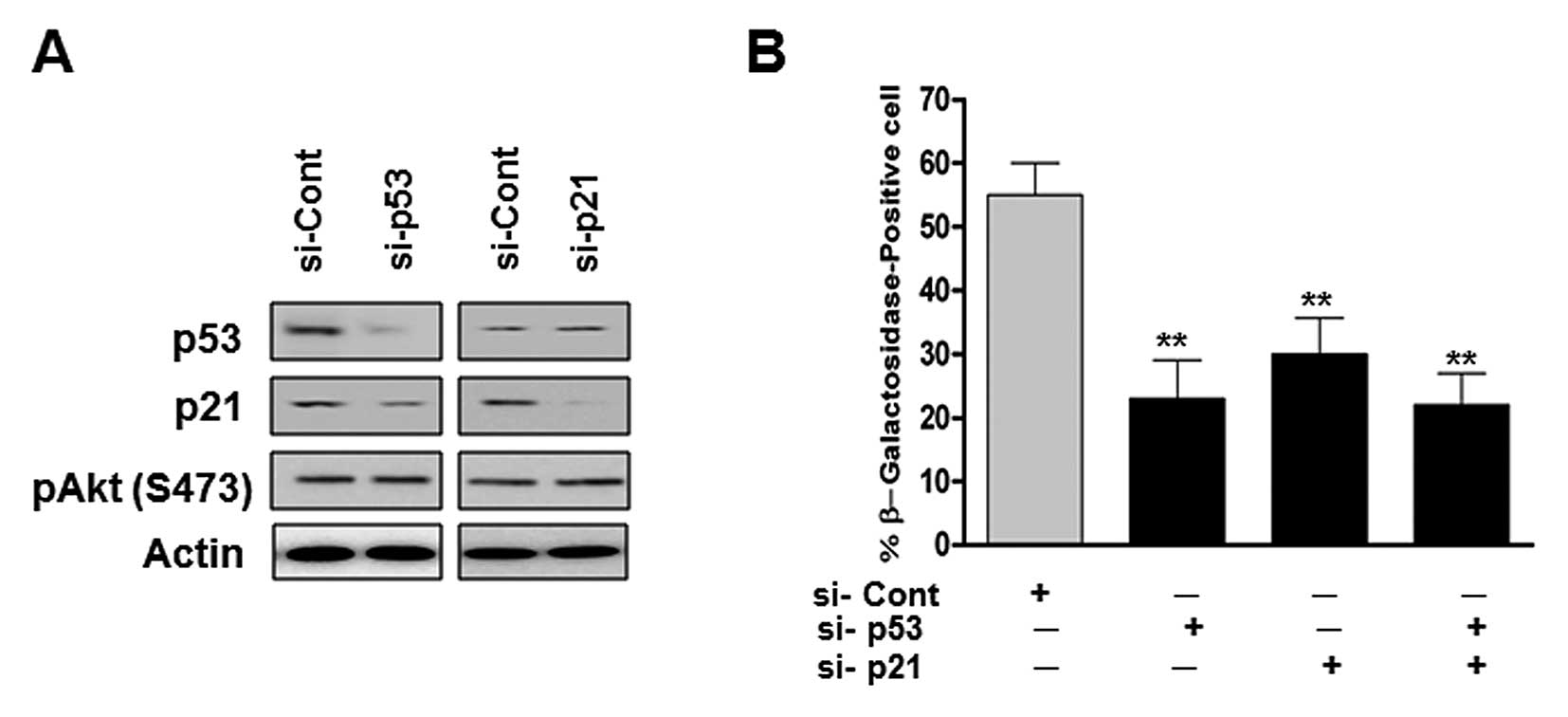
Role of the p53/p21 pathway in the
senescence of human glioma cells treated with 20(S)-Rg3
The p53/p21 signaling pathway is a major
senescence-triggering pathway in response to various stresses. As
shown in Fig. 1F, the treatment of
U87 cells with 20(S)-Rg3 significantly increased the expression
levels of p53 and p21. Therefore, we examined whether
20(S)-Rg3-mediated senescence requires signaling of the p53/p21
pathway in U87 cells. We transfected U87 cells with p53 and/or p21
si-RNA and looked for senescent phenotypes. As shown in Fig. 4B, p53 siRNA transfected cells
showed a significant decrease in SA-β-Gal activity. Cells
transfected with p21 siRNA or co-transfected p53 siRNA showed
similar results with p53 depleted cells (Fig. 4B). These data suggest that the
p53/p21 plays an essential role in the maintenance of senescence
triggered by 20(S)-Rg3.
Discussion
The induction of apoptosis was previously defined as
the mechanism of 20(S)-Rg3 action for the growth arrest of various
tumor cells (9,10,12).
In agreement with these reports, our data showed that a high
concentration of 20(S)-Rg3 could induce apoptosis in glioma cells.
Here, we showed that 20(S)-Rg3 also induces cellular senescence
after prolonged treatment at sub-lethal drug concentrations.
Cellular senescence, along with mitotic catastrophe
and apoptosis, has been proposed as one of the mechanisms mediating
the antitumor effects of chemotherapies (2–5).
Tumor cells were considered to have lost the ability to senesce, as
cellular senescence would provide an important barrier to
tumorigenesis (17,18). Previous data have shown that cancer
cell senescence can be induced by treatment with chemotherapeutic
drugs, radiation, genetic manipulation, or other agents (19). The induction of senescence by
chemotherapeutic drugs was dose-dependent and correlated with the
growth arrest observed in various cells (2,20–22).
In contrast to cell death, however, senescence leaves tumor cells
alive and physiologically active (3). In human tumor cell lines treated
in vitro and in vivo with differentiating agents,
terminal proliferative arrest with minimal toxicity to normal cells
has been observed (1,23). Therefore, understanding the
mechanism of premature senescence is crucial for the development of
safer and more effective cancer treatment strategies.
Gain-of-function mutations in the PI3K/Akt signaling
pathway are frequently found in human glioblastomas (14). In mammalian cells, the activation
of Akt has been reported to induce proliferation and survival,
thereby promoting tumorigenesis (24–26).
However, previous studies demonstrated that the constitutive
activation of Akt promotes senescence-like arrest of cell growth
through the regulation of intracellular ROS levels (15,16,27,28).
Thus, Akt-induced growth arrest may be another antitumorigenic
mechanism, similar to Ras-induced senescence (29,30).
In this study, we revealed that 20(S)-Rg3-induced senescence is
associated with Akt activation, and depletion of Akt partially
prevented Rg3-induced ROS generation and senescence induction in
glioma cells. Elevated intracellular ROS levels have been widely
accepted as triggers of cellular senescence (31–33).
Senescent cells increase ROS generation (34,35),
and premature cellular senescence can be readily induced by
sub-lethal doses of pro-oxidants (36,37).
In the present study, ROS levels were elevated during
20(S)-Rg3-induced senescence, and the ROS scavenger NAC reverted
20(S)-Rg3-induced senescent phenotypes. However, decreases in
intracellular ROS levels by NAC did not affect 20(S)-Rg3-induced
Akt activation (data not shown). These data suggest that 20(S)-Rg3
induces senescence by a mechanism largely dependent on Akt/ROS
signaling. Furthermore, 20(S)-Rg3-induced arrest was accompanied by
substantial increases in p53 and p21, and the depletion of p53 or
p21 prevented 20(S)-Rg3-induced premature senescence in U87 cells.
Thus, senescence may occur in a manner dependent on the p53/p21
pathway. However, further studies are required to determine the
exact mechanism involved.
In summary, our study presented a new anticancer
mechanism of 20(S)-Rg3 action by inducing senescence. Chronic
treatment with 20(S)-Rg3 at a sub-apoptotic concentration elicits
ROS generation via Akt activation and p53/p21-dependent
senescence-like growth arrest in glioma cells. Our study provides
useful insight for the future development of 20(S)-Rg3 as a novel
class of anticancer agents.
Acknowledgements
This project was supported by a
research grant from the Soram Cancer and Immunotherapy Research
Center (Republic of Korea).
References
|
1.
|
Vergel M, Marin JJ, Estevez P and Carnero
A: Cellular senescence as a target in cancer control. J Aging Res.
2011:7253652010.PubMed/NCBI
|
|
2.
|
Schmitt CA: Senescence, apoptosis and
therapy – cutting the lifelines of cancer. Nat Rev Cancer.
3:286–295. 2003.
|
|
3.
|
Shay JW and Roninson IB: Hallmarks of
senescence in carcinogenesis and cancer therapy. Oncogene.
23:2919–2933. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Hornsby PJ: Senescence as an anticancer
mechanism. J Clin Oncol. 25:1852–1857. 2007. View Article : Google Scholar
|
|
5.
|
Ventura A, Kirsch DG, McLaughlin ME, et
al: Restoration of p53 function leads to tumour regression in vivo.
Nature. 445:661–665. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Ohgaki H and Kleihues P: Genetic pathways
to primary and secondary glioblastoma. Am J Pathol. 170:1445–1453.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Kitagawa I, Yoshikawa M, Yoshihara M,
Hayashi T and Taniyama T: Chemical studies of crude drugs (1).
Constituents of Ginseng radix rubra. Yakugaku Zasshi. 103:612–622.
1983.(In Japanese).
|
|
8.
|
Helms S: Cancer prevention and
therapeutics: Panax ginseng. Altern Med Rev. 9:259–274.
2004.PubMed/NCBI
|
|
9.
|
Yuan HD, Quan HY, Zhang Y, Kim SH and
Chung SH: 20(S)-Ginsenoside Rg3-induced apoptosis in HT-29 colon
cancer cells is associated with AMPK signaling pathway. Mol Med
Rep. 3:825–831. 2010.PubMed/NCBI
|
|
10.
|
Wang BS, Zhang LS, Song DM, Zhang JH and
Liu YM: Effect of gensenoside Rg3 on apoptosis of Hep-2 and
expression of HIF-1alha in human laryngeal cancer cell line under
anoxic conditions. Zhong Yao Cai. 32:102–106. 2009.(In
Chinese).
|
|
11.
|
Kim HS, Lee EH, Ko SR, Choi KJ, Park JH
and Im DS: Effects of ginsenosides Rg3 and Rh2 on the proliferation
of prostate cancer cells. Arch Pharm Res. 27:429–435. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Jian J, Hu ZF and Huang Y: Effect of
ginsenoside Rg3 on Pim-3 and Bad proteins in human pancreatic
cancer cell line PANC-1. Ai Zheng. 28:461–465. 2009.(In
Chinese).
|
|
13.
|
Dimri GP, Lee X, Basile G, et al: A
biomarker that identifies senescent human cells in culture and in
aging skin in vivo. Proc Natl Acad Sci USA. 92:9363–9367. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Solomon DA, Kim JS, Jenkins S, et al:
Identification of p18 INK4c as a tumor suppressor gene in
glioblastoma multiforme. Cancer Res. 68:2564–2569. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Nogueira V, Park Y, Chen CC, et al: Akt
determines replicative senescence and oxidative or oncogenic
premature senescence and sensitizes cells to oxidative apoptosis.
Cancer Cell. 14:458–470. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Dolado I and Nebreda AR: AKT and oxidative
stress team up to kill cancer cells. Cancer Cell. 14:427–429. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Campisi J and d’Adda di Fagagna F:
Cellular senescence: when bad things happen to good cells. Nat Rev
Mol Cell Biol. 8:729–740. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Schmitt CA: Cellular senescence and cancer
treatment. Biochim Biophys Acta. 1775:5–20. 2007.PubMed/NCBI
|
|
19.
|
Roninson IB: Tumor cell senescence in
cancer treatment. Cancer Res. 63:2705–2715. 2003.PubMed/NCBI
|
|
20.
|
te Poele RH, Okorokov AL, Jardine L,
Cummings J and Joel SP: DNA damage is able to induce senescence in
tumor cells in vitro and in vivo. Cancer Res. 62:1876–1883.
2002.PubMed/NCBI
|
|
21.
|
Nardella C, Clohessy JG, Alimonti A and
Pandolfi PP: Pro-senescence therapy for cancer treatment. Nat Rev
Cancer. 11:503–511. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Lleonart ME, Artero-Castro A and Kondoh H:
Senescence induction; a possible cancer therapy. Mol Cancer.
8:32009. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Chang BD, Xuan Y, Broude EV, et al: Role
of p53 and p21waf1/ cip1 in senescence-like terminal proliferation
arrest induced in human tumor cells by chemotherapeutic drugs.
Oncogene. 18:4808–4818. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Datta SR, Brunet A and Greenberg ME:
Cellular survival: a play in three Akts. Genes Dev. 13:2905–2927.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Blume-Jensen P and Hunter T: Oncogenic
kinase signalling. Nature. 411:355–365. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Testa JR and Bellacosa A: AKT plays a
central role in tumorigenesis. Proc Natl Acad Sci USA.
98:10983–10985. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Miyauchi H, Minamino T, Tateno K, Kunieda
T, Toko H and Komuro I: Akt negatively regulates the in vitro
lifespan of human endothelial cells via a p53/p21-dependent
pathway. EMBO J. 23:212–220. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Lee JJ, Kim BC, Park MJ, et al: PTEN
status switches cell fate between premature senescence and
apoptosis in glioma exposed to ionizing radiation. Cell Death
Differ. 18:666–677. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Serrano M, Lin AW, McCurrach ME, Beach D
and Lowe SW: Oncogenic ras provokes premature cell senescence
associated with accumulation of p53 and p16INK4a. Cell. 88:593–602.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Wright WE and Shay JW: Cellular senescence
as a tumor-protection mechanism: the essential role of counting.
Curr Opin Genet Dev. 11:98–103. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Passos JF and Von Zglinicki T: Oxygen free
radicals in cell senescence: are they signal transducers? Free
Radic Res. 40:1277–1283. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Beckman KB and Ames BN: The free radical
theory of aging matures. Physiol Rev. 78:547–581. 1998.PubMed/NCBI
|
|
33.
|
Ben-Porath I and Weinberg RA: The signals
and pathways activating cellular senescence. Int J Biochem Cell
Biol. 37:961–976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Hagen TM, Yowe DL, Bartholomew JC, et al:
Mitochondrial decay in hepatocytes from old rats: membrane
potential declines, heterogeneity and oxidants increase. Proc Natl
Acad Sci USA. 94:3064–3069. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Lee AC, Fenster BE, Ito H, et al: Ras
proteins induce senescence by altering the intracellular levels of
reactive oxygen species. J Biol Chem. 274:7936–7940. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Chen Q and Ames BN: Senescence-like growth
arrest induced by hydrogen peroxide in human diploid fibroblast F65
cells. Proc Natl Acad Sci USA. 91:4130–4134. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Chen QM, Bartholomew JC, Campisi J, Acosta
M, Reagan JD and Ames BN: Molecular analysis of H2O2-induced
senescent-like growth arrest in normal human fibroblasts: p53 and
Rb control G1 arrest but not cell replication. Biochem J.
332:43–50. 1998.PubMed/NCBI
|