Introduction
UV irradiation is a major environmental stressor
that results in altered cutaneous functions: for example, induction
of inflammation, dyspigmentation, premature aging, skin cancer, and
attenuation of barrier function (1). The terrestrial solar UV spectrum can
be divided into UVA (320–400 nm), UVB (280–320 nm), and UVC
(200–280 nm). Wavelengths in the UVB region are absorbed into the
epidermis of the skin and cause skin disorders such as the
formation of sunburn cells (apoptotic cells) and skin cancer
(2,3).
UVB causes DNA damage and apoptosis of epidermal
cells, and the signaling pathways associated with UVB-induced cell
death have been extensively investigated (3). However, the cellular self-protection
system in which epidermal cells respond to UVB is unclear.
Autophagy is an intracellular degrading and recycling process by
which organelles, cytoplasmic components, and invading pathogens
are delivered to lysosomes for degradation (4,5).
This process recycles cellular components and produces building
blocks under stress conditions/nutrient starvation; it also
eliminates damaged proteins/organelles (4–6).
Therefore, autophagy has been proposed to be a cellular
self-protection process in response to stress conditions (7,8).
However, some studies suggest that autophagy may contribute to
cellular damage (7).
In this study, we examined the effect of UVB on
autophagy in a mouse epidermal cell line (JB6). JB6 cells have been
extensively used to study the mechanisms of epidermal cell
transformation (9–11). UVB causes the death of JB6 cells in
the form of apoptosis (12). We
demonstrated that UVB activated autophagy in JB6 cells, and
autophagy appeared to be a protective response to UVB-induced
damage. Furthermore, we showed that GSK3β negatively regulated
autophagy and inhibition of GSK3β also offered protection against
UVB-induced cell death.
Materials and methods
Materials
LiCl, bafilomycin A1, wortmannin, rapamycin,
3-methyladenine (3-MA) and anti-actin antibody were purchased from
Sigma (St. Louis, MO, USA). Anti-beclin 1 antibody was purchased
from Abcam (Cambridge, MA, USA). Anti-LC3 antibody was purchased
from Medical and Biological Laboratories (Nagoya, Japan). The
antibodies directed against GSK3β, phospho-GSK3β, AMPK,
phospho-AMPK and p62 were obtained from Cell Signaling Technology
(Beverly, MA, USA).
Cell culture and UVB irradiation
JB6 mouse epidermal cells (CI 41) were cultured in
EMEM supplemented with 10% fetal bovine serum (FBS), 2 mM
L-glutamine, 25 μg/ml gentamicin, 100 U/ml penicillin and 100 μg/ml
streptomycin at 37°C with 5% CO2. The establishment of
JB6 cell lines stably expressing wild-type GSK3β (WT),
constitutively active GSK3β (S9A), and dominant-negative GSK3β
(K85R) have been previously described (10). The cells were irradiated with a UVB
lamp (UVP, 0–400 mJ/cm2) as previously described
(13) and then incubated at 37°C
for the indicated time.
Detection of LC3 puncta
GFP-LC3 plasmid was a generous gift from Dr
Gutterman (University of Texas MD Anderson Cancer Center, Houston,
TX). Transfections were performed using Lipofectamine™ 2000
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s
protocol. At 24 h after transfection, cells were exposed to UVB
irradiation and were examined by a fluorescence microscope at
indicated times. The GFP-LC3 puncta/cell were quantified as
previously described (14,15). For each group, twenty cells in
randomly selected visual fields were counted. The experiment was
replicated three times.
Determination of cell viability
Cells were exposed to UVB irradiation at indicated
dosages and incubated for 24 h. Cell viability was determined by
MTT assay as previously described (16).
Immunoblotting
The procedure for immunoblotting has been previously
described (10). Briefly, cells
were washed twice with PBS and lysed with RIPA buffer [150 mM NaCl,
0.1% sodium dodecyl sulfate (SDS), 50 mM Tris (pH 8.0), 0.5%
deoxycholic acid sodium, 1% Nonidet P-40 (NP-40), 0.1 mg/ml
phenylmethylsulfonyl fluoride, 3% aprotinin, and 1 mM sodium
orthovanadate] on ice for 10 min. Cell lysates were centrifuged at
12,000 rpm at 4°C for 10 min. The supernatant was then collected
and the protein concentration was measured with a protein assay kit
(Bio-Rad Laboratories, Hercules, CA, USA). An aliquot of the total
protein (40 μg) was loaded into each lane of an SDS-polyacrylamide
gel. The protein was electrophoretically transferred to
nitrocellulose membranes and blocked with 5% BSA in 0.01 M TBS (pH
7.4) and 0.05% Tween-20 (TBST) at room temperature for 1 h. The
blots were probed with primary antibodies for 2 h at room
temperature or overnight at 4°C. After three quick washes with
TBST, the membranes were incubated with horseradish
peroxidase-conjugated goat anti-rabbit or goat anti-mouse IgG
(Amersham, Arlington Heights, IL, USA) for 1 h and the bands were
visualized with the enhanced chemiluminescence method
(Amersham).
Statistical analysis
All the data are expressed as the mean ± SD from at
least three independent experiments. The statistical analysis was
performed using the analysis of variance (ANOVA) followed by post
hoc analyses. A value of p<0.05 was considered statistically
significant.
Results
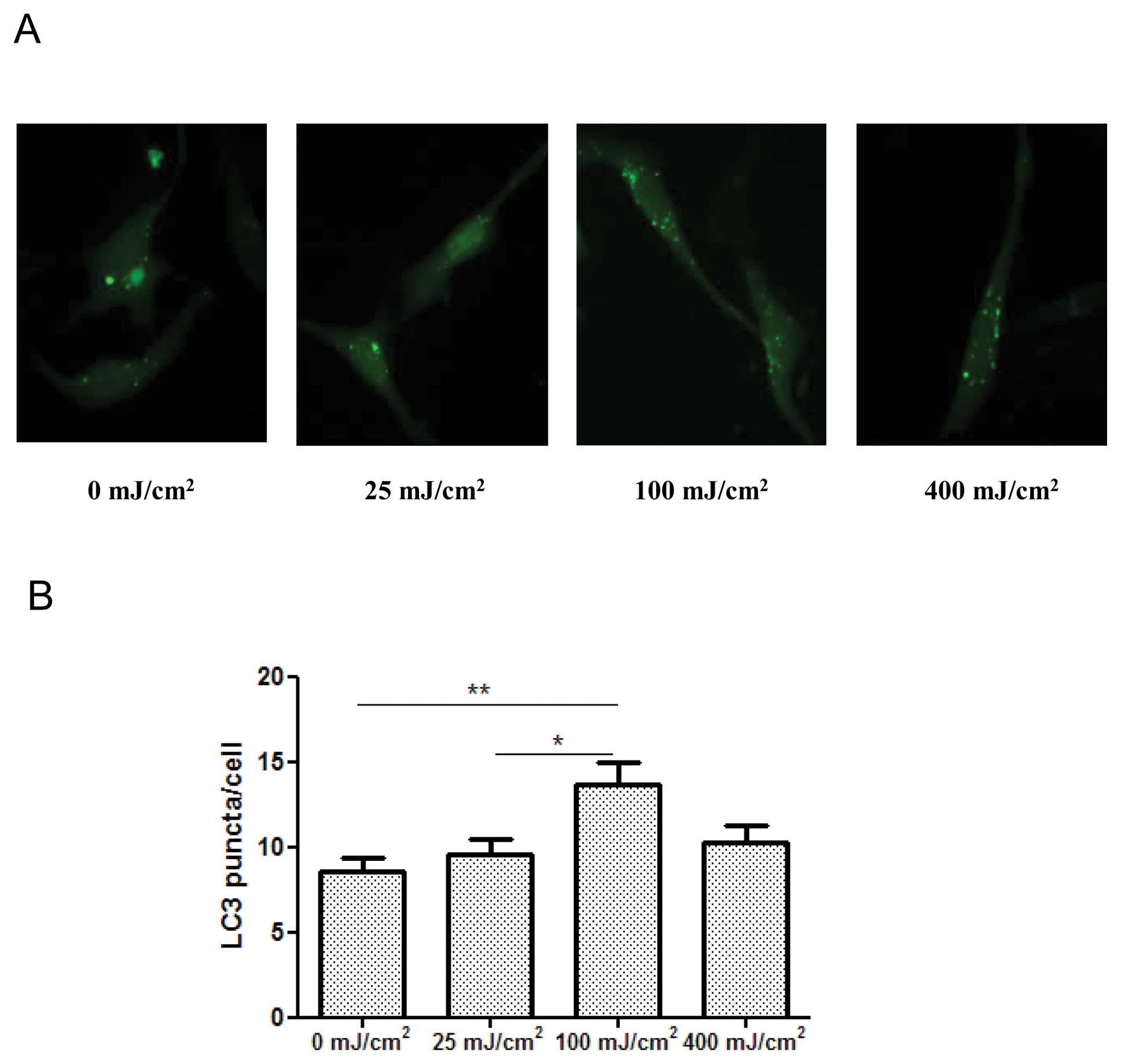
UVB irradiation activates autophagy
First, we examined the effect of UVB irradiation on
autophagy in cultured JB6 mouse epidermal cells. The synthesis and
conversion of LC3 is connected closely with the level of autophagy,
making it a key marker in cells (17). The formation of LC3 puncta is an
important indication of autophagy. We transfected JB6 cells with a
GFP-LC3 plasmid, then examined the distribution and amount of green
fluorescence LC3 puncta. We determined the effect of UVB
irradiation at 0, 25, 100, 400 mJ/cm2 after 24 h of
exposure. As shown in Fig. 1, a
significant increase of LC3-GFP puncta formation was observed after
UVB exposure at 100 mJ/cm2. UVB irradiation at 25 or 400
mJ/cm2 did not alter LC3 puncta. LC3 lipidation, which
is indicated by the formation of LC3-II, is an index of autophagy.
We demonstrated that UVB irradiation (100 mJ/cm2)
increased the level of LC3-II (induction of LC3 lipidation) in JB6
cells in a time-dependent manner (Fig.
1C). Meanwhile, the expression of beclin 1 was upregulated, but
the level of p62 was downregulated (Fig. 1C). Beclin 1, also known as Atg6, is
a protein required for the formation of the initial autophagic
structure, while p62 is regulated by autophagy-dependent
degradation. Together, these results suggested that UVB irradiation
activated autophagy.
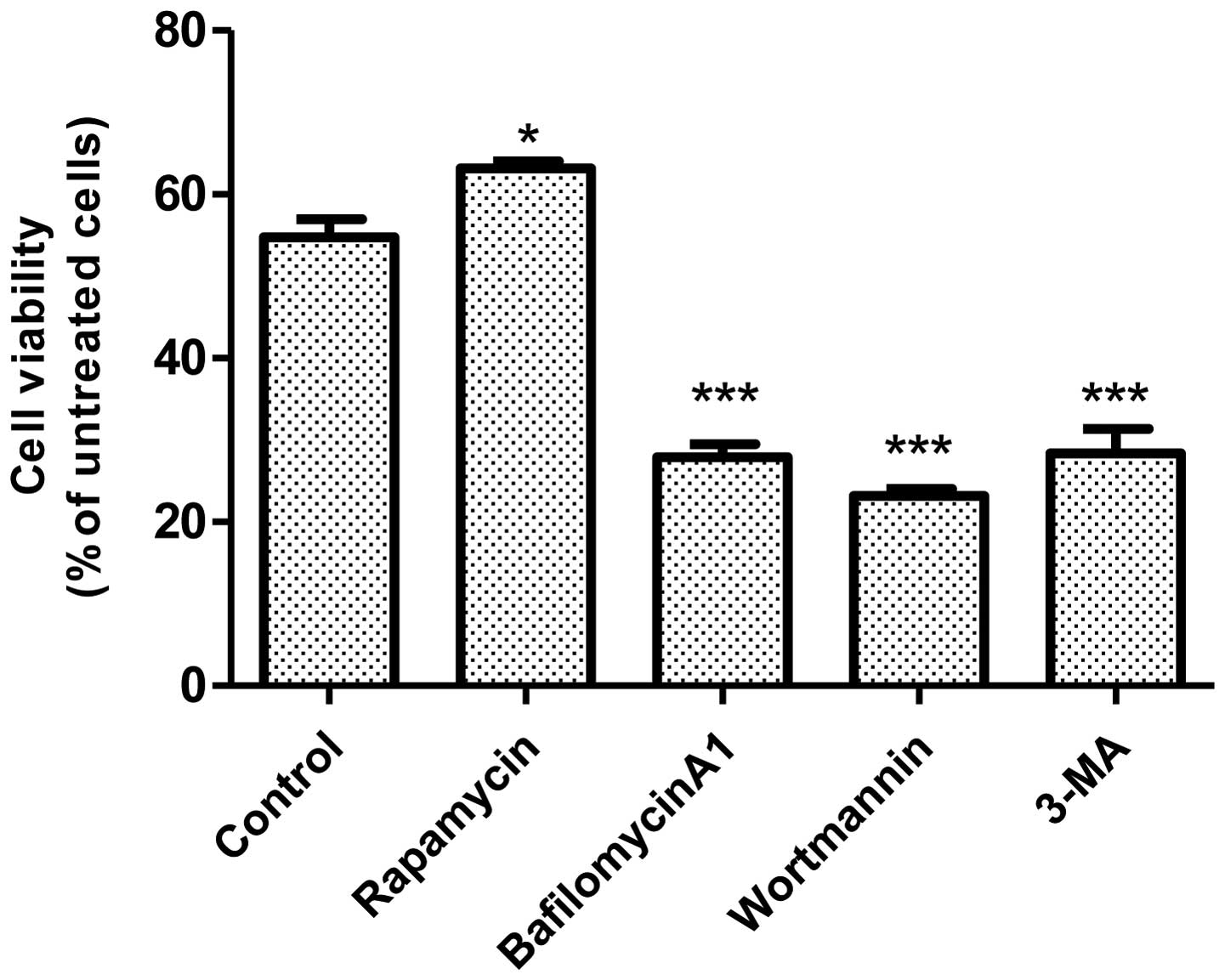
We next sought to determine the role of autophagy in
UVB irradiation-induced cell death. As shown in Fig. 2, activation of autophagy by
rapamycin offered protection, whereas inhibition of autophagy by
bafilomycin A1, wortmannin, or 3-MA exacerbated UVB
irradiation-induced cell death. These results suggested that
autophagy was a protective response to UVB irradiation-induced
damage.
The involvement of GSK3β in UVB-activated
autophagy
The activity of GSK3β is negatively regulated by the
phosphorylation at Ser9, but positively at
Tyr216(18). We showed
that UVB irradiation increased the level of pGSK3β
(Ser9) and decreased pGSK3β (Tyr216),
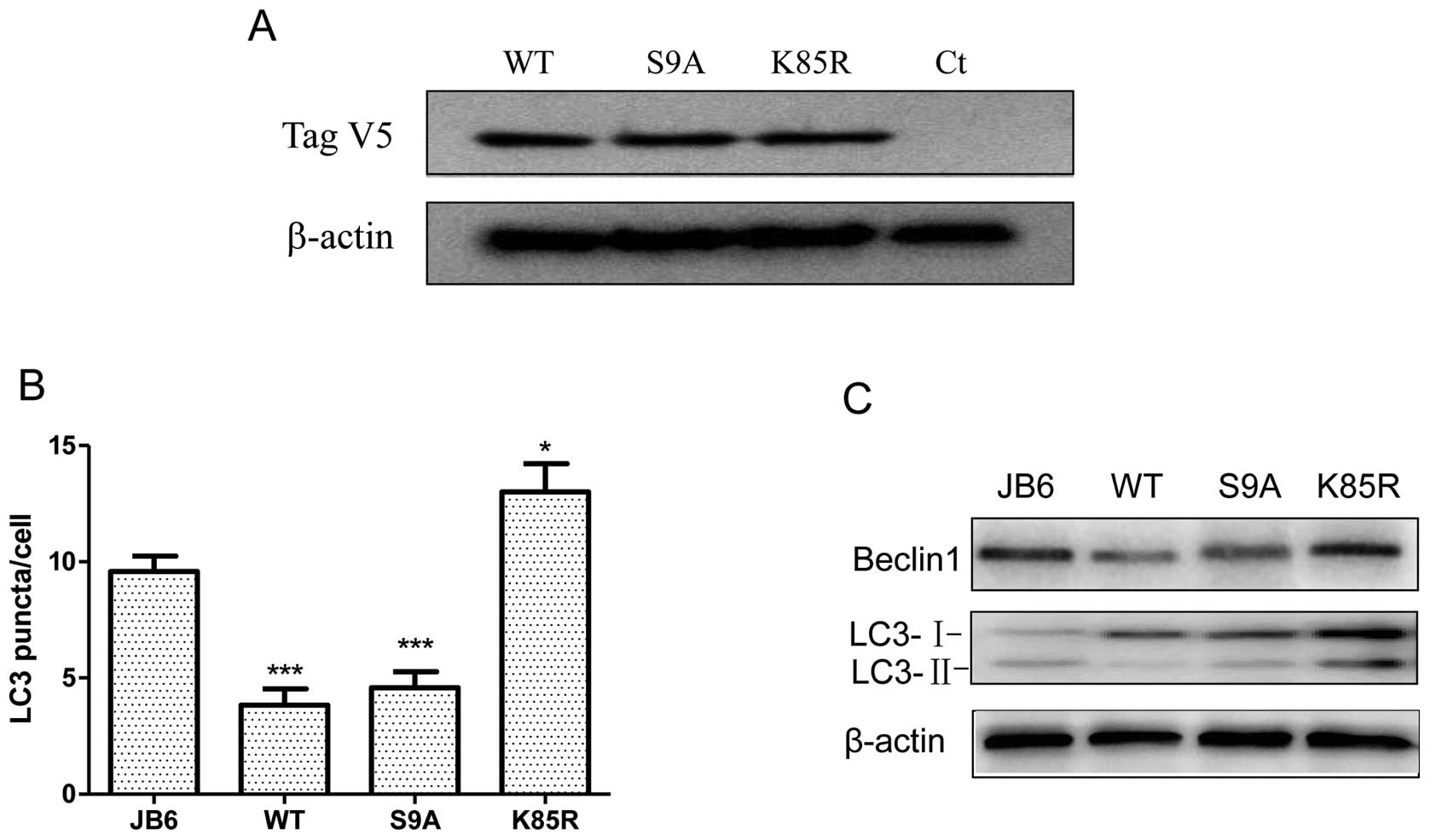
indicating it inhibited GSK3β activity (Fig. 1C). To evaluate the role of GSK3β in
UVB-induced autophagy, we established JB6 cells stably expressing
various GSK3β mutants. These constructs included wild-type (WT),
constitutive-active (S9A) and dominant-negative (K85R) GSK3β. S9A
mutant is resistant to inhibitory regulation by restraining
phosphorylation at Ser9; K85R mutant represents a
deficit kinase and functions as a dominant-negative protein. We
have previously shown they effectively stimulated or inhibited
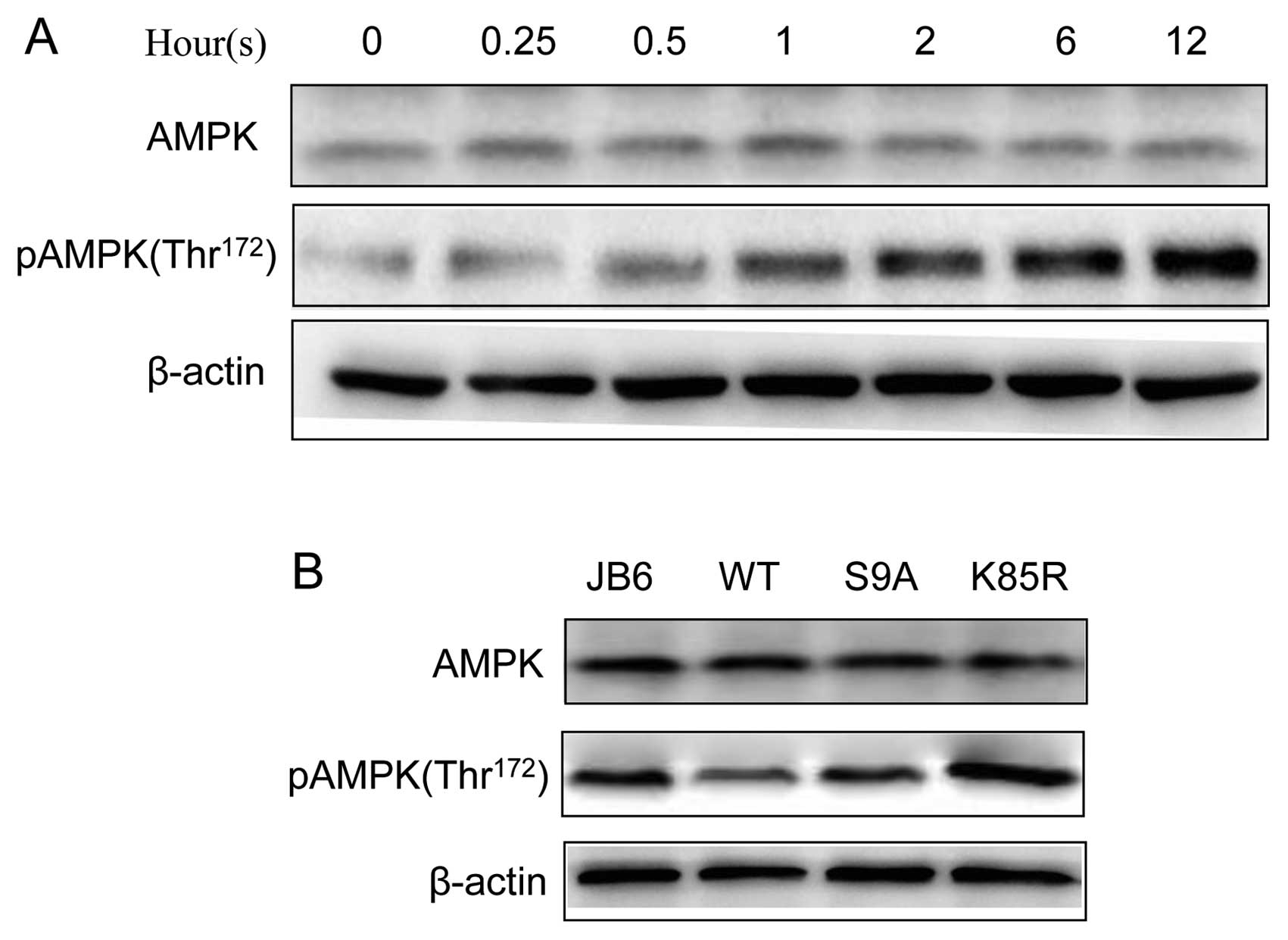
GSK3β activity, respectively (10). Overexpression of these exogenous
GSK3β proteins was verified by the expression of a V5 tag by
immunoblotting (Fig. 3A). We
demonstrated that manipulation of GSK3β activity altered
UVB-mediated autophagy. Upon UVB exposure (100 mJ/cm2),
overexpression of WT and S9A GSK3β significantly decreased the
number of LC3 puncta, whereas K85R GSK3β increased the amount of
LC3 puncta (Fig. 3B). Consistent
with this result, overexpression of WT and S9A GSK3β decreased the
level of LC3-II and beclin 1; overexpression of K85R increased the
expression of LC3-II and beclin 1. Together, these data suggested
that inhibition of GSK3β resulted in an increase in autophagy. In
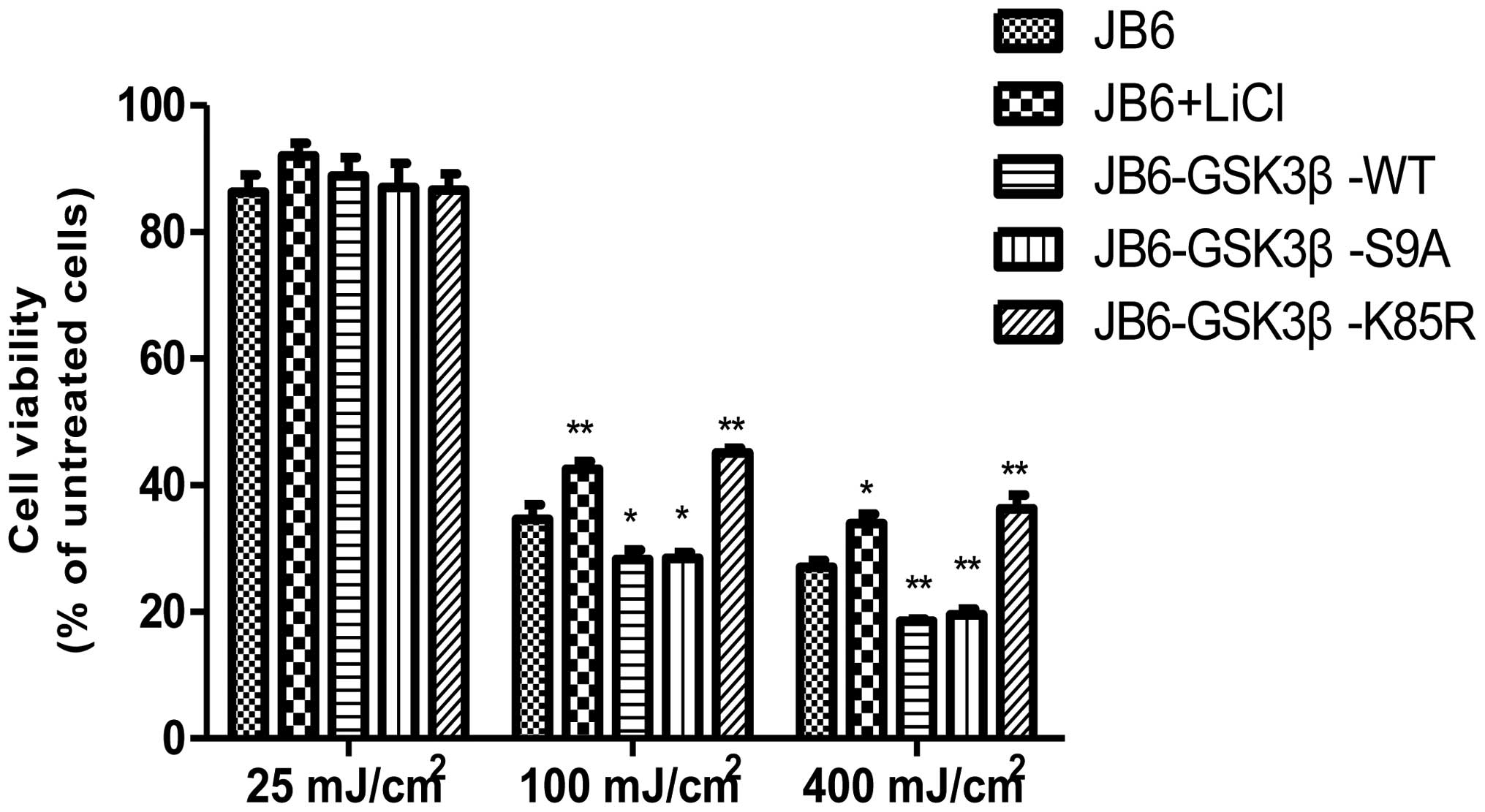
parallel, overexpression of WT and S9A GSK3β exacerbated
UVB-induced cell death, whereas overexpression of K85R GSK3β
protected cells against UVB-induced cell death (Fig. 4). Protection mediated by the
inhibition of GSK3β was further supported by a study using lithium
treatment. Lithium, an inhibitor of GSK3β, reduced UVB-mediated
cell death (Fig. 4).
Interaction between AMPK and GSK3β in
response to UVB
AMPK is a critical regulator of autophagy and
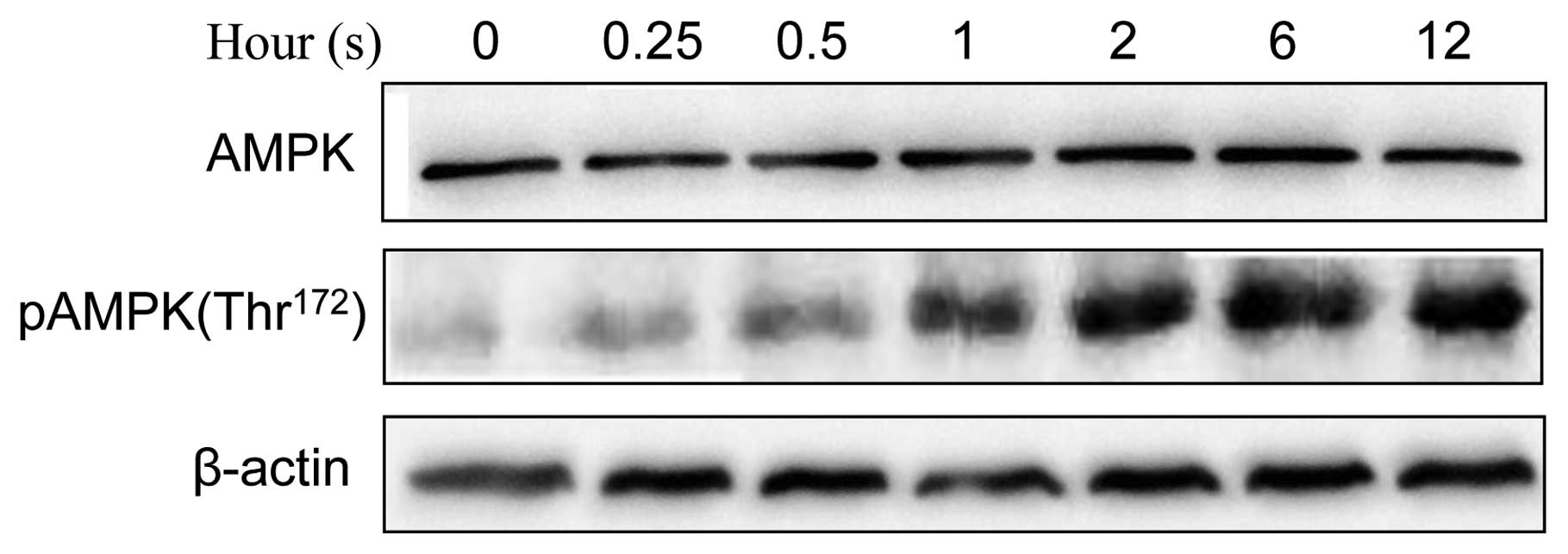
activation of AMPK results in enhanced autophagy (19). We confirmed that UVB induced the
phosphorylation of AMPK (Thr172) in JB6 cells (Fig. 5). We further investigated the
relationship between AMPK and GSK3β. Inhibition of GSK3β by lithium
was sufficient to activate AMPK (Fig.
6A). Furthermore, overexpression of dominant-negative K85R
GSK3β enhanced UVB-stimulated phosphorylation of AMPK (pAMPK),
whereas overexpression of S9A or WT GSK3β inhibited UVB-mediated
pAMPK (Fig. 6B). These results
suggested that GSK3β negatively regulated AMPK phosphorylation.
Discussion
In this study, we demonstrate for the first time
that UVB can activate autophagy in epidermal cells, and autophagy
appears to be a cytoprotective response to UVB-mediated damage.
UVB irradiation causes DNA damage and apoptosis of
epidermal cells, contributing to human skin cancer in the process
of tumor initiator and promoter (20,21).
Mouse JB6 cells are a well-established in vitro model to
study UVB-mediated damage and transformation of epidermal cells
(22–24). Using this model, we demonstrate
that UVB-induced reduction in the viability of JB6 cells is
accompanied by the increase of autophagy which is evident by the
formation of LC3 puncta, induction of LC3 lipidation, increase in
beclin 1 expression, and decrease in the level of p62. Inhibition
of autophagy by bafilomycin A1, wortmannin, or 3-MA exacerbates
UVB-induced cell death. In contrast, activation of autophagy by
rapamycin protects JB6 cells against UVB-mediated damage. This
finding is consistent with a previous study showing that UV
irradiation induced autophagy in A549 and H1299 cells (25,26).
In that study, autophagy also seemed to be cytoprotective, and
inhibition of autophagy exacerbated UV-triggered apoptotic cell
death in these cells (26).
Similarly, autophagy was shown to be cytoprotective against
apoptosis induced by DNA-damaging agents (25).
It is interesting to note that UVB induces autophagy
in a dose-dependent manner. At a low dosage, such as 25
mJ/cm2, UVB does not affect cell viability and
autophagy. At 100 mJ/cm2, it causes cell death and
activates autophagy. However, at a higher dosage, 400
mJ/cm2, it produces more cell death, but fails to
activate autophagy (Fig. 1). It is
likely that at a high dosage, UVB impairs autophagic machineries.
This possibility remains to be investigated.
Another important finding for this study is that
glycogen synthase kinase 3β (GSK3β) is involved in UVB-induced
autophagy. GSK3β, a serine/threonine protein kinase, which was
first described in glycogen metabolism and insulin signaling
(27,28), is involved in multiple biological
events such as embryonic development, stem cell survival,
differentiation, neurodegeneration, tumorigenesis, and cell death
(18,29,30).
We have previously shown that inhibition of GSK3β promotes the
transformation of epidermal cells (10). GSK3β activity is regulated by
site-specific phosphorylation. The activity of GSK3β is upregulated
by phosphorylation on the Tyr216 residue, and
conversely, phosphorylation on Ser9 inhibits GSK3β
activity. Phosphorylation of Ser9 is mediated by a
number of signaling pathways, such as PI3K/AKT, PKC, MAPK/p90RS, or
mTOR/p70S6 (18,31). The mechanism for the regulation of
phosphorylation at Tyr216 is less clear. We demonstrate
that UVB increases GSK3β phosphorylation at Ser9 but
inhibits its phosphorylation at Tyr216, indicating that
UVB inhibits GSK3β activity. UVB is shown to activate MAPK, PKC,
and PI3K/AKT signaling pathways (32). It is therefore likely that
UVB-induced phosphorylation of Ser9 is mediated by one
or some of these pathways. Regardless of the mechanisms in which
UVB inhibits GSK3β, it is likely that UVB activates autophagy
through the inhibition of GSK3β because dominant-negative GSK3β
enhances UVB-induced autophagy, whereas overexpression of GSK3β
inhibits UVB-induced autophagy (Fig.
3). These results suggest that GSK3β negatively regulates
autophagy and UVB may affect autophagy by modulating GSK3β
activity.
AMP-activated protein kinase (AMPK), a crucial
stress-sensing enzyme, is activated by a rise in the cellular
AMP/ATP ratio. AMPK is an important mediator of autophagy (19). It has been demonstrated that
activation of AMPK results in autophagy in human keratinocytes
(33). Cadmium-induced activation
of AMPK causes autophagy in JB6 cells (34). UV irradiation can regulate AMPK
activity. For example, UVB is reported to activate AMPK in murine
basal cell carcinoma and skin keratinocytes (35,36).
UVC is shown to activate AMPK in pancreatic cancer cells (37). However, Zhang and Bowden (38) suggest that UVB inhibits AMPK in
human keratinocytes. We demonstrate here that UVB activates AMPK in
JB6 cells, and therefore UVB-mediated autophagy is regulated by
AMPK (Fig. 5).
There is considerable interaction between GSK3β and
AMPK (39–42). The interaction appears to be
two-way; that is, AMPK can affect GSK3β activity and is also
regulated by GSK3β. In our system, it appears that inhibition of
GSK3β results in AMPK activation (Fig.
6). More importantly, inhibition of GSK3β by a
dominant-negative GSK3β potentiates UVB-mediated AMPK activation,
while overexpression of GSK3β inhibits UVB-induced AMPK activation.
This is consistent with the effect of UVB/GSK3β on autophagy and
supports a role of AMPK in UVB-mediated autophagy.
UVB causes apoptosis in JB6 cells through complex
mechanisms; it may be mediated by oxidative stress, PKC, or a
p53-dependent manner (12,43,44).
The signaling pathways for UVB-mediated autophagy and apoptosis may
or may not overlap. Our study indicates the GSK3β/AMPK pathway
contributes to UVB-mediated autophagy. The interaction or
cross-talk between the pathways governing autophagy and apoptosis
in response to UVB remain to be studied.
Acknowledgements
This study is supported by grants from
the National Institutes of Health (NIH) of the United States
(AA017226) and the National Natural Science Foundation of China
(NSFC 81071305).
References
|
1.
|
N PustisekM SitumUV-radiation, apoptosis
and skinColl Antropol352392412011
|
|
2.
|
Y MatsumuraHN AnanthaswamyToxic effects of
ultra-violet radiation on the skinToxicol Appl
Pharmacol195298308200410.1016/j.taap.2003.08.019
|
|
3.
|
S LippensE HosteP VandenabeeleP AgostinisW
DeclercqCell death in the
skinApoptosis14549569200910.1007/s10495-009-0324-z
|
|
4.
|
S AlersAS LöfflerS WesselborgB StorkThe
incredible ULKsCell Commun Signal107201210.1186/1478-811X-10-7
|
|
5.
|
S PalumboS CominciniAutophagy and ionizing
radiation in tumors: the ‘Survive or not Survive’ dilemmaJ Cell
PhysiolMay142012(Epub ahead of print).
|
|
6.
|
N MizushimaM KomatsuAutophagy: renovation
of cells and
tissuesCell147728741201110.1016/j.cell.2011.10.02622078875
|
|
7.
|
N ChenV Karantza-WadsworthRole and
regulation of autophagy in cancerBiochim Biophys
Acta179315161523200910.1016/j.bbamcr.2008.12.01319167434
|
|
8.
|
J Martínez-BorraC López-LarreaAutophagy
and self-defenseAdv Exp Med Biol7381691842012
|
|
9.
|
Z DongMJ BirrerRG WattsLM MatrisianNH
ColburnBlocking of tumor promoter-induced AP-1 activity inhibits
induced transformation in JB6 mouse epidermal cellsProc Natl Acad
Sci USA91609613199410.1073/pnas.91.2.6098290571
|
|
10.
|
C MaJ WangY GaoTW GaoG ChenKA BowerM
OdetallahM DingZ KeJ LuoThe role of glycogen synthase kinase 3beta
in the transformation of epidermal cellsCancer
Res6777567764200710.1158/0008-5472.CAN-06-466517699780
|
|
11.
|
D ZhangJ LiJ GaoC Huangc-Jun/AP-1
pathway-mediated cyclin D1 expression participates in low dose
arsenite-induced transformation in mouse epidermal JB6 C141
cellsToxical Appl
Pharmacol2351824200910.1016/j.taap.2008.11.00219059425
|
|
12.
|
YK WonCN OngHM ShenParthenolide sensitizes
ultraviolet (UV)-B-induced apoptosis via protein kinase C-dependent
pathwaysCarcinogenesis2621492156200510.1093/carcin/bgi19416051639
|
|
13.
|
L SongM GaoW DongM HuJ LiX ShiY HaoY LiC
Huangp85α mediates p53 K370 acetylation by p300 and regulates its
promoter-specific transactivity in the cellular UVB
responseOncogene30136013712011
|
|
14.
|
A AlexanderSL CaiJ KimA NanezM SahinKH
MacleanK InokiKL GuanJ ShenMD PersonD KusewittGB MillsMB KastanCL
WalkerATM signals to TSC2 in the cytoplasm to regulate mTORC1 in
response to ROSProc Natl Acad Sci
USA10741534158201010.1073/pnas.091386010720160076
|
|
15.
|
G ChenZ KeM XuM LiaoX WangJA FrankKA
BowerX ShiJ LuoAutophagy is a protective response to ethanol
neurotoxicityAutophagy(In press).
|
|
16.
|
M XuKA BowerS WangJA FrankG ChenM DingS
WangX ShiZ KeJ LuoCyanidin-3-glucoside inhibits ethanol-induced
invasion of breast cancer cells overexpressing ErbB2Mol
Cancer9285201010.1186/1476-4598-9-28521034468
|
|
17.
|
S BarthD GlickKF MacleodAutophagy: assays
and artifactsJ Pathol221117124201010.1002/path.269420225337
|
|
18.
|
J LuoGlycogen synthase kinase 3beta
(GSK3beta) in tumorigenesis and cancer chemotherapyCancer
Lett273194200200910.1016/j.canlet.2008.05.04518606491
|
|
19.
|
MM MihaylovaRJ ShawThe AMPK signaling
pathway coordinates cell growth, autophagy and metabolismNat Cell
Biol1310161023201110.1038/ncb232921892142
|
|
20.
|
JC EhrhartFP GosseletRM CulerrierA
SarasinUVB-induced mutations in human key gatekeeper genes
governing signalling pathways and consequences for skin
tumourigenesisPhotochem Photobiol
Sci2825834200310.1039/b302281a14521217
|
|
21.
|
Y XuY ShaoJ ZhouJJ VoorheesGJ
FisherUltraviolet irradiation-induces epidermal growth factor
receptor (EGFR) nuclear translocation in human keratinocytesJ Cell
Biochem107873880200910.1002/jcb.2219519415674
|
|
22.
|
C HuangWY MaZ DongThe
extracellular-signal-regulated protein kinases (Erks) are required
for UV-induced AP-1 activation in JB6
cellsOncogene1828282835199910.1038/sj.onc.120263910362253
|
|
23.
|
YK WonCN OngX ShiHM ShenChemopreventive
activity of parthenolide against UVB-induced skin cancer and its
mechanismsCarcinogenesis2514491458200410.1093/carcin/bgh15115033901
|
|
24.
|
S RoyG DeepC AgarwalR AgarwalSilibinin
prevents ultraviolet B radiation-induced epidermal damages in JB6
cells and mouse skin in a p53-GADD45alpha-dependent
mannerCarcinogenesis33629636201210.1093/carcin/bgr29922166495
|
|
25.
|
H Rodriguez-RochaA Garcia-GarciaMI
PanayiotidisR FrancoDNA damage and autophagyMutat
Res711158166201110.1016/j.mrfmmm.2011.03.00721419786
|
|
26.
|
LH ChenPM ChuYJ LeePH TuCW ChiHC LeeSH
ChiouTargeting protective autophagy exacerbates UV-triggered
apoptotic cell deathInt J Mol
Sci1312091224201210.3390/ijms1301120922312313
|
|
27.
|
AV OugolkovDD BilladeauTargeting GSK-3: a
promising approach for cancer therapy?Future
Oncol291100200610.2217/14796694.2.1.9116556076
|
|
28.
|
JE FordeTC DaleGlycogen synthase kinase 3:
a key regulator of cellular fateCell Mol Life
Sci6419301944200710.1007/s00018-007-7045-717530463
|
|
29.
|
JW KimJE LeeMJ KimEG ChoSG ChoEJ
ChoiGlycogen synthase kinase 3 beta is a natural activator of
mitogen-activated protein kinase/extracellular signal-regulated
kinase kinase kinase1(MEKK1)J Biol
Chem2781399514001200310.1074/jbc.M300253200
|
|
30.
|
J LuoGSK3beta in ethanol neurotoxcityMol
Neurobiol40108121200910.1007/s12035-009-8075-y19507062
|
|
31.
|
Q DingX HeW XiaJM HsuCT ChenLY LiDF LeeJY
YangX XieJC LiuMC HungMyeloid cell leukemia-1 inversely correlates
with glycogen synthase kinase-3beta activity and associates with
poor prognosis in human breast cancerCancer
Res6745644571200710.1158/0008-5472.CAN-06-178817495324
|
|
32.
|
M NomuraA KajiWY MaS ZhongG LiuGT BowdenKI
MiyamotoZ DongMitogen- and stress-activated protein kinase 1
mediates activation of Akt by ultraviolet B irradiationJ Biol
Chem2762555825567200110.1074/jbc.M10116420011350959
|
|
33.
|
X TongKA SmithJC PellingApigenin, a
chemopreventive bioflavonoid, induces AMP-activated protein kinase
activation in human keratinocytesMol
Carcinog51268279201210.1002/mc.2079321538580
|
|
34.
|
YO SonX WangJA HitronZ ZhangS ChengA
BudhrajaS DingJC LeeX ShiCadmium induces autophagy through
ROS-dependent activation of the LKB1-AMPK signaling in skin
epidermal cellsToxical Appl
Pharmacol255287296201110.1016/j.taap.2011.06.02421767558
|
|
35.
|
YA ByekovaJL HerrmannJ XuCA ElmetsM
AtharLiver kinase B1 (LKB1) in thepathogenesis of UVB-induced
murine basal cell carcinomaArch Biochem
Biophys508204211201110.1016/j.abb.2011.01.00621272562
|
|
36.
|
C CaoS LuR KivlinB WallinE CardA
BagdasarianT TamakloeWJ WangX SongWM ChuN KouttabA XuY WanSIRT1
confers protection against UVB-and H2O2-induced cell death via
modulation of p53 and JNK in cultured skin keratinocytesJ Cell Mol
Med1336323643200910.1111/j.1582-4934.2008.00453.x18681908
|
|
37.
|
S AdachiI YasudaJ KawaquchiT YamauchiM
NakashimaM ItaniM NakamuraT YoshiokaH MoriwakiO KozawaUltraviolet
enhances the sensitivity of pancreatic cancer cells to gemcitabine
by activation of 5′AMP-activated protein kinaseBiochem Biophys Res
Commun4145359201121945432
|
|
38.
|
J ZhangGT BowdenUVB irradiation regulates
Cox-2 mRNA stability through AMPK and HuR in human keratinocytesMol
Carcinog47974983200810.1002/mc.2045018449856
|
|
39.
|
SM ShinIJ ChoSG KimResveratrol protects
mitochondria against oxidative stress through AMP-activated protein
kinase-mediated glycogen synthase kinase-3beta inhibition
downstream of poly (ADP-ribose) polymerase-LKB1 pathwayMol
Pharmacol76884895200910.1124/mol.109.058479
|
|
40.
|
P de CandiaG MinopoliV VergaA GargiuloM
VanoniL AlberghinaNutritional limitation sensitizes mammalian cells
to GSK-3β inhibitors and leads to growth impairmentAm J
Pathol17818141823201121435461
|
|
41.
|
HD YuanGC PiaoAn active part of
Artemisia sacrorum Ledeb. Suppresses gluconeogenesis through
AMPK mediated GSK3β and CREB phosphorylation in human HepG2
cellsBiosci Biotechnol Biochem7510791084201121670525
|
|
42.
|
HD YuanY Kim doHY QuanSJ KimMS JungSH
ChungGinsenoside Rg2 induces orphan nuclear receptor SHP gene
expression and inactivates GSK3β via AMP-activated protein kinase
to inhibit hepatic glucose production in HepG2 cellsChem Biol
Interact1953542201222062806
|
|
43.
|
N ChenW MaC HuangZ DongTranslocation of
protein kinase Cepsilon and protein kinase Cdelta to membrane is
required for ultraviolet B-induced activation of mitogen-activated
protein kinases and apoptosisJ Biol
Chem2741538915394199910.1074/jbc.274.22.1538910336426
|
|
44.
|
S YangBJ MisnerRJ ChiuFL Meyskens JrRedox
effector factor-1, combined with reactive oxygen species, plays an
important role in the transformation of JB6
cellCarcinogenesis2823822390200710.1093/carcin/bgm12817566060
|