Introduction
Breast cancer is currently seen as a molecularly
heterogeneous disease including at least five subtypes, which are
defined by gene expression patterns (1). Among these subtypes luminal A and
luminal B cancers are defined by an estrogen receptor (ER) positive
phenotype and are more differentiated than the HER2 phenotype.
Luminal B and HER2 cancer cells show increased proliferation and
are believed to have a higher potential for drug resistance, both
contributing to a more aggressive behavior (2). The most investigated targeted therapy
is the selective estrogen receptor modulator (SERM) tamoxifen. For
ER-positive disease only, 5 years of adjuvant tamoxifen reduces the
annual breast cancer death rate by 31%. However, the majority of
patients show recurrent disease during treatment, which indicates a
primary or secondary tamoxifen resistance in more than 50% of
treated patients (3). One factor
leading to resistance may be the presentation of breast cancer as
heterogeneous tumor manifestation, as for example approximately one
third of initially ER-positive breast tumors develop ER-negative
lymph node metastases (4). In
addition, it has been shown that therapy response to tamoxifen is
also negatively affected by overexpression of HER2, co-expression
of the ER modulator Amplified In Breast Cancer-1 (AIB1), and the
epidermal growth factor receptor (EGFR) (5–7). At
the transcriptional level, the paired box 2 gene product (PAX2),
which competes with AIB1 for binding and regulation of HER2
transcription, when in complex with ERα is a crucial mediator of ER
mediated repression of HER2 by tamoxifen (8). Thus, presence or absence of PAX2
links the luminal with the HER2 dominated subtypes of breast
cancer, and decreased expression of PAX2 predicts
tamoxifen-resistance and a worse clinical outcome (8). In fact, tamoxifen acts as an agonist
and was shown to be equally effective at regulating genes at the
ERα and ERβ (9). In
tamoxifen-treated patients, the nuclear co-activator AIB1 acts as a
marker of disease relapse by reducing the antagonistic activity of
tamoxifen-bound ERα, whereas in untreated patients AIB1
overexpression is associated with decreased risk of relapse
(5,10). Besides altered nuclear co-activator
expression and overexpression of HER2 and EGFR, activation of AKT
was also shown to predict a worse outcome in tamoxifen-treated
patients (11). Approximately 19%
of tamoxifen-resistant tumors showed increased HER2 expression and
79% showed downregulation of ER expression compared to individual
primary tumors (12). Additionally
to the available clinical data allowing different hypotheses of
tamoxifen-resistance, various models of secondary resistant breast
cancer cell lines after long-term drug exposure have been
developed. Many of these models demonstrated a cross-talk mechanism
between the erbB- and the AKT/mammalian target of rapamycin (mTOR)
pathways as essential step in endocrine resistance (12–15).
In breast cancer, cells developed to be resistant to tamoxifen or
fulvestrant, antiestrogen sensitivity could be restored by
co-treatment with inhibitors of the erbB-pathway such as
trastuzumab, lapatinib or gefitinib, and resistance was due to erbB
mediated non-genomic activation of estrogen responsive elements
(EREs) in these models (12–14).
In our previously presented model of 4OH-tamoxifen (OHT)-resistant
MCF7-TR and T47D-TR cells we demonstrated expression of receptors
for Gonadotropin-Releasing Hormone (GnRH) I and GnRH-II in parental
and resistant sublines, and OHT sensitivity could be restored by
pretreatment with analogs of GnRH-I/II (16). In comparison to parental cells,
expression analysis in the sublines showed increased HER2
expression and resistant cells remained ERα/β positive. Tumor cell
analogs of GnRH-I and GnRH-II inhibited mitogenic signal
transduction of growth factor receptors via activation of a
phosphotyrosine phosphatase, and blocked the autophosphorylation of
the EGFR and activation of mitogen-activated protein kinase (MAPK,
ERK-1/2), resulting in downregulation of cancer cell proliferation
(16–19). In addition, GnRH-I inhibited AKT
activation and induced apoptosis in prostate cancer cells (20). The insulin-like growth factor
(IGFR) I receptor/phosphatidyl-inositol-3-kinase (PI3K)/AKT/mTOR
pathway has also been subject of intensive research in the field of
antiestrogen resistance (21,22).
To summarize the consistent findings of the cited studies, the
IGFR/PI3K/AKT/mTOR pathway communicates synergistically with the
erbB signal transduction through activation of AKT and MAPK
(ERK-1/2), especially in cells overexpressing HER2. Specific
inhibitors of MAPK, IGFR or mTOR increased the antiproliferative
effects of antiestrogens and restored anti-estrogen sensitivity
(21, 22). Besides the increased activation of
AKT associated with HER2 overexpression, AKT activation is found in
cells with mutated tumor suppressor gene PTEN, and its activation
is negatively regulated by the carboxy-terminal modulator protein
(CTMP) (13,23,24).
In addition, the PI3K/AKT/mTOR pathway can also be activated by
intrinsic activation of the membrane bound ER GPR30 (25).
The aim of this study was to analyze the expression
of mediators of growth factor receptor pathways focusing on
HER2/EGFR/MAPK/AIB-1, IGFR/PI3K/AKT/mTOR, and the autocrine
non-ER-mediated estrogen dependent cell growth via aromatase and
GPR30 expression in our previously described model of OHT-resistant
breast cancer cells (16). We
tried to define the essential mediators required to restore OHT
sensitivity by testing a panel of specific inhibitors of erbB and
AKT/mTOR signalling in combination with OHT. Since our in
vitro model identified analogs of GnRH-I/II as effective drugs
to restore antiestrogen sensitivity, we defined their potential
clinical relevance by comparing the expression of receptors for
GnRH-I and GnRH-II with expression of ER and HER2 in 100 breast
cancers.
Materials and methods
Cell lines and culture conditions
Human breast cancer cell lines MCF-7 and T47D were
purchased from the American Type Culture Collection (ATCC,
Manassas, VA, USA). Cells were kept in medium as previously
described (16). The resistant
sublines MCF-7-TR and T47D-TR were developed as described in detail
(16), and the medium
concentration of 4OH-tamoxifen (OHT; Sigma, Deisenhofen, Germany)
was 1.25 µM. Re-evaluation of mRNA expression of ERα/ERβ, HER2,
EGFR, and GnRH-I/II-R in the resistant sublines by PCR analysis and
immunoprecipitation of ER, as well as apoptosis assays, showed no
marked differences to results previously reported (16).
Flow cytometry analysis of surface
expression of HER2, EGFR and IGFR
Flow cytometry was carried out as previously
described (16). Antibodies:
anti-HER2 (1:10; TAB250, Zymed Laboratories, San Francisco, CA,
USA), monoclonal mouse anti-EGFR antibody (1:10; clone 29.1;
Sigma), mouse anti-IGF-1Rα (1:10; clone MOPC21, BD Biosciences,
Frankfurt, Germany), FITC-conjugated secondary goat anti-mouse
antibody (1:20; Sigma). Cells were analyzed by flow cytometry on
FACSCalibur equipment using Cellquest software (Becton Dickinson,
Mountain View, CA, USA).
Reverse transcription-PCR of gpr30, aib1,
pten and l7
Total-RNA of all cell lines was isolated using
RNeasy™ mini kit (Qiagen, Hilden, Germany), following
manufacturer's instructions. Total-RNA (1 μg) was reverse
transcribed using Superscript II (Promega, Mannheim, Germany),
following company instructions and the cDNA amplified by PCR using
Taq DNA polymerase (Roche, Mannheim, Germany). The PCR for
gpr30 consisted of 35 cycles with an annealing temperature
of 60°C, whereas the PCR conditions for aib1 and pten
were 22 cycles and an annealing temperature of 57°C and 32 cycles
with an annealing temperature of 51°C, respectively. As an internal
control, reverse transcription and amplification of the l7
housekeeping gene mRNA was performed. Specific primers sequences
for gpr30 (sense: 5′-CTCCAACAGCTGCCTA AACC-3′, antisense:
5′-ATGTGGCCAAGGCTGTCTAC-3′) for pten (sense:
5′-ACACCGCCAAATTTAATTGC-3′, antisense: 5′-ACATAGCGCCTCTGACTGG-3′),
for aib1 (sense: 5′-TCAC TGAGATCCTCCATGAG-3′, antisense:
5′-GGCATCTGTAAG CCTTGGTT-3′) and for l7 (sense:
5′-AGATGTACAGAACTGA AATTC-3′, antisense:
5′-ATTTACCAAGAGATCGAGCAA-3′), were designed using the program
Primer 3 (Whitehead Institute for Biomedical Research. Cambridge,
MA, USA).
Preparation of cellular extract and
immunoblotting
Equal amounts of protein were analyzed using
SDS-PAGE. Extract from placental tissue served as control for
analysis of cellular aromatase expression. After electrophoretic
separation proteins were blotted onto nitrocellulose membranes
(Hybond-ECL nitrocellulose membrane, GE Healthcare, Munich,
Germany). After blocking and washing the western blots were
incubated with the following antibodies: anti-Akt antibody (no.
9272), anti-phospho-Akt (Ser473) antibody (no. 4058),
anti-PTEN antibody (no. 9552), anti-phospho-p44/42 MAPK (Erk1/2)
(Thr202/Tyr204) (no. 4376), anti-CTMP antibody (no. 4612) (all Cell
Signaling Technology, Danvers, MA, USA), anti-Erk1 antibody (no.
14-6718, eBioscience, San Diego, CA, USA, recognizes also MAPK p42
Erk2), antiaromatase antibody (ab34193, Abcam, Cambridge, UK),
GPR30 (LS-A1183, GeneTex, Irvine, CA, USA) or antiactin antibody
(Sigma). Visualization of the protein bands was achieved by using
an enhanced chemiluminescence (ECL) detection system (Millipore,
Billerica, MA, USA) and a radiographic film (Kodak BioMax MR film,
Kodak, Rochester, NY, USA).
DNA sequencing of pten and exons of
pik3ca
Genomic DNA of cells was obtained using the innuPREP
DNA mini kit (Analytik Jena, Jena, Germany). Primers for
pten were specific for the 9 exons of the gene and were
designed according to Bertelsen et al (26). Two exons of the pik3ca gene,
which encodes for the catalytic subunit p110α of the PI3K, were
also sequenced. Primers for pik3ca were designed according
to Curtin et al (27). All
primers were purchased from biomers.net (Ulm, Germany) or
MWG-Biotech (Ebersberg, Germany). A total of 1 μg of each genomic
DNA-sample was used in a PCR as described above to amplify the
sequences of interest. After purification with the QIAquick 96 PCR
Purification Kit (Qiagen), 125 ng DNA of every sample in 10 mM
Tris-Cl-Puffer (pH 8.5) and 20 pmol of every forward primer were
brought to total volume of 7 μl and sequenced. Sequencing was
performed by Seqlab (Göttingen, Germany). If necessary, a second
sample with the reverse primer was sequenced. Chromatograms of the
sequenced samples were analysed with the software Chromas Lite 2.0
(Technelysium Pty Ltd, Tewantin, Queensland, Australia).
Array based comparative genomic
hybridization (aCGH)
aCGH was performed using a high density array system
with 244,000 oligonucleotides on a chip (Cat. No. G4411B, Agilent,
Waldbronn, Germany), covering the complete genome in an average of
approximately 5–13 kb. DNA of the cell lines was isolated using the
QIAamp DNA Mini kit, (Qiagen Cat. No. 51304), following the
supplier's instructions for cultured cells. Labelling and
hybridization was also done as described by the supplier. As a
reference, DNA of a female donor was taken (Cat. No. G1521,
Promega, Mannheim, Germany). Hybridization was performed for 40 h
at 65°C in a hybridization oven (Agilent G2545A). After washing
slides were scanned in a laser scanning system (Agilent microarray
scanner G2565CA). Images were imported into the software package
Genomic Workbench Standard Edition 5.0.14 (Agilent). Then raw data
were processed using the feature extraction software component of
this package and further analyzed in the DNA analytics part of the
software. Image acquisition and analysis was performed as described
in detail previously by Wilkens et al (28). At least 10 metaphase spreads were
analyzed in each case.
Proliferation assays
Cells were placed on 96-well micro-plates in 100 μl
MEM-Earl culture medium without phenol red supplemented with 10%
(v/v) charcoal-stripped FCS (Allgaeu BioTech Service, Goerisried,
Germany), 0.22% (w/v) NaHCO3 and 2% (v/v) stable L-glutamine per
well. After 24 h 100 μl of medium with respective concentrations of
drugs were added. This was repeated every 24 h. After 48, 72 and 96
h 20 μl of alamarBlue™ indicator dye (AbD Serotec, Duesseldorf,
Germany) was added to each well and microplates were incubated for
additional 4 h in the incubator at 37°C. Absorbance at 570 nM and
as a reference at 600 nM was measured in each well in a microplate
reader (BioTek, Bad Friedrichshall, Germany). To analyze
cross-resistance to OHT we tested the selective estrogen
destabilisator fulvestrant (ICI 182780 AstraZeneca, Frankfurt,
Germany). For inhibition of the erbB signal transduction we used
MEK inhibitor PD98059 (Calbiochem Merck, Darmstadt, Germany) and
EGFR tyrosine kinase inhibitor gefitinib (ZD1839/Iressa;
AstraZeneca). To inhibit the PI3K/AKT/mTOR pathway we used
rapamycin (Calbiochem Merck) and perifosine (Zentaris, Frankfurt,
Germany). We performed dose- and time-dependent proliferation
assays for each of the inhibitors. In the results we present the
data of experiments with minimum concentrations of inhibitors
yielding reproducible significant growth-inhibitory effects in
parental MCF-7 and T47D cells after 96 h only, because we found no
consistent growth-inhibitory effects after 48 and 72 h.
Apoptosis assays
For apoptosis assays, cells were placed in 6-well
plates and kept under the same conditions as in the proliferation
assays, using the same respective concentrations of drugs up to 96
h. To quantify apoptosis we used a procedure based on detecting
advanced DNA degradation as previously reported (16). Analysis was performed on a
FACSCalibur (Becton Dickinson) equipment using Cellquest software.
In each experiment 1x105 cells were counted. To verify
the results obtained by this method an APO LOGIX™ - JC-1
Mitochondrial Membrane Potential Detection Kit (Bachem, Weil am
Rhein, Germany) was used. Cells were cultured and treated as
described above and then the mitochondrial membrane potential assay
carried out following the manufacturer's instructions. The results
of the APO LOGIX-JC-1 test of early apoptosis events yielded
comparable percentages of apoptotic cells to those with delayed
advanced DNA degradation detected by the method described above. We
present the results of cells with advanced DNA degradation.
Immunoprecipitaion of inhibited AKT
activation
Resistant cell lines MCF-7TR and T47D-TR were grown
to 70% confluence and then cultured according to the proliferation-
and apoptosisassays before starting treatment with GnRH-I agonist
triptorelin (100 nM; Ferring Pharmaceuticals, Copenhagen, Denmark),
GnRH-II agonist [D-Lys6]GnRH-II (100 nM; developed in
our lab and synthesized by Peptide Specialty Laboratories GmbH,
Heidelberg, Germany), gefitinib (4.5 μM) and perifosine (30 μM).
Treatment with gefitinib, triptorelin and [D-Lys6]
GnRH-II was carried out for 15, 30, 45 and 60 min, and with
perifosine up to 72 h. After the treatment cells were lysed and the
extracts were prepared for immunoblotting as described above.
Expression of receptors for GnRH-I and
GnRH-II in breast cancers
Paraffin-embedded tumor samples of 100 patients with
early breast cancer treated at our institution in 2006 were
analyzed for expression of receptors for GnRH-I and GnRH-II by
immunohistochemistry (29). No
patients had distant metastases; 38 patients presented at stage I,
47 at stage II and 15 at stage III. Eighty-four patients had
invasive ductal cancer, 16 invasive lobular cancer. Positive
expression of receptors for GnRH-I/II, ER and progesterone receptor
(PR) was defined by an Allred score of at least 3. Expression was
compared to overexpression of HER2, defined by HercepTest score 3+
or 2+ and amplification in FISH and ER expression. All patients
gave informed consent for experimental analysis on their
specimens.
Statistical analysis
A minimum of three repetitions was performed on PCR,
immunoblotting, proliferation assays and apotosis assays. Data
obtained from the proliferation and apoptosis assays were tested
for significant differences by one-way ANOVA followed by
Student-Newman-Keuls' test for comparison of individual groups.
Mean values of receptor expression in breast cancer samples were
compared by two-tailed t-tests and a correlation matrix was
composed using Pearson's r correlation coefficients. Statistical
analyses were performed using GraphPad Prism Software 5.0.
Results
Comparative expression analysis and
sequencing of pten and pik3ca
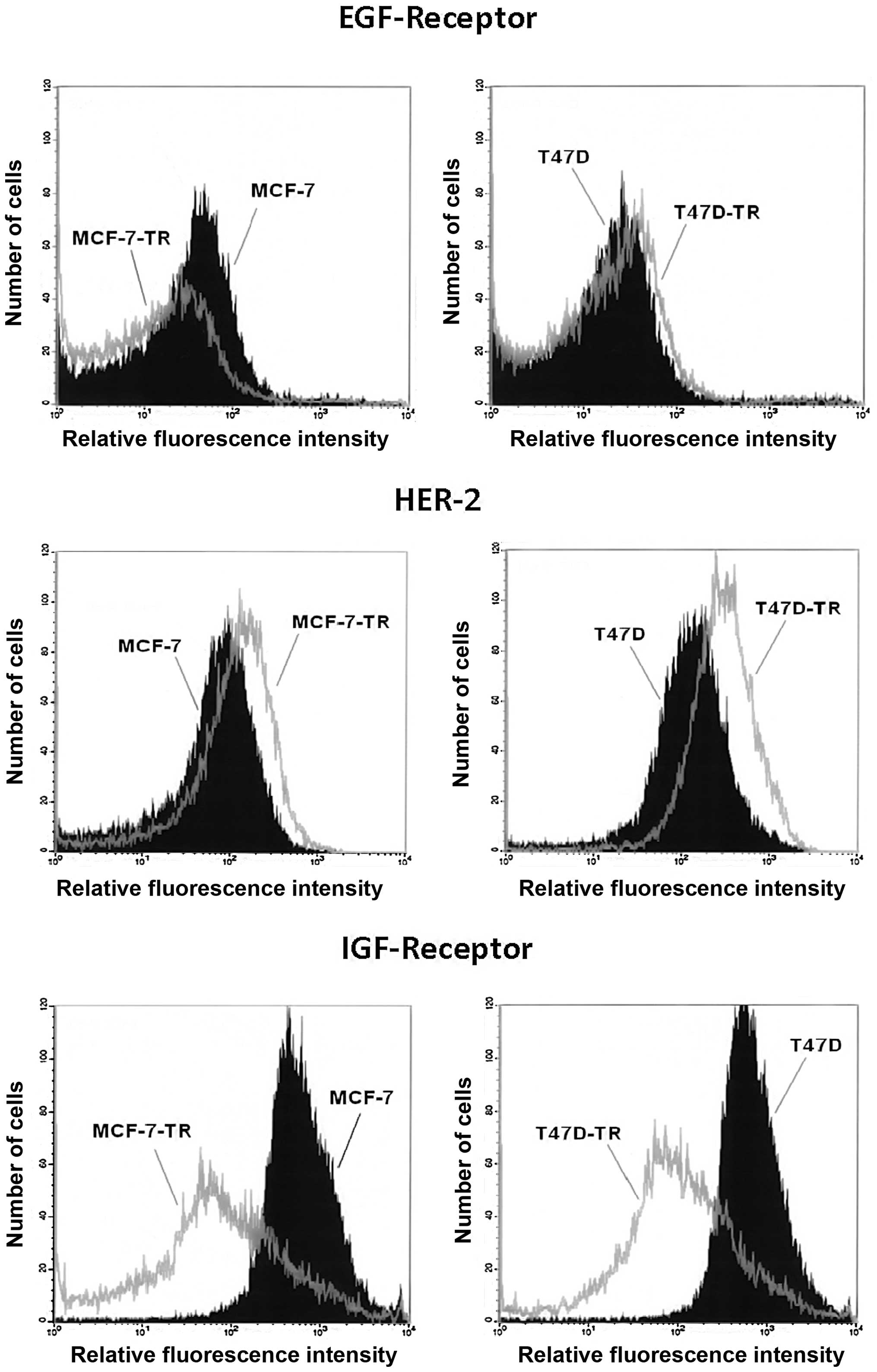
Analysis of surface expression showed similar levels
of EGFR in T47D and the resistant T47D-TR cells, but reduced EGFR
expression in MCF-7-TR cells compared to parental cells. Both
resistant cell lines showed increased expression of HER2, which was
more pronounced in T47D-TR cells. Both resistant cell lines showed
clearly reduced expression of IGFR (Fig. 1). Comparing cellular aromatase
expression to human placental tissue we could not detect aromatase
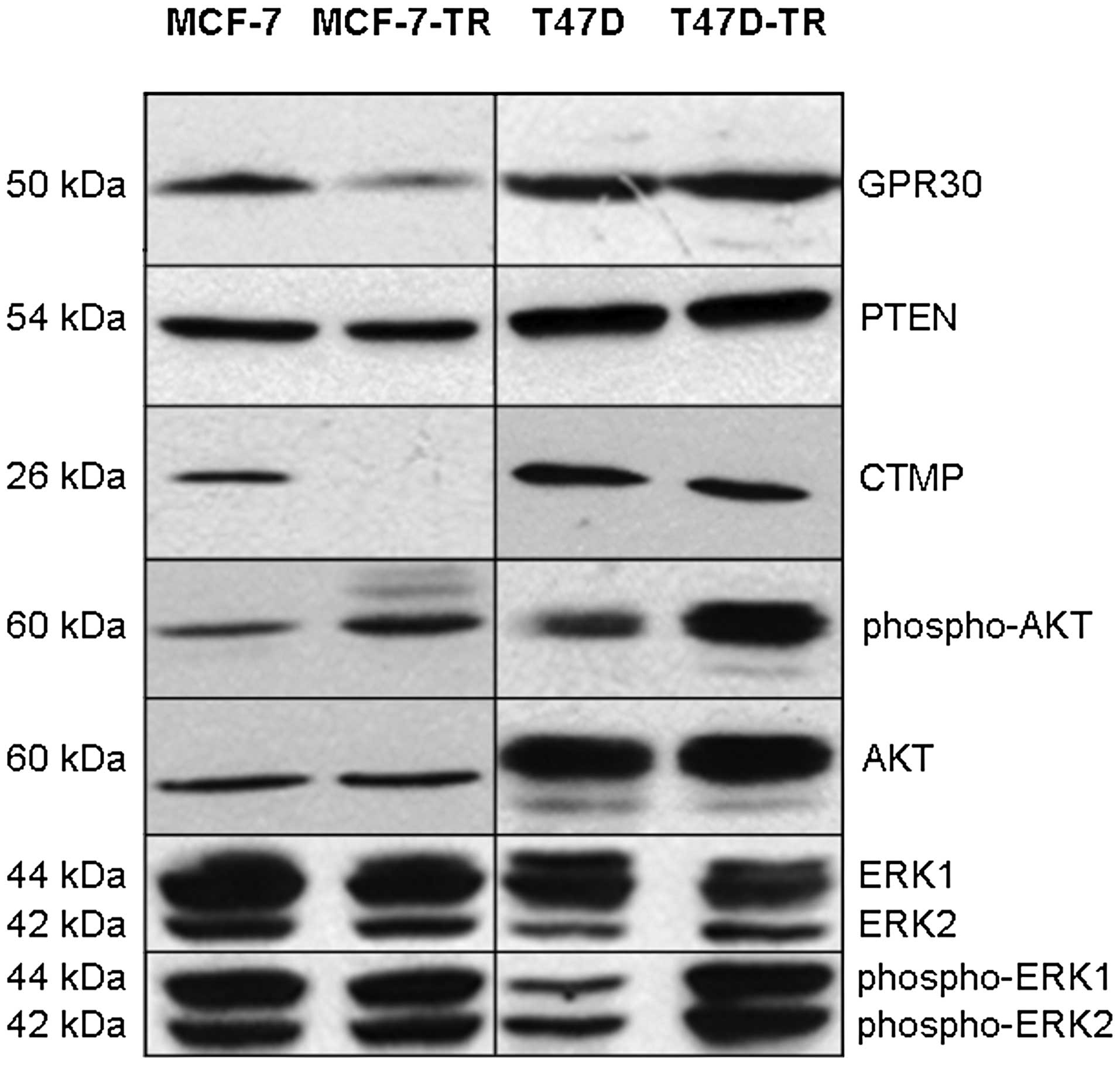
expression in any cell line (data not shown). Expression of GPR30
was slightly decreased in both resistant cell lines as shown by PCR
and immunoblotting (Fig. 2, PCR
not shown). There was no significant difference in expression of
aib1 measured by PCR (data not shown). There was no
difference in expression of PTEN (Fig.
2, PCR not shown). Compared to parental cells no difference in
basal expression of MAPK (ERK-1/2) was found in the resistant
sublines, while activated MAPK was increased in T47D-TR cells but
not in MCF-7-TR cells (Fig. 2).
Basal AKT expression was more similar in the sublines compared to
parental cells, but AKT activation was found in both resistant
sublines (Fig. 2). MCF-7-TR cells
showed complete loss of CTMP expression, while CTMP expression in
T47D-TR was comparable to parental cells (Fig. 2). Sequencing of pten and
exons of pik3ca confirmed previously reported mutations in
T47D cells, which were also found in T47D-TR cells, but not in
MCF-7 and MCF-7-TR cells.
Comparative genomic hybridization
(CGH)
CGH analyses revealed appreciable aberrations in
resistant sublines compared to parental cells, which were
predominantly gene losses. Aberrations listed in Table I refer to gains and losses of each
chromosome of each resistant subline compared to parental cells.
The only consistent aberrations for both resistant sublines were
major copy number losses on chromosomes 6q and 21q.
Detailed comparative analysis of gene losses or amplifications of
parental and resistant cell lines yielded only a few genes
previously reported to be associated to erbB, PI3K/AKT and ER
mediated growth regulation. In particular, T47D-TR cells showed
amplification of the sprouty homolog 2 gene (SPRY2) on chromosome
13 (location 13q31.1), and MCF-7-TR showed amplification of the
breast carcinoma amplified sequence-3 (BCAS3) gene on chromosome 17
(location 17q23). We found no difference in the PAX2 or CTMP gene
between the resistant cell lines.
 | Table I.Comparative genomic hybridization of
MCF-7-TR and T47D-TR cells compared to parental cell lines MCF-7
and T47D. |
Table I.
Comparative genomic hybridization of
MCF-7-TR and T47D-TR cells compared to parental cell lines MCF-7
and T47D.
| Gene gains | Gene losses | No or minor
changes |
|---|
| MCF-7-TR | 1p, 3q, 5q, 6p, 7q,
8q, 14q, 16q, 17q, 20q | 1q, 2q, 4p, 5p, 6q,
7p, 8p, 9p, 11p, 13q, 15q, 18q, 20p, 21q, 22q | 10, 12 19, X |
| T47D-TR | 13q | 5q, 6q, 14q, 17q,
21q, Xp, Xq | 1, 2, 3, 4, 7, 8,
9, 10, 11, 12, 15, 16, 18, 19, 20, 22 |
Inhibition of proliferation and induction
of apoptosis
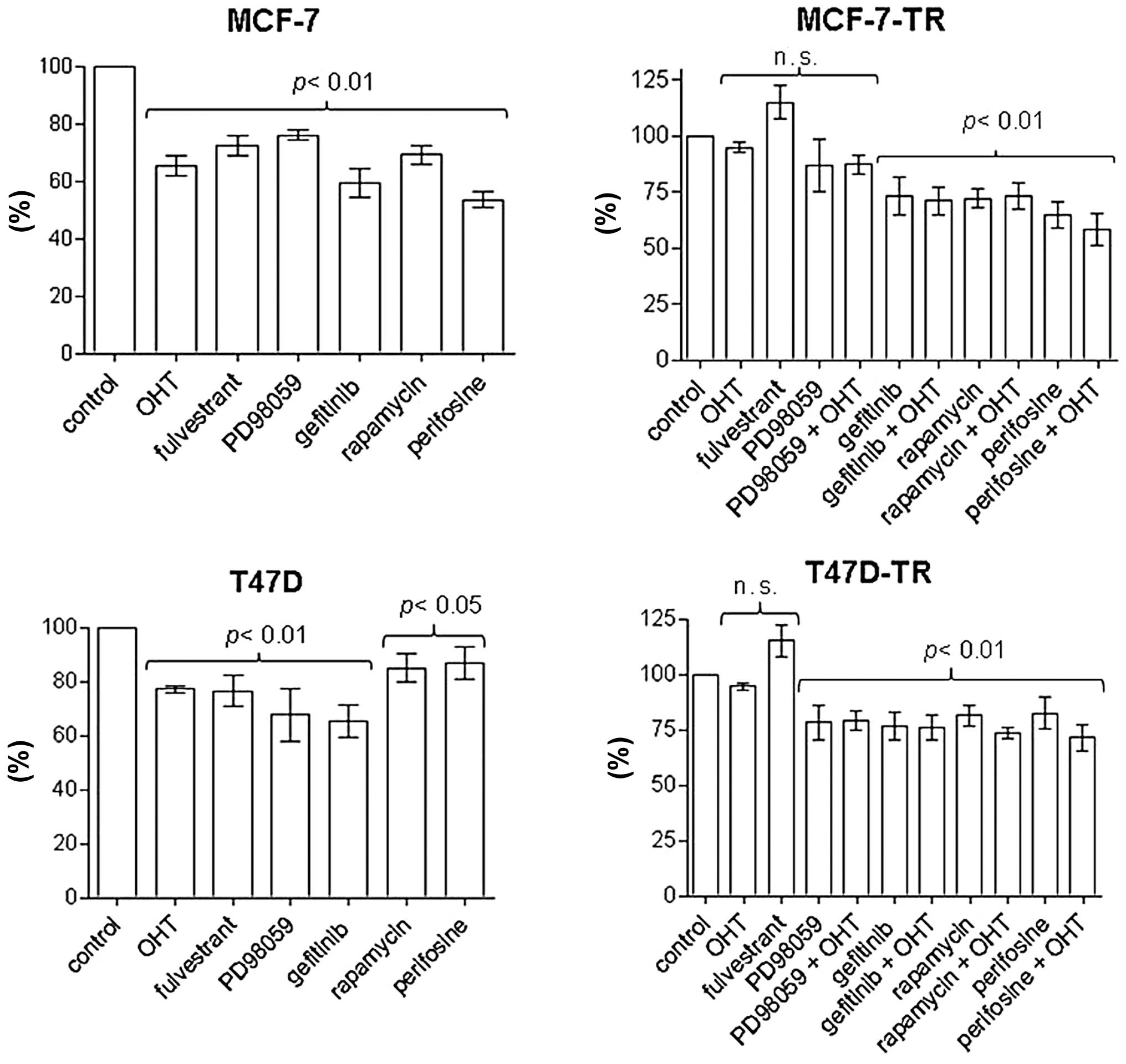
OHT (2.5 μM) and fulvestrant (100 nM) inhibited
proliferation and induced apoptosis in MCF-7 and T47D cells, but
had no significant effect on proliferation and apoptosis in the
resistant sublines; apoptosis in MCF-7 cells: control 8.1±3.4%, OHT
18.3±2.5% (p<0.01 vs. control), fulvestrant 18.7±8% (p<0.05
vs. control); apoptosis in T47D cells: control 10.2±4%, OHT
19.4±2.8%, fulvestrant 18.9±3.3% (both p<0.01 vs. control)]
(Figs. 3 and 4). Specific inhibitors of the EGFR/MAPK
and the AKT/mTOR pathways were evaluated for significant
antiproliferative effects in MCF-7 and T47D cells. After 96 h of
treatment the inhibitor of the EGFR associated tyrosine kinase
gefitinib (4.5 μM), the MEK inhibitor PD98059 (5 μM), rapamycin (20
nM) and the AKT inhibitor perifosine (30 μM) showed reproducible
antiproliferative effects. In contrast to T47D-TR cells, PD98059
had no significant antiproliferative effect on MCF-TR cells, but
gefitinib, rapamycin and perifosine showed growth-inhibitory
effects comparable to those of parental cells in both resistant
sublines (Fig. 3). Addition of OHT
to any specific inhibitor had no significant effect on
proliferation in the resistant sublines compared to single-agent
treatment (Fig. 3). After 96 h
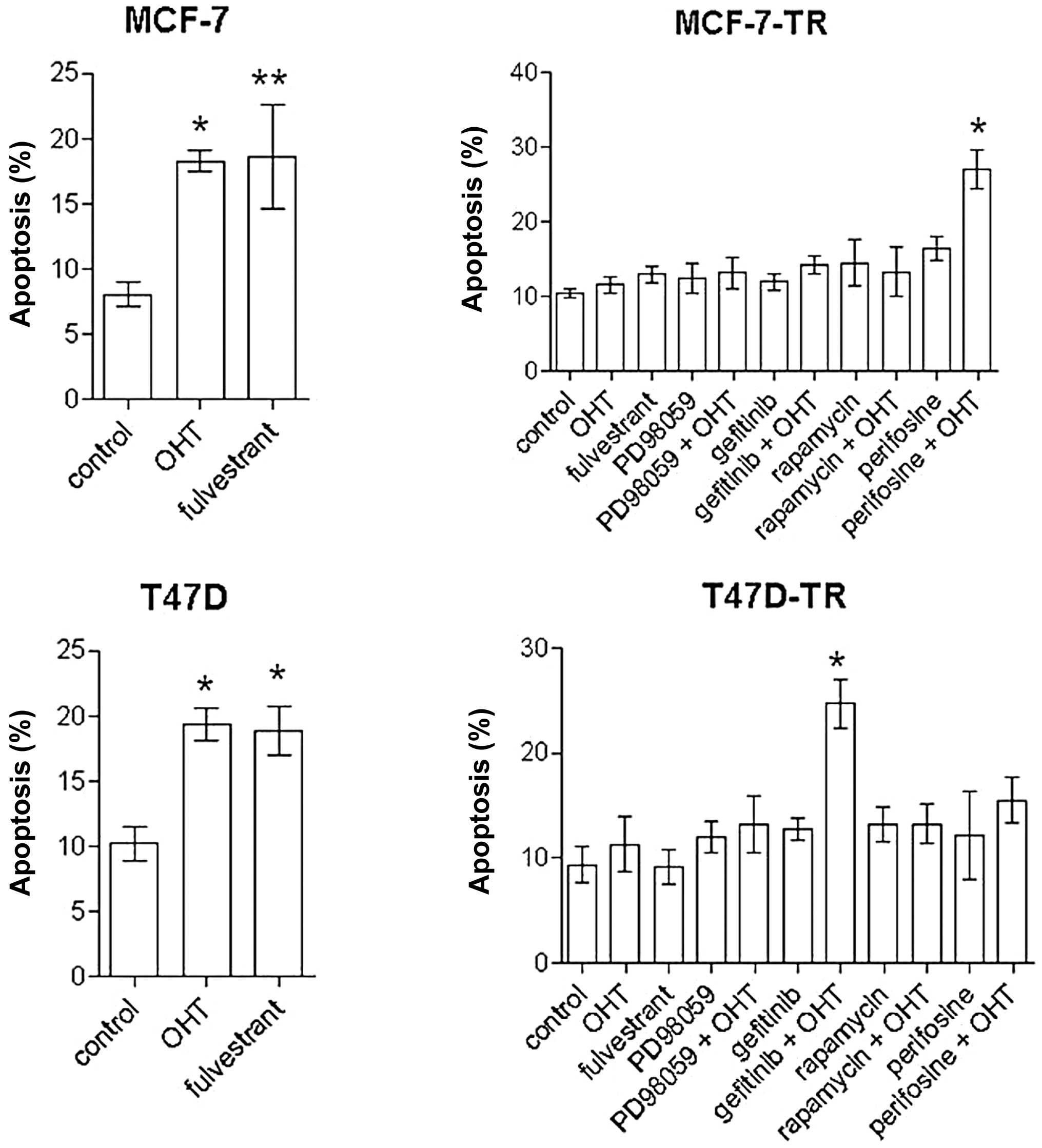
perifosine (30 μM) and rapamycin (20 nM) induced apoptosis in MCF-7
cells [spontaneous apoptosis rate in control cells 8.1±3.4%,
perifosine 19.5±2.4% (p<0.001 vs. control), rapamycin 15.7±5.7%
(p<0.01 vs. control)], but not in T47D cells. In the resistant
sublines no single-agent treatment significantly induced apoptosis
after 96 h. In MCF-7-TR cells the combination of perifosine and OHT
significantly increased apoptosis [spontaneous apoptosis rate in
control cells 10.5±3.2%, perifosine plus OHT 27±9.2% (p<0.001
vs. control, OHT and perifosine)] (Fig. 4). In T47D-TR cells the combination
of gefitinib and OHT showed significant apoptotic effects
[spontaneous apoptosis rate in control cells 9.4±5.9%, gefitinib
plus OHT 24.7±6.2% (p<0.001 vs. control and OHT, p<0.01 vs.
gefitinib)]. All other combinations with OHT showed no significant
apoptotic effects.
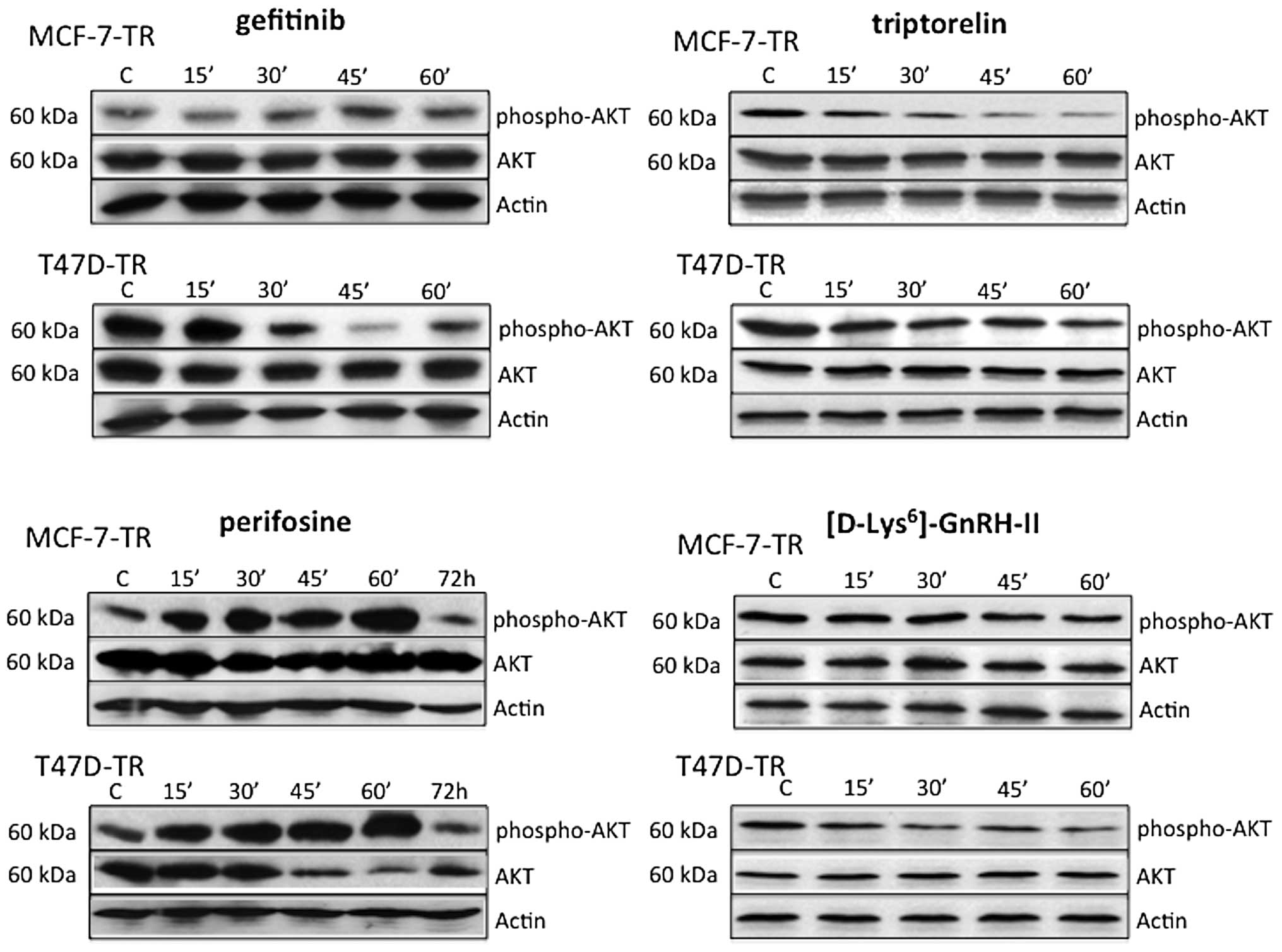
Effects of perifosine, gefitinib,
triptorelin and [D-Lys6]GnRH-II on AKT expression and
activation
In the resistant sublines MCF-7-TR and T47D-TR
perifosine activated AKT during the first hour of treatment, but
after 72 h the amount of activated AKT was lower compared to
untreated control cells. In MCF-7-TR cells perifosine had no effect
on basal AKT expression, but in T47D-TR cells basal AKT was clearly
decreased after 45 min, and levels remained low until 72 h.
Gefitinib did not affect basal expression of AKT in MCF-7-TR and
T47D-TR cells during the first hour of treatment. Gefitinib
inhibited AKT activation in T47D-TR cells, but not in MCF-7-TR
cells. In both resistant sublines activation of AKT was inhibited
during the 1 h of treatment with triptorelin or
[D-Lys6]GnRH-II (Fig.
5).
Expression of receptors for GnRH-I and
GnRH-II in human breast cancers
Specimens of 100 breast cancers showed positive
expression (Allred score >2) for GnRH-I receptor in 46%, and for
GnRH-II receptor in 53%. ER expression (Allred score >2) was
found in 63% of tumors, and HER2 overexpression (HercepTest 3+ or
2+ and amplified in FISH) was found in 38%. GnRH-I receptor
expression was positively correlated with GnRH-II receptor
expression (r=0.34; p=0.0004) and HER2 overexpression (r=0.23;
p=0.02), but not with ER expression. GnRH-II receptor expression
was also positively correlated with HER2 overexpression (r=0.2;
p=0.05), but not with ER expression. In this sample, ER expression
showed no significant correlation to HER2 overexpression. There was
no ER-positive/PR-negative tumor. Nineteen tumors were negative for
ER/PR and HER-2, and in these specimens GnRH-I and/or GnRH-II
receptor expression was found in 10 cases, of which 7 cases showed
expression of both receptors.
Discussion
In our model of OHT-resistant breast cancer cells,
antiestrogen resistance was due to numerous genomic aberrations,
which caused minor but important phenotype differences of both
compared cell lines. Both resistant sublines shared similarities,
such as increased HER2 expression, activated AKT, decreased IGFR
and GPR30 expression, but also important differences were found,
such as decreased EGFR expression and loss of CTMP expression in
MCF-7-TR cells, and erbB-associated activated MAPK (ERK-1/2) in
T47D-TR cells. The AKT-inhibitor perifosine was successful in
overcoming tamoxifen resistance in MCF-7-TR cells with loss of CTMP
expression, but not in T47D-TR cells. ErbB-inhibition by gefitinib
to overcome tamoxifen resistance was only successful in T47D-TR
cells. We found major genomic aberrations in both cell lines,
mainly gene losses, confirming not only altered expression profiles
but indeed the development of mutated subclones of parental cells.
These subclones showed cross-resistance to fulvestrant, reduced
GPR30 expression and no detectable aromatase exn our model of
OHT-resistant breast cancer cells, antiestrogen resistance was due
to numerous genomic aberrations, which caused minor but important
phenotype differences of the compared cell lines. Both resistant
sublines shared similarities, pression, therefore autocrine
estrogen-mediated growth control is unlikely.
In contrast to many previous reports on
antiestrogen-resistant breast cancer models, resistance was not due
to increased expression of AIB1, EGFR, IGFR and loss or mutation of
PTEN and PIK3CA. However, we found BCAS3 amplification in MCF-7-TR,
which was reported to act as ERα co-activator and to communicate
with metastasis-associated protein-1 (MTA1) in tumor progression
and tamoxifen resistance of breast cancer, and also to affect
non-genomic ERα activation through MAPK and PI3K/AKT signalling
pathways in combination with PELP1/MNAR (30–32).
Expression of wild-type tumor suppressor gene product hSPRY2
is frequently downregulated in breast cancer overexpressing HER2,
despite the fact that hSPRY2 does not act as general
inhibitor of erbB associated tyrosine kinases and ERK-1/2 (33). However, we found SPRY2 gene
amplification of T47D-TR cells, which may reflect a protective
effect as regulator of the Ras/MAPK pathway against hormonal
changes e.g. ER-inhibition by OHT. Nevertheless, the SPRY2 gene in
breast cancer development and drug resistance still needs to be
investigated (33). Previous, very
well described in vitro and in vivo models of breast
cancer cells with acquired antiestrogen-resistance explained
development of resistance by different mechanisms (8,12,13,15,21,34–36).
In cell models with proven agonistic activity of tamoxifen,
resistance was frequently associated with increased HER2
expression, activation of MAPK (ERK-1/2), downregulation of PAX2,
increased expression of AIB1 and/or activation of IGFR/AKT
signalling, and inhibitors of erbB signalling delayed tamoxifen
resistance or restored its antiproliferative action (1,8,13,15,21,34,36,37).
In cells with long-term estrogen-deprivation or long-term exposure
to fulvestrant IGFR downregulation and activation of
HER2/erbB3/MAPK was reported. In these cells resistance could be
delayed by anti-HER2/EGFR treatment, but reactivation of HER2 and
signalling through AKT lead to anti-ER/anti-erbB de novo
resistance (15,35,36).
Especially in ER-positive cells multi-targeting of erbB mediators
might be an option to prevent resistance, even at initially low
expression of HER2 (12,14,35,36,38).
Besides, anti-erbB treatment with trastuzumab, pertuzumab,
lapatinib and gefitinib, targeting MAPK and PI3K/AKT/mTOR signal
transduction is also an option to prevent or overcome antiestrogen
resistance (21,22). The MEK inhibitor PD98059, which was
demonstrated to be active in tamoxifen-resistant cells (21), failed to restore the effects of OHT
in both of our resistant sublines. Inhibition of mTOR by rapamycin
also failed to restore OHT sensitivity in our model. Thus, AKT
activation alone is not a predictor of response to rapamycin. The
AKT inhibitor perifosine acts by decreasing plasma membrane
localization of AKT, inhibition of AKT phosphorylation, and
reduction of the levels of total AKT (39). Both resistant sublines showed
increased AKT activity, but perifosine restored OHT sensitivity
only in MCF-7-TR cells with loss of CTMP expression. However, we
have no explanation for the flair-up effect of perifosine on AKT
activation in the resistant sublines during the first hour of
treatment, which was not found in parental cells (data not shown).
In MCF-7-TR cells gefitinib and PD98059 had no effect in
combination with OHT, which suggests an erbB-independent growth
regulation. In tamoxifen-resistant breast cancer cells with
downregulated IGFR signal transduction it has been shown that the
inhibitory effects on mediators of erbB and AKT were abrogated
(40). In comparison, in T47D-TR
cells the increase of HER2 expression was more pronounced, EGFR
expression was not decreased, and inhibition of EGFR by gefitinib
restored OHT sensitivity, which suggests a primarily erbB-mediated
cause of antiestrogen resistance.
Regarding analogs of GnRH-I/II to overcome tamoxifen
resistance, we previously showed that analogs of GnRH-I and GnRH-II
inhibited MAPK activity and restored OHT sensitivity in our
resistant sublines (16), herein
we also demonstrate the inhibition of AKT activation in these
cells. GnRH-I receptor expression was described in about 50–64% of
breast cancers (41,42). Antiproliferative effects of analogs
of GnRH-I alone or in combination with tamoxifen in breast cancer
cells were already reported 25 years ago (43,44).
In gynecologic malignancies, analogs of GnRH-I activate a G-protein
coupled phosphotyrosine phosphatase and antagonize EGFR-mediated
mitogenic signal transduction (16,18,19,45).
We detected a GnRH-II receptor-like antigen in human placenta and
gynecologic cancers including breast cancer cells, and showed that
the antiproliferative effects of GnRH-II and the GnRH-I antagonist
cetrorelix in tumor cells were not mediated through the GnRH-I
receptor (16,29,46,47).
Analogs of GnRH-I and GnRH-II inhibited activation of MAPK
(ERK-1/2) and AKT, and restored OHT sensitivity in both resistant
cell lines of our model. Here we confirm the expression of GnRH-I
receptor in approximately 50% of breast cancers and are the first
to report on GnRH-II receptor expression in more than 50% of breast
cancers, with both receptors correlating to HER2, but not to ER
expression. Buchholz et al found expression of GnRH-I
receptor in particular in triple-negative breast cancers (48). More recently, GnRH-II antagonists
induced apoptosis in human gynecologic malignancies and breast
cancers in vivo and in vitro, without any apparent
side effects in nude mice (49,50).
In endocrine-resistant, HER2 overexpressing or triple-negative
breast cancer, GnRH-I/II receptor expression provides potential
targets in multi-targeting therapy strategies to avoid or overcome
drug resistance.
Abbreviations:
|
AIB1
|
amplified in beast cancer-1;
|
|
CTMP
|
carboxy-terminal modulator
protein;
|
|
EGFR
|
epidermal growth factor receptor;
|
|
ER
|
estrogen receptor;
|
|
ERE
|
estrogen responsive elements;
|
|
ERK
|
extraregulated kinase;
|
|
GnRH
|
gonadotropin-releasing hormone;
|
|
GPR30
|
G-protein coupled receptor 30
|
|
IGF
|
insulin-like growth factor
|
|
mTOR
|
mammalian target of rapamycin
|
|
MAPK
|
mitogen-activated protein kinase
|
|
OHT
|
4-hydroxy-tamoxifen
|
|
PAX2
|
paired box 2 gene product
|
|
PI3K
|
phosphitdyl-inositole-3-kinase
|
|
SERM
|
selective estrogen receptor
|
Acknowledgements
We thank Renate Dietrich, Matthias
Läsche, Hiltrud Schulz and Sonja Blume for excellent technical
assistance. We also thank Prisca Eser, Brett McKinnon and Stefan
Aebi for proofreading the manuscript. This study was supported by
the Deutsche Forschungsgemeinschaft (DFG GU 981/1-1 to Andreas
Günthert).
References
|
1.
|
Prat A and Perou CM: Mammary development
meets cancer genomics. Nat Med. 15:842–844. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Cheang MC, Chia SK, Voduc D, Gao D, Leung
S, Snider J, Watson M, Davies S, Bernard PS, Parker JS, Perou CM,
Ellis MJ and Nielsen TO: Ki67 index, HER2 status, and prognosis of
patients with luminal B breast cancer. J Natl Cancer Inst.
101:736–750. 2009. View Article : Google Scholar
|
|
3.
|
Early Breast Cancer Trialists'
Collaborative Group (EBCTCG): Effects of chemotherapy and hormonal
therapy for early breast cancer on recurrence and 15-year survival:
an overview of the randomised trials. Lancet. 365:1687–1717.
2005.PubMed/NCBI
|
|
4.
|
Aitken SJ, Thomas JS, Langdon SP, Harrison
DJ and Faratian D: Quantitative analysis of changes in ER, PR and
HER2 expression in primary breast cancer and paired nodal
metastases. Ann Oncol. 21:1254–1261. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Osborne CK, Bardou V, Hopp TA, Chamness
GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM and Schiff
R: Role of the estrogen receptor coactivator AIB1 (SRC-3) and
HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer
Inst. 95:353–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Dowsett M, Houghton J, Iden C, Salter J,
Farndon J, A'Hern R, Sainsbury R and Baum M: Benefit from adjuvant
tamoxifen therapy in primary breast cancer patients according
oestrogen receptor, progesterone receptor, EGF receptor and HER2
status. Ann Oncol. 17:818–826. 2006. View Article : Google Scholar
|
|
7.
|
Dihge L, Bendahl PO, Grabau D, Isola J,
Lövgren K, Rydén L and Fernö M: Epidermal growth factor receptor
(EGFR) and the estrogen receptor modulator amplified in breast
cancer (AIB1) for predicting clinical outcome after adjuvant
tamoxifen in breast cancer. Breast Cancer Res Treat. 109:255–262.
2008. View Article : Google Scholar
|
|
8.
|
Hurtado A, Holmes KA, Geistlinger TR,
Hutcheson IR, Nicholson RI, Brown M, Jiang J, Howat WJ, Ali S and
Carroll JS: Regulation of ERBB2 by oestrogen receptor-PAX2
determines response to tamoxifen. Nature. 456:663–666. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Ball LJ, Levy N, Zhao X, Griffin C,
Tagliaferri M, Cohen I, Ricke WA, Speed TP, Firestone GL and
Leitman DC: Cell type-and estrogen receptor-subtype specific
regulation of selective estrogen receptor modulator regulatory
elements. Mol Cell Endocrinol. 299:204–211. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Kirkegaard T, McGlynn LM, Campbell FM,
Müller S, Tovey SM, Dunne B, Nielsen KV, Cooke TG and Bartlett JM:
Amplified in breast cancer 1 in human epidermal growth factor
receptor - positive tumors of tamoxifen-treated breast cancer
patients. Clin Cancer Res. 13:1405–1411. 2007. View Article : Google Scholar
|
|
11.
|
Pérez-Tenorio G and Stål O: Activation of
AKT/PKB in breast cancer predicts a worse outcome among endocrine
treated patients. Br J Cancer. 86:540–545. 2002.
|
|
12.
|
Leary AF, Drury S, Detre S, Pancholi S,
Lykkesfeldt AE, Martin LA, Dowsett M and Johnston SR: Lapatinib
restores hormone sensitivity with differential effects on estrogen
receptor signaling in cell models of human epidermal growth factor
receptor 2-negative breast cancer with acquired endocrine
resistance. Clin Cancer Res. 16:1486–1497. 2010. View Article : Google Scholar
|
|
13.
|
Shou J, Massarweh S, Osborne CK, Wakeling
AE, Ali S, Weiss H and Schiff R: Mechanisms of tamoxifen
resistance: increased estrogen receptor-HER2/neu cross-talk in
ER/HER2-positive breast cancer. J Natl Cancer Inst. 96:926–935.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Konecny GE, Pegram MD, Venkatesan N, Finn
R, Yang G, Rahmeh M, Untch M, Rusnak DW, Spehar G, Mullin RJ, Keith
BR, Gilmer TM, Berger M, Podratz KC and Slamon DJ: Activity of the
dual kinase inhibitor lapatinib (GW572016) against
HER-2-overexpressing and trastuzumab-treated breast cancer cells.
Cancer Res. 66:1630–1639. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Frogne T, Benjaminsen RV, Sonne-Hansen K,
Sorensen BS, Nexo E, Laenkholm AV, Rasmussen LM, Riese DJ II, de
Cremoux P, Stenvang J and Lykkesfeldt AE: Activation of ErbB3, EGFR
and Erk is essential for growth of human breast cancer cell lines
with acquired resistance to fulvestrant. Breast Cancer Res Treat.
114:263–275. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Günthert AR, Gründker C, Olota A, Läsche
J, Eicke N and Emons G: Analogs of GnRH-I and GnRH-II inhibit
epidermal growth factor-induced signal transduction and resensitize
resistant human breast cancer cells to 4OH-tamoxifen. Eur J
Endocrinol. 153:613–625. 2005.
|
|
17.
|
Emons G, Müller V, Ortmann O and Schulz
KD: Effects of LHRH-analogues on mitogenic signal transduction in
cancer cells. J Steroid Biochem Mol Biol. 65:199–206. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Gründker C, Völker P, Schulz KD and Emons
G: Luteinizing hormone-releasing hormone agonist triptorelin and
antagonist cetrorelix inhibit EGF-induced c-fos expression in human
gynecological cancers. Gynecol Oncol. 78:194–202. 2000.PubMed/NCBI
|
|
19.
|
Gründker C, Völker P and Emons G:
Antiproliferative signaling of luteinizing hormone-releasing
hormone in human endometrial and ovarian cancer cells through G
protein alpha(I)-mediated activation of phosphotyrosine
phosphatase. Endocrinology. 142:2369–2380. 2001.
|
|
20.
|
Kraus S, Levy G, Hanoch T, Naor Z and
Seger R: Gonadotropin-releasing hormone induces apoptosis of
prostate cancer cells: role of c-Jun NH2-terminal kinase, protein
kinase B, and extracellular signal-regulated kinase pathways.
Cancer Res. 64:5736–5744. 2004. View Article : Google Scholar
|
|
21.
|
Ghayad SE, Vendrell JA, Larbi SB, Dumontet
C, Bieche I and Cohen PA: Endocrine resistance associated with
activated ErbB system in breast cancer cells is reversed by
inhibiting MAPK or PI3K/Akt signaling pathways. Int J Cancer.
126:545–562. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
deGraffenried LA, Friedrichs WE, Russell
DH, Donzis EJ, Middleton AK, Silva JM, Roth RA and Hidalgo M:
Inhibition of mTOR activity restores tamoxifen response in breast
cancer cells with aberrant Akt Activity. Clin Cancer Res.
10:8059–8067. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Chow LM and Baker SJ: PTEN function in
normal and neoplastic growth. Cancer Lett. 241:184–196. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Maira SM, Galetic I, Brazil DP, Kaech S,
Ingley E, Thelen M and Hemmings BA: Carboxyl-terminal modulator
protein (CTMP), a negative regulator of PKB/Akt and v-Akt at the
plasma membrane. Science. 294:374–380. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Revankar CM, Mitchell HD, Field AS, Burai
R, Corona C, Ramesh C, Sklar LA, Arterburn JB and Prossnitz ER:
Synthetic estrogen derivatives demonstrate the functionality of
intracellular GPR30. ACS Chem Biol. 2:536–544. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Bertelsen BI, Steine SJ, Sandvei R, Molven
A and Laerum OD: Molecular analysis of the PI3K-AKT pathway in
uterine cervical neoplasia: frequent PIK3CA amplification and AKT
phosphorylation. Int J Cancer. 118:1877–1883. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Curtin JA, Stark MS, Pinkel D, Hayward NK
and Bastian BC: PI3-kinase subunits are infrequent somatic targets
in melanoma. J Invest Dermatol. 126:1660–1663. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Wilkens L, Tchinda J, Burkhardt D, Nolte
M, Werner M and Georgii A: Analysis of hematologic diseases using
conventional karyotyping, fluorescence in situ hybridization
(FISH), and comparative genomic hybridization (CGH). Hum Pathol.
29:833–839. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Eicke N, Günthert AR, Viereck V, Siebold
D, Béhé M, Becker T, Emons G and Gründker C: GnRH-II receptor-like
antigenicity in human placenta and in cancers of the human
reproductive organs. Eur J Endocrinol. 153:605–612. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Cheskis BJ, Greger J, Cooch N, McNally C,
Mclarney S, Lam HS, Rutledge S, Mekonnen B, Hauze D, Nagpal S and
Freedman LP: MNAR plays an important role in ERα activation of
Src/MAPK and PI3K/Akt signaling pathways. Steroids. 73:901–905.
2008.
|
|
31.
|
Gururaj AE, Holm C, Landberg G and Kumar
R: Breast cancer-amplified sequence 3, a target of
metastasis-associated protein 1, contributes to tamoxifen
resistance in premenopausal patients with breast cancer. Cell
Cycle. 5:1407–1410. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Kumar R, Zhang H, Holm C, Vadlamudi RK,
Landberg G and Rayala SK: Extranuclear coactivator signaling
confers insensitivity to tamoxifen. Clin Cancer Res. 15:4123–4130.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Lo TL, Yusoff P, Fong CW, Guo K, McCaw BJ,
Phillips WA, Yang H, Wong ES, Leong HF, Zeng Q, Putti TC and Guy
GR: The ras/mitogen-activated protein kinase pathway inhibitor and
likely tumor suppressor proteins, sprouty 1 and sprouty 2 are
deregulated in breast cancer. Cancer Res. 64:6127–6136. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Kurokawa H, Lenferink AE, Simpson JF,
Pisacane PI, Sliwkowski MX, Forbes JT and Arteaga CL: Inhibition of
HER2/neu (erbB-2) and mitogen-activated protein kinases enhances
tamoxifen action against HER2-overexpressing, tamoxifen-resistant
breast cancer cells. Cancer Res. 60:5887–5894. 2000.
|
|
35.
|
Massarweh S, Osborne CK, Jiang S, Wakeling
AE, Rimawi M, Mohsin SK, Hilsenbeck S and Schiff R: Mechanisms of
tumor regression and resistance to estrogen deprivation and
fulvestrant in a model of estrogen receptor-positive,
HER-2/neu-positive breast cancer. Cancer Res. 66:8266–8273. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Creighton CJ, Massarweh S, Huang S,
Tsimelzon A, Hilsenbeck SG, Osborne CK, Shou J, Malorni L and
Schiff R: Development of resistance to targeted therapies
transforms the clinically associated molecular profile subtype of
breast tumor xenografts. Cancer Res. 68:7493–7501. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Hutcheson IR, Knowlden JM, Madden TA,
Barrow D, Gee JM, Wakeling AE and Nicholson RI: Oestrogen
receptor-mediated modulation of the EGFR/MAPK pathway in
tamoxifen-resistant MCF-7 cells. Breast Cancer Res Treat. 81:81–93.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Arpino G, Gutierrez C, Weiss H, Rimawi M,
Massarweh S, Bharwani L, De Placido S, Osborne CK and Schiff R:
Treatment of human epidermal growth factor receptor
2-overexpressing breast cancer xenografts with multiagent
HER-targeted therapy. J Natl Cancer Inst. 99:694–705. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Kondapaka SB, Singh SS, Dasmahapatra GP,
Sausville EA and Roy KK: Perifosine, a novel alkylphospholipid,
inhibits protein kinase B activation. Mol Cancer Ther. 2:1093–1103.
2003.PubMed/NCBI
|
|
40.
|
Knowlden JM, Jones HE, Barrow D, Gee JM,
Nicholson RI and Hutcheson IR: Insulin receptor substrate-1
involvement in epidermal growth factor receptor and insulin-like
growth factor receptor signalling: implication for Gefitinib
(‘Iressa’) response and resistance. Breast Cancer Res Treat.
111:79–91. 2008.PubMed/NCBI
|
|
41.
|
Fekete M, Wittliff JL and Schally AV:
Characteristics and distribution of receptors for
[D-TRP6]-luteinizing hormone-releasing hormone, somatostatin,
epidermal growth factor, and sex steroids in 500 biopsy samples of
human breast cancer. J Clin Lab Anal. 3:137–147. 1989.
|
|
42.
|
Baumann KH, Kiesel L, Kaufmann M, Bastert
G and Runnebaum B: Characterization of binding sites for a
GnRH-agonist (buserelin) in human breast cancer biopsies and their
distribution in relation to tumor parameters. Breast Cancer Res
Treat. 25:37–46. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Miller WR, Scott WN, Morris R, Fraser HM
and Sharpe RM: Growth of human breast cancer cells inhibited by a
luteinizing hormone-releasing hormone agonist. Nature. 313:231–233.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Foekens JA, Henkelman MS, Fukkink JF,
Blankenstein MA and Klijn JG: Combined effects of buserelin,
estradiol and tamoxifen on the growth of MCF-7 human breast cancer
cells in vitro. Biochem Biophys Res Commun. 140:550–556. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Emons G, Ortmann O, Becker M, Irmer G,
Springer B, Laun R, Hölzel F, Schulz KD and Schally AV: High
affinity binding and direct antiproliferative effects of LHRH
analogues in human ovarian cancer cell lines. Cancer Res.
53:5439–5446. 1993.PubMed/NCBI
|
|
46.
|
Gründker C, Schlotawa L, Viereck V, Eicke
N, Horst A, Kairies B and Emons G: Antiproliferative effects of the
GnRH antagonist cetrorelix and of GnRH-II on human endometrial and
ovarian cancer cells are not mediated through the GnRH type I
receptor. Eur J Endocrinol. 151:141–149. 2004.PubMed/NCBI
|
|
47.
|
Gründker C, Günthert AR, Millar RP and
Emons G: Expression of gonadotropin-releasing hormone II (GnRH-II)
receptor in human endometrial and ovarian cancer cells and effects
of GnRH-II on tumor cell proliferation. J Clin Endocrinol Metab.
87:1427–1430. 2002.PubMed/NCBI
|
|
48.
|
Buchholz S, Seitz S, Schally AV, Engel JB,
Rick FG, Szalontay L, Hohla F, Krishan A, Papadia A, Gaiser T,
Brockhoff G, Ortmann O, Diedrich K and Köster F: Triple-negative
breast cancers express receptors for luteinizing hormone-releasing
hormone (LHRH) and respond to LHRH antagonist cetrorelix with
growth inhibition. Int J Oncol. 35:789–796. 2009.
|
|
49.
|
Gründker C, Fost C, Fister S, Nolte N,
Günthert AR and Emons G: Gonadotropin-releasing hormone type II
antagonist induces apoptosis in MCF-7 and triple-negative
MDA-MB-231 human breast cancer cells in vitro and in vivo. Breast
Cancer Res. 12:R492010.PubMed/NCBI
|
|
50.
|
Fister S, Günthert AR, Aicher B, Paulini
KW, Emons G and Gründker C: GnRH-II antagonists induce apoptosis in
human endometrial, ovarian, and breast cancer cells via activation
of stress-induced MAPKs p38 and JNK and proapoptotic protein Bax.
Cancer Res. 69:6473–6481. 2009. View Article : Google Scholar
|