Contents
Introduction
Association between hormones and DNA damage
Association between DNA damage and cancer
therapy
Involvement of hormones and their receptors in DNA
repair systems
Perspectives
Introduction
Breast and prostate cancers are known to be
hormone-responsive types of cancer (1). There is evidence supporting the role
of hormones in stimulating cancer cell growth. In addition, the
progression from hormone-dependent to hormone-independent disease
remains a critical issue in the management of these cancers.
Recently, studies have reported an association between DNA repair
deficiency and loss of hormone receptors (2). The modulation of the DNA repair
system may contribute to hormone-independent cancer development.
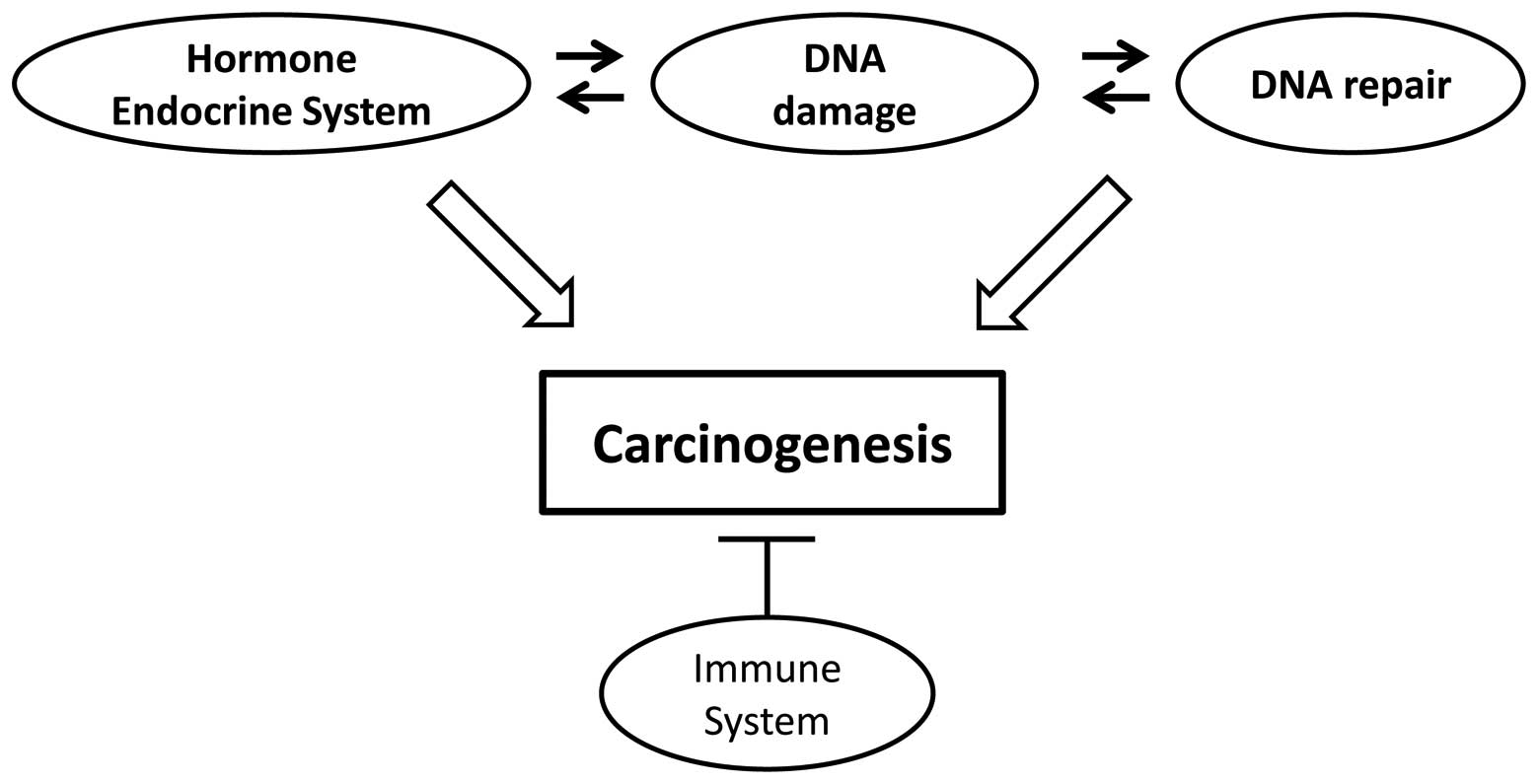
There may be connections between carcinogenesis, the hormone
endocrine system and DNA repair systems (Fig. 1). It has been suggested that the
immune system surveys the body for cancerous cells, inhibiting
carcinogenesis.
Cancer is a complex disease which can aquire a more
invasive and therapy-resistant character by a number of molecular
changes. The malignant transformation occurs in the cells with
increased proliferative activity since DNA replication errors
accumulate along with the high DNA duplication rate (3–5).
However, cells possess a machinery to maintain genomic integrity in
response to various genotoxic events. Under genotoxic conditions,
cells do not progress through the cell cycle by activating the DNA
damage checkpoint (6–8). The checkpoint acts as a process to
transmit information from damaged DNA lesions to the cell cycle
regulators. Mutations or downregulations in several genes which
make the effects of DNA damage less severe are known to predispose
to cancer development. For example, mutations in ataxia
telangiectasia-mutated (ATM) have been associated with an increased
risk of cancer (9,10). The ATM is a checkpoint kinase that
phosphorylates a number of proteins in response to DNA damage,
including the tumor suppressor gene products, p53 and BRCA1, which
have been implicated in DNA repair pathways. Normal cells show a
balance of the various mechanisms of the DNA repair machinery. The
p53 tumor suppressor regulates several DNA repair pathways involved
in the cellular response to genotoxic stresses, which can block
cell proliferation and/or induce apoptosis (11). Since p53 also plays an important
role in the transcriptional regulation of genes encoding proteins
involved in DNA repair and apoptosis, the modification of p53 may
appear to be a pivotal determinant of the fate of cells. During
carcinogenic progression, p53 is mutated or deleted in
almost half of all human cancers (12,13)
and fails to function normally. BRCA1 regulates the
expression of several genes identified as having key roles in
breast and ovarian cancer risk. BRCA1 also participates in
DNA repair and recombination processes related to the maintenance
of genomic integrity and the control of cell proliferation
(14). As most BRCA1 mutant
cancers are hormone receptor-negative, hormonal factors may
contribute to the etiology of BRCA1 mutant cancers (15,16).
Advances in the field of DNA repair biology have led
to a better understanding of the molecular events which are related
to the pathogenesis of hormone-related cancers. Indeed, an
understanding of the underlying molecular mechanisms involved in
the transition to hormone refractory disease is also essential for
the development of effective therapeutic and preventive strategies.
In the present review, we summarize the function of DNA repair
molecules from the viewpoint of carcinogenesis and hormone-related
cell modulation.
Association between hormones and DNA
damage
Since estrogens stimulate cell proliferation
(17), increased exposure to
estrogens promotes the opportunity to develop random genetic
errors. In addition, some hormone oxidative metabolites catalyzed
by cytochrome p450 enzymes can form unstable adducts with DNA
leading to mutations (18).
Furthermore, the metabolic process produces reactive oxygen species
that accounts for oxidative damage to genome DNA. The continuous
mutagenic potential may contribute to the initiation of cancer.
Prolonged exposure to estrogen is strongly associated with an
increased risk of developing breast cancer. Estrogens bind
primarily to estrogen receptors, which are believed to mediate the
various actions of estrogens. On the other hand, androgen signaling
may induce apoptosis by activating the DNA damage response
(19). The BRCA1 protein may
function to suppress mammary epithelial cell proliferation by
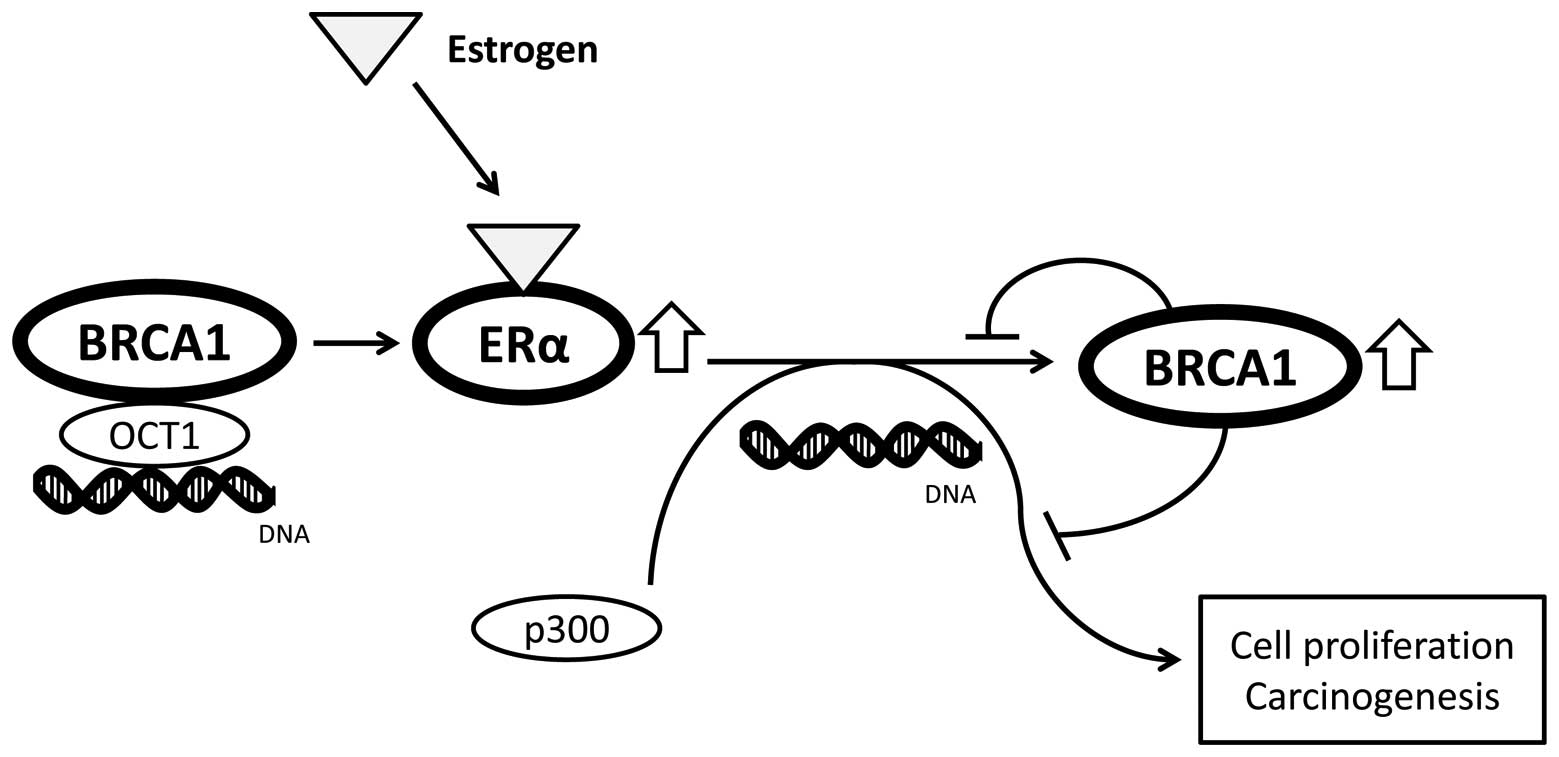
inhibiting estrogen receptor-mediated pathways (Fig. 2). In addition, an estrogen receptor
complex modulates BRCA1 transcription under conditions of estrogen
stimulation. Recent studies have shown an association between DNA
repair deficiency and loss of estrogen as well as androgen
receptors (2). In addition,
estrogens are not only essential for mammary growth and
differentiation, but also enhance the activity of the p53 tumor
suppressor protein (20). This
paradox, the bidirectional functions of some hormones in
carcinogenesis or anticarcinogenesis, should be resolved in the
near future.
p53 is a key transcription factor that regulates
several genes and protects against genomic instability. Mutations
in the p53 gene are frequent in breast cancers and are
associated with poor prognosis. The focal adhesion kinase (FAK), a
tyrosine kinase, is overexpressed in a variety of human tumors
including breast cancers (21).
FAK is a critical regulator of cell motility and its overexpression
is associated with increased metastatic potential. FAK expression
is downregulated in a p53-dependent manner in response to estrogen
(22). The loss of FAK
downregulation in p53 mutant cells correlates with increased
invasive capacity upon estrogen stimulation. In this way, p53 is an
important downregulator of FAK and the loss of p53 function in
breast cancer may contribute to the metastatic potential of
estrogen-responsive cancers. The p53 downstream transcriptional
target, p21/WAF-1, is increased in estrogen-treated cells in
response to DNA damage. The p53 gene may form a center
within a network connected by genes that are regulated by hormones
and DNA repair. In addition, the crosstalk between p53 and androgen
receptor signaling suggests that p53 activation may augment the
anticancer effects of hormonal therapy in prostate cancer (23).
The combined ingestion of soy isoflavone and
curcumin decreases the serum level of prostate-specific antigen
(PSA) (24). This combination
affects the expression and phosphorylation of ATM and inhibits
prostate cancer cell proliferation. Testosterone, an androgen,
augments the activation of the DNA damage-response, which may
suppress the initial malignant transformation of prostate cancer.
Vitamin D has also been shown to exhibit cancer-preventive
activities. There is evidence that vitamin D provides protection
against DNA damage (25). Vitamin
D enhances the expression of DNA repair genes, such as ATM and
Rad50, thereby promoting effective DNA double-strand break (DSB)
repair, protecting cells from DNA damage and stress and
consequently, carcinogenesis (26).
Association between DNA damage and cancer
therapy
Exposure to ionizing radiation or DNA-damaging
agents causes DSBs. Genetic defects in DNA damage response genes
and/or downregulation of the DNA repair mechanism promote genomic
instability, which can lead to carcinogenesis (27). Cells are equipped with multiple DNA
repair mechanisms for the maintenance of genomic stability and the
suppression of carcinogenesis. Standard DNA repair pathways
available in mammalian cells include homologous repair,
non-homologous end-joining and single-strand annealing (28). There are different pathways that
repair DSBs. The DNA repair system is essential for the survival of
normal and cancer cells. A set of signaling pathways detects the
DSBs and mediates either DNA repair survival or apoptotic cell
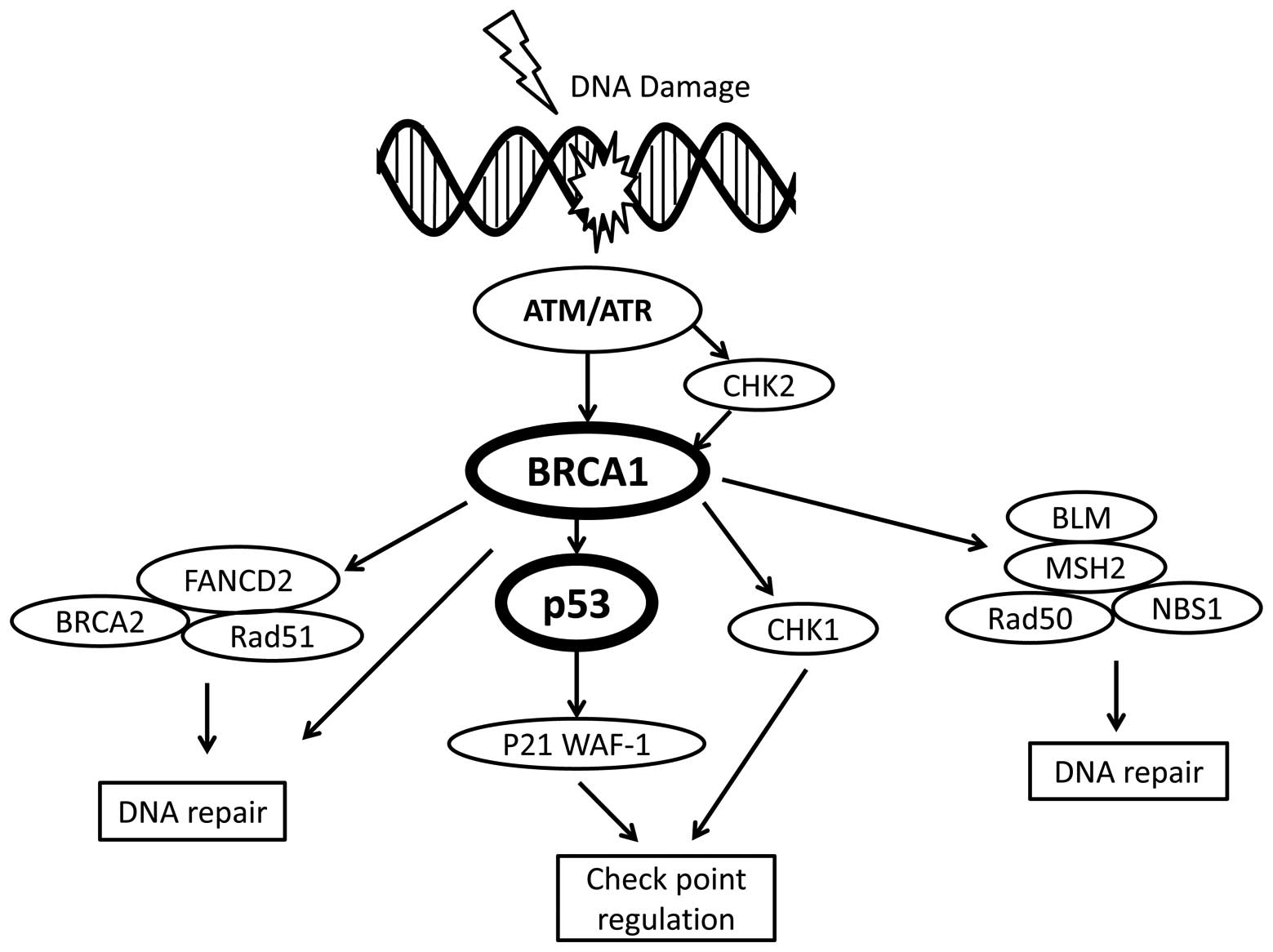
death. The main DNA damage recognition molecule is ATM (29), which is a checkpoint kinase that
phosphorylates a number of proteins in response to DNA damage,
including p53 and BRCA1 (Fig. 3).
Schematic structures of these important molecules are shown in
Fig. 4. An additional consequence
of defective DNA repair is cellular hypersensitivity to
DNA-damaging agents (30). The
inhibition of DNA repair pathways blocks the mechanisms that are
required for survival in the presence of oncogenic mutations.
DNA-damaging agents then function more effectively as a therapeutic
strategy for cancer cells with DNA repair defects (30). Epigenetic mechanisms, such as
histone modifications and DNA methylation have been evaluated with
a view of enhancing cancer therapy via the regulation of the
expression of genes involved in DNA repair (31).
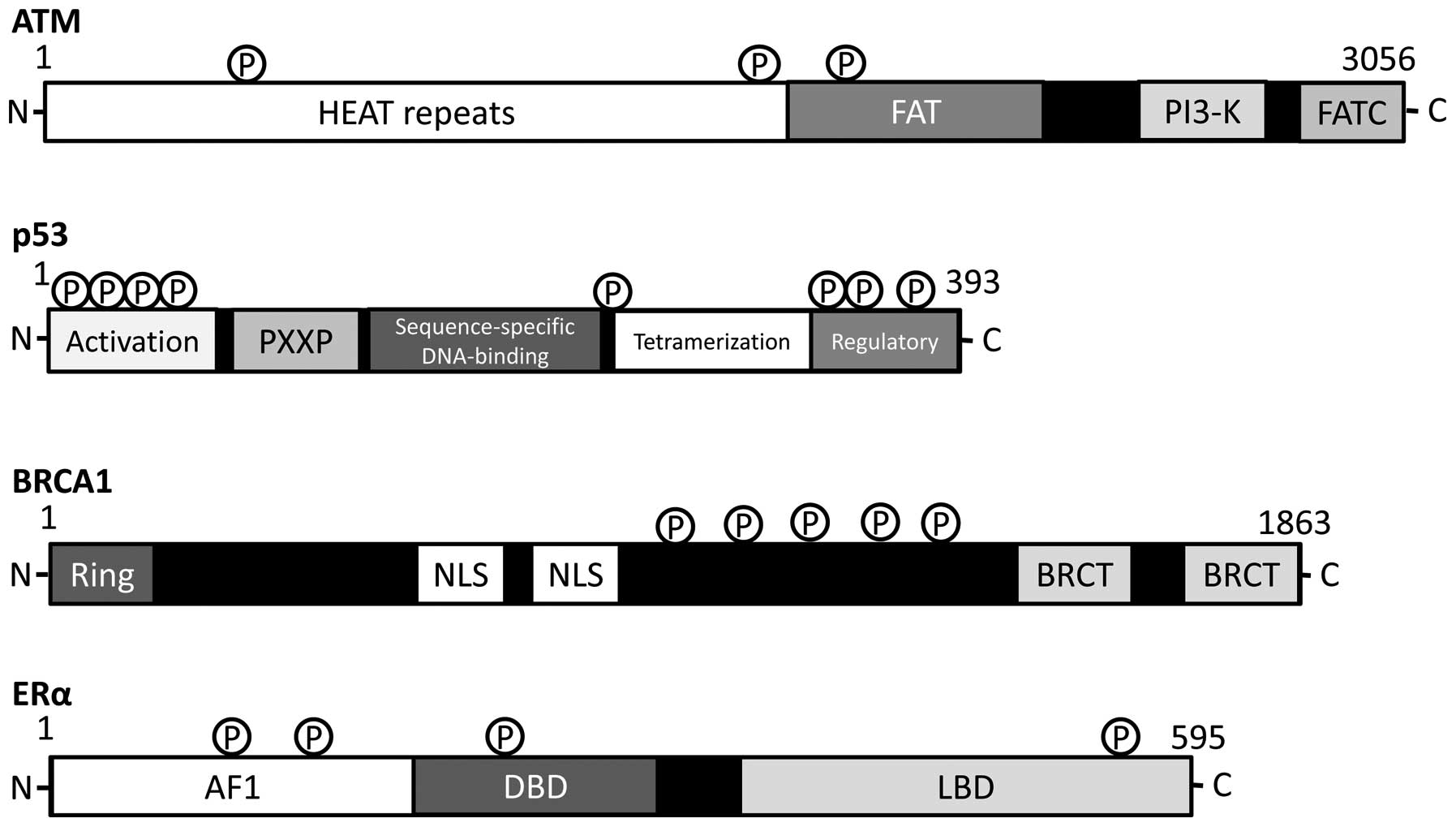 | Figure 4.Schematic diagram indicating the
domain structures of ATM, p53, BRCA1 and estrogen receptor (ER)-α
proteins. The functionally important sites including the sites of
protein phosphorylation are shown. Note that the sizes of proteins
are modified for clarity. HEAT, huntington, elongation factor 3, a
subunit of PP2A and TOR1; FAT, FRAP-ATM-TRRAP; FATC,
FAT-C-terminal; PXXP, a proline repeat, Ring, really interesting
new gene, finger domain; NLS, nuclear localization signal, BRCT,
BRCA1 C terminus; AF1, activation function 1 DBD, DNA-binding
domain; LBD, ligand-binding domain. |
Through stress-induced activation, p53 triggers the
expression of the target genes that protect the genetic integrity
of cells. The p53 gene is frequently mutated in multiple
cancer tissues, suggesting that p53 plays a critical role in
preventing cancers. Mutant p53 can be classified as a
loss-of-function or gain-of-function protein depending on the
mutation type (32). Wild-type p53
is inactive under normal physiological conditions and is activated
in response to various types of DNA-damaging stresses. p53
activation may lead to the regression of existing neoplastic
lesions and may therefore be important in developing cancer
prevention (33). Failure of the
DNA repair functions leads to p53-mediated induction of apoptotic
cell death. The p53 protein undergoes post-translational
modifications such as acetylation of lysines, nitration of
tyrosines and the phosphorylation of serine/threonine residues in
response to certain stresses (34). In addition, multiple mechanisms
have been revealed to regulate p53 activity, which determines the
selectivity of p53 for specific transcriptional targets. Among the
transcriptional targets of p53, p21WAF-1 has been shown to play an
important role in both p53-dependent (35) and -independent pathways (36). p21WAF-1 inhibits cell cycle
progression through the interaction with the cyclin-dependent
kinase (CDK) complex. In this way, p53 has been known to play a
central role in maintaining a stable genome through its role in
cell cycle checkpoints, DNA repair and apoptosis.
BRCA1 also fulfills the criteria for a tumor
suppressor gene whose function is required to block cancer
development. BRCA1 hereditary breast cancer is a type of
cancer with defects in a DNA repair pathway (37). The mutation is then associated with
increased genomic instability in cells, which accelerates the
mutation rate of other critical genes. Studies have established
functional roles for BRCA1 in DNA damage signaling, DNA repair
processes and cell cycle checkpoints (38). Consistent with these functional
roles, cells deficient in BRCA1 exhibit severe genomic
instability and chromosomal aberrations. Although BRCA1 gene
mutations are rare in sporadic breast and ovarian cancers, BRCA1
protein expression is frequently reduced in sporadic cases
(39). BRCA1 cDNA encodes
for the 1863-amino acid protein with an amino terminal zinc ring
finger motif and two putative nuclear localization signals
(Fig. 4). The amino-terminal
domain possesses E3 ubiquitin ligase activity (40) and the carboxyl-terminal domain is
involved in binding to specific phospho-proteins (41). The role of BRCA1 in cell cycle
control involves its ability to interact with various cyclins and
CDKs, activate the CDK inhibitor, p21WAF-1, and p53. BRCA1 becomes
hyperphosphorylated following exposure to DNA-damaging agents, and
the specific function of BRCA1 is regulated by phosphorylation
(42). In addition to the roles in
the regulation of DNA damage response, the BRCA1 protein interacts
with the estrogen and androgen receptor, and regulates their
activity (43), inhibiting
estrogen receptor-α activity and stimulating androgen receptor
activity. Thus, BRCA1 mutations confer a modulatory risk for
each type of hormone-responsive cancer. In other words, hormonal
factors intensely contribute to the cancer risk in BRCA1
mutation carriers. Alternatively, hormonal factors also affect
therapeutic tumor cell killing via modulating DNA repair. Hence,
the DNA repair capacity may be a novel therapeutic modality in
overcoming drug resistance in cancer. Either survival or apoptosis,
which is determined by the balance between DNA damage and DNA
repair capacity, may thus be one of the major problems of cancer
therapy.
Involvement of hormones and their receptors
in DNA repair systems
Hormones, such as estrogen, progesterone and
androgen often contribute to the initiation and promotion of
carcinogenesis via specific hormone receptors. A number of
candidate genes have been identified as biomarkers for breast and
prostate cancers such as those involved in hormone-synthesis,
activation and secretion (44).
Furthermore, hormonal therapies generally regulate cancer cell
growth and have provided improvements in survival in
hormone-related diseases. For example, anti-estrogens are effective
in estrogen receptor-positive breast cancer, and androgen
deprivation therapies are common in advanced prostate cancer.
However, transition to hormone refractory cancer cells remains an
important clinical issue, which limits the benefits of these
therapies.
Estrogens, not only play a key role in the
proliferation and differentiation of the mammary epithelium, but
also regulate diverse cellular processes in brain, cardiovascular
and bone metabolism (45). In
addition, several normal and carcinoma cells express estrogen
receptors and their proliferation is stimulated by estrogen, which
in turn increases the likelihood that DNA damage occurs. The
chronic exposure estrogen then contributes to carcinogenesis in
estrogen receptor-positive cells. However, estrogens also stimulate
the expression of genes that can repair DNA damage including BRCA1
(Fig. 2). DNA repair defects in
the progression of breast cancer may occur in the transition to
hormone-independence. The estrogen-bound receptor dimerizes and
associates with chromatin. The receptor dimers bind directly to a
DNA sequence motif, the estrogen response element. There are two,
estrogen receptor-α and estrogen receptor-β. Estrogen receptor-α is
thought to be proliferative, whereas the activation of estrogen
receptor-β is thought to induce apoptosis. A key DNA repair
protein, MSH2, has been shown to be a potent co-activator of
estrogen receptor-α (46). An
increase in estrogen receptor-β levels may be associated with a
reduction in breast cancer risk (47). There is evidence showing that the
reduced expression of estrogen receptor-β correlates with increased
prostate cancer risk. Estrogen receptor-β may inhibit cellular
proliferation by antagonizing the actions of estrogen receptor-α
(48). Exposure to genistein,
which is an estrogen-like chemical compound present in plants such
as soy, reduces breast cancer risk (49). The expression of BRCA1 is
upregulated in mammary glands prepubertally exposed to genistein,
suggesting that BRCA1 plays a role in mediating the
cancer-protective effects of genistein. However, high soy intake
does not reduce breast cancer risk if consumed in adulthood. In
addition, physiologically high levels of estrogen are believed to
stimulate carcinogenesis. Childhood and adolescence may be
particularly sensitive periods for breast cancer initiation. There
is an urgent need to further understand DNA repair pathways and
their distinct roles in childhood and adulthood.
Prostate cancer is a frequently diagnosed (∼20%)
malignancy in males (50,51). The risk factors may act through
hormonal mechanisms, since the drug that blocks androgenic hormone
activation reduces cancer risk. In addition, some sporadic prostate
cancer cases are linked to polymorphisms of genes involved in
androgen activation and its signaling pathway (50,51).
Androgens have been recognized to play a role in controlling the
growth of the normal prostate gland. The androgen receptor is a
ligand-activated transcription factor of the nuclear receptor
superfamily that plays a critical role in male physiology, whose
signaling also contributes to carcinogenesis and cancer progression
by regulating the transcription of androgen-responsive genes. One
of the androgen-responsive genes, PSA, is a clinically important
marker that is used for the diagnosis and evaluation of the
prostate cancer (52). During the
progression of androgen-sensitive prostate cancer to
androgen-refractory, the majority of cancer cells still express the
androgen receptor; however, in many cases it is mutated. The
downregulation of the androgen receptor induces apoptotic cell
death and inhibits cancer cell proliferation. Therefore, androgen
signaling has been recognized as a key target for prostate cancer
prevention and therapy.
Variations in estrogen receptor activities may also
modulate prostate cancer risk (53). The increased consumption of
phytoestrogens such as genistein, also a well-known tyrosine kinase
inhibitor, has been associated with a reduced risk of prostate
cancer (54). Phytoestrogens
protect cells against reactive oxygen species by scavenging free
radicals. In addition, phytoestrogens upregulate the expression of
glycogen synthase kinase-3β (GSK-3β), enhance GSK-3β binding to
β-catenin, and induce apoptotic cell death (55), suggesting that phytoestrogens can
induce apoptosis and inhibit prostate cancer cell growth.
Phytoestrogens have also been found to inhibit molecules in the
mitogen-activated kinase (MAPK) pathway (56). Furthermore, they can block the
activation of p38-MAPK by TGF-β in prostate cancer (57), inhibiting cancer cell invasion and
metastasis. Reduced MSH2 protein expression in prostate cancer
cells has been shown to correlate with an overall recurrence-free
interval, while the reduced protein expression may be associated
with an increased risk of prostate cancer initiation (58). MSH2 expression has been shown to
correlate with the expression of p53, while it negatively
correlates with the expression of the estrogen receptor (59). The downregulation of the
MSH2 gene has been associated with invasive breast cancer
and the hormone independence of prostate cancer. Elevated
post-meiotic segregation increased 2 (PMS2) expression also appears
to negatively correlate with prognosis in prostate cancer patients
(60,61). The overexpression of PMS2 may
confer DNA damage tolerance.
Perspectives
The hormone signaling pathway is a complex signaling
network, and further in-depth research in this area is required.
The DNA repair system is a highly conserved DNA editing process
that maintains genomic fidelity through the recognition and repair
of damaged nucleotides. Discrepancy between cell proliferation and
DNA repair functions may account for the accumulation of DNA errors
and vulnerability to carcinogenesis. The roles of human tumor
susceptibility genes in the DNA repair machinery are important for
the development of methods for the prediction of risk, diagnosis,
prevention and therapy of human cancers. For example, as hormonal
regulation contributes to the expression of the tumor
susceptibility gene product, BRCA1, the hormonal prevention of
BRCA1 mutant breast cancers may be effective. The discovery
of other molecular pathways would be helpful to further understand
the fundamental mechanisms of hormone independence. Certain
evidence supports the cancer-protective roles of dietary selenium,
vitamin D, vitamin E, lycopene and soy foods (62,63).
Lycopene, which is a carotenoid found in tomatoes, appears to be an
effective protective dietary factor for prostate cancer, and acts
as an antioxidant by scavenging reactive oxygen species to protect
against DNA damage. A significantly reduced risk of prostate cancer
following higher lycopene consumption has been shown. Further
mechanistic studies are also required in order to understand the
precise molecular mechanisms of hormonal carcinogenesis, cancer
prevention and the DNA repair system for more effective therapeutic
interventions of these human malignancies.
Acknowledgements
This study was supported by
Grants-in-Aid from the Ministry of Education, Culture, Sports,
Science and Technology of Japan. In addition, this study was
supported in part by a grant from Shin-ei Pharmaceutical Co.,
Ltd.
References
|
1.
|
Folkerd EJ and Dowsett M: Influence of sex
hormones on cancer progression. J Clin Oncol. 28:4038–4044. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Martin L, Coffey M, Lawler M, Hollywood D
and Marignol L: DNA mismatch repair and the transition to hormone
independence in breast and prostate cancer. Cancer Lett.
291:142–149. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Xu Y and Price BD: Chromatin dynamics and
the repair of DNA double strand breaks. Cell Cycle. 10:261–267.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Crasta K, Ganem NJ, Dagher R, Lantermann
AB, Ivanova EV, Pan Y, et al: DNA breaks and chromosome
pulverization from errors in mitosis. Nature. 482:53–58. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Rodriguez GP, Romanova NV, Bao G, Rouf NC,
Kow YW and Crouse GF: Mismatch repair-dependent mutagenesis in
nondividing cells. Proc Natl Acad Sci USA. 109:6153–6158. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Vurusaner B, Poli G and Basaga H: Tumor
suppressor genes and ROS: complex networks of interactions. Free
Radic Biol Med. 52:7–18. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Hanel W and Moll UM: Links between mutant
p53 and genomic instability. J Cell Biochem. 113:433–439. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Jones RM and Petermann E: Replication fork
dynamics and the DNA damage response. Biochem J. 443:13–26. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Smith J, Tho LM, Xu N and Gillespie DA:
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and
cancer. Adv Cancer Res. 108:73–112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Hennequin C, Quero L and Favaudon V: DNA
repair and tumour radiosensitivity: focus on ATM gene. Bull Cancer.
98:239–246. 2011.PubMed/NCBI
|
|
11.
|
Molchadsky A, Rivlin N, Brosh R, Rotter V
and Sarig R: p53 is balancing development, differentiation and
de-differentiation to assure cancer prevention. Carcinogenesis.
31:1501–1508. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Muller PA, Vousden KH and Norman JC: p53
and its mutants in tumor cell migration and invasion. J Cell Biol.
192:209–218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Essmann F and Schulze-Osthoff K:
Translational approaches targeting the p53 pathway for anti-cancer
therapy. Br J Pharmacol. 165:328–344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Karve TM, Li X and Saha T: BRCA1-mediated
signaling pathways in ovarian carcinogenesis. Funct Integr
Genomics. 12:63–79. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Berstein LM: Endocrinology of the wild and
mutant BRCA1 gene and types of hormonal carcinogenesis. Future
Oncol. 4:23–39. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Okumura N, Yoshida H, Kitagishi Y,
Nishimura Y and Matsuda S: Alternative splicings on p53, BRCA1 and
PTEN genes involved in breast cancer. Biochem Biophys Res Commun.
413:395–399. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Mueck AO and Sitruk-Ware R: Nomegestrol
acetate, a novel progestogen for oral contraception. Steroids.
76:531–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Bolton JL and Thatcher GR: Potential
mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol.
21:93–101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Comstock CE and Knudsen KE: The complex
role of AR signaling after cytotoxic insult: implications for
cell-cycle-based chemotherapeutics. Cell Cycle. 6:1307–1313. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Lu S, Becker KA, Hagen MJ, Yan H, Roberts
AL, Mathews LA, et al: Transcriptional responses to estrogen and
progesterone in mammary gland identify networks regulating p53
activity. Endocrinology. 149:4809–4820. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Golubovskaya VM, Conway-Dorsey K, Edmiston
SN, Tse CK, Lark AA, Livasy CA, et al: FAK overexpression and p53
mutations are highly correlated in human breast cancer. Int J
Cancer. 125:1735–1738. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Anaganti S, Fernández-Cuesta L, Langerød
A, Hainaut P and Olivier M: p53-Dependent repression of focal
adhesion kinase in response to estradiol in breast cancer
cell-lines. Cancer Lett. 300:215–224. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Lee HJ, Chattopadhyay S, Yoon WH, Bahk JY,
Kim TH, Kang HS, et al: Overexpression of hepatocyte nuclear
factor-3alpha induces apoptosis through the upregulation and
accumulation of cytoplasmic p53 in prostate cancer cells. Prostate.
70:353–361. 2010.PubMed/NCBI
|
|
24.
|
Ide H, Tokiwa S, Sakamaki K, Nishio K,
Isotani S, Muto S, et al: Combined inhibitory effects of soy
isoflavones and curcumin on the production of prostate-specific
antigen. Prostate. 70:1127–1133. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Welsh J: Cellular and molecular effects of
vitamin D on carcinogenesis. Arch Biochem Biophys. 523:107–114.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Ting HJ, Yasmin-Karim S, Yan SJ, Hsu JW,
Lin TH, Zeng W, et al: A positive feedback signaling loop between
ATM and the vitamin D receptor is critical for cancer
chemoprevention by vitamin D. Cancer Res. 72:958–968. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Kelly GL and Strasser A: The essential
role of evasion from cell death in cancer. Adv Cancer Res.
111:39–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Natarajan AT and Palitti F: DNA repair and
chromosomal alterations. Mutat Res. 657:3–7. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Bhatti S, Kozlov S, Farooqi AA, Naqi A,
Lavin M and Khanna KK: ATM protein kinase: the linchpin of cellular
defenses to stress. Cell Mol Life Sci. 68:2977–3006. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Okumura N, Yoshida H, Kitagishi Y,
Nishimura Y, Iseki S and Matsuda S: Against lung cancer cells: to
be, or not to be, that is the problem. Lung Cancer Int. View Article : Google Scholar : 2012. View Article : Google Scholar
|
|
31.
|
Gigek CO, Chen ES, Calcagno DQ, Wisnieski
F, Burbano RR and Smith MA: Epigenetic mechanisms in gastric
cancer. Epigenomics. 4:279–294. 2012. View Article : Google Scholar
|
|
32.
|
Martinez-Rivera M and Siddik ZH:
Resistance and gain-of-resistance phenotypes in cancers harboring
wild-type p53. Biochem Pharmacol. 83:1049–1062. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Stegh AH: Targeting the p53 signaling
pathway in cancer therapy - the promises, challenges and perils.
Expert Opin Ther Targets. 16:67–83. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Kim DH, Kundu JK and Surh YJ: Redox
modulation of p53: mechanisms and functional significance. Mol
Carcinog. 50:222–234. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Gartel AL: p21(WAF1/CIP1) and cancer: a
shifting paradigm? Biofactors. 35:161–164. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Ocker M and Schneider-Stock R: Histone
deacetylase inhibitors: signalling towards p21cip1/waf1. Int J
Biochem Cell Biol. 39:1367–1374. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Osborne MP: Chemoprevention of breast
cancer. Surg Clin North Am. 79:1207–1221. 1999. View Article : Google Scholar
|
|
38.
|
Roy R, Chun J and Powell SN: BRCA1 and
BRCA2: different roles in a common pathway of genome protection.
Nat Rev Cancer. 12:68–78. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Prado A, Andrades P and Parada F: Recent
developments in the ability to predict and modify breast cancer
risk. J Plast Reconstr Aesthet Surg. 63:1581–1587. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Ohta T, Sato K and Wu W: The BRCA1
ubiquitin ligase and homologous recombination repair. FEBS Lett.
585:2836–2844. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Leung CC and Glover JN: BRCT domains: easy
as one, two, three. Cell Cycle. 10:2461–2470. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Ouchi T: BRCA1 phosphorylation: biological
consequences. Cancer Biol Ther. 5:470–475. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Kotsopoulos J and Narod SA: Androgens and
breast cancer. Steroids. 77:1–9. 2012. View Article : Google Scholar
|
|
44.
|
Dumitrescu RG: Epigenetic markers of early
tumor development. Methods Mol Biol. 863:3–14. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Centrella M and McCarthy TL: Estrogen
receptor dependent gene expression by osteoblasts - direct,
indirect, circumspect, and speculative effects. Steroids.
77:174–184. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Wada-Hiraike O, Yano T, Nei T, Matsumoto
Y, Nagasaka K, Takizawa S, et al: The DNA mismatch repair gene
hMSH2 is a potent coactivator of oestrogen receptor alpha. Br J
Cancer. 92:2286–2291. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Murphy LC and Leygue E: The role of
estrogen receptor-β in breast cancer. Semin Reprod Med. 30:5–13.
2012.
|
|
48.
|
Warner M and Gustafsson JA: The role of
estrogen receptor beta (ERbeta) in malignant diseases - a new
potential target for antiproliferative drugs in prevention and
treatment of cancer. Biochem Biophys Res Commun. 396:63–66. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Lamartiniere CA: Timing of exposure and
mammary cancer risk. J Mammary Gland Biol Neoplasia. 7:67–76. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Shaik AP, Jamil K and Das P: CYP1A1
polymorphisms and risk of prostate cancer: a meta-analysis. Urol J.
6:78–86. 2009.PubMed/NCBI
|
|
51.
|
Cancel-Tassin G and Cussenot O: Prostate
cancer genetics. Minerva Urol Nefrol. 57:289–300. 2005.
|
|
52.
|
Agoulnik IU and Weigel NL: Androgen
receptor action in hormone-dependent and recurrent prostate cancer.
J Cell Biochem. 99:362–372. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Purohit A and Foster PA: Steroid sulfatase
inhibitors for estrogen- and androgen-dependent cancers. J
Endocrinol. 212:99–110. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Banerjee S, Li Y, Wang Z and Sarkar FH:
Multi-targeted therapy of cancer by genistein. Cancer Lett.
69:226–242. 2008. View Article : Google Scholar
|
|
55.
|
Park S and Choi J: Inhibition of
beta-catenin/Tcf signaling by flavonoids. J Cell Biochem.
110:1376–1385. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Power KA and Thompson LU: Can the
combination of flaxseed and its lignans with soy and its
isoflavones reduce the growth stimulatory effect of soy and its
isoflavones on established breast cancer? Mol Nutr Food Res.
51:845–856. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Sánchez Y, Amrán D, Fernández C, de Blas E
and Aller P: Genistein selectively potentiates arsenic
trioxide-induced apoptosis in human leukemia cells via reactive
oxygen species generation and activation of reactive oxygen
species-inducible protein kinases (p38-MAPK, AMPK). Int J Cancer.
123:1205–1214. 2008.
|
|
58.
|
Langeberg WJ, Kwon EM, Koopmeiners JS,
Ostrander EA and Stanford JL: Population-based study of the
association of variants in mismatch repair genes with prostate
cancer risk and outcomes. Cancer Epidemiol Biomarkers Prev.
19:258–264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Lacroix-Triki M, Lambros MB, Geyer FC,
Suarez PH, Reis-Filho JS and Weigelt B: Absence of microsatellite
instability in mucinous carcinomas of the breast. Int J Clin Exp
Pathol. 27:22–31. 2010.
|
|
60.
|
Norris AM, Woodruff RD, D’Agostino RB Jr,
Clodfelter JE and Scarpinato KD: Elevated levels of the mismatch
repair protein PMS2 are associated with prostate cancer. Prostate.
67:214–225. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Chen Y, Wang J, Fraig MM, Henderson K,
Bissada NK, Watson DK, et al: Alterations in PMS2, MSH2 and MLH1
expression in human prostate cancer. Int J Oncol. 22:1033–1043.
2003.PubMed/NCBI
|
|
62.
|
Van Poppel H and Tombal B: Chemoprevention
of prostate cancer with nutrients and supplements. Cancer Manag
Res. 3:91–100. 2011.PubMed/NCBI
|
|
63.
|
Kristal AR, Arnold KB, Neuhouser ML,
Goodman P, Platz EA, Albanes D, et al: Diet, supplement use, and
prostate cancer risk: results from the prostate cancer prevention
trial. Am J Epidemiol. 172:566–577. 2010. View Article : Google Scholar : PubMed/NCBI
|