Introduction
Colorectal cancer (CRC) is the third most common
epithelial malignancy worldwide: approximately 1,000,000 new cases
and 500,000 deaths occur each year (1). Liver metastases develop in 40–50% of
patients with CRC and represent one of the most common causes of
death. Surgical resection remains an expected procedure to ensure
long-term survival or cure (2).
After hepatectomy to treat metastatic liver tumor, activation of
the signaling pathway from c-Met-related hepatocyte growth factor
(HGF) becomes important in the progress of liver regeneration.
Because the HGF/c-Met pathway also plays a critical role in the
carcinogenesis of CRC (3), an
increased level of serum HGF following hepatectomy has been feared
to prompt cancer growth. We previously reported that c-Met
overexpression was closely associated with liver metastasis, but
c-Met expression is reduced in liver metastatic lesions compared
with that seen in primary lesions (4). Therefore, surgical resection might
unfavorably affect cancer cell progression, but the mechanism and
outcome of c-Met expression remain unclear.
During key biological processes such as embryonic
development, tissue remodeling, restitution, or wound repair,
epithelial cells must escape from their rigid structural
constraints through a well-known process termed
epithelial-mesenchymal transition (EMT) (5). As an inducer of EMT in normal mammary
epithelial cells, transforming growth factor-β (TGF-β) was first
described and evaluated as a key factor (6). The TGF-β family of polypeptides is
associated with a wide variety of biological functions, and its
effect is elicited through activation of Smad2/3/4 for
translocation from receptor to the nucleus. In CRC cells, loss of
TGF-β sensitivity is frequently due to loss of or mutation in the
signaling pathway, notably to its receptor and to the Smad process
(7,8). TGF-β induces non-Smad pathways
including those of mitogen-activated protein kinase (MAPK) or
phosphoinositide 3-kinase (PI3K) (9). Substantial activation of the
HGF/c-Met pathway also leads to scattering and invasion of cancer
cells through activation of the cell signaling pathway, and it may
regulate EMT (10).
The purpose of the present study was to further
develop our previous study on c-Met expression in CRC (4), to investigate EMT in the process of
liver metastases, and to evaluate the effects of chemotherapy on
EMT cells as a therapeutic strategy for colorectal liver
metastasis.
Materials and methods
Cell lines and culture conditions
Cells from the CT26 murine colorectal carcinoma cell
line were obtained from the American Type Culture Collection (ATCC,
Manassas, VA). Cells were cultured in RPMI-1640 medium (Wako,
Osaka, Japan) supplemented with 10% heat-inactivated fetal bovine
serum (FBS), 1 mM HEPES buffer, 1 mM sodium pyruvate solution, and
1% penicillin-streptomycin-amphotericin solution (all from
Sigma-Aldrich, St. Louis, MO) in a humidified atmosphere of 5%
CO2/95% air at 37°C. Cells were passaged twice a
week.
Animals housing and in vivo
experiments
Male 5-week-old BALB/c mice were purchased from SRL
(Hamamatsu, Japan) and housed in the animal facilities of the
Division of Animal Experiment, Life Science Research Center, Gifu
University with free access to water and food. A liver metastatic
model of CRC was created by injection of 1.0×106 CT26
cells into the spleen of BALB/c mice as described previously
(4). At 21 days after injection,
murine spleen and liver were removed and evaluated by western blot
analysis, in an immunohistochemical study. Animal experiments in
this study were performed in compliance with the guidelines of the
Institute for Laboratory Animal Research, Gifu University Graduate
School of Medicine, and the UCCCR Guidelines for the Welfare of
Animals in Experimental Neoplasia.
Cell proliferation assay
Cell growth was assessed by a standard
3-(4,5-dimethyl-thiazol-2-yl)-2,5-dephenyltetrazolium bromide (MTT)
assay (11,12), which detects the dehydrogenase
activity in viable cells. A total of 3×103 CT26 cells
were seeded into each of the 96-well culture plates or the same
density of cells was seeded in 6-cm dish plates overnight and kept
in a humidified atmosphere of 5% CO2 and 95% air at
37°C. The medium was exchanged for serum-free RPMI-1640 medium, and
after 48-h incubation, growth stimulation by growth factors was
started by adding 5 ng/ml of TGF-β and 20 or 40 ng/ml of HGF to
each well in the same condition. Recombinant TGF-β1 and recombinant
HGF were purchased from R&D Systems (Minneapolis, MN). After 72
h, the culture medium was removed, and 100 μl of a 0.5-mg/ml
solution of MTT (Sigma-Aldrich) was added to each well. The plates
were then incubated for 4 h at 37°C. The culture medium was
replaced with 100 μl of dimethyl sulfoxide (Wako) per well,
and the absorbance at the 540-nm wavelength was measured using a
2104 EnVision Multilabel Reader (Perkin-Elmer, Waltham, MA).
In preparing the protein samples, cells were treated
with Akt inhibitor (BioVision, Inc., Mountain View, CA), and U-0126
(Cayman Chemical, Ann Arbor, MI) for 24 h before administration of
growth factors. 5-FU (Kyowa-Kirin, Tokyo, Japan) was administered
after 96 h of contact with growth factors.
Western blot analysis and antibodies
Treatment of the specimens was as described
previously (13–15). Cell lysates were boiled in Sample
Buffer Solution (Wako). Total cell protein extracts (20
μg/lane) were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis using SuperSep™ (Wako)
and were electrophoretically transfected onto polyvinyl difluoride
membranes. The membranes were blocked with PVDF blocking reagent
(Toyobo, Osaka, Japan) for 1 h. The membranes were then incubated
with primary antibodies against β-actin, E-cadherin, vimentin,
Snail (Snail1), Slug (Snail2), Smad pathway proteins (p-Smad2,
p-Smad3), p-ERK (extracellular signal-regulated kinase), ERK,
p-Akt, Akt, p-JNK (c-jun N-terminal kinase), JNK, and caspase-3
(1:5,000; Cell Signaling Technology, Danvers, MA) overnight at 4°C.
The primary antibodies were diluted with Can Get Signal Solution 1
(Toyobo). The membranes were then washed with Dako Washing Buffer
(Dako, Glostrup, Denmark) and incubated with the appropriate
secondary antibodies (1:25,000; Millipore, Darmstadt, Germany),
which were diluted with Can Get Signal Solution 2 (Toyobo). The
immunoreactive proteins were visualized by chemiluminescence using
ImmunoStar LD reagents (Wako), and images were captured by a
LAS-4000 (Fuji film, Tokyo, Japan).
Immunohistochemistry
An LSAB kit (Dako) was used for immunohistochemical
analysis (16,17). In brief, sections were pretreated
by microwave treatment in citrate buffer for 15 min to retrieve
antigenicity. After peroxidase activity was blocked with 3%
H2O2/methanol for 10 min, sections were
incubated with normal goat serum (Dako) for 20 min to block
non-specific antibody binding sites. Sections were incubated with
the following primary antibodies: c-Met, 1:200 dilution (Santa Cruz
Biotechnology, Santa Cruz, CA) and E-cadherin, 1:100 dilution (Cell
Signaling Technology). Sections were incubated with primary
antibody for 1 h at 25°C followed by incubations with biotinylated
anti-rabbit/mouse IgG and peroxidase-labelled streptavidin for 10
min each. Staining was completed by incubation for 10 min with
substrate-chromogen solution. Sections were counterstained with
0.1% hematoxylin. No specific staining was observed in the negative
control slides prepared without primary antibody.
Transfection and small interfering RNA
experiments for c-Met
CT26 cells were cultured in a medium without
antibiotics for 24 h before transfection to 50–70% confluence.
Cells were transfected with a small interfering RNA (siRNA)
oligonucleotide using Lipofectamine RNAiMAX (Invitrogen, Carlsbad,
CA) in a final siRNA concentration of 40 nmol/l in serum-free
Opti-MEM (Invitrogen) according to the manufacturer’s instructions.
At 6 h after transfection, the medium was replaced with RPMI-1640
medium supplemented with 10% FBS. The total proteins were extracted
48 h later, and expression levels of the c-Met protein were
analyzed by western blotting. siRNA oligonucleotides for c-Met were
purchased from Invitrogen.
Statistical analysis
The data were examined using the Student’s t-test,
χ2 test, and ANOVA or Kruskal-Wallis test (with
appropriate post hoc analysis for multiple comparisons) to
determine statistical significance. p-value of <0.05 was
regarded as statistically significant.
Results
Effect of c-Met on cancer
progression
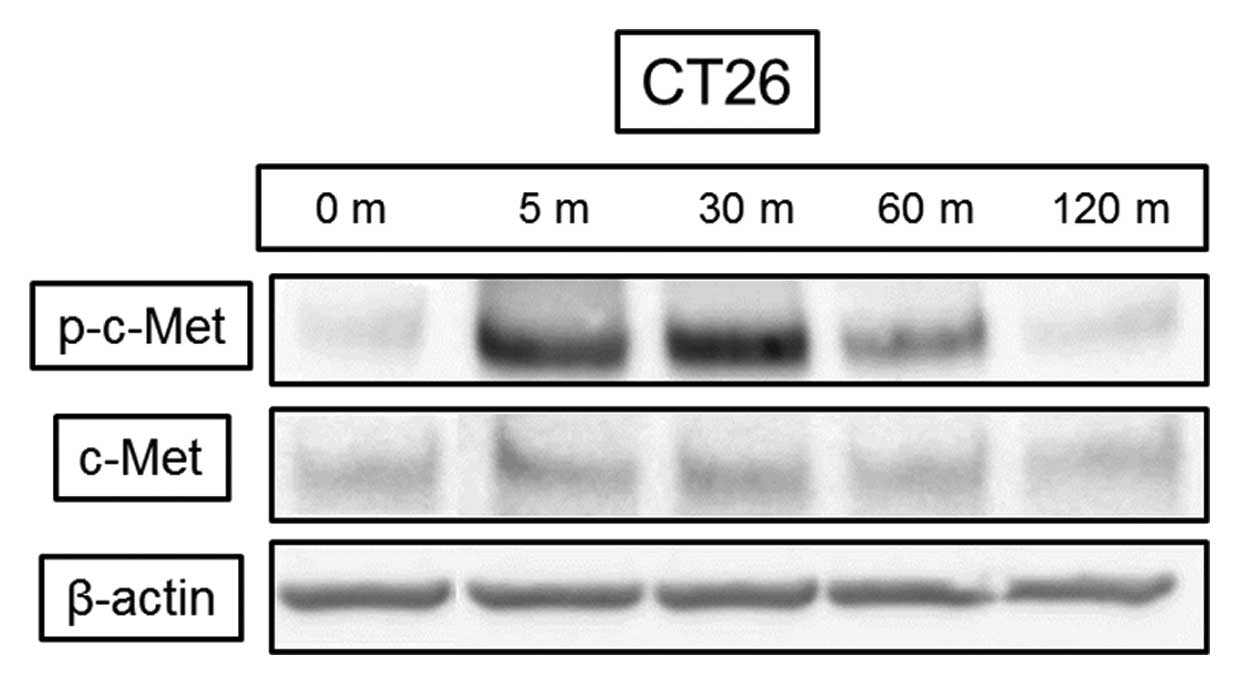
HGF induced c-Met phosphorylation in the CT26
colorectal cancer cell line after 5 min with no change in the total
amount of protein (Fig. 1). HGF at
both the 20- and 40-ng/ml concentrations resulted in significant
cell proliferation of 110% and 115%, respectively, (p<0.05)
compared with control at 72 h (data not shown).
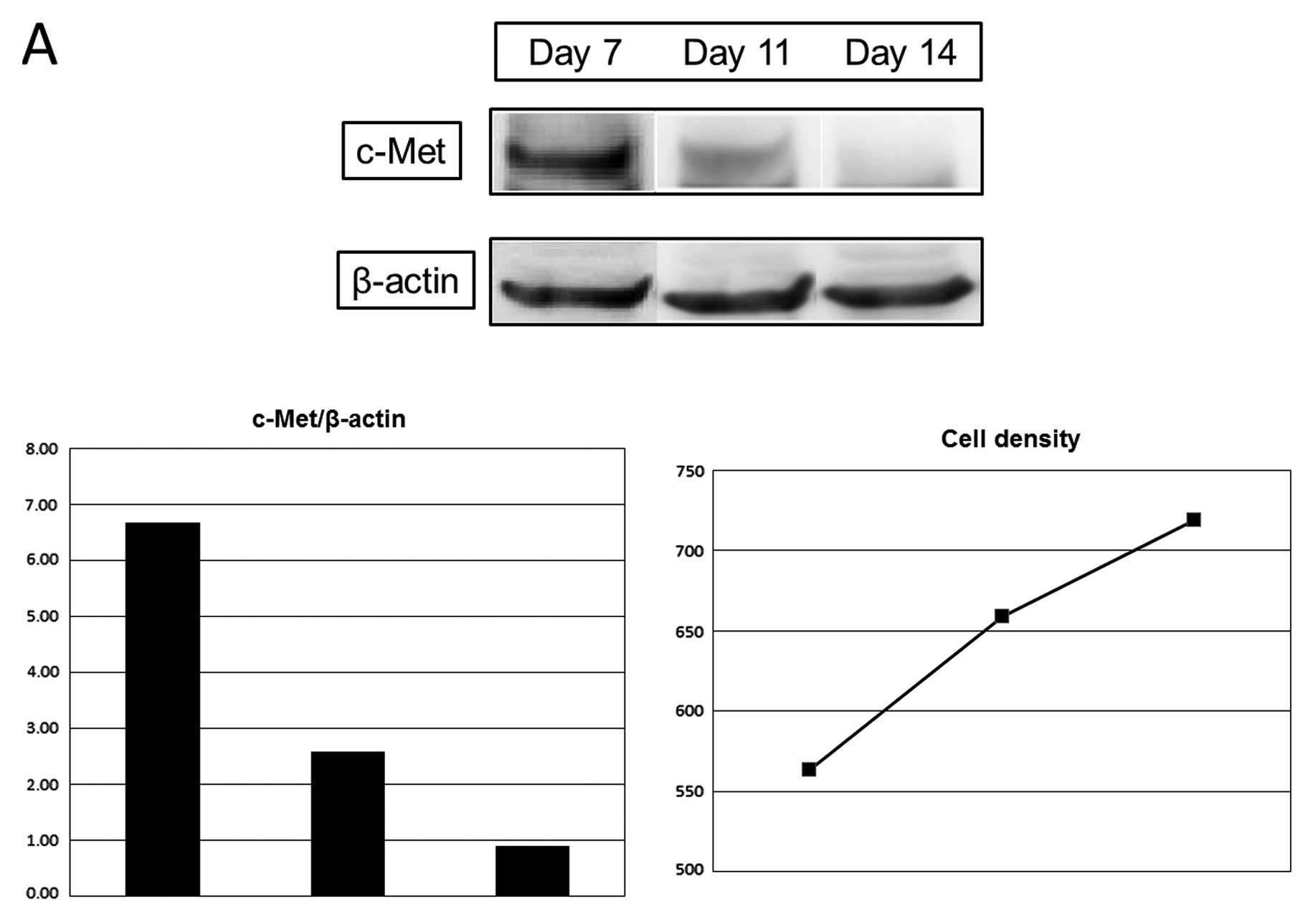
Expression of c-Met was decreased with the increase
of cell density, 39% on day 11 and 13% on day 14, compared with day
7, and E-cadherin expression was increased, 104% on day 11 and 139%
on day 14 (Fig. 2).
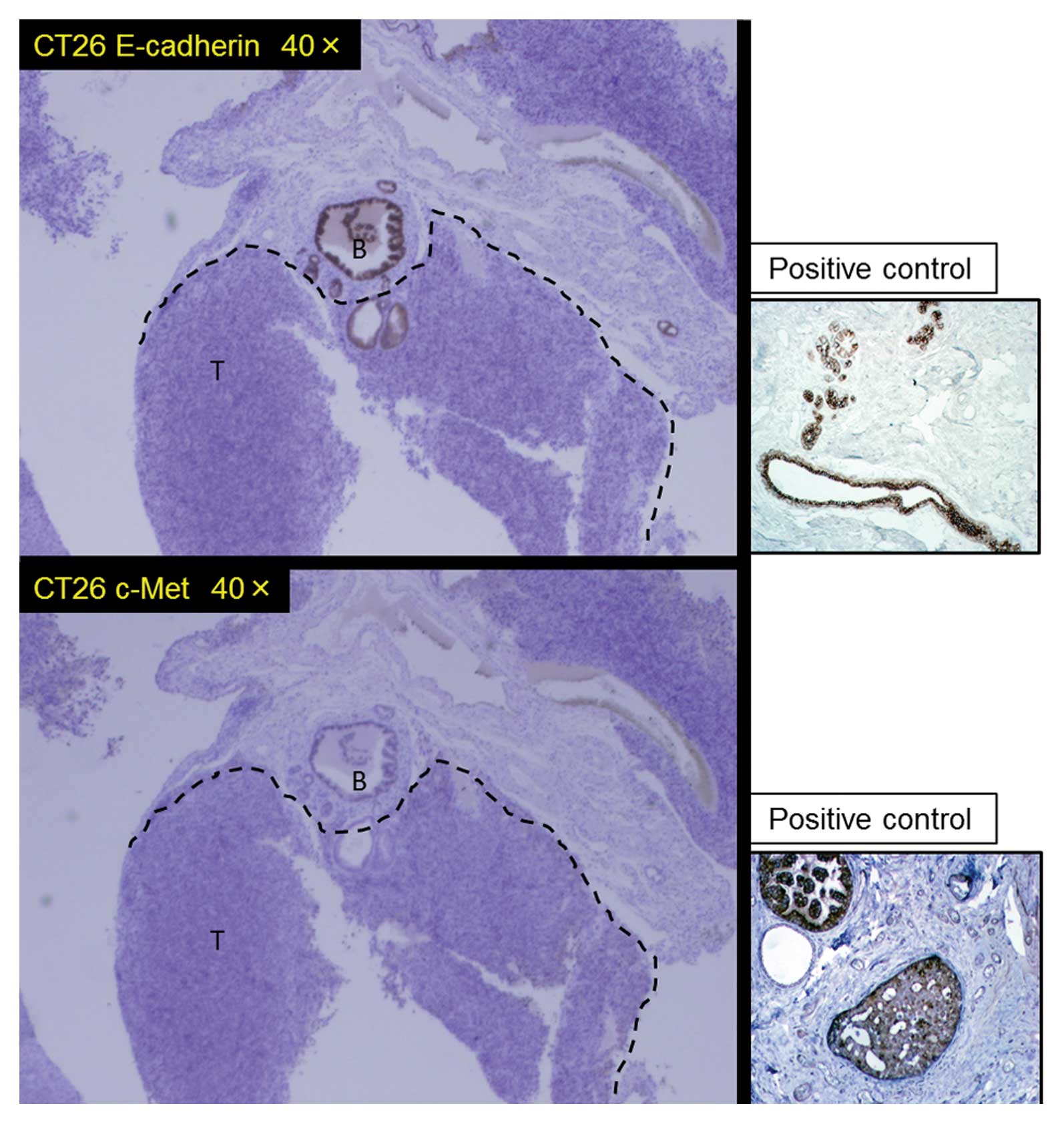
Immunohistochemical study revealed the expression of both c-Met and
E-cadherin to diminish in the metastatic liver tumors as shown in
Fig. 3. Cells in which c-Met mRNA
was knocked down by siRNA techniques (Fig. 4A) clearly showed reduced liver
metastases compared with regular cells at day 21 in the in
vivo BALB/c mouse model (Fig.
4B).
Signal pathway to EMT by TGF-β and
HGF
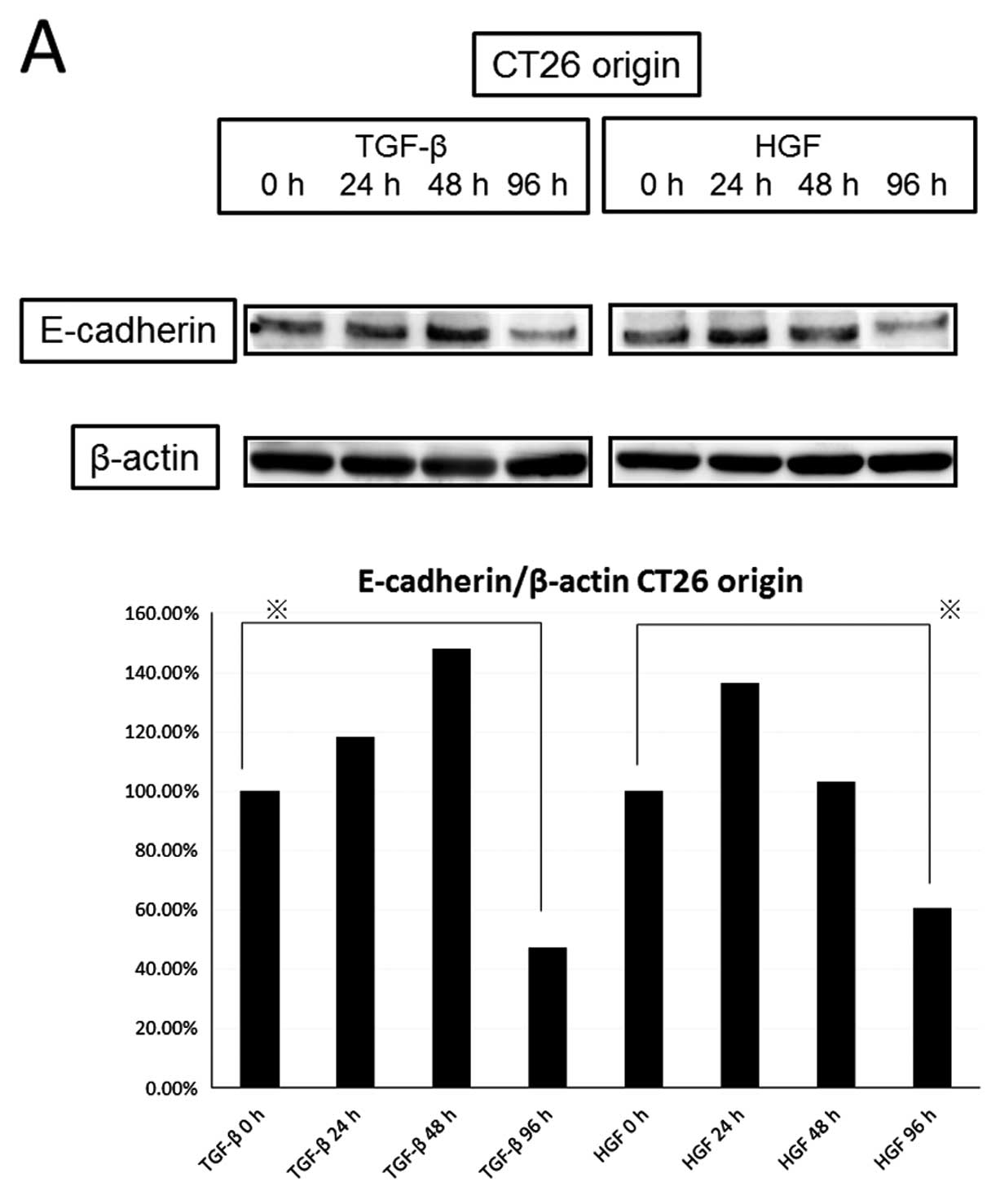
We compared the expression of E-cadherin and
vimentin by HGF or TGF-β (Fig. 5).
The 20-ng/ml concentration of HGF reduced the level of E-cadherin
as well as the 5-ng/ml concentration of TGF-β in a time-dependent
manner (TGF-β decreased to 47.3% and HGF decreased to 60.8% at 96
h), but knockdown of c-Met diminished the decrease of E-cadherin by
HGF. HGF increased the expression of vimentin similarly to TGF-β in
a time-dependent manner (increased to 308% by TGF-β and 221% by HGF
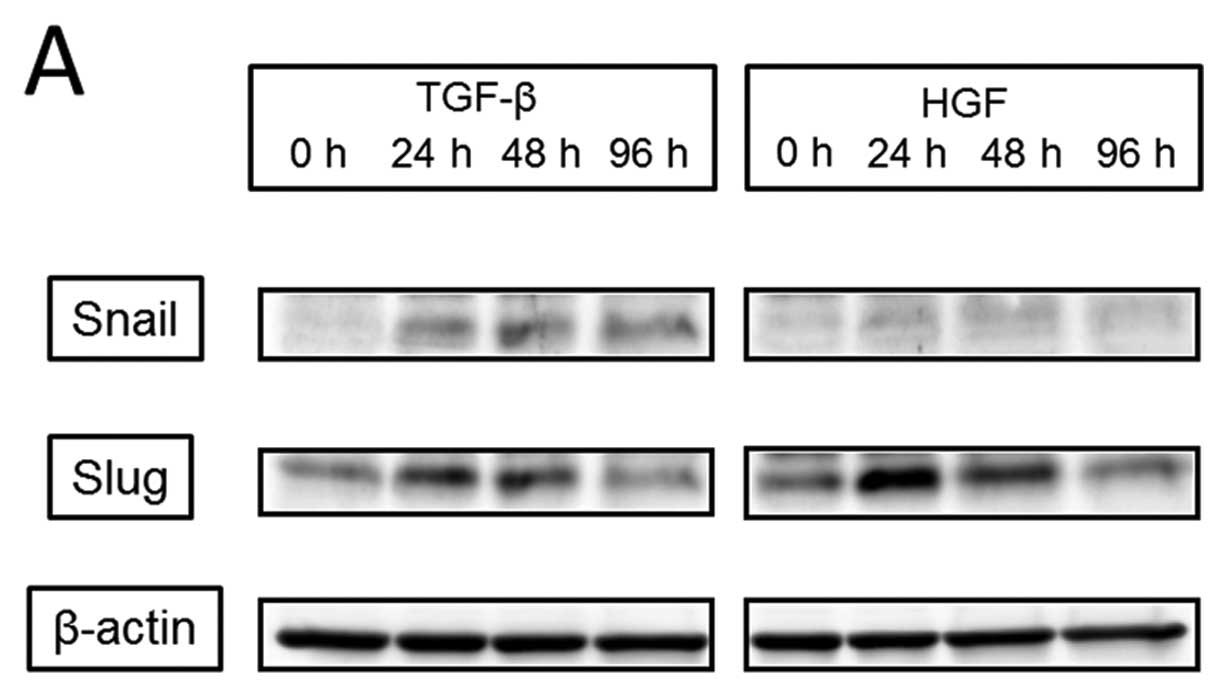
at 96 h). Next, we observed activation of EMT transcription factors
(Fig. 6). Expression of Snail was
detected with the addition of TGF-β starting at 24 h; it peaked at
48 h and remained peaked continuously to 96 h. However, expression
of Snail was not detected at any time after the addition of HGF.
Slug expression was increased at 24 h by the addition of both TGF-β
and HGF. With TGF-β, the peak was detected at 24 h and continued to
48 h, and with HGF, the peak was detected at 24 h and slightly
decreased at 48 h. The increase in Slug expression was diminished
at 96 h for both TGF-β and HGF. TGF-β induced the activation of
Smad2 at 30 min, which continued to 90 min, and Smad3, which peaked
at 30 min, despite no differences in the total amounts of Smad2/3,
Smad4, and Smad7 present (data not shown). In contrast, HGF exerted
no action on these factors, and c-Met knockdown had no effect on
the TGF-β-induced cell signal pathway (data not shown).
 | Figure 6.Differences in the EMT signaling
pathway between TGF-β and HGF. (A, upper panel), Snail was detected
by addition of TGF-β starting from 24 h, peaked at 48 h, and
continued to 96 h, but was not detected at any time by addition of
HGF. (A, lower panel), Slug was increased by both TGF-β and HGF.
With TGF-β, the peak was detected at 24 h and continued to 48 h;
with HGF, the peak was detected at 24 h with a slight decrease at
48 h. (B), TGF-β induced the activation of Smad2 for 30 min, which
continued to 90 min, and Smad3, which peaked at 30 min; however, no
activation was induced by HGF. |
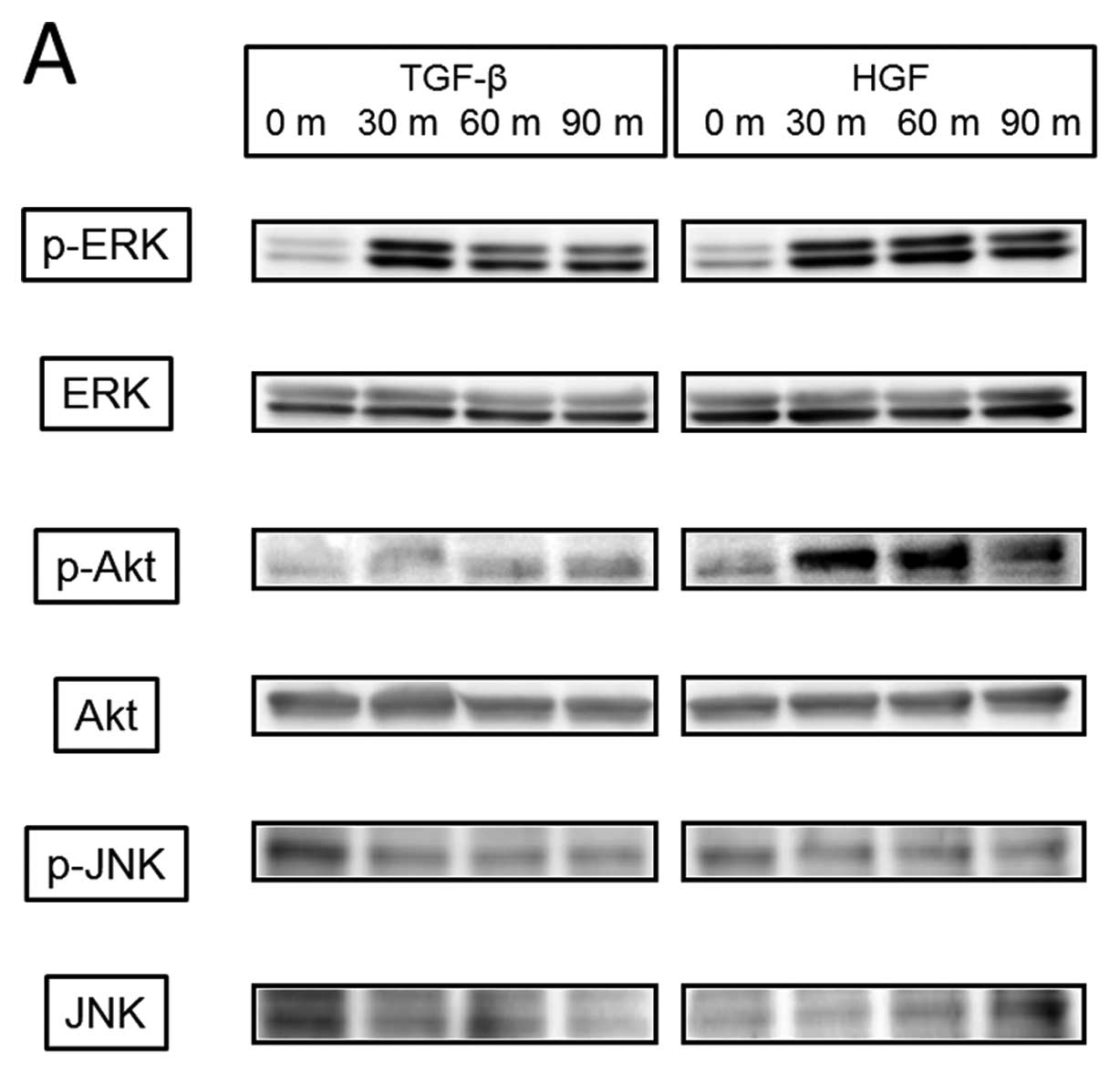
We then examined the cellular signaling pathway
induced by TGF-β or HGF (Fig. 7A)
and found that TGF-β induced phosphorylation of ERK from 30 min
with weak phosphorylation continuing to 90 min, but no
phosphorylation of Akt and JNK. HGF mediated phosphorylation of
both ERK and Akt from 30 min, which continued over 90 min, but not
JNK. Akt inhibitor blocked phosphorylation of Akt but had no effect
on TGF-β-induced activation of ERK, Snail, and Slug. U-0126, which
is a MAPK kinase inhibitor, did not reduce Snail activity either by
TGF-β at a concentration that blocked ERK phosphorylation (Fig. 7B). In contrast, HGF-induced Slug
activation was completely inhibited by both Akt inhibitor and
U-0126 (Fig. 7C), and changes in
E-cadherin and vimentin phosphorylation by HGF were also blocked by
these inhibitors (data not shown).
Effect of chemotherapeutic agent on the
EMT process
5-Fluorouracil (5-FU), one of the most common and
basic of chemotherapeutic agents, mediated cell death of the
present CT26 cell line (IC50: 4.87±0.61 μM) for
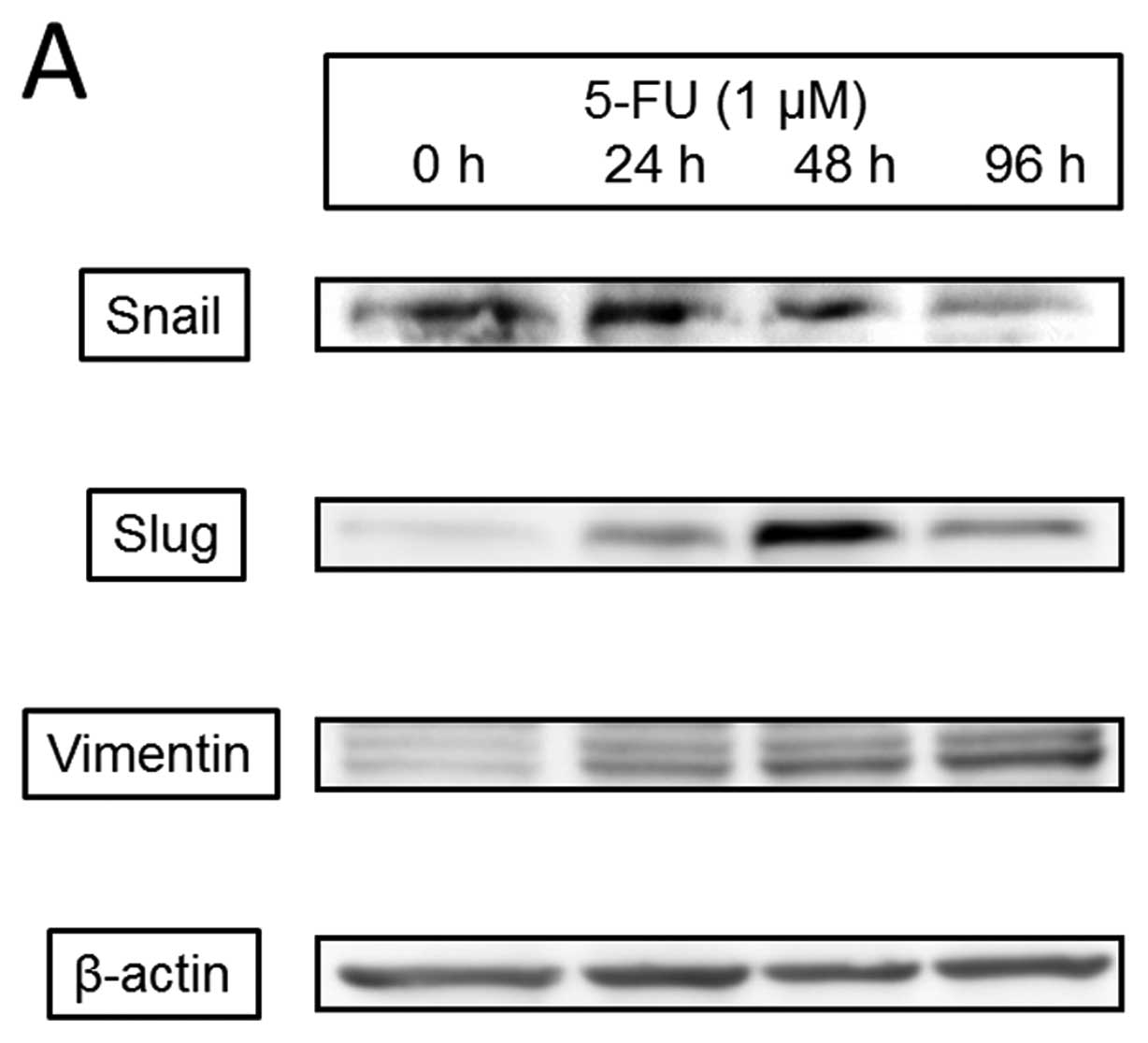
72 h. We evaluated the effects of 5-FU on EMT transcription factors
(Fig. 8) and found that 1
μM 5-FU increased expression of Snail, which peaked at 24 h
and gradually decreased at 96 h; Slug, which began at 24 h and
clearly peaked at 48 h; and vimentin, which increased at 24 h and
continued to 96 h. 5-FU phosphorylated ERK and Akt after 30 min but
had no effect on Smad2/3. 5-FU also induced caspase-3 activation at
24 h that peaked at 48 h.
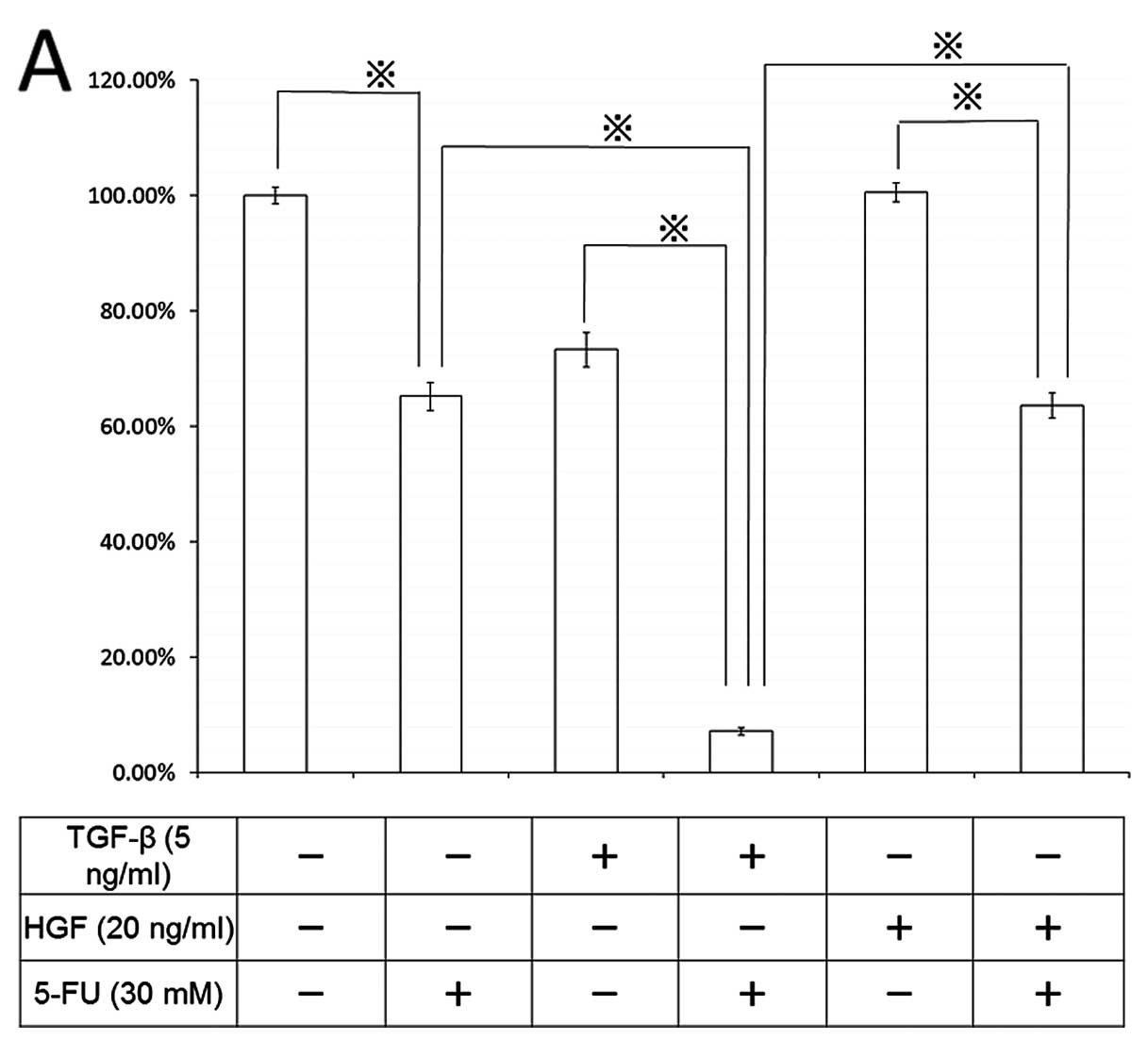
Finally, we studied the chemotherapeutic effect of
5-FU on cell death and signal transduction in the EMT process
(Fig. 9). Pretreatment of CT26
cells with 5 ng/ml TGF-β for 96 h enhanced 5-FU-induced cell death
by 10%, compared with the control, and 20 ng/ml HGF also augmented
the chemotherapeutic effect of 5-FU by 63%. During EMT induced by
both TGF-β and HGF, 5-FU-induced stronger ERK phosphorylation than
that in non-EMT-induced cells; however, no effect on caspase-3 was
detected (data not shown).
Discussion
In recent years, EMT has been the focus of
investigation into the mechanisms of the liver metastatic process
(5,18,19).
EMT changes the morphology of the cancer cell to that of a spindle
shape with mediating migratory competence and invasive capacity
overflow (20). Following
metastasis of cancer cells promoted by EMT, the induction of MET
(mesenchymalepithelial transition) occurs, and cancer cells build
up in a distant organ (18). This
regulation is controlled by already known and newly discovered
signaling pathways (18). Among
factors such as EMT that stimulate scattering of epidermal cells,
the HGF/c-Met pathway is also enhanced (10,21).
The present study evaluated both HGF action on the process of liver
metastasis and estimated which therapeutic procedure, hepatectomy
and/or chemotherapy, is more beneficial for the treatment of liver
metastases.
A hallmark of EMT represents the loss of E-cadherin,
an important caretaker of the epithelial phenotype (18,22).
E-cadherin is a cell-cell adhesion molecule, and its loss is
consistently observed at sites of EMT during cancer metastases,
indicating that its level of expression correlates with cancer
progression (23). A recent study
showed E-cadherin itself to interact with receptor tyrosine kinases
such as c-Met through cell-cell adhesion (24). The present study found that with
the decreased expression of c-Met, both cell density and E-cadherin
increased. Vimentin, another commonly used molecular marker for
EMT, is well known (25), and its
increase with the loss of expression of E-cadherin by HGF described
in the present study may be involved in EMT for colon cancer cells.
HGF is also a major driver of cancer progression (26) and regulates regular signaling
pathways, such as those of Akt or ERK, to promote carcinogenesis
(27). Because TGF-β, one of the
most essential inducers of EMT, is important in the progression of
carcinoma to an invasive state (28), we evaluated differences in the
signal pathway.
Among the molecular factors related to EMT
induction, such as Snail, Slug, Twist, EF1/ZEB1, and SIP1/ZEB2
(21), Snail is a zinc finger
transcription factor that induces EMT by directly repressing
E-cadherin expression and confers epithelial cells with migratory
and invasive properties as an important step for metastasis
(6,29). Slug, another zinc finger protein,
is closely related to the Snail pathway and regulation of
E-cadherin gene transcription (30). Furthermore, other cross-talk
pathways have also been focused on (31), and the synergy between Ras
signaling and Smad signaling was found to be critical in the
induction of EMT. In fact, the receptor of TGF-β is well known to
activate MAPKs, such as ERK, JNK, and p38 MAP kinases, PI3 kinase,
and small GTPases (31). The
present study showed that ERK/Akt signaling, but not the Smad
pathway, might be the main pathway in HGF-induced EMT, despite the
fact that the Smad pathway, but not ERK/Akt, was critical for
induction of EMT by TGF-β. The MAPK/Akt pathway is indispensable in
HGF/c-Met signaling (32), and
activated Akt was reported to induce loss of cell-cell adhesion,
morphological changes, and induction of cell motility (33). Additionally, HGF-induced cell
scattering or invasive action is abrogated due to down-regulation
of phosphorylated Akt (10). In
contrast, in the induction of EMT, TGF-β cooperates with other
signaling pathways, such as Wnt (34), Hedgehog (35), and Notch (36), which are all pathways linked to the
stem cell renewal pathway (37).
TGF-β-induced EMT or its repressors, such as Twist or Snail, also
confer stem cell-like properties to non-carcinogenic, immortalized
human mammary epithelial cells, providing the first link between
EMT and ‘stemness’. Indeed, the EMT process was found to generate
stem-like properties in breast cancer cells (38). As a concept of cancer stemness,
Snail includes not only stem cell-like properties but also
resistance to chemo/radiation therapy under the self-renewal
process (39). Namely, there is a
possibility for cancer therapy through inhibition of EMT not only
to reduce metastasis but also to improve drug sensitivity (40). Therefore, we are planning
additional study of chemotherapeutic agents to evaluate a novel
concept for therapeutic strategy.
Chemotherapeutic agents such as paclitaxel (41) or oxaliplatin (42) make cancer cells susceptible to EMT.
The present study showed 5-FU to phosphorylate ERK/Akt and activate
Snail and Slug, but not Smad, in an EMT-like manner. Because the
ERK pathway has dual actions related to both cell proliferation and
growth inhibition (12,13,43),
it is still unclear whether 5-FU-induced ERK activation itself is
directly related with EMT. However, there is another possibility,
that 5-FU produces reactive oxygen species (ROS) and that these ROS
inhibit phosphatase action of cell signaling-related proteins to
lead to phosphorylation of ERK accordingly (11,12).
ROS itself was shown to lead to EMT though the expression of Snail
and to cause genomic instability and oxidative damage to DNA
(44). Taken together, EMT was
evaluated in relation to drug resistance of anticancer agents
(41). However, the early
response, but not the long-term reaction, to these agents is still
unclear and controversial. Some reports showed high expression of
E-cadherin to relate to higher chemosensitivity (45,46),
whereas its expression has also been related to lower
chemosensitivity (47). Further,
expression of E-cadherin does not correlate with the effect of
chemotherapy or reflect patient prognosis (48). Commonly, loss or low expression of
E-cadherin in liver metastasis occurs more frequently in CRC
related to poor patient prognosis (49). In the present study, low expression
of E-cadherin was detected in the EMT process and high
chemosensitivity for 5-FU was shown. In the signalling pathway as
well, ERK activation by 5-FU-increased more when EMT was present,
indicating ROS expression to be higher. Although the connection
between E-cadherin expression and drug-sensitivity in the present
study was unclear, EMT and the signaling thereof were affected by
different agents, TGF-β and HGF. In particular, the ERK/Akt pathway
might be critical in the process of HGF-induced EMT.
After hepatectomy for liver metastasis from CRC,
serum HGF elevates for liver regeneration (50), but this does not increase the risk
of new metastasis and aggressive tumor formation. As demonstrated
in the present study, the reason was due to diminished c-Met
expression at the metastatic site. Chemotherapeutic agents should
be effective even if c-Met expression results in regeneration of
cancer cells. However, long-term use of chemotherapeutic agents
might induce drug-resistant and distant metastases through the
activation of the EMT-related signaling pathway. Further
investigation will be necessary to determine therapeutic strategy
with the use of anticancer agents.
References
|
1.
|
Bingham S and Riboli E: Diet and cancer -
the European prospective investigation into cancer and nutrition.
Nat Rev Cancer. 4:206–215. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Benoist S and Nordlinger B: The role of
preoperative chemotherapy in patients with resectable colorectal
liver metastases. Ann Surg Oncol. 16:2385–2390. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Takeuchi H, Bilchik A, Saha S, et al:
c-MET expression level in primary colon cancer: a predictor of
tumor invasion and lymph node metastases. Clin Cancer Res.
9:1480–1488. 2003.PubMed/NCBI
|
|
4.
|
Matsui S, Osada S, Tomita H, et al:
Clinical significance of aggressive hepatectomy for colorectal
liver metastasis, evaluated from the HGF/c-Met pathway. Int J
Oncol. 37:289–297. 2010.PubMed/NCBI
|
|
5.
|
Bates RC and Mercurio AM: The
epithelial-mesenchymal transition (EMT) and colorectal cancer
progression. Cancer Biol Ther. 4:365–370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Zavadil J and Bottinger EP: TGF-beta and
epithelial-to-mesenchymal transitions. Oncogene. 24:5764–5774.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Bierie B and Moses HL: Tumour
microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer.
Nat Rev Cancer. 6:506–520. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Zhang B, Halder SK, Zhang S and Datta PK:
Targeting transforming growth factor-beta signaling in liver
metastasis of colon cancer. Cancer Lett. 277:114–120. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Chang HY, Kao MC, Way TD, Ho CT and Fu E:
Diosgenin suppresses hepatocyte growth factor (HGF)-induced
epithelialmesenchymal transition by down-regulation of Mdm2 and
vimentin. J Agric Food Chem. 59:5357–5363. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Osada S, Tomita H, Tanaka Y, et al: The
utility of vitamin K3 (menadione) against pancreatic cancer.
Anticancer Res. 28:45–50. 2008.PubMed/NCBI
|
|
12.
|
Osada S, Sakashita F, Hosono Y, et al:
Extracellular signal-regulated kinase phosphorylation due to
menadione-induced arylation mediates growth inhibition of pancreas
cancer cells. Cancer Chemother Pharmacol. 62:315–320. 2008.
View Article : Google Scholar
|
|
13.
|
Osada S, Saji S and Osada K: Critical role
of extracellular signal-regulated kinase phosphorylation on
menadione (vitamin K3) induced growth inhibition. Cancer.
91:1156–1165. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Osada S and Carr BI: Mechanism of novel
vitamin K analog induced growth inhibition in human hepatoma cell
line. J Hepatol. 34:676–682. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Osada S, Osada K and Carr BI: Tumor cell
growth inhibition and extracellular signal-regulated kinase (ERK)
phosphorylation by novel K vitamins. J Mol Biol. 314:765–772. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Osada S, Sakashita F, Katoh H, Sugiyama Y
and Adachi Y: Identification of an immune tolerance reaction in
response to pretreatment with frozen pancreatic tissue in islet
cell transplantation in rats. Pancreas. 30:e29–e33. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Osada S, Imai H, Tomita H, et al: Vascular
endothelial growth factor protects hepatoma cells against oxidative
stress-induced cell death. J Gastroenterol Hepatol. 21:988–993.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Wilmanns C, Steinhauer S, Grossmann J and
Ruf G: Site-dependent differences in clinical, pathohistological,
and molecular parameters in metastatic colon cancer. Int J Biol
Sci. 5:458–465. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Tsuji T, Ibaragi S, Shima K, et al:
Epithelial-mesenchymal transition induced by growth suppressor
p12CDK2-AP1 promotes tumor cell local invasion but suppresses
distant colony growth. Cancer Res. 68:10377–10386. 2008. View Article : Google Scholar
|
|
21.
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelialmesenchymal transition: new insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Kang Y and Massague J:
Epithelial-mesenchymal transitions: twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Wu Y and Zhou BP: New insights of
epithelial-mesenchymal transition in cancer metastasis. Acta
Biochim Biophys Sin. 40:643–650. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Xie LQ, Bian LJ, Li Z, Li Y, Li ZX and Li
B: Altered expression of E-cadherin by hepatocyte growth factor and
effect on the prognosis of nasopharyngeal carcinoma. Ann Surg
Oncol. 17:1927–1936. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Walsh LA and Damjanovski S: IGF-1
increases invasive potential of MCF 7 breast cancer cells and
induces activation of latent TGF-beta1 resulting in epithelial to
mesenchymal transition. Cell Commun Signal. 9:102011. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Ding W, You H, Dang H, et al:
Epithelial-to-mesenchymal transition of murine liver tumor cells
promotes invasion. Hepatology. 52:945–953. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Ogunwobi OO and Liu C: Hepatocyte growth
factor upregulation promotes carcinogenesis and
epithelial-mesenchymal transition in hepatocellular carcinoma via
Akt and COX-2 pathways. Clin Exp Metastasis. 28:721–731. 2011.
View Article : Google Scholar
|
|
28.
|
Nawshad A, Lagamba D, Polad A and Hay ED:
Transforming growth factor-beta signaling during
epithelial-mesenchymal transformation: implications for
embryogenesis and tumor metastasis. Cells Tissues Organs.
179:11–23. 2005. View Article : Google Scholar
|
|
29.
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: an alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Pon YL, Zhou HY, Cheung AN, Ngan HY and
Wong AS: p70 S6 kinase promotes epithelial to mesenchymal
transition through snail induction in ovarian cancer cells. Cancer
Res. 68:6524–6532. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Miyazono K: Transforming growth factor-β
signaling in epithelialmesenchymal transition and progression of
cancer. Proc the Japan Academy, Series B. 85:314–323. 2009.
|
|
32.
|
Trusolino L, Bertotti A and Comoglio PM:
MET signalling: principles and functions in development, organ
regeneration and cancer. Nat Rev Mol Cell Biol. 11:834–848. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Larue L and Bellacosa A:
Epithelial-mesenchymal transition in development and cancer: role
of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene.
24:7443–7454. 2005.
|
|
34.
|
Vincan E and Barker N: The upstream
components of the Wnt signalling pathway in the dynamic EMT and MET
associated with colorectal cancer progression. Clin Exp Metastasis.
25:657–663. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Karhadkar SS, Bova GS, Abdallah N, et al:
Hedgehog signalling in prostate regeneration, neoplasia and
metastasis. Nature. 431:707–712. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Wang Z, Banerjee S, Li Y, Rahman KM, Zhang
Y and Sarkar FH: Down-regulation of notch-1 inhibits invasion by
inactivation of nuclear factor-kappaB, vascular endothelial growth
factor, and matrix metalloproteinase-9 in pancreatic cancer cells.
Cancer Res. 66:2778–2784. 2006. View Article : Google Scholar
|
|
37.
|
Fuxe J, Vincent T and Garcia de Herreros
A: Transcriptional crosstalk between TGF-beta and stem cell
pathways in tumor cell invasion: role of EMT promoting Smad
complexes. Cell Cycle. 9:2363–2374. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Creighton CJ, Chang JC and Rosen JM:
Epithelial-mesenchymal transition (EMT) in tumor-initiating cells
and its clinical implications in breast cancer. J Mammary Gland
Biol Neoplasia. 15:253–260. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Mani SA, Guo W, Liao MJ, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: an emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Kajiyama H, Shibata K, Terauchi M, et al:
Chemoresistance to paclitaxel induces epithelial-mesenchymal
transition and enhances metastatic potential for epithelial ovarian
carcinoma cells. Int J Oncol. 31:277–283. 2007.
|
|
42.
|
Yang AD, Fan F, Camp ER, et al: Chronic
oxaliplatin resistance induces epithelial-to-mesenchymal transition
in colorectal cancer cell lines. Clin Cancer Res. 12:4147–4153.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Tanahashi T, Osada S, Imai H, et al:
Signal transduction of vitamin K3 for pancreas cancer therapy.
Oncol Rev. 5:57–60. 2010. View Article : Google Scholar
|
|
44.
|
Radisky DC, Levy DD, Littlepage LE, et al:
Rac1b and reactive oxygen species mediate MMP-3-induced EMT and
genomic instability. Nature. 436:123–127. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Matsubara D, Ishikawa S, Oguni S,
Aburatani H, Fukayama M and Niki T: Molecular predictors of
sensitivity to the MET inhibitor PHA665752 in lung carcinoma cells.
J Thorac Oncol. 5:1317–1324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Koo JS, Jung W and Jeong J: The predictive
role of E-cadherin and androgen receptor on in vitro
chemosensitivity in triple-negative breast cancer. Jpn J Clin
Oncol. 39:560–568. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Nakamura T, Kato Y, Fuji H, Horiuchi T,
Chiba Y and Tanaka K: E-cadherin-dependent intercellular adhesion
enhances chemoresistance. Int J Mol Med. 12:693–700.
2003.PubMed/NCBI
|
|
48.
|
Graziano F, Mandolesi A, Ruzzo A, et al:
Predictive and prognostic role of E-cadherin protein expression in
patients with advanced gastric carcinomas treated with palliative
chemotherapy. Tumour Biol. 25:106–110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Yu SJ, Yu JK, Ge WT, Hu HG, Yuan Y and
Zheng S: SPARCL1, Shp2, MSH2, E-cadherin, p53, ADCY-2 and MAPK are
prognosis-related in colorectal cancer. World J Gastroenterol.
17:2028–2036. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Osada S, Kanematsu M, Imai H and Goshima
S: Clinical significance of serum HGF and c-Met expression in tumor
tissue for evaluation of properties and treatment of hepatocellular
carcinoma. Hepatogastroenterology. 55:544–549. 2008.PubMed/NCBI
|