Introduction
Tumor necrosis factor (TNF)-related
apoptosis-inducing ligand (TRAIL) is a member of the TNF family
(1,2). Recombinant TRAIL (rhTRAIL) and its
agonist antibodies are capable of inducing apoptosis in human
cancer cells and sparing most normal human cells (3,4) and
are, therefore, currently under clinical trials as therapeutic
agents for human cancers (3,5).
TRAIL-induced apoptosis is mediated by the transmembrane receptor
death receptor 4 (DR4) (TRAIL-R1) (6) and DR5 (TRAIL-R2) (7) located on the target cell surface.
Each receptor contains a cytoplasmic region designated the ‘death
domain’ responsible for transducing the death signal. On ligand
binding, DR4 or DR5 initiates apoptosis by assembling death induced
signaling complex (DISC) components at the inner surface of the
plasma membrane (8). This is
achieved by their death domains recruiting the adaptor molecule
Fas-associated death domain (FADD) and the apoptosis initiating
protease pro-caspase-8 (or pro-caspase-10). Within the DISC,
pro-caspase-8 or pro-caspase-10 is autocatalytically cleaved and in
turn, subsequently cleaves and activates effector caspases, such as
caspase-3, caspase-6 and caspase-7 in an extrinsic apoptotic
pathway. At the same time, caspase-8 stimulates the cleavage of
Bid, thus amplifying caspase activation via mitochondrial
activation of the intrinsic apoptotic pathway (9).
TRAIL has been shown to induce apoptosis of lung
cancer cell lines, but recent studies have shown that some cancer
cells, including lung cancer cells, can acquire resistance during
TRAIL treatment (10–13). Patients with tumors which acquire
TRAIL-resistance during treatment probably suffer from the
potential side-effects rather than benefit from TRAIL treatment. Up
to now, little is known about the molecular mechanisms that
contribute to the development of acquired resistance during
treatment with TRAIL. Consequently, understanding acquired
TRAIL-resistance may enable us to design better strategies to
prevent its occurrence and to improve the therapeutic potential of
TRAIL. So far, intrinsic TRAIL-resistance has been attributed to
the loss of TRAIL death receptors, upregulation of TRAIL decoy
receptors, downregulation of caspase-8 or caspase-10, enhanced
expression of cellular FLICE-like inhibitory protein (cFLIP) and
cIAP or alterations in expression of the Bcl-2 family proteins
(14,15). However, none of these factors show
a consistent correlation with acquired TRAIL resistance in multiple
cancer cells.
For TRAIL-induced apoptosis, TRAIL binding to its
death receptors is an upstream event during caspase activation and
the receptors are crucial in initialing apoptosis. Accordingly,
downregulation of TRAIL death receptors and upregulation of TRAIL
decoy receptors both lead to TRAIL-resistance, also confirmed by a
previous study (17). However,
there seems to be no evidence of a direct correlation between the
expression level of a receptor and the sensitivity of cancer cells
to TRAIL (3,18). Recent studies suggest that mRNA and
protein expression levels of death receptors do not reflect their
functional protein levels due to post-translational regulation
(16). Moreover, other studies
indicate that the functional status of death receptors correlates
with the death receptor expression levels on the cell surface in
some colon cancer cells (19) and
leukemia cells (20).
Lipid rafts are plasma membrane microdomains
enriched in cholesterol and glycosphingolipids (21). They have an important role in
clustering or aggregating surface receptors into membrane complexes
at specific sites and this is essential for initiating signaling
from several receptors (11,22,23).
Several studies report that the distribution of receptors in lipid
rafts is related to their sensitivity to their respective ligands
(24). Drugs or interventions that
can interact with lipid rafts are capable of affecting the
sensitivity to TRAIL by altering receptor distribution in the
plasma membrane (25).
Consequently, death receptor distribution in lipid rafts might play
an important role in the development of acquired TRAIL-resistance
of cancer cells. In this study, we established the isogenic
TRAIL-resistant cell line H460R from the TRAIL-sensitive human lung
cancer cell line H460 to examine the potential mechanisms of
acquired resistance to TRAIL.
Materials and methods
Cell culture and acquired TRAIL-resistant
lung cancer cell line
The human non-small cell lung carcinoma (NSCLC) cell
line NCI-H460 (H460) was obtained directly from the Type Culture
Collection of the Chinese Academy of Sciences (Shanghai, China),
where cell lines have been tested and authenticated, and the H460
cell line was passaged in our laboratory for less than 6 months
after resuscitation. The cells were cultured in RPMI-1640 medium
(HyClone, Logan, UT, USA) containing 10% heat-inactivated fetal
bovine serum (FBS), penicillin (100 U/ml) and streptomycin (100
mg/ml) at 37°C in a humidified atmosphere of 5% CO2.
To establish an acquired TRAIL-resistant lung cancer
cell line, parental H460 cells were plated at clonal density in
24-well plates to ensure there was no more than one cell in each
well. H460 is a TRAIL-sensitive sub-line which is lethal under 80
ng/ml rhTRAIL treatment and it was exposed to stepwise increases in
rhTRAIL concentrations (10–128 ng/ml) over a period of 2 months to
induce cells capable of growing at high concentrations of rhTRAIL.
The resulting cell line was designated as H460R. H460R cells were
cultured in medium with continuous exposure to 50 ng/ml rhTRAIL to
maintain their TRAIL resistance.
Antibodies and reagents
rhTRAIL, designed for clinical use, was obtained
from Shanghai Bio-Tech Co. Ltd. (Shanghai, China). Monoclonal
antibodies to human DR4 and DR5, and caspase-3, and caspase-8 were
purchased from Abcam. Antibodies to caveolin-1, FADD, Bid, cFLIP
and GAPDH were from Santa Cruz Biotechnology. Fluorescein
(FITC)-conjugated goat anti-mouse/rabbit IgG and rhodamine
(TRITC)-conjugated goat anti-rabbit IgG were from Jackson
ImmunoResearch Laboratories. Horseradish peroxidase
(HRP)-conjugated goat anti-mouse IgG and goat anti-rabbit IgG were
from Southern Biotech. The broad-spectrum caspase inhibitor
ZVAD-fmk, nystatin, Triton X-100, Tween-20 and other chemicals of
analytical grade were purchased from Sigma-Aldrich (Oakville, ON,
Canada).
Plasmids, siRNA and transfection
For plasmid construction, DR4/DR5 cDNA was amplified
from the total-mRNA of H460 cells by RT-PCR and was subcloned into
pRetroQ-DsRed Monomer-C1 vectors/pEGFP-C1 vectors (Clontech
Laboratories, Mountain View, CA, USA), yielding a construct for
expression of a fusion protein with a DsRed-monomer/GFP tag at the
C-terminus of DR4/DR5. All constructs were verified by automated
DNA sequencing.
For small interfering RNA (siRNA) oligonucleotides,
DR4, DR5 and negative control FAM siRNA oligonucleotides were
obtained from Shanghai GenePharma Co. Ltd. (Shanghai, China). Three
pairs of siRNA oligonucleotides each were designed for DR4 and DR5.
The negative control FAM siRNA with green fluorescence was used as
a control.
For transfection, cells were seeded into 6-well
plates without antibiotics. After 24 h, transfections were carried
out using the Lipo2000 reagent (Invitrogen, Carlsbad, CA, USA)
according to the manufacturer’s instructions. The efficiency of
transfection was evaluated by fluorescence intensity using inverted
fluorescence microscopy (Nikon, Japan).
Cytotoxicity assays
Cytotoxicity and cell survival were determined by
the cell counting kit-8 (CCK-8) assay. Briefly, cells were plated
at 20,000 per well in 96-well plates (Corning Inc., Corning, NY,
USA). The next day, cells (confluence 80–90%) were treated with
indicated concentrations of rhTRAIL or cisplain and incubated for 4
h. In some experiments, cells were transfected with DR4/DR5
plasmid/siRNA before adding rhTRAIL. At the end of the experiment,
the culture medium was removed and a mixture of fresh medium
(without phenol red) containing CCK-8 (Sigma-Aldrich) was added for
1 h. The absorbance was determined using a microplate reader at 450
nm (Tecan, Port Melbourne, VIC, Australia). Cell viability was
defined as the relative absorbance of treated versus untreated
cells. All assays were performed in five replicates and repeated at
least three times.
Apoptosis assays
Cells were grown on 6-well plates to 70–80%
confluence and treated with transfection and/or indicated
concentrations of rhTRAIL. First, cells were pretreated with the
caspase inhibitor ZVAD-fmk for 2 h to block endogenous
TRAIL-induced apoptosis, then at selected time points, cells were
collected and incubated with 5 μl Annexin V-FTIC (BestBio,
Shanghai, China) for 15 min and 10 μl propidium iodide
(BestBio) for 5 min in the dark at 4°C. The apoptotic profile was
obtained by flow cytometry immediately afterwards (Beckman Coulter,
Miami, FL, USA).
Immunofluorescence flow cytometry
The cell-surface expression of death receptors was
analyzed by flow cytometry. First, cells were pretreated with
ZVAD-fmk to block TRAIL-induced apoptosis and then cells were
treated with 50 ng/ml rhTRAIL for the time indicated. Cells were
harvested and incubated with unlabeled primary antibodies (dilution
ratio 1:100) for 1 h at 4°C. The respective binding sites of
rhTRAIL and primary antibodies to DR4/DR5 are different, which
obviates the undetectability of TRAIL-occupied DR4/DR5. After three
washes with PBS, cells were incubated with FITC-conjugated goat
anti-rabbit IgG antibody (dilution ratio 1:50) for 45 min in the
dark at 4°C. An isotype-matched FITC-conjugated non-relevant IgG
monoclonal antibody was used as a negative control, leading to
virtually identical background values. Finally, cells were
resuspended in 1% paraformaldehyde in PBS for flow cytometric
analysis and both the percentage of antigen-positive cells and the
mean fluorescence intensity were measured.
Reverse transcription-PCR
RNA was isolated using the TRIzol reagent
(Invitrogen). RT-PCR was carried out using the high-fidelity RT-PCR
kit (Invitrogen) as per the manufacturer’s instructions. Primer
pairs used for detection of DR4 are 5′-agaga gaagtccctgcacca-3′ and
5′-gtcactccagggcgtacaat-3′ (57°C), primer pairs for DR5 are
5′-caccaggtgtgattcaggtg-3′ and 5′-ccccactgtg ctttgtacct-3′ (61°C),
primer pairs for GAPDH are 5′-tggaaggact catgaccaca-3′ and
5′-ttcagctcagggatgacc tt-3′ (56°C). Amplification products were
electrophoresed on 1.8% agarose gels containing ethidium bromide
for visualization under UV light. All PCR reactions were performed
at least three times. PCR products were normalized to GAPDH
expression.
Western blot analysis
Whole-cell extracts were prepared in RIPA lysis
buffer (Biyuntian, Shanghai, China) containing protease inhibitors
and were separated by 10–12% SDS-PAGE electrophoresis under
denaturing conditions, then transferred to PVDF membranes by
electroblotting. The membrane was blocked with 5% skim milk in TBST
buffer, incubated with the primary antibodies and reacted with
HRP-conjugated secondary antibodies. The immunoreactive proteins
were visualized with chemiluminescence reagent (Biyuntian). GAPDH
was used to normalize protein levels.
Confocal microscopy
Cells were cultured on glass chamber slides to
50–60% confluence and treated with rhTRAIL and/or nystatin. First,
cells were fixed with 4% PBS-buffered paraformaldehyde for 20 min,
washed with PBS three times, and blocked with 1% goat serum for 1
h. Afterwards, cells were counterstained with DR4/DR5 rabbit
monoclonal antibody and caveolin-1 mouse monoclonal antibody at 4°C
overnight, then counterstained with TRITC-conjugated goat
anti-rabbit antibody and FITC-conjugated goat anti-mouse antibody
at room temperature in the dark for 1 h. Immunofluorescence was
analyzed using a confocal laser-scanning microscope (Leica,
Wetzlar, Germany). The respective binding sites of rhTRAIL and
primary antibodies to DR4/DR5 are different, which obviates the
undetectability of TRAIL-occupied DR4/DR5.
Lipid raft and non-raft isolation
Lipid raft and non-raft-soluble fractions were
separated by discontinuous sucrose density gradients of Triton
X-100 cell lysates from treated and untreated cells. Briefly, cells
from 10 flasks of 15 cm culture dishes (1×108 cells)
were homogenized on ice for 30 min in 1 ml of MNX buffer (1% Triton
X-100 in 25 mmol/l MES, 150 mmol/l NaCl, pH 6.5) supplemented with
1 mmol/l of phenylmethylsulfonyl fluoride and protease inhibitor
cocktail. The homogenates were mixed with 1 ml of 80% sucrose made
with MNX buffer and placed on the bottom of a centrifuge tube. The
samples were then overlaid with 3.5 ml of 35% sucrose and 3.5 ml of
5% sucrose and centrifuged at 175,000 × g (Beckman Coulter) for 20
h at 4°C. Nine fractions (1 ml) were collected from the top to the
bottom of the gradient and analyzed. To identify lipid raft
fractions, the fractions were examined by western blot analysis
with antibody to the lipid rafts marker caveolin-1 (26). The protein in each fraction was
analyzed by western blot analysis.
Results
Characterization of TRAIL-resistant
cells
To examine the molecular alterations that can lead
to acquired TRAIL-resistance, we developed an isogenic
TRAIL-resistant H460R cell line from a TRAIL-sensitive parental
H460 cell line. At a TRAIL concentration of 50 ng/ml, some H460
cells were rounded and floating in the medium, suggesting cell
death, whereas H460R cells formed a typical epithelioid monolayer
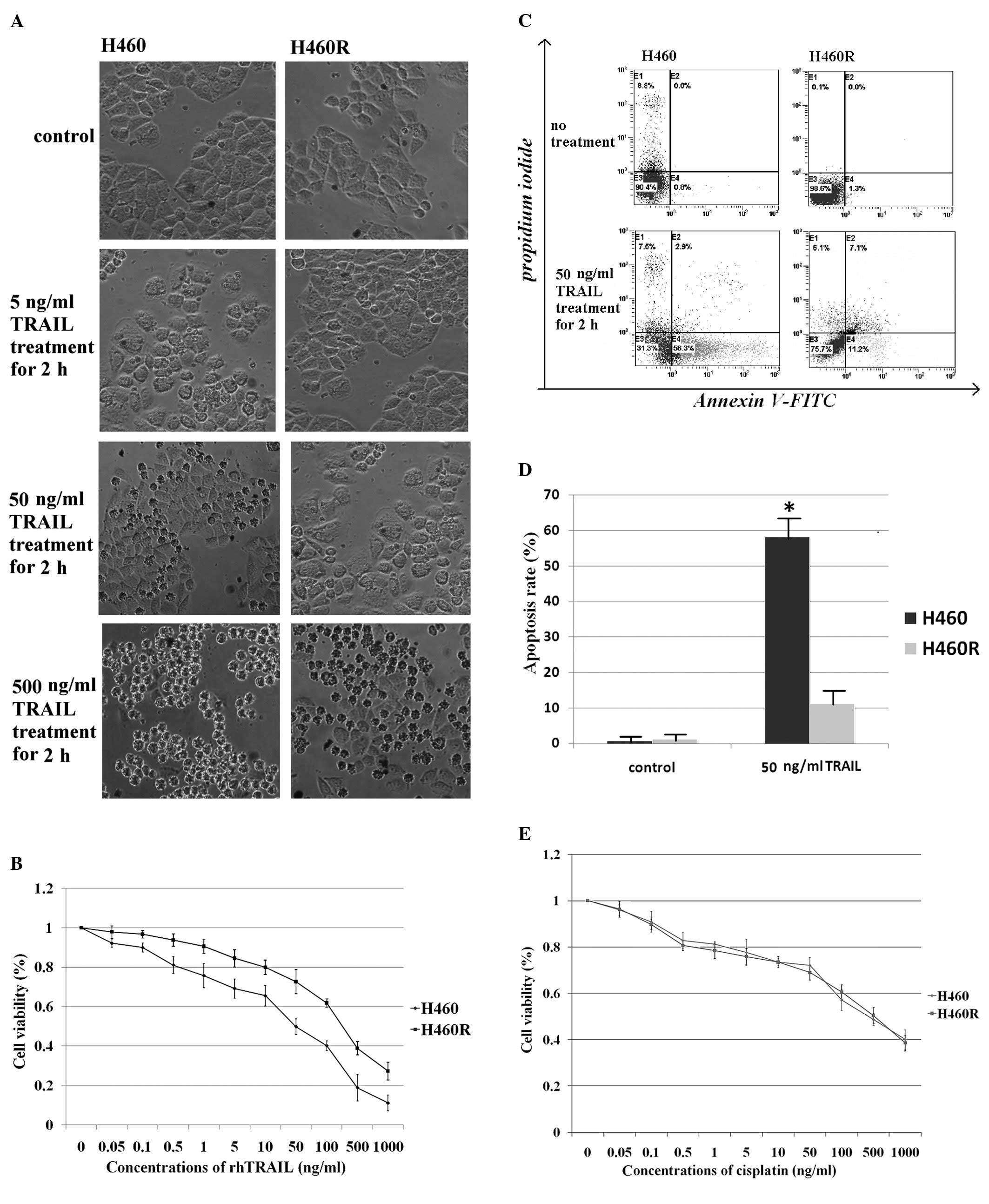
(Fig. 1A). The ability of TRAIL
induced cell death was determined by measuring the percentage of
viable cells using a CCK-8 assay. The CCK-8 assay confirmed that
H460 cells (IC50 = 50 ng/ml) were TRAIL sensitive,
whereas H460R cells (IC50 = 250 ng/ml) were TRAIL
resistant (Fig. 1B). Consistent
with these results, flow cytometric analysis showed that H460 cells
exhibited a higher apoptotic rate after rhTRAIL treatment (Fig. 1C and D). To assess whether H460R
cells were cross-resistant to cytotoxic drugs, such as cisplatin,
they and the parental H460 cells were exposed to increasing
concentrations of cisplatin and cell survival was determined by the
CKK-8 assay. We found that both TRAIL-resistant cells and
TRAIL-sensitive cells were efficiently killed by the drug (Fig. 1E). Less than 20% cell survival was
observed at high concentrations of cisplatin. Flow cytometric
analysis of apoptosis in H460 and H460R cells indicated that both
cell lines were similarly susceptible to cisplatin (data not
shown). The data show that TRAIL-resistant H460R cells remained
sensitive to cytotoxic drugs. This is significant for the
management of platinum-based chemotherapeutic regimens.
Expression of death receptors is not
altered in H460R cells
TRAIL-induced apoptosis occurs through its binding
to receptors DR4 and DR5 (27).
The receptors are crucial in initialing apoptosis. The expression
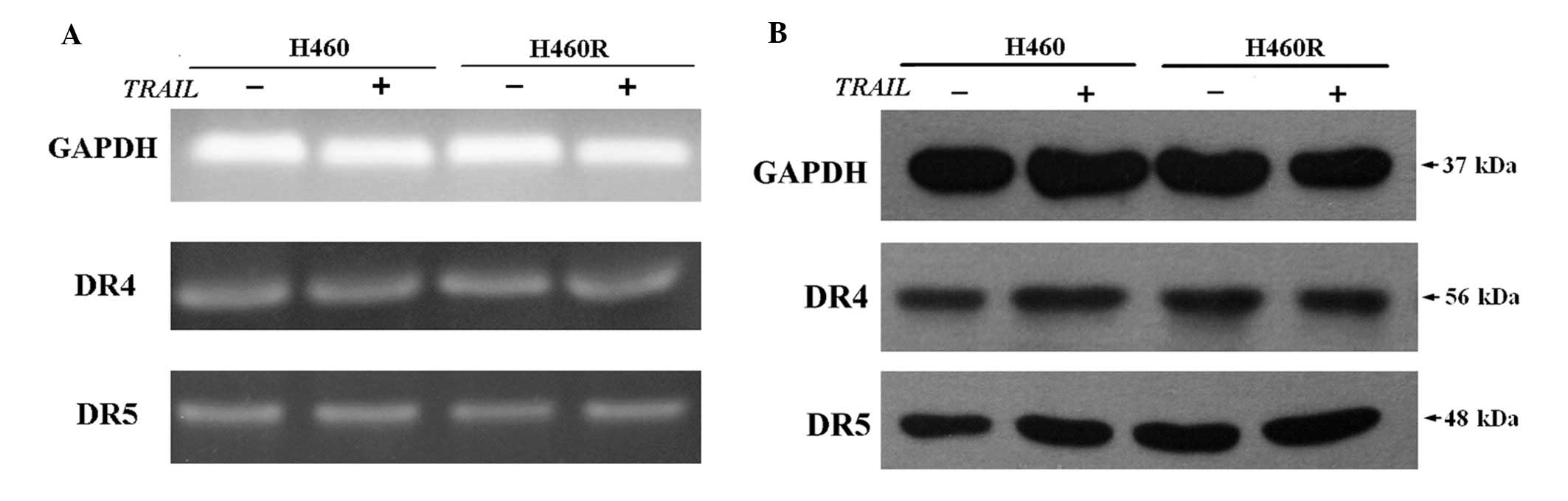
of DR4 and DR5 was evaluated by RT-PCR (Fig. 2A) and western blot analysis
(Fig. 2B). We did not observe any
marked distinction between DR4/DR5 expression in H460R cells and
H460 cells, even when cells were pretreated with 50 ng/ml rhTRAIL
for 2 h. Our results confirm that there is no correlation between
the mRNA and protein expression levels of death receptors and
sensitivity to TRAIL.
Overexpression of death receptors
upregulates sensitivity to TRAIL
To further explore the role of death receptors in
TRAIL-induced apoptosis, overexpression of DR4/DR5 was performed in
the parental H460 cells and the acquired TRAIL-resistant H460R
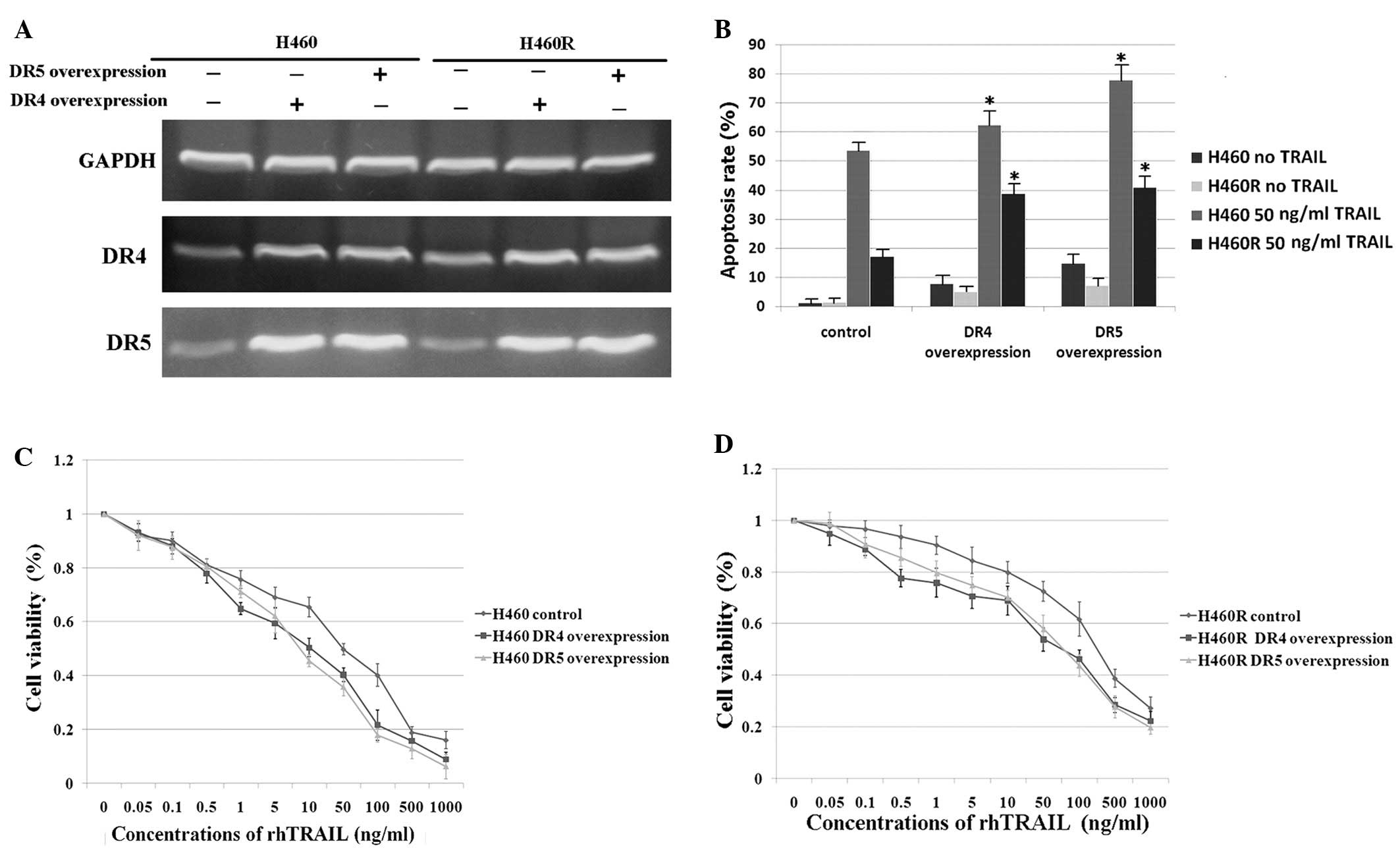
cells. After transfection with a DR4/DR5 overexpression plasmid,
the expression of DR4/DR5 markedly increased as detected by RT-PCR
(Fig. 3A) and this was also
confirmed by western blot analysis (data not shown). Moreover, we
found that overexpression of DR4 also elevated mRNA expression of
DR5 and vice versa (Fig. 3A). This
has not been previously reported and is worthy of further
investigation. DR4/DR5 overexpression in H460 and H460R cells
significantly elevated TRAIL induced-apoptosis (Fig. 3B) and the CKK-8 assay further
showed that the overexpression of death receptors could overcome
the acquired TRAIL-resistance of H460R cells (Fig. 3D). Interestingly, equivalent
overexpression levels of death receptors led to a higher apoptosis
in H460 cells than H460R cells, which also indicated that the
expression levels of death receptor mRNA and protein did not
directly correlate with their functionality.
Silencing of death receptors
downregulates sensitivity to TRAIL
Silencing of DR4/DR5 was performed in parental H460
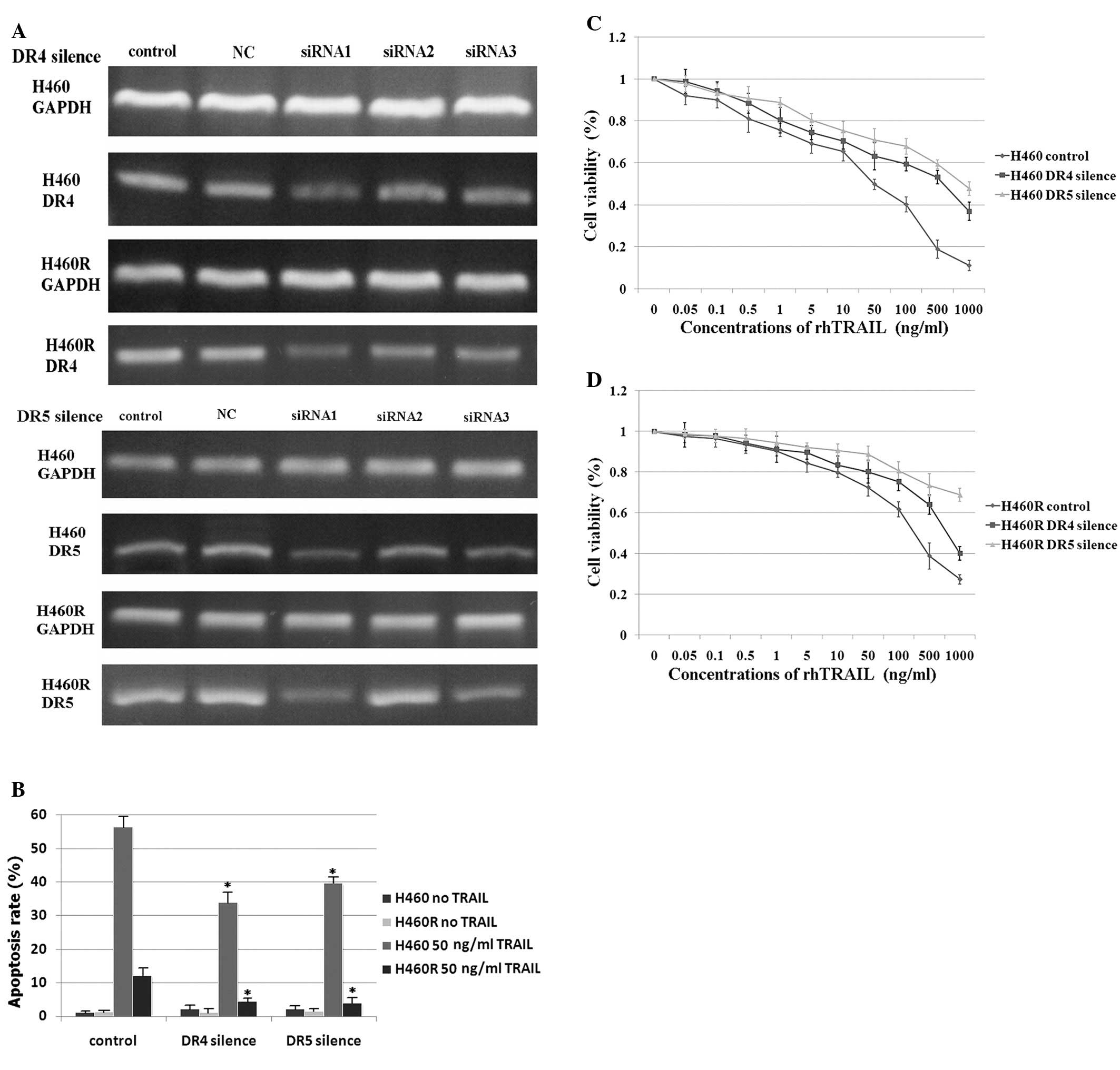
cells and acquired TRAIL-resistant H460R cells. After DR4/DR5 siRNA
(three siRNA oligonucleotides each for DR4 and DR5) transfection,
the expression of DR4/DR5 significantly reduced as detected by
RT-PCR (Fig. 4A). DR4-siRNA1 and
DR5-siRNA1 showed the highest efficiency of gene silencing and
consequently all further experiments were conducted with DR4-siRNA1
and DR5-siRNA1. The silencing of death receptors led to resistance
to TRAIL, which was shown by detection of apoptosis by flow
cytometry (Fig. 4B) and viability
by CCK-8 (Fig. 4C and D). These
results suggest that the expression of DR4 and DR5 still plays an
important role in TRAIL-induced apoptosis.
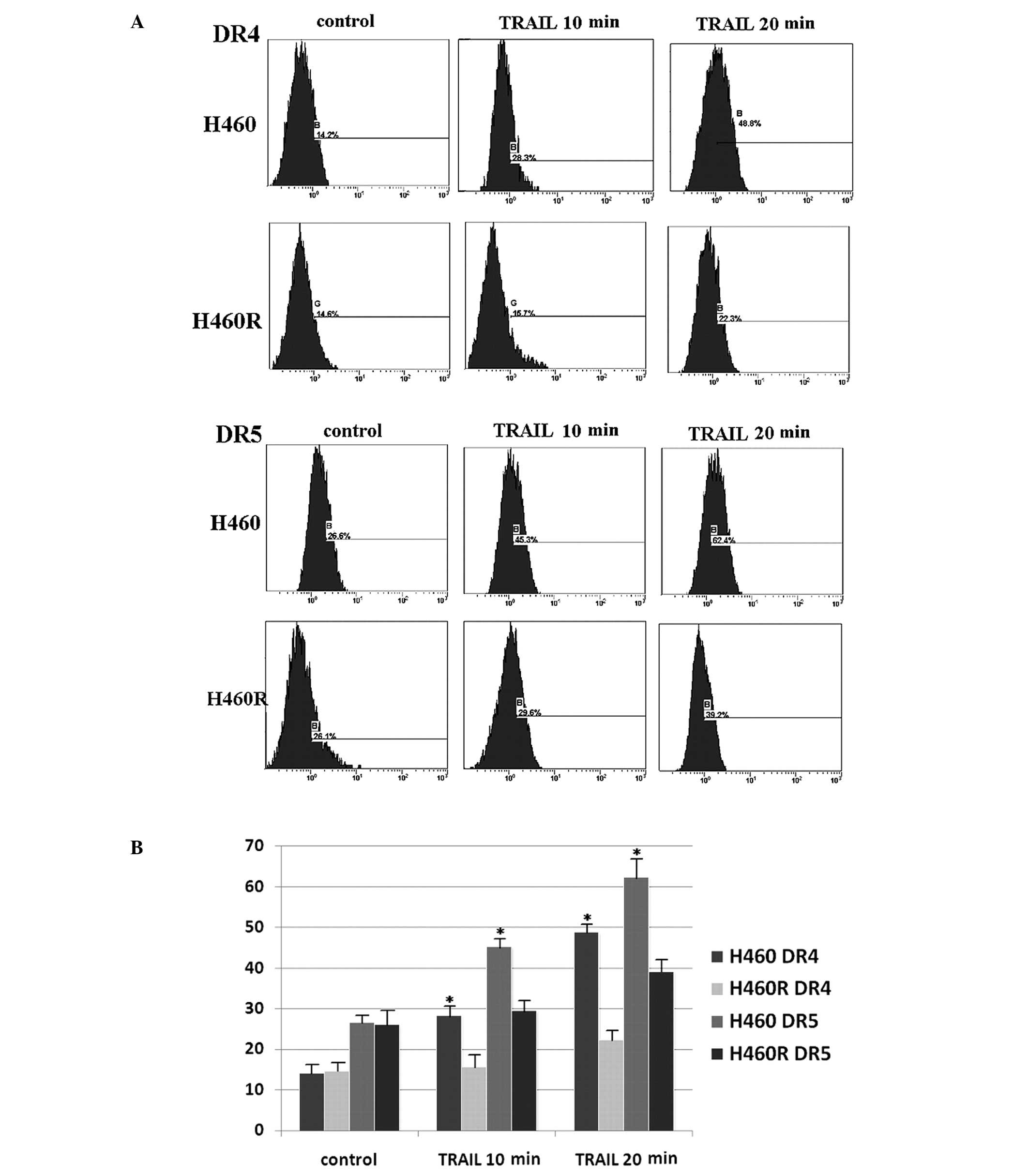
TRAIL-induced death receptor accumulation
on the cell surface
It has been reported that the functional status of
death receptors may correlate with the death receptors expression
levels on the cell surface in cancer cells (19). We examined the cell surface
expression levels of death receptors by flow cytometry using
FITC-conjugated antibodies. Nevertheless, H460 and H460R cells
showed similar cell surface expression levels of DR4/DR5 (Fig. 5), which confirms that there is no
direct correlation between the expression level of a death receptor
and the sensitivity of cancer cells to TRAIL again. However, after
50 ng/ml rhTRAIL treatment, parental H460 cells showed a
progressive increase of DR4 and DR5 cell surface expression in a
time dependent manner (Fig. 5),
but the accumulation of death receptors on cell surface in acquired
TRAIL-resistant H460R cells was very slight. In view of the
unaltered mRNA and protein expression of death receptors after
TRAIL treatment, it is possible that the lack of death receptor
accumulation on the cell surface contributes to development of
acquired resistance to TRAIL in H460R cells.
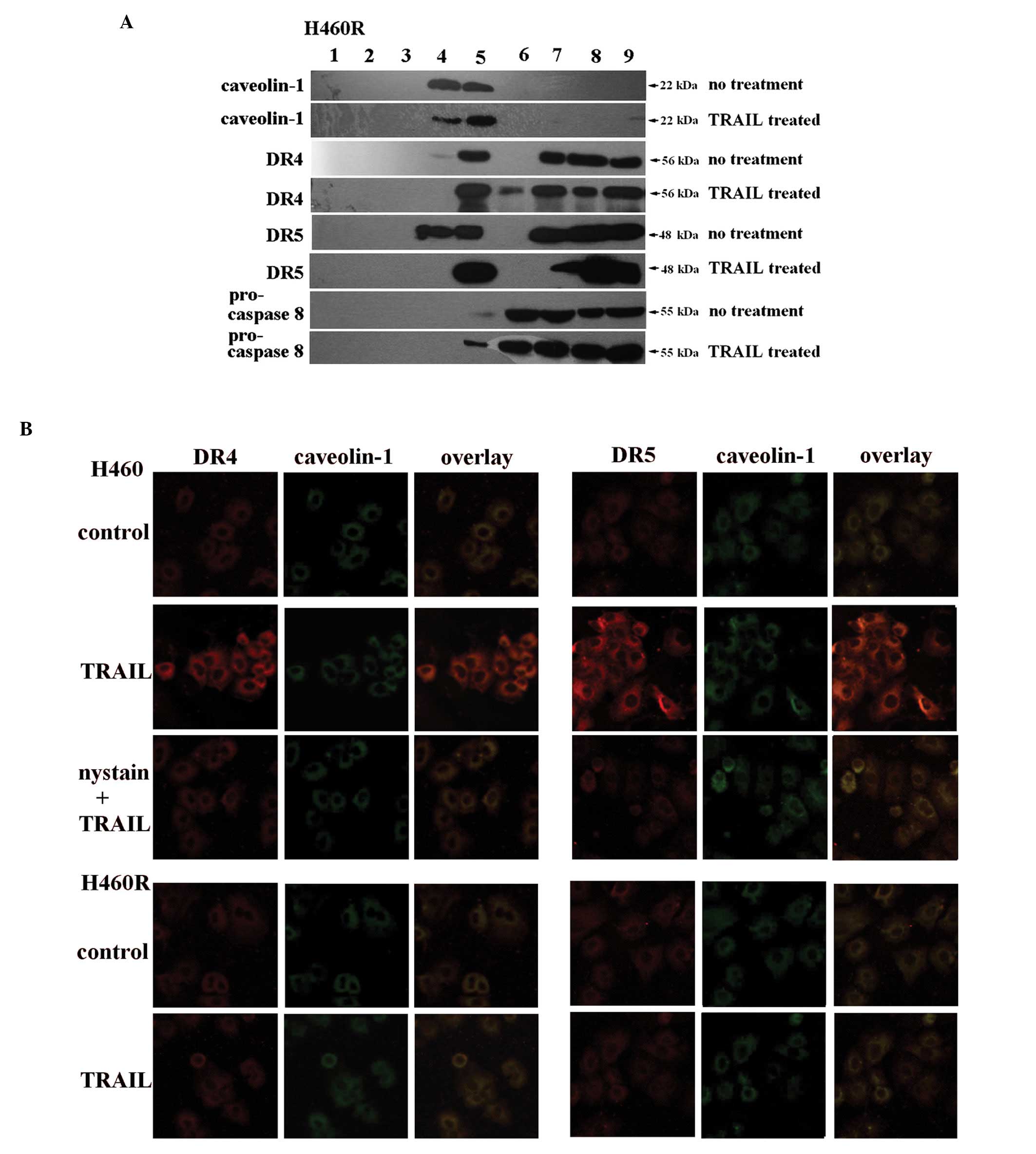
TRAIL promotes death receptor aggregation
in lipid rafts
Recent studies have highlighted the role of lipid
rafts in the initiation of death receptor-induced apoptosis. We
therefore investigated whether TRAIL sensitivity arises from
TRAIL-induced death receptor relocation to lipid rafts. In this
study, upon ultracentrifugation of extracts from H460R cell line on
a linear sucrose gradient, lipid raft marker caveolin-1 (26) was detected in fractions 4 and 5
(Fig. 6A). Combined with the
corresponding results of H460 cell line in our previous study
(28), it is shown that DR4 and
DR5 were detected in both the lipid raft and non-raft fractions of
both cell lines without rhTRAIL pretreatment, whereas pro-caspase-8
was found in non-raft fractions only (Fig. 6A) (28). The results suggested that there was
no correlation between TRAIL sensitivity and DR4/DR5 initial
distribution in lipid rafts without any treatment of the cell
lines. However, H460 cells pretreated with rhTRAIL showed a
redistribution of DR4, DR5 and pro-caspase-8 from the non-raft into
the lipid raft fractions (28),
which did not occur in H460R cells (Fig. 6A). The observations of H460 cells
in our previous study is about intrinsic TRAIL-resistance by
comparing with A549 (28). But in
this study, combined with the observations of H460R cells, it
suggested that the failure of TRAIL-induced death receptor
redistribution into lipid rafts contributes to the development of
acquired resistance to TRAIL in H460R cells and the migration of
pro-caspase-8 may be related to the formation of DISC in lipid
rafts.
In order to confirm the above results, we examined
DR4 and DR5 localization in H460 and H460R cells by confocal
microscopy. Caveolin-1 was visualized with FITC-conjugated
antibodies and DR4/DR5 were visualized with TRITC-conjugated
antibodies. In TRAIL-sensitive H460 cells, TRAIL significantly
promoted the colocalization of DR4/DR5 patches with caveolin-1
(Fig. 6B), whereas acquired
TRAIL-resistant H460R cells exposed to rhTRAIL did not induce
obvious DR4 or DR5 clustering (Fig.
6B). Taken together, our results indicate that death receptors
do not redistribute into lipid rafts in H460R cells, possibly
resulting in acquired TRAIL-resistance.
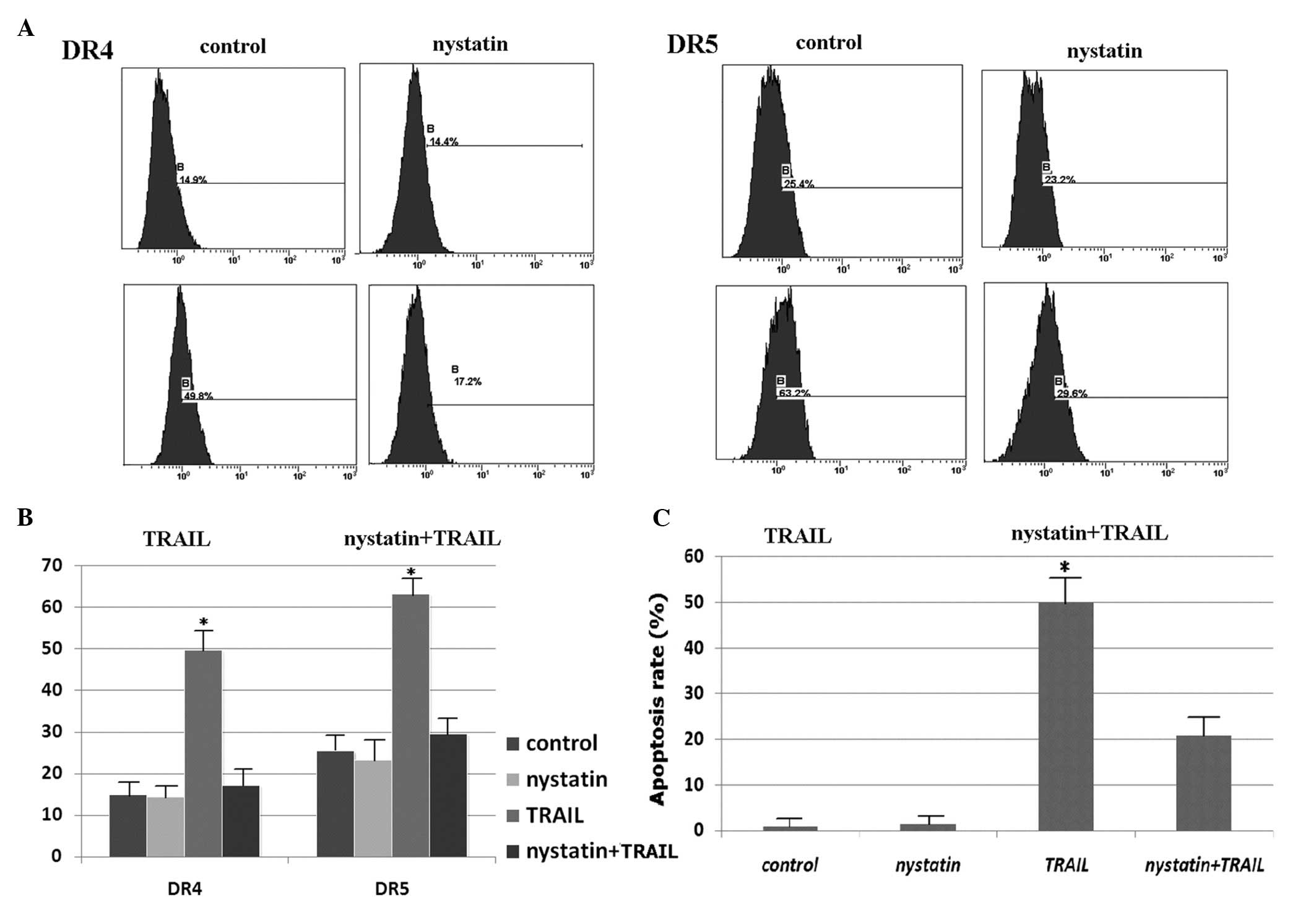
Nystatin prevents death receptor
aggregation in lipid rafts
Nystatin, a cholesterol-sequestering agent that
disrupts lipid rafts was used to investigate the effect of death
receptor redistribution on apoptosis. A total of 50 ng/ml nystatin
pretreatment of H460 cells for the last 1 h did not affect the
initial distribution of DR4 and DR5 on the membrane surface
(Fig. 7A and B) or in lipid rafts
(see our previous results in reference 28), and the combination of nystatin and
rhTRAIL did not induce obvious DR4 or DR5 clustering on the
membrane surface (Fig. 7A and B)
or in lipid rafts (Fig. 6B)
(28). Similar observations were
made for procaspase-8 (28).
Nystatin treatment significantly suppressed TRAIL-induced apoptosis
(Fig. 7C). Similar results of H460
cells were also observed in our previous study on intrinsic
TRAIL-resistance by comparing with A549 and H460 cells (28). Taken together, our results suggest
that TRAIL-induced DR4 and DR5 clustering in aggregated lipid rafts
facilitates TRAIL-sensitive apoptosis in H460 cells, which confirms
our above conclusion.
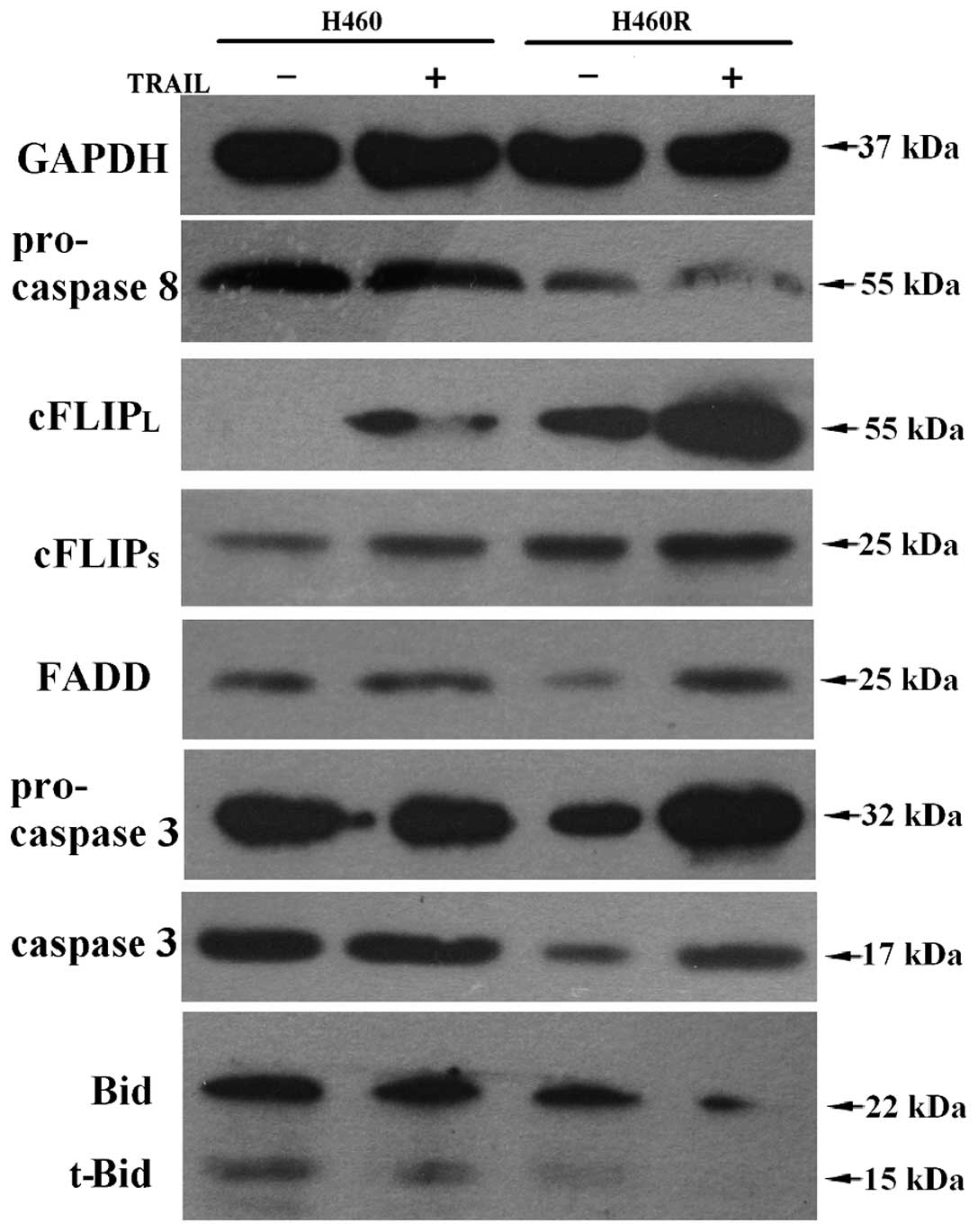
The distinct expression of apoptotic
molecules
We also determined the expression of some apoptotic
molecules by western blot analysis (Fig. 8). Pro-caspase-8, FADD,
pro-caspase-3, caspase-3 and Bid or truncated Bid showed higher
expression levels in H460 cells, whereas cFLIP showed higher
expression level in H460R cells. The result suggest that H460 cells
possess a higher activity of apoptosis not only in a
receptor-mediated extrinsic pathway, but also in an intrinsic
pathway involving mitochondria.
Discussion
Resistance to TRAIL is a major limitation for its
clinical use in the treatment of human cancers. TRAIL-resistance
may be intrinsic or acquired during a course of therapy. Intrinsic
resistance is observed when tumors are first exposed to TRAIL,
whereas acquired resistance is seen in tumors that no longer
respond to TRAIL to which they were initially sensitive. The
molecular alterations leading to TRAIL-resistance have been mostly
studied in the context of cell lines that are intrinsically
resistant to TRAIL. However, little is known about the molecular
alterations that contribute to the development of acquired
resistance during treatment with TRAIL. Understanding the basis of
the acquired resistance of cancer cells to TRAIL-mediated apoptosis
is obviously important as TRAIL may be used for the treatment of
human cancers. To address the possible mechanisms of acquired
TRAIL-resistance in lung cancer cell lines, we established an
isogenic acquired TRAIL-resistant cell line and identified a new
mechanism by which cancer cells can develop TRAIL-resistance.
Cisplatin is an irreplaceable drug of currently
prevailing platinum-based regimens for the treatment of NSCLC. It
is widely accepted that cisplatin causes cross-linking in
intra-strands and interstrands of DNA, ultimately leading to cell
death (29). We have shown that
H460R cells are resistant to TRAIL but remain sensitive to
chemotherapeutic agents, such as cisplatin (Fig. 1E), most likely because the
apoptotic pathway induced by TRAIL is different from that induced
by cisplatin. TRAIL-induced apoptosis is caspase-dependent, whereas
cisplatin-induced DNA damage activates several pathways that may be
caspase-independent. For example, activation of the mitogen
activated protein kinase-extracellular signal-regulated kinase
pathway facilitates apoptosis independently of caspases (29). Consequently, a patient with a tumor
which has acquired resistance to TRAIL may still benefit from the
management of platinum-based chemotherapeutic regimens and vice
versa.
TRAIL-induced apoptosis occurs through its binding
to its receptors DR4 and DR5 (27). In addition to DR4 and DR5, TRAIL
interacts with two other receptors, decoy receptor 1
(DcR1/TRAIL-R3/TRID) and DcR2 (TRAIL-R4). DcR1 lacks a cytoplasmic
death domain and DcR2 has a truncated death domain and they
therefore inhibit TRAIL-induced apoptosis by competing with DR4 and
DR5 for binding to TRAIL (30,31).
This is also considered to be the reason why normal cells escape
TRAIL-induced apoptosis. However, many reports have indicated that
the expression of decoy receptors does not correlate with
resistance to TRAIL of cancer cell lines (24,32,33).
Consequently, we did not detect the expression of decoy receptors
(DcR1 and DcR2) in this study.
In our study, analysis of mRNA and total protein
expression of death receptors did not provide any explanation for
acquired TRAIL-resistance in H460R cells (Fig. 2). Several recent studies suggest
that the mRNA and protein expression of death receptors does not
reflect their functional protein levels due to post-translational
regulation. For example, O-glycosylation of death receptors
correlates with TRAIL sensitivity in pancreatic carcinoma,
non-small cell lung carcinoma and melanoma cell lines.
Unfortunately, this is only found in a small portion of cancers
(16). To further explore the role
of death receptors in TRAIL-induced apoptosis, DR4/DR5
overexpression or silencing was performed in parental H460 cells
and acquired TRAIL-resistant H460R cells. We showed that the
overexpression of death receptors could overcome the resistance of
H460R cells (Fig. 3) and that the
silencing of death receptors could lead to resistance of H460 cells
to TRAIL (Fig. 4). These results
suggest that the expression of DR4/DR5 is necessary but not
sufficient for TRAIL-induced apoptosis.
The effects of receptors appear to be upstream
events during apoptosis initiation, and only cell membrane surface
DR4 and DR5 are able to bind with TRAIL and transduce the apoptotic
signal. It is reported that the functional status of death
receptors correlates with death receptor expression levels on the
cell surface in some colon cancer cells (19) and leukemia cells (20). In our previous study, SW480 and
Hep-2R cell lines upregulated their sensitivity to TRAIL by
elevating the cell surface expression of death receptors (17). The lack of expression of DR4 and
DR5 on the surface of some cancer cells is therefore a resistance
mechanism (34), regardless of
total DR4 and DR5 protein levels. Absence of DR4/DR5 on the cell
surface is sufficient to account for the failure to form DISC and
undergo later events of apoptosis. In this study, without any
treatment, the expression level of DR4/DR5 on the H460R cell
surface is identical to that of the parental line H460, which seems
contradictory to the view above. But the expression level of
DR4/DR5 on the H460 cell surface is elevated by TRAIL treatment in
a time-dependent manner (Fig. 5).
At the same time, it is worth noting that the mRNA and total
protein expression level of DR4/DR5 could not be affected by TRAIL
(Fig. 2). Taken together, it
suggested that the death receptors could migrate onto the cell
surface in H460 cells but not in H460R cells after TRAIL treatment.
Accordingly, the failure of death receptors to translocate to the
cell surface is likely to contribute to the development of acquired
resistance to TRAIL in H460R cells.
From the above we can come to the conclusion that
death receptor redistribution to the cell surface is an important
process for TRAIL-mediated apoptosis. Lipid rafts serve as plasma
membrane platforms for death receptor redistribution and death
receptor-initiated signals (26,35,36).
Several models for signal initiation in rafts have been proposed
(37,38). Redistribution of receptors in lipid
rafts is related to the sensitivity of their respective ligands by
regulating the efficacy of signaling (24). TRAIL also is capable of inducing
DR4 and DR5 migration into lipid rafts as well as the formation of
DISC (28) and agents can
sensitize cells to TRAIL by upregulating the death receptors in
lipid rafts, such as oxaliplatin (39), resveratrol (26), depsipeptide (25), and quercetin (36). Consistent with these reports, when
compared with parental H460 cells, acquired TRAIL-resistant H460R
cells display a decreased TRAIL-induced DR4/DR5 relocation to lipid
rafts, demonstrated in this study by western blot analysis of lipid
isolation (Fig. 6A) (28) and confocal microscopy (Fig. 6B). Besides DR4 and DR5,
pro-caspase-8 is also recruited into lipid rafts in H460 cells
(28).
Nystatin, a cholesterol-sequestering agent that
disrupts lipid rafts was used in our studies to investigate the
effect of lipid raft aggregation on apoptosis. Nystatin partially
prevented lipid raft aggregation and DR4 and DR5 clustering
(Figs. 6B and 7), and reduced apoptosis in parental H460
cells (Fig. 7). Similar
observations were also found in our previous study about intrinsic
TRAIL-resistance by comparing with A549 and H460 cells (28). These data suggest that the
integrity of lipid rafts is necessary for death receptor clustering
and that the difference in DR4/DR5-redistribution upon TRAIL
treatment might explain the acquired TRAIL-resistance in H460R
cells. However, the molecular mechanisms of death receptor
redistribution in lipid rafts remained unclear. It may involve the
protein sorting machinery. Recent studies suggest that protein
transport and endocytosis pathways might play important roles in
the regulation of cell surface death receptor expression (19,40,41).
Several studies have shown that the ubiquitin ligases c-Cbl and
Cblb are negative regulators of lipid rafts (39,42,43).
Others have reported that clathrin is a key component in the
endocytosis pathways (44–46). Additional studies are required to
characterize the signaling pathways that are responsible for
TRAIL-induced redistribution of its death receptors and
pro-caspase-8.
TRAIL is able to trigger the redistribution of death
receptors as well as pro-caspase-8 into lipid rafts. This
relocation in lipid rafts can subsequently induce the formation of
a functional DISC. In the DISC, pro-caspase-8 is cleaved and
initiates TRAIL-induced apoptosis. Consequently, more death
receptor and pro-caspase-8 aggregation in lipid rafts of H460 cells
might lead to higher expression levels of FADD, caspase-3 and Bid.
Otherwise, acquired TRAIL-resistant H460R cells showed a higher
expression of cFLIP in DISC (Fig.
8). cFLIP is a competitive inhibitor of caspase-8, blocks
TRAIL-induced apoptosis by being recruited to the DISC and
inhibiting of pro-caspase-8 cleavage and activation, thereby
preventing activation of the caspase cascade (47). Our results seemed not to rule out
the possibility of cFLIP overexpression resulting in
TRAIL-resistance, but compared with the event of TRAIL binding to
its death receptors, the expression of cFLIP is a downstream event
during caspase activation in isogenic H460R and H460 cells, so the
translocation of death receptor into lipid rafts remains crucial in
initialing apoptosis. An alternative explanation was also provided
where more c-FLIP and RIP mediate the unfunctional DISCs assembly
in the non-raft phase of the plasma membrane in TRAIL-resistant
cancer cells, leading to the inhibition of pro-caspase-8 cleavage
and TRAIL-resistance (26). Our
results are in line with the view that the expression levels of
pro-caspase-8, FADD, and cFLIP are closely correlated with the
status of death receptor redistribution into lipid rafts.
Additional and further studies are required to confirm the views
above. In addition to this, we also found that the expression level
of Bid, which acts as a bridge between death receptor signaling and
mitochondria signaling (9,48), was also correlated with the
redistribution of death receptors.
In conclusion, our studies indicate that TRAIL does
not increase the total expression levels of death receptors but
induces their redistribution into lipid rafts in TRAIL-sensitive
H460 cells, and that death receptors must be properly expressed on
the cell surface to recruit the components of DISC into lipid rafts
before transmitting an apoptotic signal from their ligands, and
that the development of acquired TRAIL-resistance is caused, at
least in part, by the absence of TRAIL-induced redistribution of
death receptors ino lipid rafts. Targeting the molecular mechanism
which modulates death receptor relocation to lipid rafts may
generate novel therapeutic strategies in overcoming
TRAIL-resistance and thus provide an effective therapeutic approach
in TRAIL-based combination treatments of NSCLC and possibly in
other human cancers.
Acknowledgements
This study was supported by grants
from National Natural Science Foundation of China (no.
81071908).
References
|
1.
|
Wiley SR, Schooley K, Smolak PJ, Din WS,
Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA,
et al: Identification and characterization of a new member of the
TNF family that induces apoptosis. Immunity. 3:673–682. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Pitti RM, Marsters SA, Ruppert S, Donahue
CJ, Moore A and Ashkenazi A: Induction of apoptosis by Apo-2
ligand, a new member of the tumor necrosis factor cytokine family.
J Biol Chem. 271:12687–12690. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Ichikawa K, Liu W, Zhao L, Wang Z, Liu D,
Ohtsuka T, Zhang H, Mountz JD, Koopman WJ, Kimberly RP and Zhou T:
Tumoricidal activity of a novel anti-human DR5 monoclonal antibody
without hepatocyte cytotoxicity. Nat Med. 7:954–960. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Hao C, Song JH, Hsi B, Lewis J, Song DK,
Petruk KC, Tyrrell DL and Kneteman NM: TRAIL inhibits tumor growth
but is nontoxic to human hepatocytes in chimeric mice. Cancer Res.
64:8502–8506. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Gajewski TF: On the TRAIL toward death
receptor-based cancer therapeutics. J Clin Oncol. 25:1305–1307.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Pan G, O’Rourke K, Chinnaiyan AM, Gentz R,
Ebner R, Ni J and Dixit VM: The receptor for the cytotoxic ligand
TRAIL. Science. 276:111–113. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Pan G, Ni J, Wei YF, Yu G, Gentz R and
Dixit VM: An antagonist decoy receptor and a death
domain-containing receptor for TRAIL. Science. 277:815–818. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Johnstone RW, Frew AJ and Smyth MJ: The
TRAIL apoptotic pathway in cancer onset, progression and therapy.
Nat Rev Cancer. 8:782–798. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Luo X, Budihardjo I, Zou H, Slaughter C
and Wang X: Bid, a Bcl2 interacting protein, mediates cytochrome c
release from mitochondria in response to activation of cell surface
death receptors. Cell. 94:481–490. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Ivanov VN, Bhoumik A and Ronai Z: Death
receptors and melanoma resistance to apoptosis. Oncogene.
22:3152–3161. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Lane D, Cote M, Grondin R, Couture MC and
Piche A: Acquired resistance to TRAIL-induced apoptosis in human
ovarian cancer cells is conferred by increased turnover of mature
caspase-3. Mol Cancer Ther. 5:509–521. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Lee TJ, Lee JT, Park JW and Kwon TK:
Acquired TRAIL resistance in human breast cancer cells are caused
by the sustained cFLIP(L) and XIAP protein levels and ERK
activation. Biochem Biophys Res Commun. 351:1024–1030. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Wang X, Chen W, Zeng W, Bai L, Tesfaigzi
Y, Belinsky SA and Lin Y: Akt-mediated eminent expression of c-FLIP
and Mcl-1 confers acquired resistanceto TRAIL-induced cytotoxicity
to lung cancer cells. Mol Cancer Ther. 7:1156–1163. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Zhang Land and Fang B: Mechanisms of
resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther.
12:228–237. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Jonsson G, Paulie S and Grandien A: High
level of cFLIP correlates with resistance to death receptor-induced
apoptosis in bladder carcinoma cells. Anticancer Res. 23:1213–1218.
2003.PubMed/NCBI
|
|
16.
|
Wagner KW, Punnoose EA, Januario T,
Lawrence DA, Pitti RM, Lancaster K, Lee D, von Goetz M, Yee SF,
Totpal K, Huw L, Katta V, Cavet G, Hymowitz SG, Amler L and
Ashkenazi A: Death-receptor O-glycosylation controls tumor-cell
sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med.
13:1070–1077. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Wu F, Hu Y, Long J, Zhou YJ, Zhong YH,
Liao ZK, Liu SQ, Zhou FX, Zhou YF and Xie CH: Cytotoxicity and
radiosensitization effect of TRA-8 on radioresistant human larynx
squamous carcinoma cells. Oncol Rep. 21:461–465. 2009.PubMed/NCBI
|
|
18.
|
Griffith TS, Rauch CT, Smolak PJ, Waugh
JY, Boiani N, Lynch DH, Smith CA, Goodwin RG and Kubin MZ:
Functional analysis of TRAIL receptors using monoclonal antibodies.
J Immunol. 162:2597–2605. 1999.PubMed/NCBI
|
|
19.
|
Jin Z, McDonald ER III, Dicker DT and
El-Deiry WS: Deficient tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) death receptor transport to the
cell surface in human colon cancer cells selected for resistance to
TRAIL-induced apoptosis. J Biol Chem. 279:35829–35839. 2004.
View Article : Google Scholar
|
|
20.
|
Cheng J, Hylander BL, Baer MR, Chen X and
Repasky EA: Multiple mechanisms underlie resistance of leukemia
cells to Apo2 Ligand/TRAIL. Mol Cancer Ther. 5:1844–1853. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Kand Simons and Vaz WL: Model systems,
lipid rafts, and cell membranes. Annu Rev Biophys Biomol Struct.
33:269–295. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Cummins JM, Kohli M, Rago C, Kinzler KW,
Vogelstein B and Bunz F: X-linked inhibitor of apoptosis protein
(XIAP) is a nonredundant modulator of tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human
cancer cells. Cancer Res. 64:3006–3008. 2004. View Article : Google Scholar
|
|
23.
|
Soderstrom TS, Poukkula M, Holmstrom TH,
Heiskanen KM and Eriksson JE: Mitogen-activated protein
kinase/extracellular signal-regulated kinase signaling in activated
T cells abrogates TRAIL-induced apoptosis upstream of the
mitochondrial amplification loop and caspase-8. J Immunol.
169:2851–2860. 2002. View Article : Google Scholar
|
|
24.
|
Song JH, Tse MC, Bellail A, Phuphanich S,
Khuri F, Kneteman NM and Hao C: Lipid rafts and nonrafts mediate
tumor necrosis factor related apoptosis-inducing ligand induced
apoptotic and nonapoptotic signals in non small cell lung carcinoma
cells. Cancer Res. 67:6946–6955. 2007. View Article : Google Scholar
|
|
25.
|
Vanoosten RL, Moore JM, Ludwig AT and
Griffith TS: Depsipeptide (FR901228) enhances the cytotoxic
activity of TRAIL by redistributing TRAIL receptor to membrane
lipid rafts. Mol Ther. 11:542–552. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Delmas D, Rebe C, Micheau O, Athias A,
Gambert P, Grazide S, Laurent G, Latruffe N and Solary E:
Redistribution of CD95, DR4 and DR5 in rafts accounts for the
synergistic toxicity of resveratrol and death receptor ligands in
colon carcinoma cells. Oncogene. 23:8979–8986. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Hao C, Song JH, Vilimanovich U and
Kneteman NM: Modulation of TRAIL signaling complex. Vitam Horm.
67:81–99. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Ouyang W, Yang C, Liu Y, Xiong J, Zhang J,
Zhong Y, Zhang G, Zhou F, Zhou Y and Xie C: Redistribution of DR4
and DR5 in lipid rafts accounts for the sensitivity to TRAIL in
NSCLC cells. Int J Oncol. 39:1577–1586. 2011.PubMed/NCBI
|
|
29.
|
Siddik ZH: Cisplatin: mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Sheikh MS, Huang Y, Fernandez-Salas EA,
El-Deiry WS, Friess H, Amundson S, Yin J, Meltzer SJ, Holbrook NJ
and Fornace AJ Jr: The antiapoptotic decoy receptor TRID/TRAIL-R3
is a p53-regulated DNA damage-inducible gene that is overexpressed
in primary tumors of the gastrointestinal tract. Oncogene.
18:4153–4159. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Liu X, Yue P, Khuri FR and Sun SY: Decoy
receptor 2 (DcR2) is a p53 target gene and regulates
chemosensitivity. Cancer Res. 65:9169–9175. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Zhang Yand and Zhang B: TRAIL resistance
of breast cancer cells is associated with constitutive endocytosis
of death receptors 4 and 5. Mol Cancer Res. 6:1861–1871. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Gomez-Benito M, Martinez-Lorenzo MJ, Anel
A, Marzo I and Naval J: Membrane expression of DR4, DR5 and
caspase-8 levels, but not Mcl-1, determine sensitivity of human
myeloma cells to Apo2L/TRAIL. Exp Cell Res. 313:2378–2388. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Horak P, Pils D, Haller G, Pribill I,
Roessler M, Tomek S, Horvat R, Zeillinger R, Zielinski C and
Krainer M: Contribution of epigenetic silencing of tumor necrosis
factor-related apoptosis inducing ligand receptor 1 (DR4) to TRAIL
resistance and ovarian cancer. Mol Cancer Res. 3:335–343. 2005.
View Article : Google Scholar
|
|
35.
|
Muppidi JR, Tschopp J and Siegel RM: Life
and death decisions: secondary complexes and lipid rafts in TNF
receptor family signal transduction. Immunity. 21:461–465. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Psahoulia FH, Drosopoulos KG, Doubravska
L, Andera L and Pintzas A: Quercetin enhances TRAIL-mediated
apoptosis in colon cancer cells by inducing the accumulation of
death receptors in lipid rafts. Mol Cancer Ther. 6:2591–2599. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Kand Simons and Toomre D: Lipid rafts and
signal transduction. Nat Rev Mol Cell Biol. 1:31–39. 2000.
View Article : Google Scholar
|
|
38.
|
Dimanche-Boitrel MT, Meurette O, Rebillard
A and Lacour S: Role of early plasma membrane events in
chemotherapy-induced cell death. Drug Resist Updat. 8:5–14. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Xu L, Qu X, Zhang Y, Hu X, Yang X, Hou K,
Teng Y, Zhang J, Sada K and Liu Y: Oxaliplatin enhances
TRAIL-induced apoptosis in gastric cancer cells by CBL-regulated
death receptor redistribution in lipid rafts. FEBS Lett.
583:943–948. 2009. View Article : Google Scholar
|
|
40.
|
Austin CD, Lawrence DA, Peden AA,
Varfolomeev EE, Totpal K, De Maziere AM, Klumperman J, Arnott D,
Pham V, Scheller RH and Ashkenazi A: Death-receptor activation
halts clathrin-dependent endocytosis. Proc Natl Acad Sci USA.
103:10283–10288. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Ren YG, Wagner KW, Knee DA, Aza-Blanc P,
Nasoff M and Deveraux QL: Differential regulation of the TRAIL
death receptors DR4 and DR5 by the signal recognition particle. Mol
Biol Cell. 15:5064–5074. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Hawash IY, Kesavan KP, Magee AI, Geahlen
RL and Harrison ML: The Lck SH3 domain negatively regulates
localization to lipid rafts through an interaction with c-Cbl. J
Biol Chem. 277:5683–5691. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Qu X, Miah SM, Hatani T, Okazaki M,
Hori-Tamura N, Yamamura H, Hotta H and Sada K: Selective inhibition
of Fcepsilon RI-mediated mast cell activation by a truncated
variant of Cbl-b related to the rat model of type 1 diabetes
mellitus. J Biochem. 137:711–720. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Lee KH, Feig C, Tchikov V, Schickel R,
Hallas C, Schutze S, Peter ME and Chan AC: The role of receptor
internalization in CD95 signaling. EMBO J. 25:1009–1023. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Schneider-Brachert W, Tchikov V, Neumeyer
J, Jakob M, Winoto-Morbach S, Held-Feindt J, Heinrich M, Merkel O,
Ehrenschwender M, Adam D, Mentlein R, Kabelitz D and Schutze S:
Compartmentalization of TNF receptor 1 signaling: internalized TNF
receptosomes as death signaling vesicles. Immunity. 21:415–428.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Zhang Y, Yoshida T and Zhang B: TRAIL
induces endocytosis of its death receptors in MDA-MB-231 breast
cancer cells. Cancer Biol Ther. 8:917–922. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Nam SY, Jung GA, Hur GC, Chung HY, Kim WH,
Seol DW and Lee BL: Upregulation of FLIP(S) by Akt, a possible
inhibition mechanism of TRAIL-induced apoptosis in human gastric
cancers. Cancer Sci. 94:1066–1073. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Li H, Zhu H, Xu CJ and Yuan J: Cleavage of
BID by caspase 8 mediates the mitochondrial damage in the Fas
pathway of apoptosis. Cell. 94:491–501. 1998. View Article : Google Scholar : PubMed/NCBI
|