Introduction
Prostate cancer is the most common cancer and the
second leading cause of cancer-related deaths in men in the US
(1). It is estimated that 217,730
new cases of prostate cancer were diagnosed in 2010 alone (2). The therapeutic options for patients
with prostate cancer include surgery, radiotherapy and chemotherapy
with cytotoxic agents. Despite a palliative benefit, these
approaches do not engender a long-term beneficial effect on the
overall survival of patients (3).
In this context, there is a pressing need to develop more effective
therapeutic approaches for end-stage prostate cancer patients and
genetic therapies represent promising approaches for the treatment
of this neoplasm (4).
Adenovirus-based vectors are the most widely used
cancer gene delivery platforms (5); however, specificity and efficacy are
major challenges for this therapeutic strategy (5). Of the existing adenovirus
technologies, the utility of conditional replication-competent
adenoviruses (CRCAs) provides an optimum approach. In our previous
studies, using the RAPAd.I system, we constructed a dual-specific
antitumor CRCA, designated Ad-hTERTp-E1a-Apoptin, incorporating the
tumor-specific promoter hTERTp and the specific antitumor gene
apoptin (6). This CRCA has the
ability of both tumor-specific growth inhibition and tumor-specific
replication. Further investigation showed that
Ad-hTERTp-E1a-Apoptin had a significantly greater antitumor
activity than replication-defective adenoviruses (Ad-CMV-Apoptin
and Ad-CMV-EGFP) (5).
Apoptosis is frequently impaired in many human
tumors, and is also an important mechanism in chemotherapy-induced
tumor cell death. Therefore, the modulation of apoptosis by
targeting pro-apoptotic and anti-apoptotic proteins may be a
powerful and effective method for treating cancer (5). Apoptin, a protein derived from
chicken anemia virus (CAV), selectively induces apoptosis in a wide
variety of transformed cells, but not in primary cells (6–9).
In this study, we used a recombinant adenovirus
expressing the CAV apoptin (Ad-hTERTp-E1a-Apoptin) to infect
prostatic carcinoma PC-3 and RM-1 cells, and prostatic carcinoma
models with RM-1 cells in C57BL/6 mice. We then tested the
lethality and effects of Ad-hTERTp-E1a-Apoptin on PC-3 and RM-1
cells in vitro and investigated the antitumor effect of
Ad-hTERTp-E1a-Apoptin on solid tumors in vivo. Our study
provided a new strategy for research on gene therapy in prostatic
carcinoma.
Materials and methods
Materials
The human prostate cell line PC-3, and the murine
cell line RM-1, were obtained from the Cell Bank of Type Culture
Collection, Chinese Academy of Sciences, Shanghai, China. Fetal
bovine serum, Dulbecco’s modified Eagle’s medium (DMEM), and
Roswell Park Memorial Institute medium 1640 (RPMI-1640) were bought
from Gibco, USA;
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT),
ethidium bromide (EB), and acridine orange (AO), from Sigma, USA;
and 4′-6-diamidino-2-phenylindole (DAPI) and Annexin V apoptosis
assay, from BioVision, USA; six-week-old C57BL/6 mice were obtained
from the Laboratory Animal Center of the Academy of Military
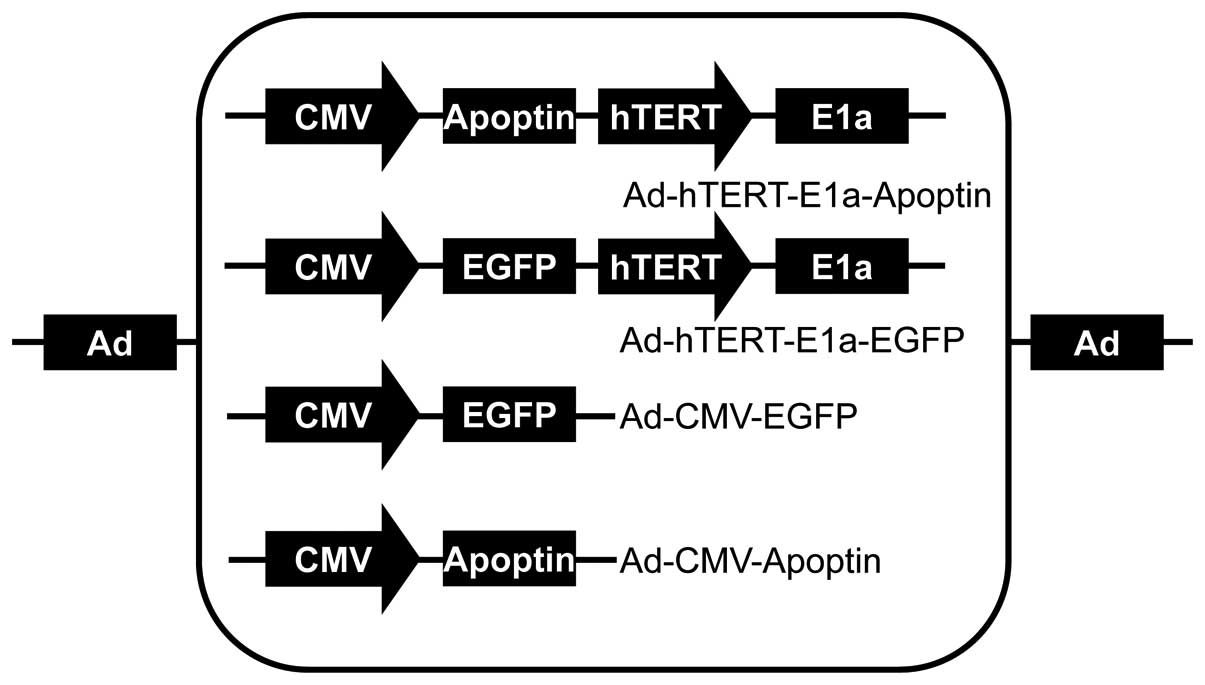
Medical Sciences, Beijing, China. The recombinant adenoviruses
Ad-hTERTp-E1a-Apoptin, Ad-hTERTp-E1a-EGFP, Ad-CMV-Apoptin, and
Ad-CMV-EGFP were constructed and saved in our laboratory (5) (Fig.
1).
Cell culture and viral infection
We incubated 5×105 PC-3 cells in
RPMI-1640 and RM-1 cells in DMEM at 37°C in 5% CO2; both
culture media were supplemented with 10% fetal bovine serum to form
complete media. Recombinant adenoviruses were diluted to
5×107 pfu/ml with either DMEM or antibiotic. Infection
was performed at a multiplicity of infection (MOI) of 100. The
diluted recombinant adenoviruses were inoculated on the cell
cultures and maintained at 37°C in 5% CO2 for 4 h and
then added to their respective complete medium, in which
cultivation continued for 48 h.
MTT colorimetric assay
PC-3 and RM-1 cells were seeded in 96-well plates
(5×103 cells/well) and infected with various
concentrations (1, 10 and 100 MOI) of recombinant adeno-viruses.
Viability was measured after 12, 24, 36, 48, 60, 72, 84 and 96 h by
treating cells with 20 μl/well MTT (5 mg/ml) and incubating
at 37°C in 5% CO2 for 4 h. The culture media were
removed, and the crystals formed were dissolved by adding 150
μl/well dimethylsulfoxide. Untreated PC-3 and RM-1 cells
were used as controls and all measurements were performed in
triplicate. The absorbance at 490 nm (A) was measured; untreated
cells were used as controls. The percent cell survival was
expressed using the following formula: (average absorbance value of
control well - average absorbance value of the experimental
well)/average absorbance value of control well (5).
AO/EB staining
After a 48-h incubation period, recombinant
adenovirus-infected PC-3 and RM-1 cells (1×106 cells;
MOI 100) were trypsinized, washed 2 times in phosphate-buffered
saline (PBS), and the cell pellet obtained was resuspended in PBS.
To the resuspended solution, we added 2 μl AO/EB solution
(AO, 100 μg/ml; EB, 100 μg/ml; dissolved in PBS) and
vortexed the resulting sample. Next, 20 μl of the sample was
placed on a microscope slide with a cover slip, and images of
representative cells were obtained with a digital video camera
connected to the 100X objective lens of a fluorescence microscope.
The images from the microscope were processed with the Image-Pro
Plus (IPP, version 5.0.2) software program.
Annexin V apoptosis assay
After a 48-h incubation period, recombinant
adenovirus-infected PC-3 and RM-1 cells (1×106 cells;
MOI, 100) were trypsinized, washed once in PBS, and the cell pellet
obtained was resuspended in 200 μl binding buffer. To the
resuspended solution, 2 μl fluorescein isothiocyanate
(FITC)-labeled Annexin V and 2 μl propidium iodide (PI) were
added. The resulting mixtures were incubated in the dark for 5 min
at room temperature and examined under a laser scanning confocal
microscope.
DAPI staining
After a 48-h incubation period, recombinant
adenovirus-infected PC-3 and RM-1 cells (1×106 cells;
100 MOI) were trypsinized and washed once in PBS as above. The cell
pellet obtained was resuspended in 200 μl 25%
glutaraldehyde, washed 3 times in PBS, resuspended again in 200
μl 100 ng/ml DAPI and a 20 μl aliquot of the
resulting solution was placed on a microscope slide, coverslipped
and imaged as described previously.
Animal experiments
RM-1 cells were harvested by trypsinization and
resuspended in serum-free DMEM after being washed with PBS. The
cell concentration was adjusted to 5×107 cells/ml.
Within 1 h of harvesting, 100 μl of cell suspension was
injected either subcutaneously into the right flank or into the
caudal vein of C57BL/6 mice. When the resulting tumors reached a
diameter of 2–5 mm (8 days), the mice were randomly divided into
six groups of five mice each. Each mouse in five of these groups
received treatment consisting of a single intratumoral and a caudal
vein injection; the treatments were repeated two times a week for
three weeks and then changed to once a week for three weeks. The
five treatments used were 100 μl injections of: i)
Ad-hTERT-E1a-Apoptin alone (1011 pfu/mouse in saline),
ii) Ad-CMV-Apoptin (1011 pfu/mouse in saline), iii)
Ad-hTERT-E1a-EGFP (1011 pfu/mouse in saline), iv)
Ad-CMV-EGFP (1011 pfu/mouse in saline) and v) saline.
The mice in the sixth group were untreated and served as controls.
Tumor size was measured using calipers every 2 days. Tumor volumes
were calculated as follows: [0.52 (smallest diameter of
tumor)2 (largest diameter of tumor)] (9). After 63 days, all mice were
sacrificed and their cumulative survival was calculated.
Statistical analysis
The statistical significance of differences was
determined using one-way analysis of variance (ANOVA), and
statistical significance was accepted as P<0.05. Log-rank tests
were used for survival analysis. Data from all animals are
presented in Kaplan-Meier plots.
Results
Lethal effect of Ad-hTERT-E1a-Apoptin on
PC-3 and RM-1 cells in vitro
Cell viability was assessed using the MTT
colorimetric assay. MTT is taken up into cells by endocytosis or by
a protein-facilitated mechanism and reduced, mainly by
mitochondrial enzymes, to yield a purple formazan product, which is
largely impermeable to cell membranes and therefore accumulates
within living cells. Solubilization of the cells liberates the
purple product, which can be detected using a colorimetric
measurement. The ability of cells to reduce MTT provides an
indication of mitochondrial integrity and activity, which in turn
may be interpreted as a measure of cell
number/proliferation/viability/survival/toxicity (10).
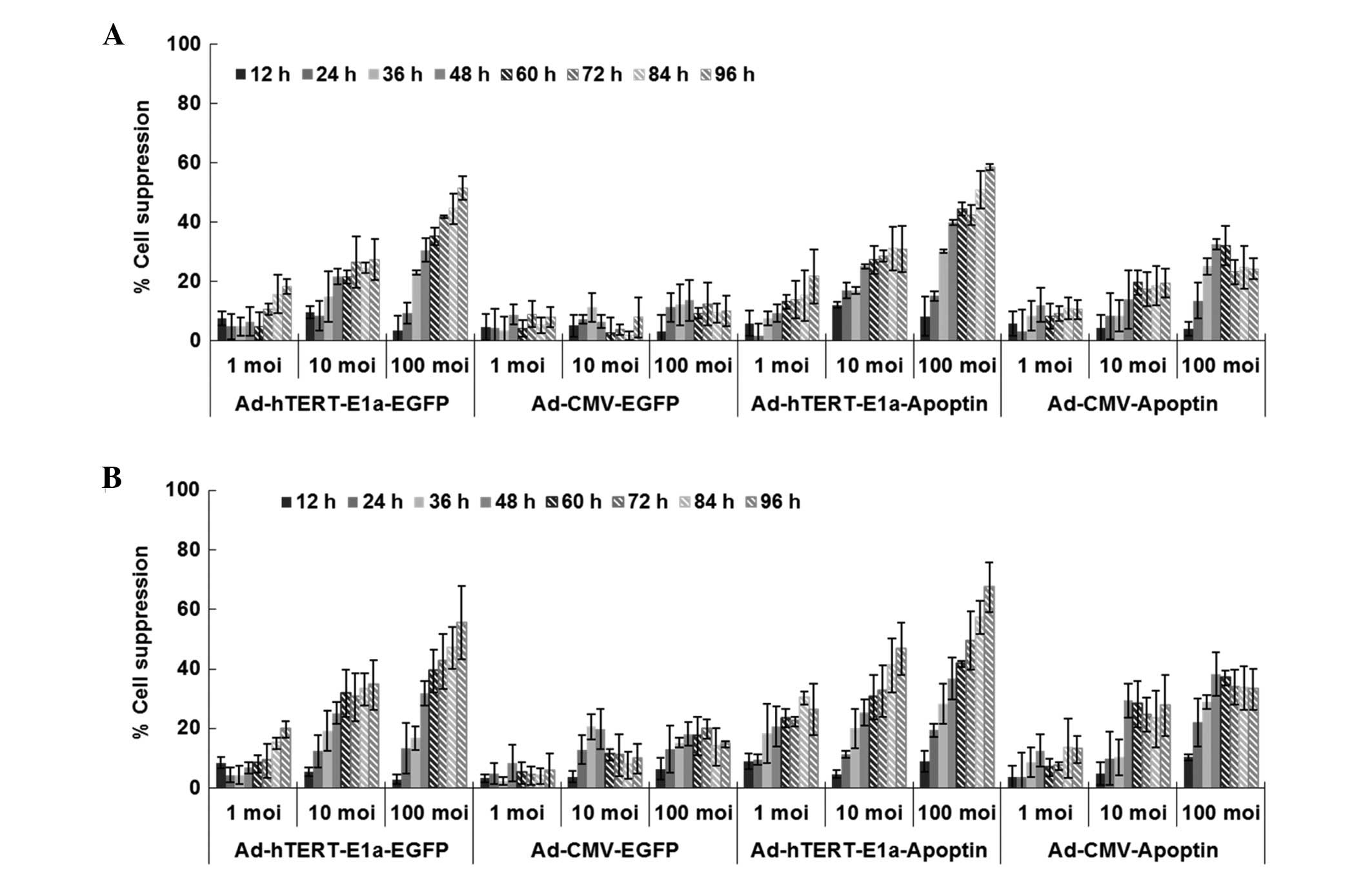
As shown in Fig. 2,
with longer infection times, the growth of PC-3 and RM-1 cells
infected with Ad-CMV-Apoptin, Ad-hTERT-E1a-EGFP, Ad-CMV-EGFP and
Ad-hTERT-E1a-Apoptin was inhibited. However, cells infected with
replication-incompetent adenoviruses (Ad-CMV-Apoptin and
Ad-CMV-EGFP) gradually resumed their growth after 48 h. In
contrast, Ad-hTERT-E1a-Apoptin and Ad-hTERT-E1a-EGFP were more
effective in inhibiting cell growth. Cell viability depended on the
MOI of the recombinant adeno-viruses to some extent. There was no
significant difference in the growth of cells at different
infection doses in the first 36 h (P>0.05). In contrast, after
48 h, the 100 MOI group showed significantly increased inhibition
compared with the 1 MOI and 10 MOI groups. In the 100 MOI group,
obvious suppression was seen after 24 h (P<0.05). With longer
infection times, both Ad-hTERT-E1a-Apoptin and Ad-hTERT-E1a-EGFP
were more effective in inhibiting cell growth, but the former was
more effective than the latter. In addition, Ad-CMV-Apoptin was
more effective than Ad-CMV-EGFP. In PC-3 and RM-1 cells, infection
with Ad-CMV-Apoptin at a MOI of 10 or 100 inhibited cell growth by
30-35% after 4 days. Infection with 1 MOI or 10 MOI of
Ad-hTERT-E1a-EGFP and Ad-hTERT-E1a-Apoptin inhibited cell growth by
20–30 and 40–50% after 4 days, respectively and that with 100 MOI
almost blocked cell growth (60–70%). Ad-CMV-EGFP, however, did not
significantly inhibit cell growth. In conclusion,
Ad-hTERT-E1a-Apoptin effectively restricts the growth of cultured
PC-3 and RM-1 cells. The interaction between infection time and MOI
was complex and synergistic and cell viability showed a
non-rigorous dependent relationship with both factors. Therefore,
we performed the following in vitro experiments 48 h after
infection at 100 MOI.
Morphological changes in the recombinant
adenovirus-infected PC-3 and RM-1 cells
For the analysis of cell death, we used fluorescent
assays of AO/EB double staining. AO is taken up by both viable and
non-viable cells, which emit green fluorescence if the dye is
intercalated into double-stranded nucleic acid (DNA) and red
fluorescence if it is bound to single-stranded nucleic acid (RNA).
EB is taken up by only non-viable cells, which emit red
fluorescence because of dye intercalation into DNA (11).
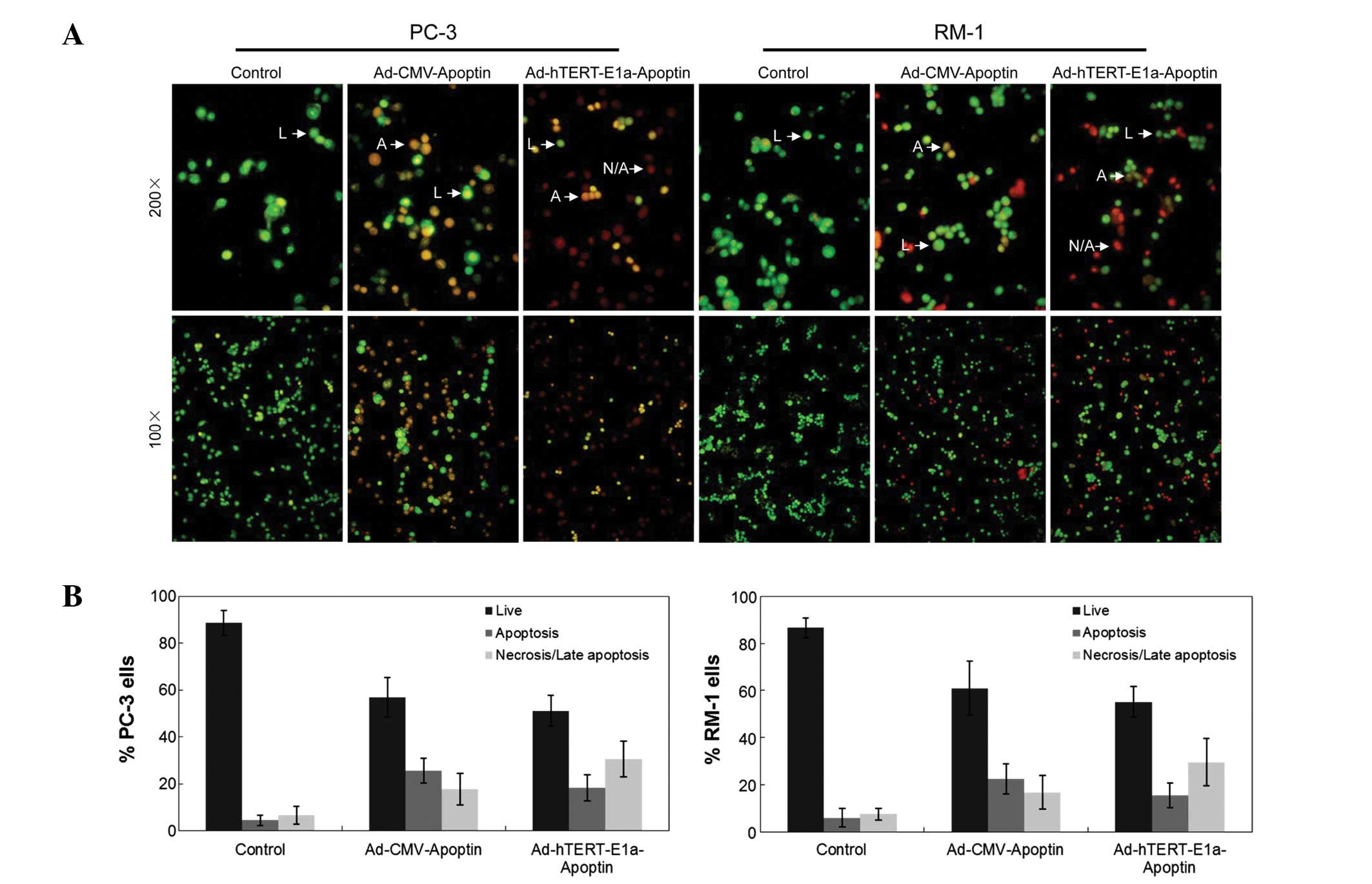
Chromatin condensation, nuclear fragmentation and
membrane destruction are the hallmarks of apoptotic cells (8). Using AO/EB staining, we analyzed the
effects of Ad-hTERTp-E1a-Apoptin and Ad-CMV-Apoptin infections on
the nuclear and the membranes of PC-3 and RM-1 cells. As shown in
Fig. 3A, normal cell membranes of
PC-3 and RM-1 cells were intact and stained bright green with AO.
Loss of cytoplasmic membrane integrity resulted in the uptake of EB
by Ad-hTERT-E1a-Apoptin- and Ad-CMV-Apoptin-infected PC-3 and RM-1
cells, with orange EB-stained cells dominating over bright green
AO-stained cells. Using the AO/EB method, we also quantified the
percentage of live, necrotic and apoptotic cells after
Ad-hTERT-E1a-Apoptin and Ad-CMV-Apoptin treatment (Fig. 3B). Infection with Ad-CMV-Apoptin
was slightly cytotoxic and the main change in morphology indicated
apoptosis more than necrosis (Fig.
3B, middle panels). In contrast, infection with
Ad-hTERT-E1a-Apoptin was strongly cytotoxic and apoptosis occurred
very quickly, so that the main change in morphology was necrosis
rather than apoptosis (Fig. 3B,
right panels).
Ability of Ad-hTERT-E1a-Apoptin to induce
tumor-specific apoptosis
This assay is based on the ability of the protein
Annexin V to bind to phosphatidylserine (PS) exposed on the outer
membrane leaflet of apoptotic cells (PS also appears on the
necrotic cell surface). In viable cells, PS is located in the inner
membrane leaflet, but upon induction of apoptosis, it is
translocated to the outer membrane leaflet and becomes available
for Annexin V binding. The addition of phosphatidylinositol (PI)
enabled viable apoptotic cells to be distinguished from necrotic
cells (12).
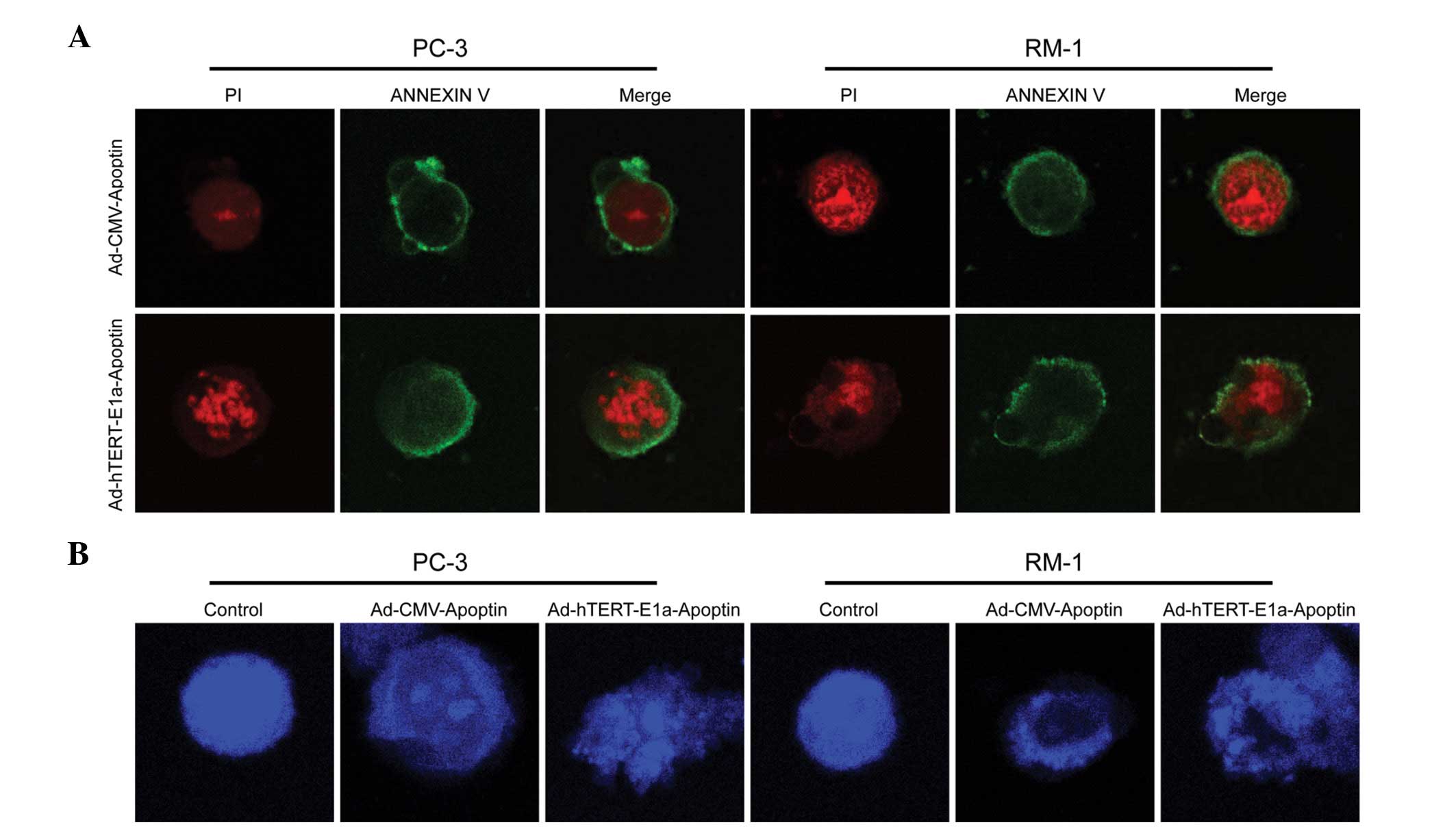
PC-3 and RM-1 cells infected with
Ad-hTERT-E1a-Apoptin and Ad-CMV-Apoptin were stained with Annexin
V-FITC/PI and observed under a laser scanning confocal microscope.
Ad-hTERT-E1a-Apoptin- and Ad-CMV-Apoptin-infected cells displayed
red fluorescence and fragmented chromatin when stained with PI
(Fig. 4A, left panels) and green
fluorescence when stained with Annexin V-FITC (Fig. 4A, middle panels). The green
fluorescence was mainly concentrated in the cell membrane, a
characteristic of phospholipid membranes valgus. PC-3 and RM-1
cells stained with Annexin V-FITC/PI showed a red nucleus (PI) and
a halo of green (FITC) on the cell surface, which are indicative of
phospholipid membranes valgus and fragmented chromatin. These
results indicated that Ad-hTERT-E1a-Apoptin and Ad-CMV-Apoptin
induced apoptosis in PC-3 and RM-1 cells.
The blue fluorescent DAPI nucleic acid stain
preferentially stains double-stranded DNA (dsDNA). The stain
appears to associate with A/T clusters in the minor groove. Binding
of DAPI to dsDNA produces ∼20-fold fluorescence enhancement,
apparently due to the displacement of water molecules from both
DAPI and the minor groove. DAPI also binds RNA but through a
different mechanism, which is thought to involve A/U-selective
intercalation. The DAPI/RNA complex exhibits a longer-wavelength
fluorescence emission maximum than the DAPI/dsDNA complex (∼500 vs.
∼460 nm) and a quantum yield that is only ∼20% as high (13).
PC-3 and RM-1 cells infected with
Ad-hTERT-E1a-Apoptin and Ad-CMV-Apoptin were stained with DAPI and
observed under a laser scanning confocal microscope. The nuclei of
uninfected cells (controls) showed a uniform blue fluorescence and
were structurally normal, while those of Ad-hTERT-E1a-Apoptin- and
Ad-CMV-Apoptin-infected cells displayed light blue fluorescence and
condensed and fragmented chromatin (Fig. 4B). These results indicated that
Ad-hTERT-E1a-Apoptin and Ad-CMV-Apoptin induced apoptosis in PC-3
and RM-1 cells.
Antitumor effect of Ad-hTERT-E1a-Apoptin
in vivo
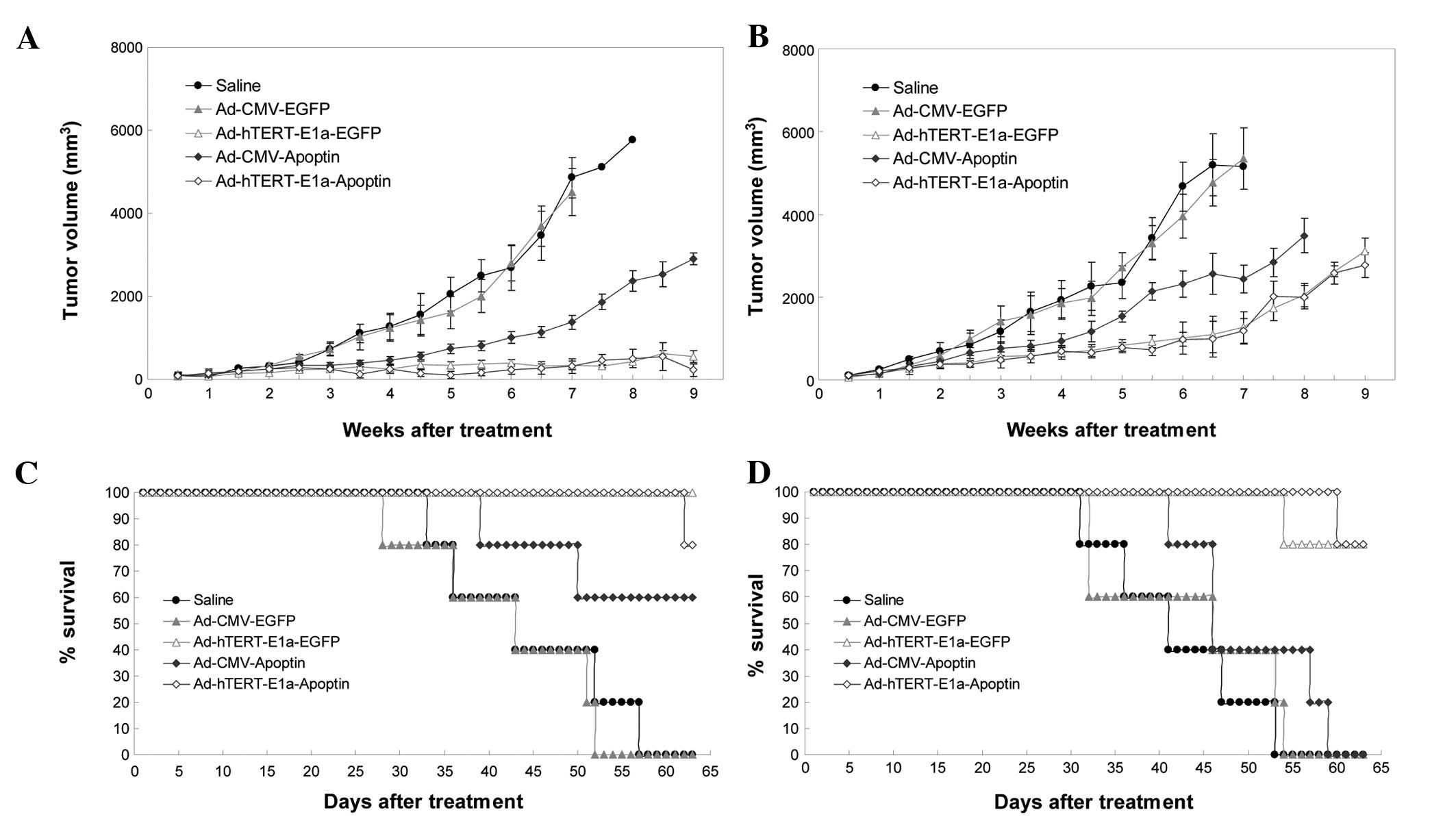
We next examined the antitumor potential of
Ad-hTERT-E1a-Apoptin in the RM-1 tumor model. The growth kinetics
of the tumors treated with intratumoral injections are shown in
Fig. 5A. Compared with the saline
control and Ad-CMV-EGFP groups, the recombinant adenovirus groups
showed suppression of tumor growth; this effect was seen after the
first three injections and continued up to the end of the treatment
period. However, soon after the last injection, tumor growth
gradually resumed in the recombinant virus groups, but was slowest
in the Ad-hTERT-E1a-Apoptin and Ad-hTERT-E1a-EGFP groups. The
growth kinetics of the tumors treated with intravenous injections
is shown in Fig. 5B. Compared with
the tumors in the saline controls and Ad-CMV-EGFP-infected groups,
those in the recombinant adenovirus groups were suppressed after
the first three injections. Tumor suppression continued up to the
end of the treatment period. However, soon after the last
injection, the Ad-CMV-Apoptin- and Ad-CMV-EGFP-infected tumors
gradually resumed their growth. Most of the Ad-hTERT-E1a-Apoptin-
and Ad-hTERT-E1a-EGFP-infected tumors also resumed growth, but
these grew more slowly. The tumors in the intravenous injection
groups grew more rapidly than those in the intratumoral injection
groups. The main cause of this difference may be the faster effect
of direct intratumoral injection than of intravenous injection. We
also evaluated the ability of the recombinant adenoviruses to
prolong the survival of the tumor-bearing mice (Fig. 5C). All saline-, Ad-CMV-Apoptin- and
Ad-CMV-EGFP-treated animals died between 26 and 52 days after
intratumoral injection. In contrast, 60 and 80% of
Ad-hTERT-E1a-EGFP- and Ad-hTERT-E1a-Apoptin-infected animals,
respectively, were still alive at that point (Fig. 5C). Mouse survival analysis showed
that Ad-hTERT-E1a-Apoptin and Ad-hTERT-E1a-EGFP treatments
significantly increased mouse survival in the RM-1 tumor model in
comparison with the other recombinant adenovirus treatments and
saline treatment (Fig. 5D).
When the experiment was terminated on day 63, 80% of
Ad-hTERT-E1a-Apoptin- and Ad-hTERT-E1a-EGFP-infected animals were
alive and the median survival time did not differ significantly
between these two groups. None of the mice in the other groups were
alive at the end of the experiment. In conclusion, inoculation with
Ad-hTERT-E1a-Apoptin had significant survival benefits and reduced
tumor size in vivo.
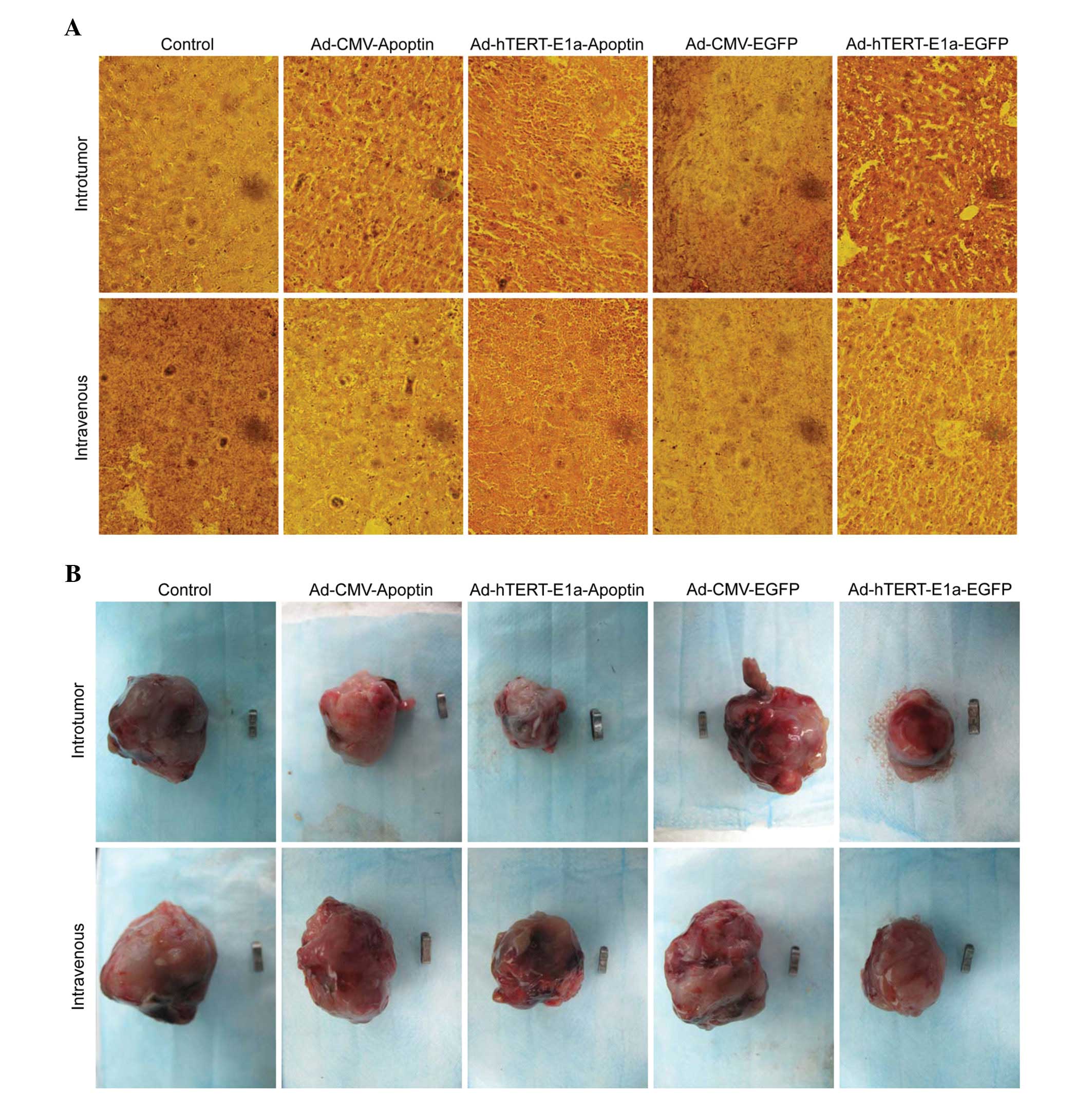
Pathological examination
In the Ad-hTERT-E1a-Apoptin- and
Ad-hTERT-E1a-EGFP-treated groups, tumors lost tissue integrity and
showed increased intercellular spaces containing remnants of
disintegrating cells (Fig. 6A).
None of these changes were seen in the Ad-CMV-EGFP-treated,
Ad-CMV-Apoptin-treated, and saline control groups. These results
indicated that Ad-hTERT-E1a-Apoptin had anti-tumor effects on solid
tumors. This recombinant adenovirus also significantly decreased
the tumor burden of the mice. Ad-hTERT-E1a-Apoptin-infected tumors
showed minimal metastatic nodules, unlike the other treatment
groups and the control group, which showed severe metastasis. Taken
together, the systemic delivery of Ad-hTERT-E1a-Apoptin
significantly reduced tumor burden and provided survival benefits
in the RM-1 tumor model.
Discussion
Like many cancers, prostate cancer is a complex
disease, and different types of therapeutic strategies are required
to demonstrate a benefit in a particular patient cohort. Most of
the ∼29,000 men who succumb to prostate cancer each year in the
United States die of metastatic disease, and this highlights the
need for better systemic therapies (14). In recent years, with the
development of molecular biology, immunology and other related
subjects, gene therapy has gradually emerged as a novel antitumor
treatment that has a huge advantage (14). Viral vectors are used in the field
of gene therapy for their simplicity, stability, ease of operation,
efficient capacity and safety without integration and have
increasingly attracted the attention and favor of researchers.
Apoptin has gained significant attention in recent
years, both as a lead for the development of cancer-specific
therapeutics and for its potential use as an indicator of cellular
transformation processes (15).
Apoptin is a 13.6 kDa viral protein encoded by the VP3 gene of
chicken anemia virus and is composed of 121 amino acids (15,16).
Because of its small size, the apoptin gene can be inserted into
various vectors such as parvoviruses, papovaviruses and
adenoviruses (17–20). It induces apoptosis independently
of death receptor pathways in a broad range of transformed and
cancer cells. Apoptin localizes in the nucleus in cancer cells;
however, in non-transformed or primary cells, it is localized to
the cytoplasm (20–22). The cellular localization of apoptin
is influenced by its phosphorylation status at threonine-108.
Phosphorylated T-108 inhibits nearby nuclear export signals,
leading to nuclear accumulation of apoptin (7,23–25).
Apoptin phosphorylation has been proposed to be regulated by
Akt-activated cyclin-dependent kinase (CDK)-2 and protein kinase C
(PKC) (26–28). Thus, nuclear localization of
apoptin and its interaction with specific signaling proteins plays
a crucial role in its selective toxicity (6,8,25).
Furthermore, apoptin does not induce apoptosis in normal,
non-transformed cells such as fibroblasts, keratinocytes, or smooth
muscle cells (7).
The hTERT promoter displays high activity in a
majority of human cancers but not in most host tissues (29,30)
and is considered a good tumor-specific regulator for oncolytic
adenoviruses (31). The hTERT
promoter can be used to control viral regulatory genes, such as
adenoviral E1A, to restrict the replication of oncolytic
adenoviruses to malignant cells and tissues. Dual-specificity
adenoviral promoters that regulate E1A expression in response to
multiple stimuli, e.g., estrogens and hypoxia, have also been
described (32). The
cancer-specific promoter hTERT can both confer tumor-specific
replication and regulate E1A expression and several tumor
cell-replicating, hTERT-driven adenoviruses have been described
(31,33). However, none of these viruses
combines both promoter elements into a single virus to regulate E1A
expression and viral replication. As hTERT is expressed in >90%
of cancers (27,28), an oncolytic virus that combines
both of these features has the potential to induce oncolytic
activity across a broad range of human tumors and tumor cell
populations. Cancer gene therapy based on oncolytic adenoviruses
has been widely studied in pre-clinical and clinical trials in
recent years. In our previous studies, using the RAPAd.I system, we
constructed the CRCA Ad-hTERT-E1a-Apoptin incorporating hTERTp and
the specific antitumor gene apoptin, which demonstrated
tumor-specific growth inhibition (5).
In this study, we described the generation of a
recombinant adenovirus vector expressing apoptin and its effects on
PC-3 and RM-1 cells in vitro and in vivo based on its
tumor-specific apoptosis-inducing activity. MTT assays indicated
that infection with Ad-hTERT-E1a-Apoptin at 100 MOI significantly
inhibited the growth of PC-3 and RM-1 cells after 48 h and that the
inhibitory effect of Ad-hTERT-E1a-Apoptin was dose- and
time-dependent. Infections at 1 or 10 MOI had less effective
growth-inhibitory effects. These data indicated that the growth
inhibition of PC-3 and RM-1 cells is related to the MOI of
Ad-hTERT-E1a-Apoptin and the time period after transduction. In
contrast, Ad-CMV-Apoptin- and Ad-CMV-EGFP-infected tumor cells
resumed proliferation after 48-h treatment at all MOI doses tested.
AO/EB, DAPI, and Annexin V assays indicated that
Ad-hTERT-E1a-Apoptin could suppress the growth of PC-3 and RM-1
cells through the induction of apoptosis. Consistent with the MTT
assay, the AO/EB, DAPI and Annexin V staining assays demonstrated
that Ad-hTERT-E1a-Apoptin and Ad-CMV-Apoptin had the most
significant growth-inhibitory effect on PC-3 and RM-1 cells and
that Ad-hTERT-E1a-Apoptin was significantly stronger than
Ad-CMV-Apoptin.
Analysis of survival and growth tendency of tumors
in animal models showed that the tumors in the Ad-hTERT-E1a-Apoptin
and Ad-hTERT-E1a-EGFP groups grew more slowly than those in the
other groups. All saline-, Ad-CMV-Apoptin- and Ad-CMV-EGFP-treated
animals died between 26 and 52 days after the last injection,
whereas Ad-hTERT-E1a-Apoptin- and Ad-hTERT-E1a-EGFP-treated mice
were still alive at this time point, indicating that
Ad-hTERT-E1a-Apoptin could significantly extend the lifespan of
animals. Moreover, the tumor size in the Ad-CMV-Apoptin-,
Ad-CMV-EGFP-, and saline-treated groups was significantly greater
than that in the Ad-hTERT-E1a-Apoptin- and
Ad-hTERT-E1a-EGFP-treated groups, indicating that
Ad-hTERT-E1a-Apoptin could suppress tumor growth in animal models.
In conclusion, Ad-hTERT-E1a-Apoptin was able to inhibit the growth
of tumor cells, extend the lifespan of animals and improve survival
and quality of life in animal models and has a potential
application in tumor gene therapy.
Taken together, gene therapy with apoptin offers
unique advantages over current approaches for cancer therapy. The
dual-specific recombinant adenovirus Ad-hTERT-E1a-Apoptin induced
significant apoptosis of PC-3 and RM-1 cells. The unique action of
the Ad-hTERT-E1a-Apoptin may provide a novel and promising
candidate for cancer gene therapy in clinical trials for prostate
cancer.
Acknowledgements
This study was supported in part by
The National Science and Technology Major Projects for ‘Major New
Drugs Innovation and Development’ (no. 2010ZX09401-305-14), The
National Natural Science Foundation of China (nos. 81072210 and
81101140) and the Key Technologies R&D Programme of Jilin
Province (nos. 10ZDGG007, 201015166 and 201101066).
References
|
1
|
Damber JE and Aus G: Prostate cancer.
Lancet. 371:1710–1721. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Burton AJ, Tilling KM, Holly JM, Hamdy FC,
Rowlands MA, Donovan JL and Martin RM: Metabolic imbalance and
prostate cancer progression. Int J Mol Epidemiol Genet. 1:248–271.
2010.PubMed/NCBI
|
|
3
|
Di Lorenzo G and De Placido S: Hormone
refractory prostate cancer (HRPC): present and future approaches of
therapy. Int J Immunopathol Pharmacol. 19:11–34. 2006.PubMed/NCBI
|
|
4
|
Dash R, Azab B, Shen XN, Sokhi UK, Sarkar
S, Su ZZ, Wang XY, Claudio PP, Dent P, Dmitriev IP, Curiel DT,
Grant S, Sarkar D and Fisher PB: Developing an effective gene
therapy for prostate cancer: new technologies with potential to
translate from the laboratory into the clinic. Discov Med.
11:46–56. 2011.PubMed/NCBI
|
|
5
|
Hu W and Kavanagh JJ: Anticancer therapy
targeting the apoptotic pathway. Lancet Oncol. 4:721–729. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Danen-Van Oorschot AA, Zhang YH, Leliveld
SR, Rohn JL, Seelen MC, Bolk MW, Van Zon A, Erkeland SJ, Abrahams
JP, Mumberg D and Noteborn MH: Importance of nuclear localization
of apoptin for tumor-specific induction of apoptosis. J Biol Chem.
278:27729–27736. 2003.PubMed/NCBI
|
|
7
|
Oro C and Jans DA: The tumour specific
pro-apoptotic factor apoptin (Vp3) from chicken anaemia virus. Curr
Drug Targets. 5:179–190. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Poon IK, Oro C, Dias MM, Zhang J and Jans
DA: Apoptin nuclear accumulation is modulated by a CRM1-recognized
nuclear export signal that is active in normal but not in tumor
cells. Cancer Res. 65:7059–7064. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li X, Jin N, Mi Z, Lian H, Sun L and Zheng
H: Antitumor effects of a recombinant fowlpox virus expressing
Apoptin in vivo and in vitro. Int J Cancer. 119:2948–2957. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maioli E, Torricelli C, Fortino V,
Carlucci F, Tommassini V and Pacini A: Critical appraisal of the
MTT assay in the presence of rottlerin and uncouplers. Biol Proced
Online. 11:227–240. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mitrovic T, Stamenkovic S, Cvetkovic V,
Tosic S, Stankovic M, Radojevic I, Stefanovic O, Comic L, Dacic D,
Curcic M and Markovic S: Antioxidant, antimicrobial and
antiproliferative activities of five lichen species. Int J Mol Sci.
12:5428–5448. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baskic D, Popovic S, Ristic P and
Arsenijevic NN: Analysis of cycloheximide-induced apoptosis in
human leukocytes: fluorescence microscopy using Annexin V/propidium
iodide versus acridin orange/ethidium bromide. Cell Biol Int.
30:924–932. 2006. View Article : Google Scholar
|
|
13
|
Ding W, Ju S, Jiang S, Zhu L, Wang Y and
Wang H: Reduced APRIL expression induces cellular senescence via a
HSPG-dependent pathway. Pathol Oncol Res. 15:693–701. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Candal E, Anadon R, DeGrip WJ and
Rodriguez-Moldes I: Patterns of cell proliferation and cell death
in the developing retina and optic tectum of the brown trout. Brain
Res Dev Brain Res. 154:101–119. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Panigrahi S, Stetefeld J, Jangamreddy JR,
Mandal S, Mandal SK and Los M: Modeling of molecular interaction
between apoptin, BCR-Abl and CrkL - an alternative approach to
conventional rational drug design. PLoS One. 7:e283952012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Adair BM: Immunopathogenesis of chicken
anemia virus infection. Dev Comp Immunol. 24:247–255. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Los M, Panigrahi S, Rashedi I, Mandal S,
Stetefeld J, Essmann F and Schulze-Osthoff K: Apoptin, a
tumor-selective killer. Biochim Biophys Acta. 1793:1335–1342. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van der Eb MM, Pietersen AM, Speetjens FM,
Kuppen PJ, van de Velde CJ, Noteborn MH and Hoeben RC: Gene therapy
with apoptin induces regression of xenografted human hepatomas.
Cancer Gene Ther. 9:53–61. 2002.PubMed/NCBI
|
|
19
|
Olijslagers S, Dege AY, Dinsart C,
Voorhoeve M, Rommelaere J, Noteborn MH and Cornelis JJ:
Potentiation of a recombinant oncolytic parvovirus by expression of
Apoptin. Cancer Gene Ther. 8:958–965. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pietersen AM, van der Eb MM, Rademaker HJ,
van den Wollenberg DJ, Rabelink MJ, Kuppen PJ, van Dierendonck JH,
van Ormondt H, Masman D, van de Velde CJ, van der Eb AJ, Hoeben RC
and Noteborn MH: Specific tumor-cell killing with adenovirus
vectors containing the apoptin gene. Gene Ther. 6:882–892. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Heilman DW, Teodoro JG and Green MR:
Apoptin nucleocytoplasmic shuttling is required for cell
type-specific localization, apoptosis, and recruitment of the
anaphase-promoting complex/cyclosome to PML bodies. J Virol.
80:7535–7545. 2006. View Article : Google Scholar
|
|
22
|
Maddika S, Booy EP, Johar D, Gibson SB,
Ghavami S and Los M: Cancer-specific toxicity of apoptin is
independent of death receptors but involves the loss of
mitochondrial membrane potential and the release of mitochondrial
cell-death mediators by a Nur77-dependent pathway. J Cell Sci.
118:4485–4493. 2005. View Article : Google Scholar
|
|
23
|
Maddika S, Mendoza FJ, Hauff K, Zamzow CR,
Paranjothy T and Los M: Cancer-selective therapy of the future:
apoptin and its mechanism of action. Cancer Biol Ther. 5:10–19.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maddika S, Wiechec E, Ande SR, Poon IK,
Fischer U, Wesselborg S, Jans DA, Schulze-Osthoff K and Los M:
Interaction with PI3-kinase contributes to the cytotoxic activity
of apoptin. Oncogene. 27:3060–3065. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wagstaff KM and Jans DA: Nuclear drug
delivery to target tumour cells. Eur J Pharmacol. 625:174–180.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang J, Cole D, Westwood N, Macpherson L,
Farzaneh F, Mufti G, Tavassoli M and Gaken J: Crucial roles for
protein kinase C isoforms in tumor-specific killing by apoptin.
Cancer Res. 70:7242–7252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Los M, Maddika S, Erb B and
Schulze-Osthoff K: Switching Akt: from survival signaling to deadly
response. Bioessays. 31:492–495. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maddika S, Panigrahi S, Wiechec E,
Wesselborg S, Fischer U, Schulze-Osthoff K and Los M: Unscheduled
Akt-triggered activation of cyclin-dependent kinase 2 as a key
effector mechanism of apoptin’s anticancer toxicity. Mol Cell Biol.
29:1235–1248. 2009.PubMed/NCBI
|
|
29
|
Kim NW, Piatyszek MA, Prowse KR, Harley
CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL and Shay
JW: Specific association of human telomerase activity with immortal
cells and cancer. Science. 266:2011–2015. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mo Y, Gan Y, Song S, Johnston J, Xiao X,
Wientjes MG and Au JL: Simultaneous targeting of telomeres and
telomerase as a cancer therapeutic approach. Cancer Res.
63:579–585. 2003.PubMed/NCBI
|
|
31
|
Wirth T, Kuhnel F and Kubicka S:
Telomerase-dependent gene therapy. Curr Mol Med. 5:243–251. 2005.
View Article : Google Scholar
|
|
32
|
Hernandez-Alcoceba R, Pihalja M, Qian D
and Clarke MF: New oncolytic adenoviruses with hypoxia- and
estrogen receptor-regulated replication. Hum Gene Ther.
13:1737–1750. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kurihara T, Brough DE, Kovesdi I and Kufe
DW: Selectivity of a replication-competent adenovirus for human
breast carcinoma cells expressing the MUC1 antigen. J Clin Invest.
106:763–771. 2000. View Article : Google Scholar : PubMed/NCBI
|