Introduction
Historically, the Chinese herb, Tripterygium
wilfordii Hook F., has been used in Chinese medicine for
centuries and triptolide which is a diterpenoid triepoxide
extracted from this herb has been shown to possess
immunosuppressive, anti-inflammatory and anti-fertility properties
(1–3). In addition, triptolide has been shown
to have promising anticancer effects on various human cancer models
in vitro and in vivo(4–6),
with potency in the nanomolar range. Given the promising aspects in
anticancer therapy, a water-soluble analogue of triptolide,
PG490-88 Na, has been developed with improved toxicity profile and
has entered into a phase I clinical trial for the treatment of
prostate cancer.
Through the research effort since its first
isolation from the herb, triptolide has been shown to be a
multi-target anticancer agent. The molecular pathways modulated by
trip-tolide that result in antiproliferative and pro-apoptotic
effects include i) inhibition of the transcriptional activity of
NF-κB and AP-1 (5,6); ii) blocking of TNF-α mediated
induction of c-IAP1 and c-IAP2 (5,6);
iii) suppression of p21 and PI3K expression (7–9); iv)
inhibition of HSP70 (10); v)
reduction of XIAP and Mcl-1 (11);
vi) inhibition of global transcription via RNA polymerase II
degradation (12); and vii)
downregulation of SUMO-specific protease 1 (13). Furthermore, triptolide can
sensitize cancer cells to Apo2L/Trail, TNF-α and chemotherapeutic
agents-induced apoptosis (4,6,7,14).
Mitogen-activated protein kinases (MAPK) are well
known to be involved in mediating cell survival. The role of ERK
has been controversial in which its activation could result in cell
proliferation and cell death. It was reported that the activation
of ERK could be a result of DNA damage that subsequently leads to
cell cycle arrest and apoptosis (15,16).
In addition, ERK activation has been suggested to be a response to
counteract endoplasmic reticulum (ER) stress-induced apoptosis
(17,18), which could be induced by external
stimuli such as cytotoxic agents. While there are studies on the
ability of triptolide to modulate the MAPK signaling pathways
(9,14,19),
data on the effect of triptolide on ERK activation and subsequent
cellular responses remain limited. Recently, triptolide has been
shown to induce the generation of reactive oxygen species (ROS) and
nitric oxide leading to apoptosis in macrophage-like cell lines
(20) as well as in colorectal
cancer cell lines (21). Given the
ability of intracellular ROS to activate ERK and subsequent
apoptosis (22–25), it would be of interest to
investigate if triptolide-induced apoptosis involves ROS generation
and ERK activation.
In view of the above findings, it was hypothesized
that triptolide could induce ROS generation, ER stress and ERK
activation, all of which were novel cellular events leading to
apoptosis in cancer cells. Using a variety of strategies in the
present study, we have shown for the first time that ERK activation
occurred in a dose- and time-dependent manner following triptolide
treatment and is an important mediator of triptolide-induced
apoptosis. Furthermore, triptolide could induce oxidative stress
and ER stress predominantly via the PERK-eIF2α pathway which acted
upstream of ERK activation.
Materials and methods
Chemicals and antibodies
Triptolide, Bapta-AM, N-acetylcysteine (NAC),
2′7′-dichlorodihydrofluorescein diacetate (H2DCFDA) and antibody
for β-actin were purchased from Sigma-Aldrich (St. Louis, MO, USA).
FR180204, SP600125, SB203580 and zVAD-fmk were purchased from
Calbiochem (San Diego, CA, USA). U0126 and antibodies for
p-ERK42/44, p-eIF2α, cleaved PARP, Bax, Bcl-xL and Ire1α were
purchased from Cell Signaling Technology (Beverly, MA, USA).
Anti-p-PERK T981 was purchased from Santa Cruz Biotechnology (Santa
Cruz, CA, USA). Anti-Bip was purchased from BD Transduction
Laboratories (Franklin Lakes, NJ, USA). Goat anti-mouse IgG and
goat anti-rabbit IgG secondary antibodies conjugated with
horseradish peroxidase were from Thermo Scientific (Rockford, IL,
USA). Dominant negative PERK plasmid was kindly provided by
Professor Wong Nai Sum (University of Hong Kong, Hong Kong,
China).
Cell culture
The human breast carcinoma cell line MDA-MB-231 was
obtained from American Type Cell Culture Collection (Manassas, VA,
USA). The cells were grown in RPMI-1640 (Sigma-Aldrich) containing
10% (v/v) fetal bovine serum (HyClone, Logan, UT, USA) supplemented
with 100 U/ml each of penicillin and streptomycin (Invitrogen,
Carlsbad, CA, USA) at 37°C in a humidified 5% CO2
atmosphere. Cells from less than 20 passages were used for the
experiments.
MTT viability assay
For MTT assay, cells were seeded in 96-well plates
at a density of 1.3±104, 9±103 and
5±103 cells per well for the 24, 48 and 72-h treatment,
respectively. The medium was changed to 200 μl of medium
containing 0.31–40 ng/ml triptolide the following day. Dimethyl
sulf-oxide (DMSO) 0.04% in culture medium was used as the vehicle
control. At the end of treatment, 50 μl of 1 mg/ml MTT (MP
Biomedicals, Solon, OH, USA), prepared in cell culture medium, was
added to each well and the plate was incubated at 37°C for 4 h.
Subsequently, the medium was aspirated and 150 μl of DMSO
was added and the plate was shaken at 200 rpm until the purple-blue
formazan solubilized. Absorbance at 570 nm was then measured using
Tecan Sunrise plate reader. For the effect of various MAPK
inhibitors on cell viability, cells were seeded at a density of
2±105 per well in a 96-well plate. On the following day,
the cells were serum-starved for 24 h before treatment with 40
ng/ml triptolide in the presence or absence of the inhibitor for 48
h. Cell viability was determined using MTT as described above.
Caspase activity assay
Cells were seeded in 96-well plate at a density of
2±105 cells/well and allowed to attach overnight. On the
following day, the medium was removed and replaced with serum-free
media, and the cells were incubated for 24 h. Subsequently,
triptolide (40 ng/ml) with and without 20 μM U0126 was added
to each well and the cells were treated for 48 h. At the end of
treatment, caspase activity was measured using the Apo-One
Homogeneous Caspase-3/7 assay kit (Promega, Madison, WI, USA)
according to the manufacturer’s instructions. The fluorescence
signal was measured using an excitation wavelength of 499 nm and an
emission wavelength of 521 nm after incubation at room temperature
for 5 h.
DNA content determination by flow
cytometry
Cells with or without treatment were harvested with
trypsin, washed with ice-cold PBS and pelleted at 10,000 rpm for 1
min. Next, the cells were resuspended and fixed in 70% ice-cold
ethanol overnight at −20°C after which the cells were collected by
centrifugation at 10,000 rpm for 1 min and the supernatant was
aspirated. The pellet was then resuspended in propidium iodide (PI)
staining buffer consisted of 50 μg/ml PI (Invitrogen), 1
mg/ml DNase-free RNase (Applichem, Darmstadt, Germany) and 0.1%
Triton X-100 prepared in PBS before incubation at 37°C for 15 min
followed by 1 h on ice. The samples were then analyzed with Beckman
Altra Flow Cytometry with 10,000 events taken. Apoptotic cells
represented by the fraction at the sub-G0/G1
phase were evaluated.
Western blot analysis
Cells were seeded in a 6-well plate at a density of
7×105 cells per well. After treatment, floating cells
and attached cells were harvested and lysed using ice-cold lysis
buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 10 mM EDTA, 1% (v/v)
NP-40, 20 mM sodium fluoride, 5 mM sodium pyrophosphate, 1 mM
sodium vanadate, 10% (v/v) glycerol, protease inhibitor cocktail)
and cleared by centrifugation at 20,000 x g for 20 min at 4°C.
Protein concentrations were determined by Bio-Rad Protein Assay,
and equal amount of protein (50 μg) was electrophoresed on
SDS-polyacrylamide gels and transferred onto nitrocellulose
membrane. Membranes were blocked with 5% BSA or 5% milk in
Tris-buffered saline with Tween-20-buffered solution (150 mM NaCl,
10 mM Tris-HCl, pH 7.5, 0.1% Tween-20) before probing with primary
antibody according to the instructions of the manufacturer.
Subsequently, the membranes were incubated with the corresponding
horseradish peroxidase conjugated secondary antibody for 1 h.
Protein bands were detected by enhanced ECL reagent (Thermo
Scientific), and visualized by CL-Xposure film (Thermo Scientific).
For reprobing, blots were stripped with Restore Western Blot
stripping buffer (Thermo Scientific).
Cell transfection
MDA-MB-231 cells were seeded in 6-well plates 24 h
before transfection. For each well, transient transfection was done
by mixing 2.5 μg plasmid DNA and 2.5 μλ PLUS™ Reagent
(Invitrogen) in 500 μl of Opti-MEM® I reduced serum media
(Invitrogen) and incubated at room temperature for 15 min. A total
of 10 μl of Lipofectamine LTX™ reagent (Invitrogen) was then
added to the mixture with gentle mixing. The mixture was incubated
at room temperature for 30 min before being added to a single well
of cells containing 2 ml fresh complete medium. After 30 h
post-transfection, the complete medium was removed, and the cells
were treated with 40 ng/ml triptolide in serum-free media for 4 h.
The expression of p-eIF2α and p-ERK were analyzed via western blot
analysis.
Intracellular ROS detection
Cell culture medium without phenol red was used for
the following experiment. MDA-MB-231 cells were seeded in a 96-well
plate at a density of 2×104 cells per well. On the
following day, the cells were serum-starved for 24 h before
treatment with 200 μl of 40 ng/ml of triptolide for 2 and 24
h in serum-free media with and without 1 mM NAC, respectively.
After treatment, the medium was aspirated and intracellular ROS was
detected using H2DCFDA. H2DCFDA in serum-free medium was
added to the cells at a final concentration of 5 μM and
incubated at 37°C in CO2 incubator for 30 min. After 30
min, the medium containing H2DCFDA was removed and the
cells were washed three times with serum-free medium. Intracellular
ROS was detected via fluorescence microscope using Nikon eclipse TE
2000-U.
Results
Triptolide induced time-, concentration-
and caspase-dependent cell death
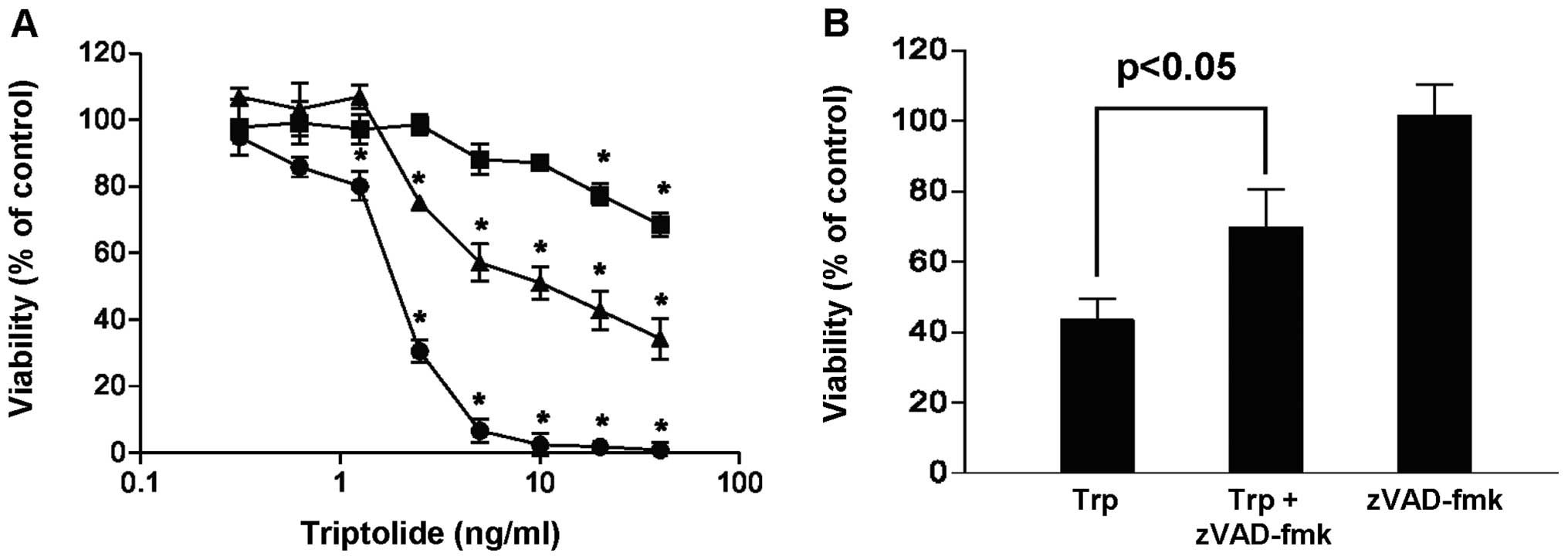
The effect of triptolide on the viability of
MDA-MB-231 cells was investigated as a function of exposure time
and drug concentration. Fig. 1A
shows that the addition of triptolide reduced the viability of
MDA-MB-231 cells in a concentration- and time-dependent manner when
compared with the vehicle control. At the maximum concentration
tested (40 ng/ml triptolide), viability of cells decreased to 68,
34 and 0% when treated for 24, 48 and 72 h, respectively.
Furthermore, when cells were treated with triptolide in the
presence of the broad caspase inhibitor, zVAD-fmk, more cells
survived (Fig. 1B), suggesting
that triptolide induced cell death via caspase activation. These
results are in line with previous study reporting the
apoptosis-inducing effect of triptolide in cancer cells.
ERK activation rather than inhibition was
critical to triptolide-induced cell death
MAP kinase pathways are known to play important
roles in the regulation of cancer cell survival and proliferation.
Yet, it remains unclear whether triptolide-induced cell death was
caused by the modulation of any of these pathways. It was thus
hypothesized that inhibiting the MAP kinase pathways during
triptolide exposure would sensitize the cancer cells to triptolide
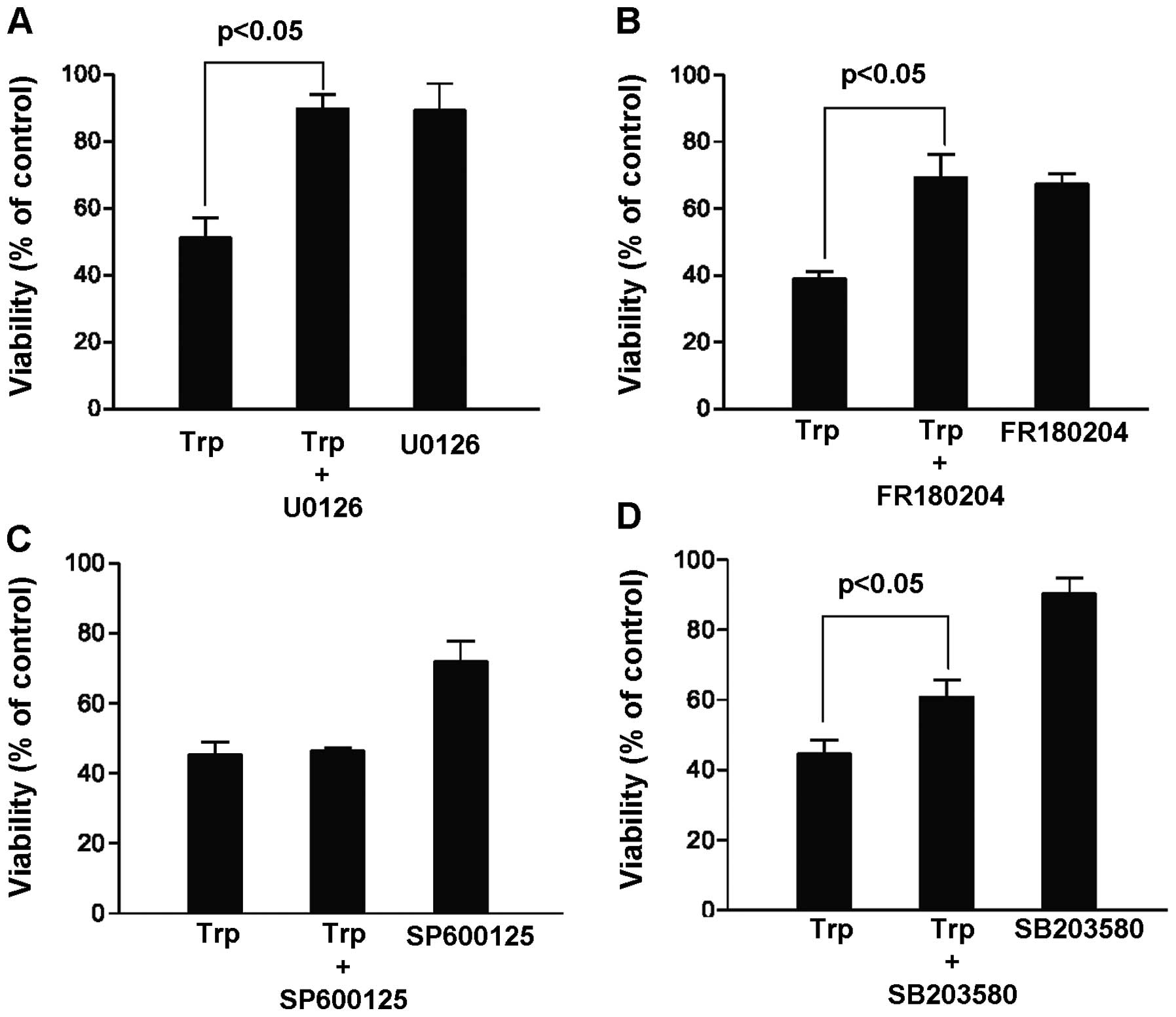
and result in more cancer cell kill. In this case, MDA-MB-231 cells
were treated for 48 h with 40 ng/ml triptolide in the presence and
absence of various MAP kinase inhibitors, including MEK inhibitor
U0126 (20 μM), ERK inhibitor FR180204 (100 μM), JNK
inhibitor SP600125 (20 μM) and p38 kinase inhibitor SB203580
(20 μM). Contrary to our hypothesis, our results summarized
in Fig. 2 indicated that the cell
killing effect of triptolide could be reversed in the presence of
the MEK, ERK and p38 kinase inhibitors. In particular, among the
three MAP kinase pathways, inhibition of the MEK/ERK pathway
appeared to give the highest increase in the percentage of viable
cells upon triptolide exposure. This finding suggests that ERK
activation rather than inhibition was critical to
triptolide-induced cell death, and our subsequent experiments were
focused on the role of ERK activation in hope to elucidate novel
mechanisms mediating the cytotoxicity of triptolide.
Triptolide-induced caspase activation is
downstream of ERK activation
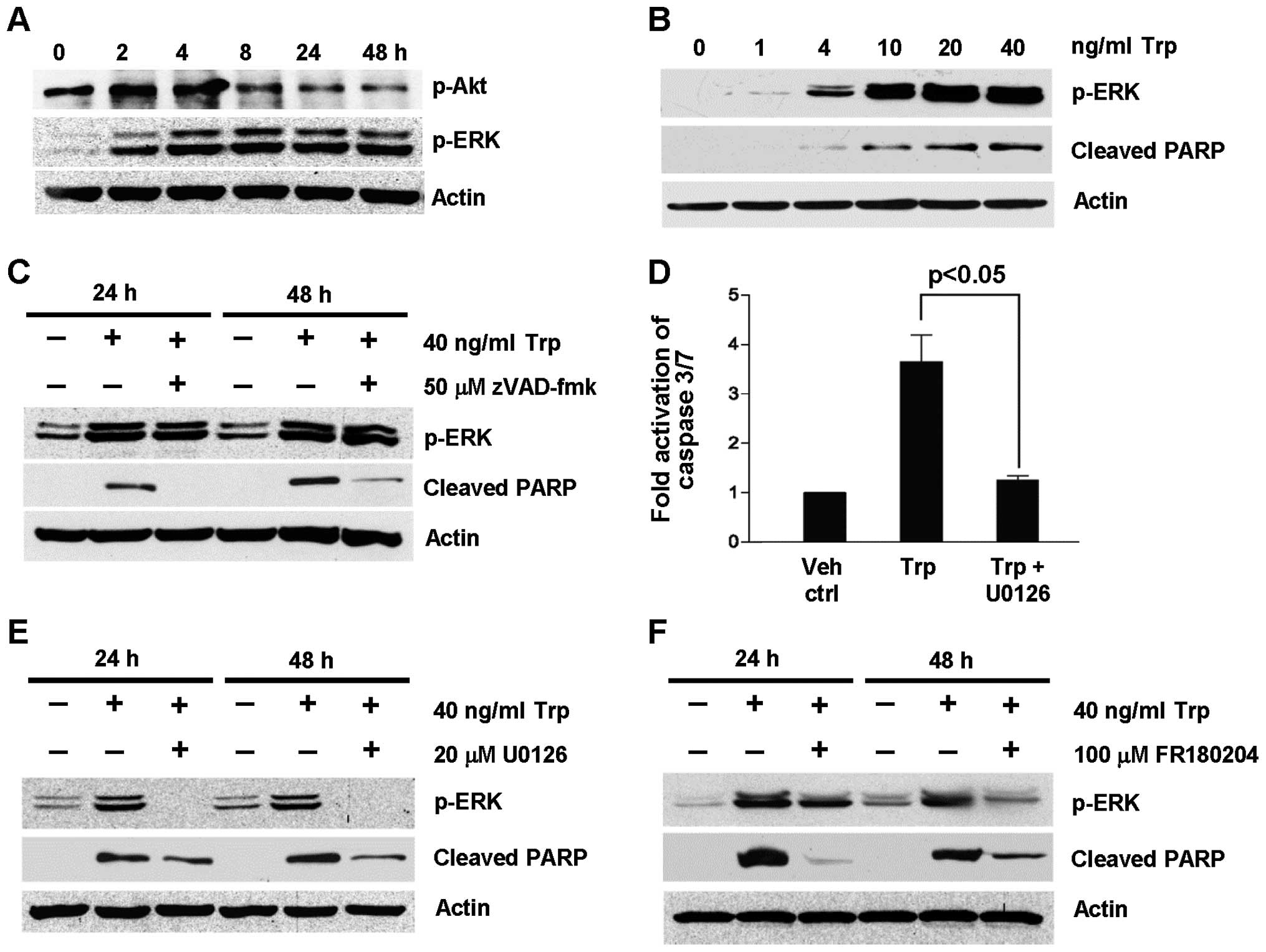
To further support that ERK activation was induced
by triptolide treatment, the expression of p-ERK was probed as a
function of triptolide exposure time (Fig. 3A) and concentration (Fig. 3B). As shown in Fig. 3A, ERK was phosphorylated as early
as 2 h after triptolide treatment and remained phosphorylated for
48 h. This was accompanied by the concomitant reduction in
phosphorylated Akt expression. Furthermore, ERK phosphorylation was
dependent on trip-tolide concentration (Fig. 3B). To determine if ERK activation
took place upstream or downstream of caspase activation to exert
its apoptotic effect, the expression levels of p-ERK and cleaved
PARP were determined under the inhibition of caspases using
broad-spectrum caspase inhibitor, zVAD-fmk, as well as the
inhibition of the MEK/ERK pathway U0126 and FR180204. As shown in
Fig. 3C, zVAD-fmk did not inhibit
triptolide-induced ERK activation at 24 and 48 h despite the
inhibition of PARP cleavage. On the contrary, the fold activation
of caspase 3/7 induced by triptolide treatment was significantly
reduced in the presence of U0126 (Fig.
3D). These results indicate that ERK activation was upstream of
caspase activation. To further confirm this observation, the effect
of U0126 and FR180204 on caspase activation was evaluated by
probing the expression of cleaved PARP in triptolide-treated cells
at 24 and 48 h. As shown in Fig. 3E
and F, triptolide-induced PARP cleavage was reduced in the
presence of the MEK/ERK pathway inhibitors, U0126 and FR180204,
with the concomitant reduction in p-ERK expression. Collectively,
these findings provide further evidence for the role of ERK
activation in triptolide-induced apoptosis.
Triptolide-induced ERK activation
modulated the expression of Bcl-2 protein members
The mitochondrial death pathway is known to be
controlled by the members of the Bcl-2 family, and it has been
reported that triptolide initiates apoptosis via the
mitochondria-mediated intrinsic apoptotic pathway (11,26).
It has also been demonstrated that Bax expression could be mediated
by ERK activation upon cisplatin treatment (27). In view of the ability of triptolide
to induce ERK activation and subsequent caspase-dependent cell
death, it is of interest to determine if ERK activation modulates
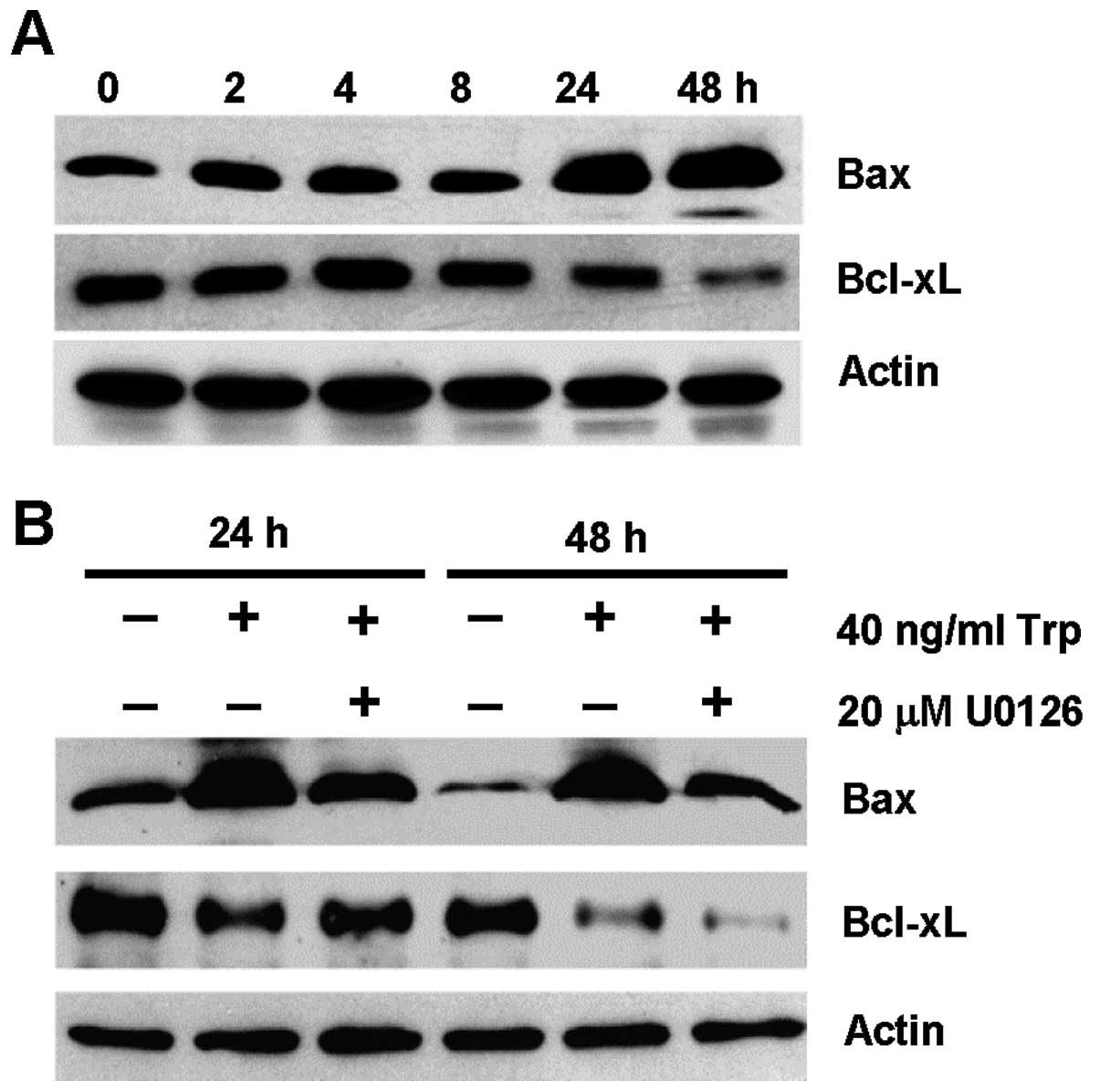
the expression of Bcl-2 family members. Fig. 4A shows the expression of the
pro-apoptotic Bax protein and the anti-apoptotic Bcl-xL protein in
MDA-MB-231 cells over a time course of 48 h upon triptolide
treatment. Bax was upregulated after 24 and 48 h of triptolide
treatment, with concomitant downregulation of Bcl-xL at 24 and 48
h. These results suggest that changes in Bax and Bcl-xL protein
expression occurred downstream of ERK activation, which occurred as
early as 2 h (Fig. 3A). To further
support this notion, the expression of Bax and Bcl-xL in
triptolide-treated cells was evaluated in the presence of U0126
inhibitor. As shown in Fig. 4B,
triptolide-induced Bax expression was inhibited partially in the
presence of U0126. However, the reduction in Bcl-xL expression
induced by triptolide treatment could be independent of ERK
activation.
Triptolide induces ER stress with
PERK-eIF2α pathway acting upstream of ERK activation
From the data collected thus far, ERK activation
induced by triptolide treatment was linked to downstream pathways
involving Bax, caspase activation and subsequent cell death. It is
of interest to determine the possible upstream signaling pathways
that lead to ERK activation upon triptolide treatment. ERK
activation has been implicated as a response to counteract ER
stress (17,18), and it was thus hypothesized that
triptolide could induce ER stress in treated cells resulting in ERK
activation. MDA-MB-231 cells were treated with 40 ng/ml triptolide
over a time course of 48 h, and the expression levels of common
markers of ER stress, including PERK, eIF2α, Ire1α and Bip, were
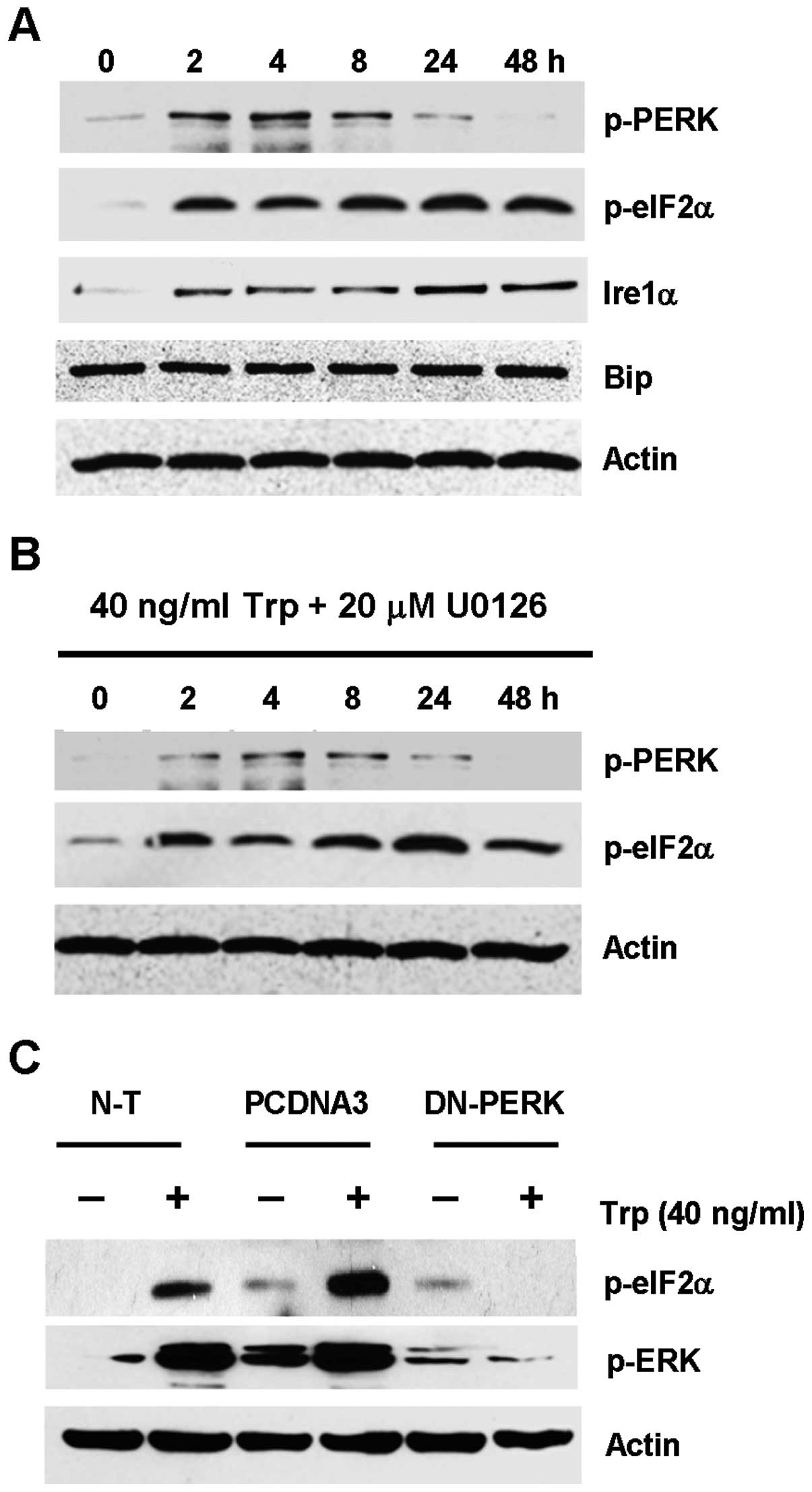
evaluated. As shown in Fig. 5A,
p-PERK in triptolide-treated cells was transiently upregulated from
2 to 8 h, which gradually decreased to basal level after 24 h. The
upregulation of p-PERK was seen with the concomitant upregulation
of its downstream effector, eIF2α, which remained phosphorylated
from 2 h to 48 h. Upregulation of Ire1α was also observed, while no
change in Bip expression was seen for all the time points
tested.
To further test the hypothesis that ER stress could
lead to ERK activation in triptolide-treated cells, ERK and PERK
were inhibited by the MEK inhibitor U0126 and by transfecting the
cells with the dominant negative PERK plasmid, respectively, so as
to dissect which kinase was acting more upstream. As shown in
Fig. 5B, the use of U0126 had no
inhibitory effect on the expression of p-PERK and p-eIF2α. This
finding indicated that ERK activation seemed to act downstream of
the activation of ER stress markers. On the contrary, triptolide
treatment in the MDA-MB-231 cells that were transfected with the
dominant negative PERK plasmid showed substantial reduction in ERK
activation, as compared to the non-transfected cells or cells
transfected with empty plasmid (Fig.
5C). Taken together, these results indicated that triptolide
induced ER stress presumably through the PERK-eIF2α pathway, which
would result in downstream ERK activation.
The role of reactive oxygen species in
triptolide-induced ERK activation and cell death
Accumulating lines of evidence suggest that ER
stress could be associated with the generation of ROS and induce
apoptosis (28,29). Since it was shown previously that
triptolide can induce ER stress, we sought to determine if ROS was
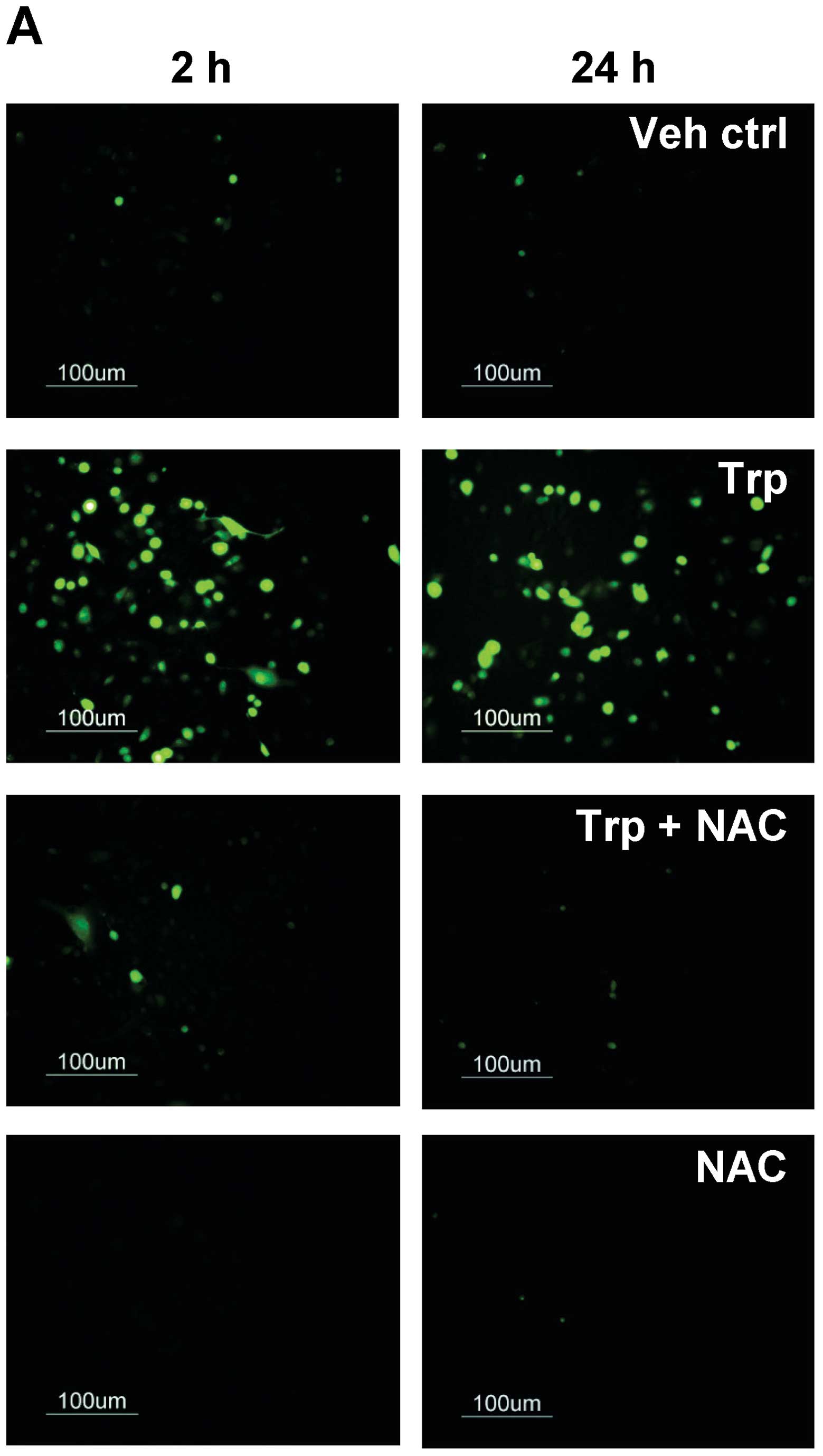
generated in triptolide-treated cells during ER stress. As shown in
Fig. 6A, ROS were generated as
early as 2 h when cells were treated with triptolide, as reflected
by the increase in fluorescent signal. Co-treatment with 1 mM NAC
abolished the fluorescent signals in triptolide-treated cells at 2
and 24 h, providing further evidence for the ability of triptolide
to induce ROS generation. To further determine the involvement of
ROS in triptolide-induced ERK activation and cell viability, p-ERK
and cell viability were evaluated. With the co-treatment of NAC,
the phosphorylation of ERK was substantially reduced in
triptolide-treated cells (Fig.
6B), and the percentage of apoptotic cells was also
significantly reduced, as reflected by the reduction in the
percentage of cells in the sub-G0/G1 phase
from 18.2±2.0 to 11.0±0.7% at 48 h post-triptolide treatment
(Fig. 6C). These findings show
that ROS was upstream of triptolide-induced ERK activation and
subsequent apoptosis.
Discussion
Triptolide, a diterpene triepoxide isolated from the
medicinal plant Tripterygium wilfordii Hook F., was first
reported to have anti-leukemic properties in 1972 (30). Over the last few decades, intensive
research efforts have been devoted to the elucidation of the
molecular effects underlying the anticancer effects of triptolide.
Lines of evidence accumulated from in vitro and in
vivo studies pointed to the ability of triptolide to directly
induce apoptosis in various cancer types via multiple targets
(31). Our current study is the
first to report yet another novel mechanism by which triptolide
induced cancer cell apoptosis. Specifically, triptolide was shown
to induce ROS generation and ER stress via the PERK-eIF2α pathway
that subsequently activated ERK and resulted in caspase-dependent
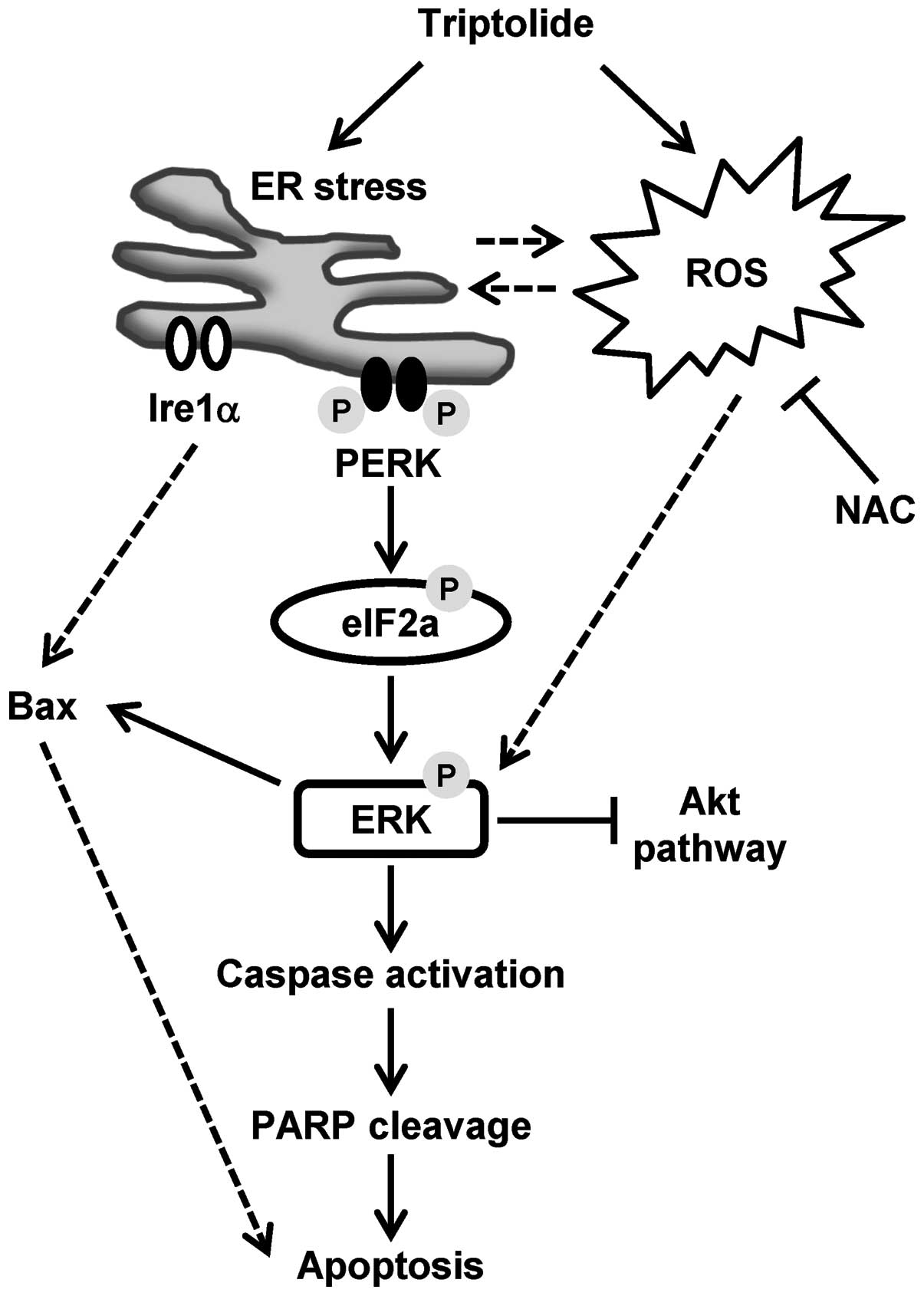
apoptosis. Our proposed model of triptolide-induced
caspase-dependent apoptosis is presented in Fig. 7, with the following sections
focused on the discussion of the role of ERK activation in relation
to ROS generation, ER stress and eventual apoptosis.
ERK signaling is part of the MAPK superfamily, and
is well known for its ability to modulate cell survival in response
to external stimuli. Recent studies have suggested a more
complicated role of ERK in which its activation could promote cell
death in some cell types under certain conditions. In particular,
ERK was selectively activated in neuronal and renal epithelial
cells upon exposure to oxidative stress and toxicants such as
cisplatin, and inhibition of the ERK pathway blocks apoptosis
(32). As triptolide is known to
cause ROS generation (20,21), it is interesting to note that
triptolide-induced ERK activation was partly mediated through ROS
generation, and this observation, as summarized in Fig. 6, is similar to those reported for
cisplatin (24). In view of the
synergistic, anticancer effect of the triptolide/cisplatin
combination (8,33), ERK activation could be another
molecular mechanism underlying the synergism of this drug
combination that is yet to be further characterized using in
vitro and in vivo models.
The ability of triptolide to induce persistent ROS
generation could trigger the unfolded protein response which is a
cellular mechanism for the adaptation to homeostatic changes such
as redox status alteration. Protein misfolding as a result of
oxidative stress, which is indicative of perturbation of ER
homeostasis, has been implicated to initiate apoptosis (29). Our data demonstrate that triptolide
was able to induce ER stress selectively via the PERK-eIF2α pathway
which is often activated by oxidant stimuli. Furthermore, ERK
activation was shown to act downstream of the PERK-eIF2α pathway.
These observations are in line with previous studies that
demonstrate the activation of ERK by ER stress inducers and
subsequent apoptosis (17,34). While JNK could be activated during
ER stress that was mediated by Ire-1α (34,35),
our data did not support the notion that JNK was activated as a
result of the activation of Ire-1α, as the JNK inhibitor, SP600125,
did not reverse triptolide-induced cancer cell killing.
In our study, inhibiting ERK activation by U0126
partially inhibited triptolide-induced Bax expression but had no
significant effect on Bcl-xL expression (Fig. 4). Furthermore, inhibition of Bax
overexpression with U0126 inhibitor was sufficient to overcome
triptolide-induced cancer cell killing. This suggests that
triptolide-induced ERK activation initiates apoptosis rather than
survival, since it only regulates the pro-apoptotic protein Bax
without substantial influence on the anti-apoptotic protein Bcl-xL.
This is consistent with a previous report that ERK may act upstream
of the Bcl-2 family (32,36). In addition to the association of
ERK with the pro-apoptotic Bax that regulates apoptosis, it has
been suggested that promotion of cell death by ERK activation could
be related to the suppression of the PI3K/Akt pathway which is the
critical survival signaling pathway in cells (32). The results presented in Fig. 3A are consistent with this notion,
and ERK activation could serve as a negative feedback signal to
downregulate p-Akt to suppress its anti-apoptotic effects, as ERK
activation occurred earlier than downregulation of p-Akt. This
notion is further supported by Dai et al that there was
cross-talk between PI3K/Akt and MEK/ERK pathways when mediating ER
stress-induced cell cycle progression and cell death in human
hepatocellular carcinoma cells (37).
In summary, we have shown that ERK activation is a
novel mechanism by which triptolide induced apoptosis in MDA-MB-231
breast cancer cells. In addition to ROS generation, we have shown
for the first time that triptolide induced ER stress via the
PERK/eIF2α pathway, which could have important implications in the
design and further development of triptolide as an ER stress
inducer for anticancer therapy.
Acknowledgements
This study was supported by the
National Medical Research Council of Singapore through an IRG
research grant (NMRC/1109/2007).
References
|
1.
|
Chen BJ: Triptolide, a novel
immunosuppressive and anti-inflammatory agent purified from a
Chinese herb Tripterygium wilfordii Hook F. Leuk Lymphoma.
42:253–265. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Lue Y, Sinha Hikim AP, Wang C, et al:
Triptolide: a potential male contraceptive. J Androl. 19:479–486.
1998.PubMed/NCBI
|
|
3.
|
Qiu D, Zhao G, Aoki Y, et al:
Immunosuppressant PG490 (trip-tolide) inhibits T-cell interleukin-2
expression at the level of purine-box/nuclear factor of activated
T-cells and NF-kappaB transcriptional activation. J Biol Chem.
274:13443–13450. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Fidler JM, Li K, Chung C, et al: PG490-88,
a derivative of triptolide, causes tumor regression and sensitizes
tumors to chemotherapy. Mol Cancer Ther. 2:855–862. 2003.PubMed/NCBI
|
|
5.
|
Jiang XH, Wong BC, Lin MC, et al:
Functional p53 is required for triptolide-induced apoptosis and
AP-1 and nuclear factor-kappaB activation in gastric cancer cells.
Oncogene. 20:8009–8018. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Lee KY, Chang W, Qiu D, et al: PG490
(triptolide) cooperates with tumor necrosis factor-alpha to induce
apoptosis in tumor cells. J Biol Chem. 274:13451–13455. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Chang WT, Kang JJ, Lee KY, et al:
Triptolide and chemotherapy cooperate in tumor cell apoptosis. A
role for the p53 pathway. J Biol Chem. 276:2221–2227. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Matsui Y, Watanabe J, Ikegawa M, et al:
Cancer-specific enhancement of cisplatin-induced cytotoxicity with
triptolide through an interaction of inactivated glycogen synthase
kinase-3beta with p53. Oncogene. 27:4603–4614. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Miyata Y, Sato T and Ito A: Triptolide, a
diterpenoid triepoxide, induces antitumor proliferation via
activation of c-Jun NH2-terminal kinase 1 by decreasing
phosphatidylinositol 3-kinase activity in human tumor cells.
Biochem Biophys Res Commun. 336:1081–1086. 2005. View Article : Google Scholar
|
|
10.
|
Phillips PA, Dudeja V, McCarroll JA, et
al: Triptolide induces pancreatic cancer cell death via inhibition
of heat shock protein 70. Cancer Res. 67:9407–9416. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Carter BZ, Mak DH, Schober WD, et al:
Triptolide induces caspase-dependent cell death mediated via the
mitochondrial pathway in leukemic cells. Blood. 108:630–637. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Wang Y, Lu JJ, He L, et al: Triptolide
(TPL) inhibits global transcription by inducing
proteasome-dependent degradation of RNA polymerase II (Pol II).
PLoS One. 6:e239932011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Huang W, He T, Chai C, et al: Triptolide
inhibits the proliferation of prostate cancer cells and
down-regulates SUMO-specific protease 1 expression. PLoS One.
7:e376932012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Frese S, Pirnia F, Miescher D, et al:
PG490-mediated sensitization of lung cancer cells to
Apo2L/TRAIL-induced apoptosis requires activation of ERK2.
Oncogene. 22:5427–5435. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Bacus SS, Gudkov AV, Lowe M, et al:
Taxol-induced apoptosis depends on MAP kinase pathways (ERK and
p38) and is independent of p53. Oncogene. 20:147–155. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Tang D, Wu D, Hirao A, et al: ERK
activation mediates cell cycle arrest and apoptosis after DNA
damage independently of p53. J Biol Chem. 277:12710–12717. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Arai K, Lee SR, van Leyen K, et al:
Involvement of ERK MAP kinase in endoplasmic reticulum stress in
SH-SY5Y human neuroblastoma cells. J Neurochem. 89:232–239. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Hu P, Han Z, Couvillon AD, et al: Critical
role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting
endoplasmic reticulum stress-induced cell death. J Biol Chem.
279:49420–49429. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Wan CK, Wang C, Cheung HY, et al:
Triptolide induces Bcl-2 cleavage and mitochondria dependent
apoptosis in p53-deficient HL-60 cells. Cancer Lett. 241:31–41.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Bao X, Cui J, Wu Y, et al: The roles of
endogenous reactive oxygen species and nitric oxide in
triptolide-induced apoptotic cell death in macrophages. J Mol Med
(Berl). 85:85–98. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Xu B, Guo X, Mathew S, et al: Triptolide
simultaneously induces reactive oxygen species, inhibits NF-kappaB
activity and sensitizes 5-fluorouracil in colorectal cancer cell
lines. Cancer Lett. 291:200–208. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Dong J, Ramachandiran S, Tikoo K, et al:
EGFR-independent activation of p38 MAPK and EGFR-dependent
activation of ERK1/2 are required for ROS-induced renal cell death.
Am J Physiol Renal Physiol. 287:F1049–F1058. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ramachandiran S, Huang Q, Dong J, et al:
Mitogen-activated protein kinases contribute to reactive oxygen
species-induced cell death in renal proximal tubule epithelial
cells. Chem Res Toxicol. 15:1635–1642. 2002. View Article : Google Scholar
|
|
24.
|
Wang X, Martindale JL and Holbrook NJ:
Requirement for ERK activation in cisplatin-induced apoptosis. J
Biol Chem. 275:39435–39443. 2000. View Article : Google Scholar
|
|
25.
|
Zhuang S, Yan Y, Daubert RA, et al: ERK
promotes hydrogen peroxide-induced apoptosis through caspase-3
activation and inhibition of Akt in renal epithelial cells. Am J
Physiol Renal Physiol. 292:F440–F447. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Yao J, Jiang Z, Duan W, et al: Involvement
of mitochondrial pathway in triptolide-induced cytotoxicity in
human normal liver L-02 cells. Biol Pharm Bull. 31:592–597. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Kim YK, Kim HJ, Kwon CH, et al: Role of
ERK activation in cisplatin-induced apoptosis in OK renal
epithelial cells. J Appl Toxicol. 25:374–382. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Hsieh YH, Su IJ, Lei HY, et al:
Differential endoplasmic reticulum stress signaling pathways
mediated by iNOS. Biochem Biophys Res Commun. 359:643–648. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Malhotra JD and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress: a vicious cycle or a
double-edged sword? Antioxid Redox Signal. 9:2277–2293. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Kupchan SM and Schubert RM: Selective
alkylation: a biomimetic reaction of the antileukemic triptolides?
Science. 185:791–793. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Liu Q: Triptolide and its expanding
multiple pharmacological functions. Int Immunopharmacol.
11:377–383. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Zhuang S and Schnellmann RG: A
death-promoting role for extracellular signal-regulated kinase. J
Pharmacol Exp Ther. 319:991–997. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Li CJ, Chu CY, Huang LH, et al:
Synergistic anticancer activity of triptolide combined with
cisplatin enhances apoptosis in gastric cancer in vitro and in
vivo. Cancer Lett. 319:203–213. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Urano F, Wang X, Bertolotti A, et al:
Coupling of stress in the ER to activation of JNK protein kinases
by transmembrane protein kinase IRE1. Science. 287:664–666. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Mhaidat NM, Thorne R, Zhang XD, et al:
Involvement of endoplasmic reticulum stress in Docetaxel-induced
JNK-dependent apoptosis of human melanoma. Apoptosis. 13:1505–1512.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Boucher MJ, Morisset J, Vachon PH, et al:
MEK/ERK signaling pathway regulates the expression of Bcl-2,
Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic
cancer cells. J Cell Biochem. 79:355–369. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Dai R, Chen R and Li H: Cross-talk between
PI3K/Akt and MEK/ERK pathways mediates endoplasmic reticulum
stress-induced cell cycle progression and cell death in human
hepatocellular carcinoma cells. Int J Oncol. 34:1749–1757.
2009.
|