Introduction
Renal cell carcinoma (RCC), which accounts for ∼2–3%
of all adult malignant neoplasm, is the most lethal of the common
urologic cancers (1,2). Traditionally, 30–40% of patients with
RCC die of their cancer, in contrast to the 20% mortality rates
associated with prostate and bladder carcinomas (3,4).
Over the past three decades, there has been an increase in the
detection of renal tumors due to the widespread use of noninvasive
imaging techniques such as ultrasound and computed tomography scan
in the investigation of various non-specific symptoms (5). This trend has correlated with an
increased proportion of incidentally discovered and localized
tumors and with improved 5-year survival rates for patients with
this stage of disease (6,7). However, other factors must also be at
play because Chow and colleagues have documented a steadily
increasing mortality rate from RCC per unit population since the
1980s and this was observed in all ethnic and both gerders
(3,8). It has been reported that the
incidence of advanced tumors per unit population has also
increased; and the mortality rate per unit population has still
been negatively affected (3,8–10).
In the European Union and the United States, between 46,000 and
63,000 new cases of RCC are diagnosed annually and ∼40% of RCC
patients die from RCC-related causes each year (7). In the treatment of renal cell
carcinoma, localized disease is curable by surgery, however,
metastatic and locally advanced disease are considerably more
difficult to treat (11,12). Historically, therapeutic options
for metastatic or for locally advanced RCC, primarily cytokine
therapy, produced low response rates and considerable toxicity
(13,14). Moreover, it is well known that RCC
is one of the most vascular of cancers as reflected by the
distinctive neovascular pattern exhibited on renal angiography.
Given the dependence of RCC on angiogenesis and the absence of
generally effective forms of systemic therapies, it is not
surprising that RCC has been targeted for anti-angiogenic
approaches. Targeted agents (such as sorafenib, sunitinib malate,
pazopanib, bevacizumab) are now well established for the treatment
of patients with advanced or metastatic renal cell carcinoma
(7,15,16).
Although the targeted therapy is spread among a
variety of new active drugs, so far complete cure is achieved very
rarely (17). Experience has shown
that the targeted therapies control tumor growth only temporarily
and after a latency period that may vary according to the molecular
characteristics of the tumor, the malignancy reappears. Resistance
to targeted therapy may be caused by activation of alternative
proangiogenic pathways, recruitment of bone marrow-derived
proangiogenic cells, increased pericyte coverage of tumor
vasculature and enhancement of invasion and metastasis (18–20).
Additionally, host genomics may adversely affect drug metabolism,
leading to poor activity or toxicities. However, there does not
appear to be cross-resistance among these therapies and subsequent
treatment lines can be beneficial. Therefore, finding a new target
for anti-angiogenesis could be helpful for treating advanced and
metastatic RCC (16).
Vascular endothelial growth inhibitor (VEGI) [also
known as tumor necrosis factor superfamily member 15 (TNFSF15) and
TNF ligand related molecule 1 (TL1)], a natural anti-angiogenic
factor, was first reported in 1999 from human umbilical vein
endothelial cells (21–23). The full gene of VEGI is ∼17 kb,
which consists of four exons and three introns and is mapped to
human chromosome 9q32. Three splicing variants of VEGI have been
reported. Its transcript was found to be expressed in the placenta,
lung, kidney, skeletal muscle, pancreas, spleen, small intestine,
prostate and colon (23). The
secreted soluble form of VEGI has been demonstrated as a potent
anti-angiogenic factor through inhibiting proliferation of
endothelial cells (24,25). Previous studies have also shown
that VEGI expression could significantly reduce motility and
adhesion of prostate and bladder cancer cells (26,27).
Zhai et al and Xiao et al reported that VEGI markedly
suppressed the growth of colon carcinoma cells (murine colon cancer
cells, MC-38) both in vitro and in vivo(23,28).
Systemic administration of VEGI also markedly inhibited tumor
growth and increased survival time in a Lewis lung cancer (LLC)
murine tumor model suggesting that pro-apoptotic effect of soluble
VEGI in endothelial cells is critical for its antitumor activity
(28–32).
Despite these observations of VEGI in solid tumors,
its role in RCC remains unknown. In the present study, the
expression of VEGI was examined in human RCC and normal renal
tissues and in RCC cell lines. The biological function of this
molecule was investigated in vitro and in vivo, in
which the expression of VEGI was manipulated by genetic
methods.
Materials and methods
Materials
Mice
All female BALB/c mice (6–8 weeks, 18–22 g) were
supplied by Shanghai Laboratory Animal Center [SLAC, Shanghai,
China, Animal Certificate No. SCXK (Hu) 2007-0005]. All the
procedures related to animal handling, care and the treatment in
this study were performed according to the guidelines approved by
the Institutional Animal Care and Use Committee (IACUC) of CrownBio
following the guidance of the Association for Assessment and
Accreditation of Laboratory Animal Care (AAALAC).
Cell lines
All cell lines used in this study were purchased
from the American Type Culture Collection (ATCC, Rockville, MD,
USA). This study used human renal cell carcinoma cells (786-O,
OS-RC-2, A-498, Caki-1), a human adrenal gland small cell carcinoma
cell line (SW-13) and a human umbilical vein endothelial cell line
(HUVEC). Cells were routinely cultured with Dubecco’s modified
Eagle’s medium (DMEM) supplemented with 10% fetal calf serum,
penicillin and streptomycin (Gibco BRC, Paisley, UK).
Human renal cell carcinoma
specimens
A total of 23 pairs renal cell carcinoma and normal
renal tissue samples were snap-frozen in liquid nitrogen
immediately after open radical nephrectomy. The pathologist
verified normal and cancer specimens. All protocols were reviewed
and approved by the local ethics committee and all patients gave
written informed consent.
RNA isolation and reverse
transcription PCR
RNA was isolated using Total RNA Isolation reagent
(ABgene, Epsom, UK). First strand cDNA was synthesized from 0.5
μg RNA using a reverse transcription kit (Sigma, Poole,
Dorset, UK). The quality of cDNA was verified through the
amplification and detection of the GAPDH housekeeping gene. VEGI
forward and reverse primers (Table
I) were designed based on the human VEGI sequence (GeneBank
accession no. BD131562). PCR was performed in a GeneAmp PCR system
2400 thermocycler (Perkin-Elmer, Norwalk, CT, USA). Conditions for
PCR were 40 sec at 94°C, 60 sec at 55°C, 60 sec at 72°C (35
cycles). PCR products were separated on a 1.4% agarose gel.
 | Table IPrimer sequences for PCR. |
Table I
Primer sequences for PCR.
| Primer | Forward | Reverse |
|---|
| hGAPDH |
5′-AGCTTGTCATCAATGGAAAT-3′ |
5′-CTTCACCACCTTCTTGATGT-3′ |
| hGAPDH (qPCR) |
5′-CTGAGTACGTCGTGGAGTC-3′ |
5′-ACTGAACCTGACCGTACA |
| |
CAGAGATGATGACCCTTTTG-3′ |
| | Z-sequence |
| VEGI |
5′-ATGAGACGCTTTTTAAGCAA-3′ |
5′-CTATAGTAAGAAGGCTCCAAAG-3′ |
| VEGI (qPCR) |
5′-CAAAGTCTACAGTTTCCCAAT-3′ |
5′-ACTGAACCTGACCGTACA |
| |
TGATTTTTAAAGTGCTGTGTG-3′ |
| | Z-sequence |
| VEGI (ribozyme
2) |
5′-CTGCAGTCTCACAACTGGAAACT |
5′-ACTAGTTAATCCTCTTTCTTGTTT |
|
GATGAGTCCGTGAGGA-3′ |
CGTCCTCACGGACT-3′ |
| VEGI
(expression) |
5′-ATGAGACGCTTTTTAAGCAA-3′ |
5′-CTATAGTAAGAAGGCTCCAAAGA-3′ |
| Primers for
detecting plasmid | T7F:
TAATACGACTCACTATAGGG | BGHR:
TAGAAGGCACAGTCGAGG |
| Primers to detect
ribozymes | RBTPF:
CTGATGAGTCCGTGAGGACGAA | RBBMR:
TTCGTCCTCACGGACTCATCAG |
IHC staining of human renal and
xenograft tumor specimens
Frozen specimens of renal cell carcinoma (n=23),
normal renal tissue (n=23) and xenograft tumors (n=24) were cut at
a thickness of 6 μm using a cryostat (Leica CM 1900, Leica
Microsystems UK Ltd., Buckinghamshire, UK). The nature of the
samples was independently verified by two pathologists. After
fixation, the sections were blocked with horse serum and probed
with or without VEGI antibody (SC-53975) for 1 h. The secondary
biotinylated antibody and the avidin-biotin complex were
subsequently applied to detect VEGI expression in accordance with
the Vectastain Universal Elite ABC kit protocol (Vector
Laboratories, Peterborough, UK). After developing color with DAB,
the sections were counterstained with Gill’s hematoxylin. Staining
was independently assessed by the authors. The monoclonal mouse
anti-human-VEGI and anti-human-VEGFR2 antibodies were purchased
from LSBio Inc. (LS-C76815 and LS-C92359, respectively, LSBio Inc.,
Seattle, WA, USA). Monoclonal mouse anti-human-Ki67 and
anti-human-CD31 (PECAM) antibodies were purchased from Santa Cruz
Biotechnology, Inc. (SC-56320 and SC-133091 respectively, Santa
Cruz, CA, USA).
Western blot analysis of VEGI
expression
Protein concentration in cell lysates were
determined using the DC Protein Assay kit (Bio-Rad) and an ELx800
spectrophotometer (Bio-Tek™). Equal amount of proteins were
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and blotted onto nitrocellulose sheets.
Proteins were then probed with the VEGI antibody (1:1,500) and
peroxidase-conjugated secondary antibodies. Monoclonal mouse
anti-human-β-actin (1:1,000, SC-130301, Santa Cruz) was used as a
housekeeping control. Protein bands were visualized using the
Supersignal™ West Dura system (Pierce Biotechnology, Inc.,
Rockford, IL, USA) and photographed using an UVITech imager
(UVITech, g5286Inc., Cambridge, UK).
Construction of VEGI expressing and
ribosome transgenes and transfection
The full-length human VEGI coding sequence and
hammer-head ribozymes targeting human VEGI was cloned into a
mammalian expression plasmid vector (pEF/His TOPO TA, Invitrogen,
Inc., Paisley, UK). Full primer sequences are provided in Table I. The recombinant plasmid vectors
were transformed into chemically competent TOP10 E. coli
(Invitrogen). After verification and amplification, empty control
plasmids or plasmids containing VEGI expression sequence or
ribozyme transgenes were then transfected into 786-O, SW-13 and
SC-OR-2 cells using electroporation (Easyjet, EquiBio Ltd., Kent,
UK). After selection with blasticidin, the transfectants were used
used for subsequent analysis.
Real-time quantitative polymerase
chain reaction (qPCR)
Real-time quantitative PCR was carried out using the
iCycleriQ5 system (Bio-Rad, Hemel Hemstead, UK) to
determine the level of expression of the VEGI transcripts in the
cell lines. GAPDH was used as a housekeeping control.
Cell growth assay
This was based on a method we previously described
(33). Briefly, cells were plated
into a 96-well plate (2,500 cells/well). Cell growth was assessed
after 1, 3 and 5 days. Crystal violet was used to stain cells and
absorbance was determined at a wavelength of 540 nm using a
spectrophotometer (Bio-Tek, Elx800, UK).
Flow cytometric analysis of
apoptosis
All cells including those floating in the culture
medium were harvested after a period of incubation. The apoptotic
population of the cells was determined using Vybrant®
Apoptosis Assay kit (Invitrogen) and flow cytometry
(CyFlow® SL, Partec GmbH, Münster, Germany).
Wounding assay
The migration of cells in a monolayer across a
wounded surface of a near-confluent cell monolayer was examined.
Cells at a density of 50,000/well were seeded into a chamber slide
and allowed to reach near confluence. The monolayer of cells was
then scraped with a fine gauge needle to create a wound of ∼200
μm. The movement of cells to close the wound was recorded
using a time lapse video recorder and analysed using the motion
analysis feature of the Image J software (NIH, USA).
Motility assay using Cytodex-2
beads
Cells (1×106) were incubated with 100
μl of cytocarrier beads overnight. The beads were washed
twice to remove dead cells and then resuspended. Beads/cells (100
μl) were transferred into each well of a 24-well plate.
After incubation for 4 h, the cells were fixed in 4% formalin and
stained with 0.5% crystal violet before counting.
Invasion assay
This was modified from a method we prevously
described (33). Transwell inserts
with 8-μm pore size were coated with 50 μg Matrigel
(BD Matrigel™ basement membrane matrix) and air-dried. After
rehydration, 20,000 cells were added to each well. After 96 h cells
that had migrated through the matrix to the other side of the
insert were fixed, stained and then counted under a microscope.
Cell-matrix adhesion assay
Cell-matrix adhesion was assessed in a standard
manner. Cells (40,000) were added in each well of 96-well plate,
previously coated with Matrigel (5 μg/well). After 40 min of
incubation, non-adherent cells were washed off using BSS buffer.
The remaining adherent cells were then fixed and stained with
crystal violet. The number of adherent cells was then counted.
Endothelial cell differentiation
assay: tube formation on Matrigel (34)
This was based on a Matrigel-sandwich tubule forming
assay developed in our laboratory. Briefly, 200 μg of cold
Matrigel solution dispersed in 100 μl (Becton-Dickinson,
Bristol, UK) was added to a 96-well plate and allowed to air-dry at
37°C. Following rehydration of Matrigel, it formed a thin bottom
layer. HUVEC cells (2.5×104) per well were seeded onto a
96-well plate and allowed to attach to the bottom of the well (4–6
h). After that, 200 μg of cold Matrigel solution dispersed
in 100 μl was loaded to each well again, followed by
incubation at 37°C for 3 h when the second layer of Matrigel
solidified. Finally, 10,000 tested cells (different renal cell
carcinoma cell lines) were added and incubated at 37°C in a
humidified chamber with 5% CO2. Following incubation for
7 or 20 h, cultures were observed and photographed (Nikon, Tokyo,
Japan).
Tumor xenografts in Nu/Nu nude
mice
Twenty-four mice were divided into three groups
(786-Owt Group, 786-OpEF/His Group and
786-OVEGIexp4 Group). Each mouse was inoculated
subcutaneously at the right flank with 786-O cells
(5×106) in 0.1 ml of PBS for tumor development. The mice
were kept in individually ventilated cage (IVC) systems at constant
temperature and humidity with 4 animals in each cage. The date of
tumor cell inoculation was denoted as day 0. The data of tumor
growth and normal behavior such as mobility, visual estimation of
food and water consumption, body weight gain/ loss (body weights
were measured twice weekly), eye/hair matting and any other
abnormal effect were collected every three days. Tumor sizes were
measured each three days in two dimensions using a caliper and the
volume was expressed in mm3 using the formula: V = 0.5
a x b2 where a and b were
the long and short diameters of the tumor, respectively.
Statistical analysis and software
The mean optical density (MOD) feature of RT-PCR and
western blot analysis was analysed using software package Image-Pro
Plus 6.0 (Media Cybernetics, USA). The method was as follows: every
image prepared for comparison was changed to grayscale image. Then,
selecting ten points in the comparison region randomly, the MOD of
the point was analyzed using the software. The length of cell
migration and HUVEC was measured by software Image J (NIH). The
distance of HUVEC cells was calculated along the long axes in each
compared image.
All statistical analysis was performed using the
SPSS 16.0 software. Two sample t-tests was used for normally
distributed data. Fisher’s exact test was used for analyzing
immunohistochemical staining in renal tissues. Differences were
considered to be statistically significant at P<0.05.
Results
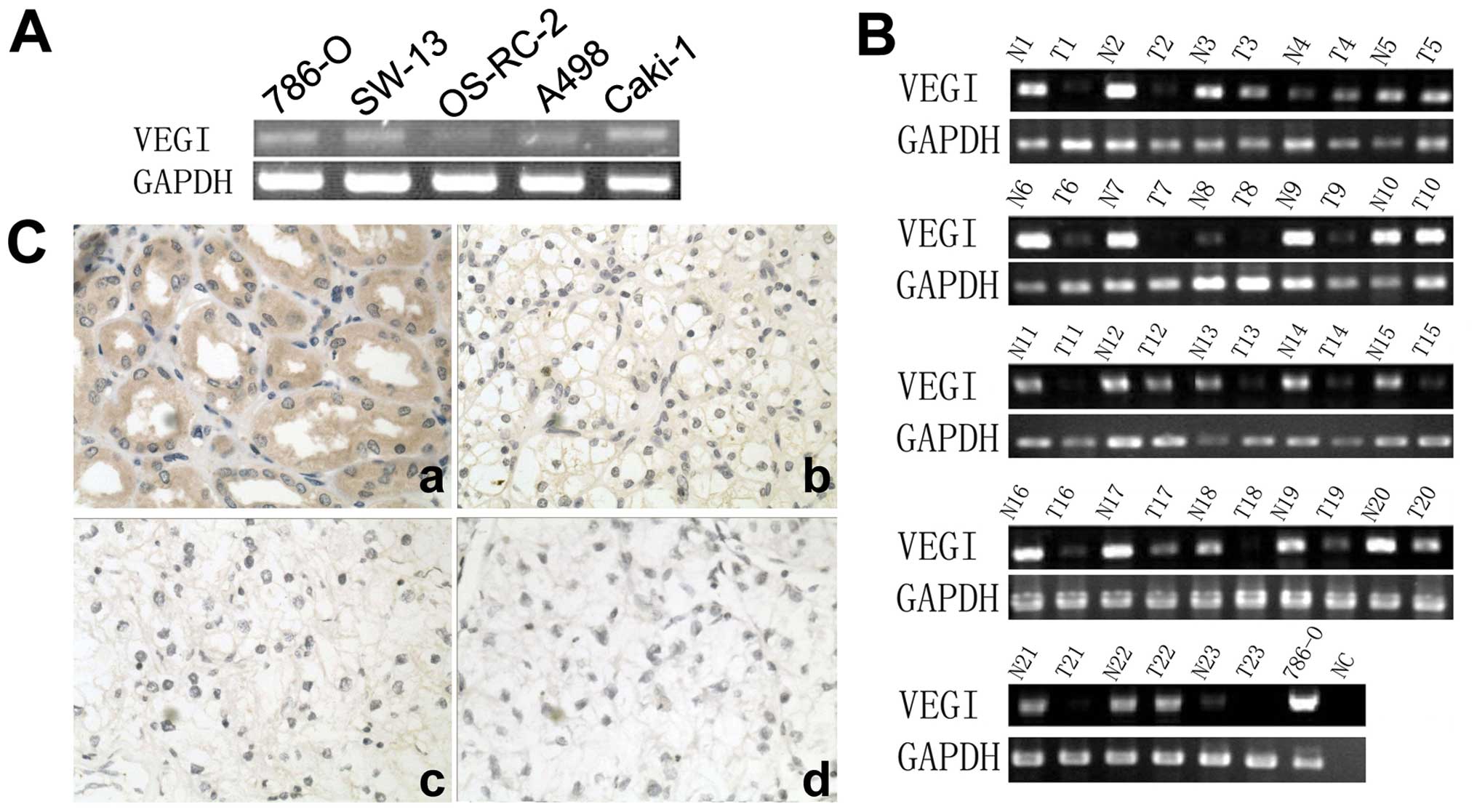
The expression of VEGI in renal cell
carcinoma cell lines and renal tissues
The expression of VEGI was examined in four renal
cell carcinoma cell lines, adrenal gland cell line and human renal
tissues using conventional RT-PCR. VEGI transcript was detectable
in all five cell lines (Fig. 1A).
It was also detected more frequently in normal renal tissues
(23/23) than that in renal cell carcinoma tissues (15/23),
P<0.01 (Fig. 1B). Moreover, the
expression of VEGI transcript was stronger in normal renal tissues
(MOD=0.87±0.13) than that in renal cell carcinoma tissues
(MOD=0.24±0.17), P<0.01. Furthermore, in immunohistochemical
staining, VEGI was seen in normal renal tubular epithelia cells,
but the staining was decreased or absent in renal cell carcinoma
cells, particularly in the specimen with higher grade (Fig. 1C). The positive staining of normal
tissue (100%, 23/23) was significantly higher than that of renal
cell carcinoma tissues (34.78%, 8/23), P<0.01. The MOD of normal
renal tissues (0.49±0.15) was higher than that of renal cell
carcinoma tissues (0.11±0.05) either, P<0.01.
Genetic manipulation of VEGI levels in
RCC cell lines
786-O, which expressed modest level of VEGI
transcript, were transfected with a VEGI expression construct and
anti-VEGI ribozyme transgenes, to respectively create sublines
showing enhanced or suppressed levels of VEGI expression. OS-RC-2
and SW-13, which expressed lower level of VEGI transcript, was only
transfected with the VEGI expression construct. As shown by RT-PCR
analysis, VEGI mRNA expression was increased in
786-OVEGIexp4, OS-RC-2VEGIexp4 and
SW-13VEGIexp4 cells, compared to wild-type
(786-Owt, OS-RC-2wt and SW-13wt)
and plasmid control (786-OpEF/His,
OS-RC-2pEF/His and SW-13pEF/His) cells
(Fig. 2A). However, VEGI mRNA
expression in the 786-OVEGIrib2 cells was lower than
that in 786-Owt cells, but was similar to that in
786-OpEF/His cells, suggesting that the ribozyme is
ineffective (Fig. 2A). Moreover,
according to the results of qPCR, VEGI mRNA expression was
dramatically increased in 786-OVEGIexp4,
OS-RC-2VEGIexp4 and SW-13VEGIexp4 cells, but
did not decrease in 786-OVEGIrib2 cells, compared with
corresponding controls (Fig. 2B).
Comparing with controls, an increase in the VEGI protein level was
also seen in 786-OVEGIexp4 cells in western blot
analysis (Fig. 2C). However, there
was no significant difference in VEGI protein level between
SW-13VEGIexp4 and OS-RC-2VEGIexp4 cells and
their controls. Therefore, we chose the 786-O cell lines to perform
our following test.
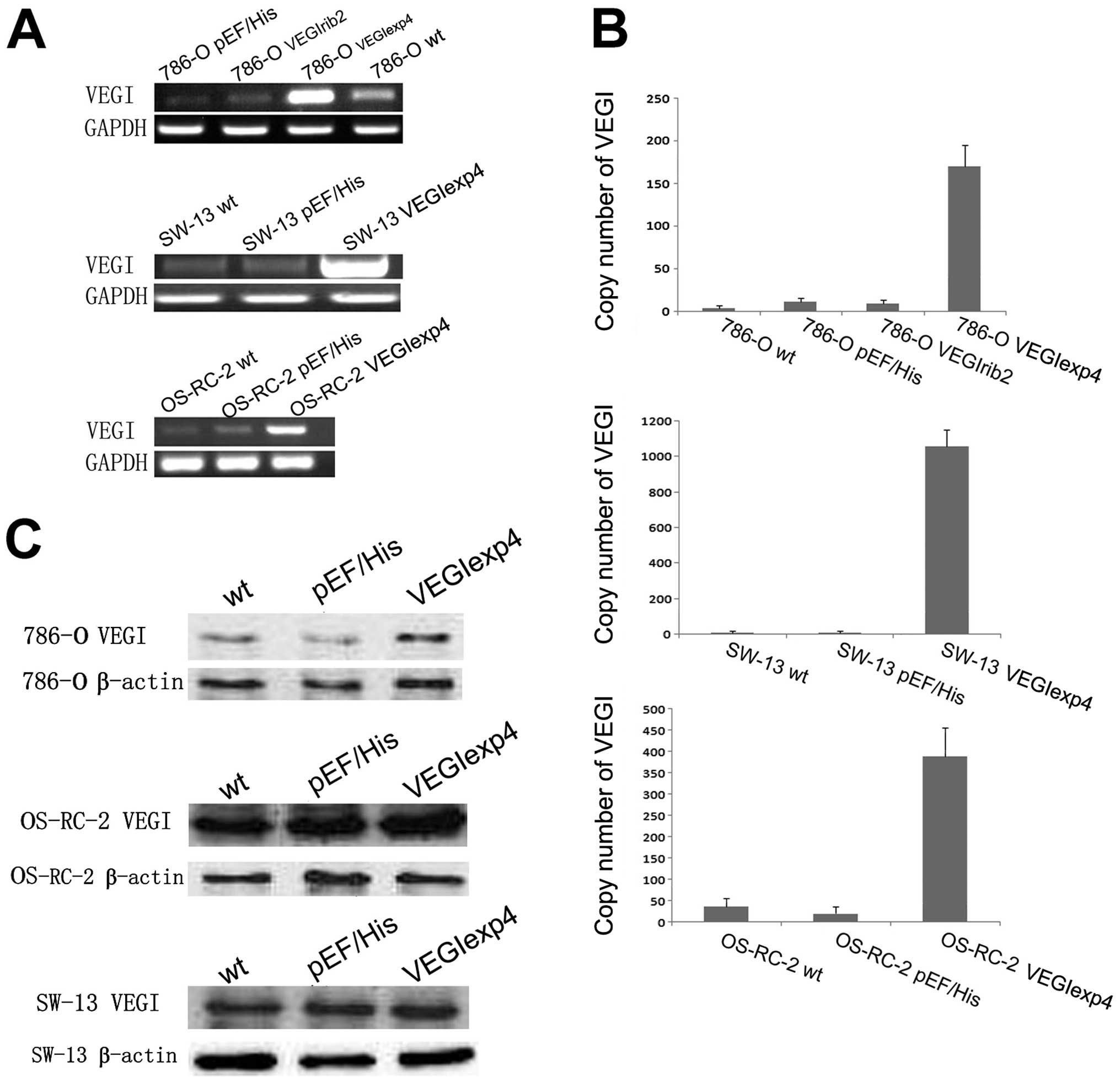 | Figure 2Confirmation of manipulation of VEGI
expression in renal cell carcinoma cells. (A) Verification of
forced expression of the VEGI transcript in 786-O, SW-13 and
OS-RC-2 cells. VEGI mRNA expression in the 786-OVEGIrib2
cells was lower than that in 786-Owt cell, but was
similar to that in 786-OpEF/His cells, suggesting that
the ribozyme is ineffective. Representative image from 4
experiments show results of the RT-PCR. (B) Verification of forced
expression of VEGI transcript in 786-O, SW-13 and OS-RC-2 cells,
using Q-PCR. VEGI mRNA expression was dramatically increased in
786-OVEGIexp4, OS-RC-2VEGIexp4 and
SW-13VEGIexp4 cells, but did not decrease in
786-OVEGIrib2 cells, compared with the wild-type and
empty plasmid control cells. The error bars represent standard
deviations. (C) Force expression of VEGI at protein level using
western blot analysis for 786-O, SW-13 and OS-RC-2 cells. VEGI
protein level was increased in 786-OVEGIexp4 cells
compared to that of wild-type and empty plasmid control cells.
However, there was no significant difference in VEGI protein level
between SW-13VEGIexp4 and OS-RC-2VEGIexp4
cells and their controls. |
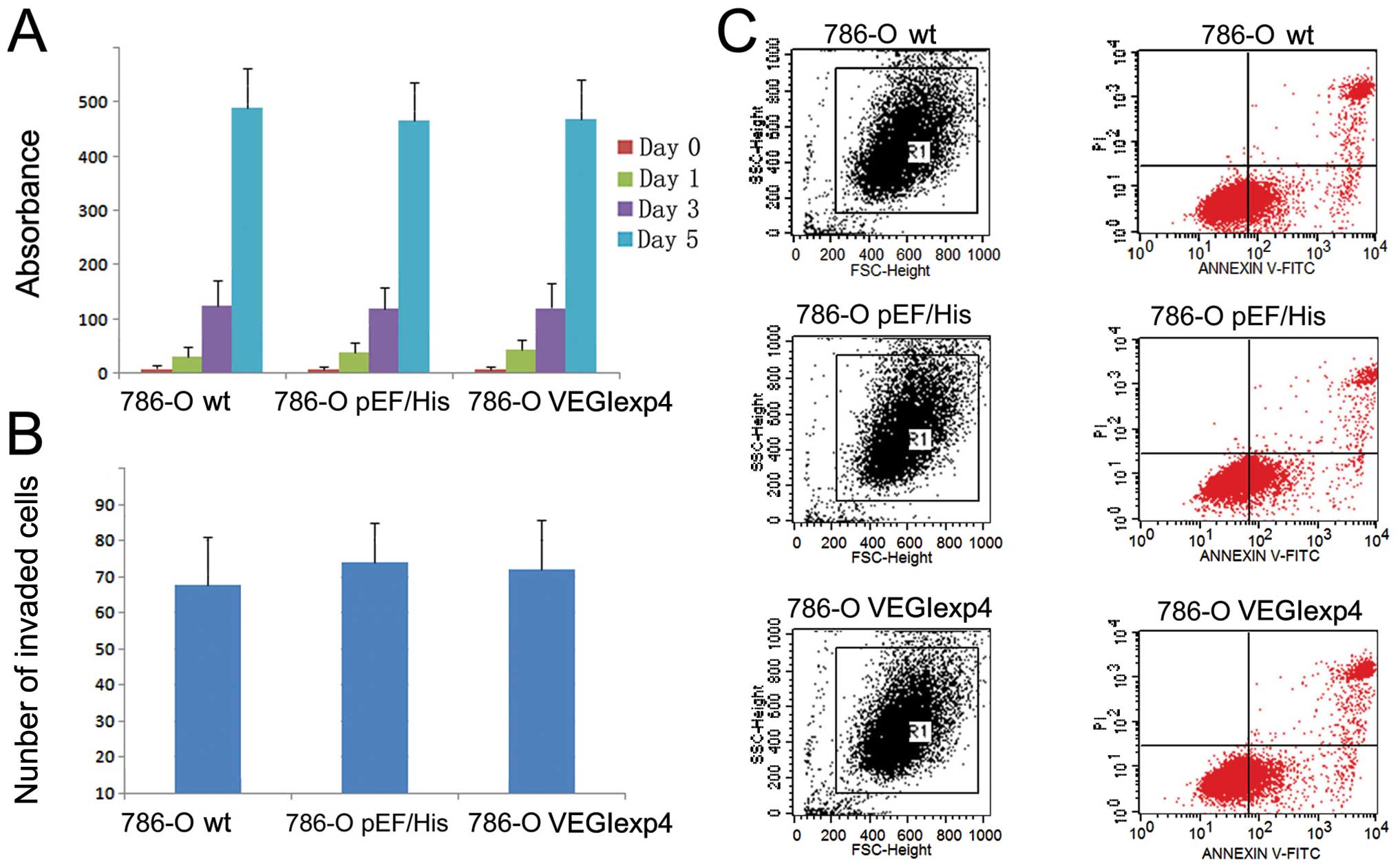
Manipulation of VEGI expression had no
impact on growth, apoptosis and invasiveness of RCC cells
First, we examined the effect on growth, apoptosis
and invasiveness of 786-O cell lines by VEGI. The growth (Fig. 3A) and invasion (Fig. 3B) of 786-OVEGIexp4 cells
did not show any difference from that of wild-type and empty
plasmid control cells (P>0.05). Furthermore, we did not find a
difference in the apoptotic populations between cells with VEGI
forced expression and control cells (P>0.05) (Fig. 3C).
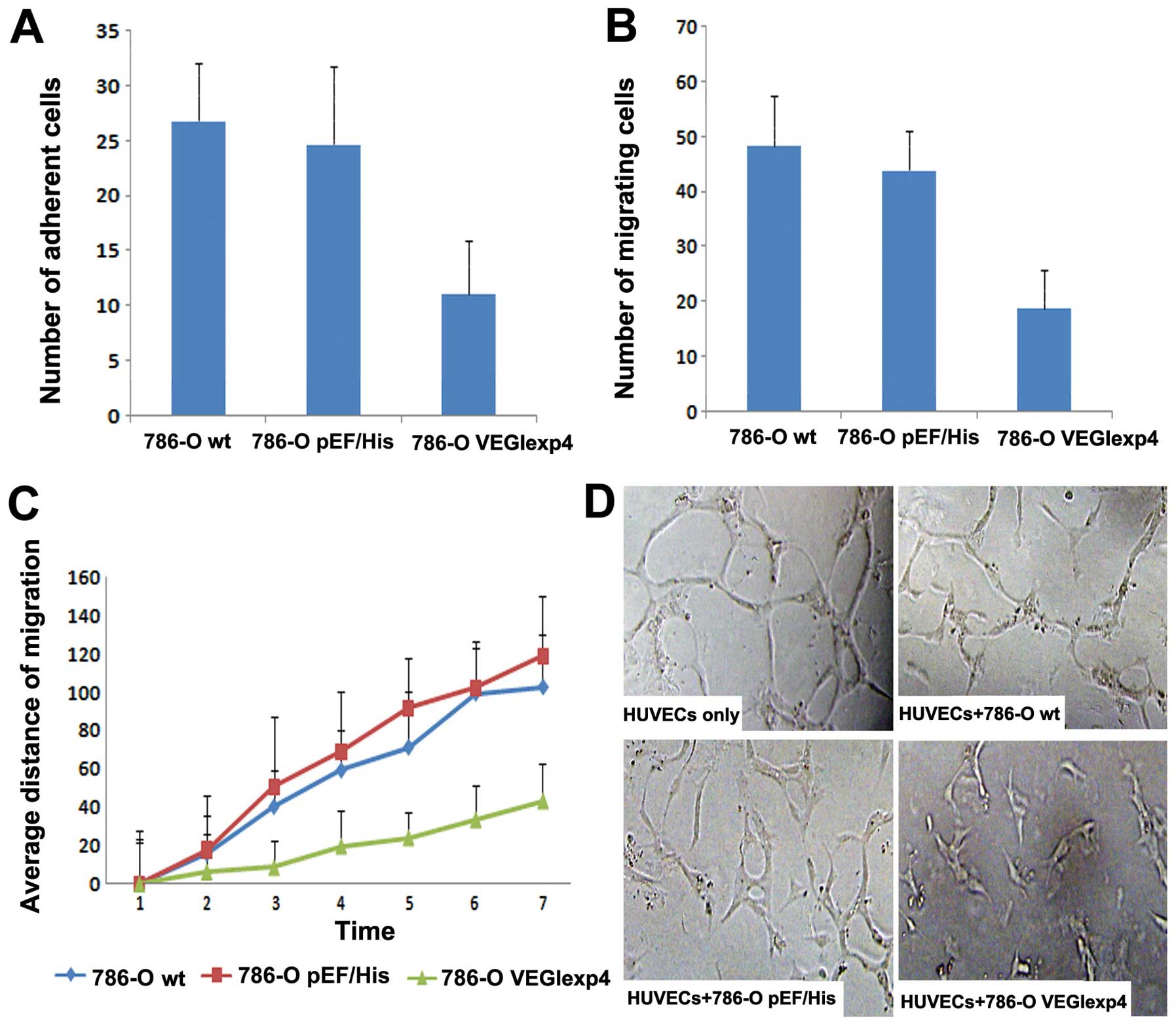
The influence of VEGI expression on
cell matrix adhesion and motility of 786-O cells
In this step, we examined the effect on cell-matrix
adhesion of 786-O cell lines by VEGI. Overexpression of VEGI
exhibited a significant inhibitory effect on cell-matrix adhesion
of the cells. Compared with 786-Owt (26.84±5.19) and
786-OpEF/His (24.72±7.01), the number of adherent cells
for 786-OVEGIexp4 (11.13±4.88) was significantly reduced
(P<0.001 vs both controls) (Fig.
4A).
In the cytocarrier based cell motility assay, we
found that cell motility was reduced significantly in
786-OVEGIexp4 cells. The number of migrating
786-OVEGIexp4 cells was 18.92±6.88 compared with
48.31±9.19 for 786-Owt cells and 43.97±7.01 for
786-OpEF/His cells (Fig.
4B) (both P<0.01). To further investigate the effect of
forced expression of VEGI on cell motility, wounding assay was
used. We also found that the motility was reduced significantly in
cells containing the VEGI expression construct. The average
migrating distance of 786-OVEGIexp4 was 43.35±19.65
μm, P<0.01 compared to both 786-Owt
(102.39±27.42 μm) and 786-OpEF/His (118.71±31.81
μm) cells (Fig. 4C).
Inhibition of vascular endothelial
tube formation by forced expression of VEGI
Single HUVECs, when cultured between the Matrigel
layers, were seen to form typical tube-like structures after 20 h.
Fig. 4D shows that the lumen was
composed of several endothelial cells linked to each other. When
HUVECs with 786-Owt/786-OpEF/His were
co-cultured, a proportion of cells formed an incomplete lumen. The
cells still had sufficient ductibility. However, when HUVECs with
786-OVEGIexp4 co-cultured, the tube formation decreased
significantly in comparison with controls (Fig. 4D). The cells collected together
with poor ductibility. The distance of HUVECs in each field
co-cultured with 786-OVEGIexp4 (1.29E-04±5.5E-06) was
significantly decreased, compared with HUVECs co-cultured with
786-Owt/786-OpEF/His and HUVECs only
(2.44E-04±9.1E-06, 2.48E-04±9.9E-06, 2.58E-04±1.8E-06,
respectively) (P<0.01).
Reduced tumor growth in xenograft
models from 786-O cells with forced expression of VEGI
To examine the effect of VEGI on tumor growth,
xenograt models were successfully established in BALB/c nude mice
by subcutaneous injection of 786-Owt,
786-OpEF/His and 786-OVEGIexp4 cells. Tumor
volume was found significantly reduced in the group of BALB/c nude
mice injected with 786-OVEGIexp4 cells (216±146
mm2), compared with 786-Owt,
786-OpEF/His cells (473±298 and 475±115 mm2,
respectively), P<0.01 (Fig.
5A).
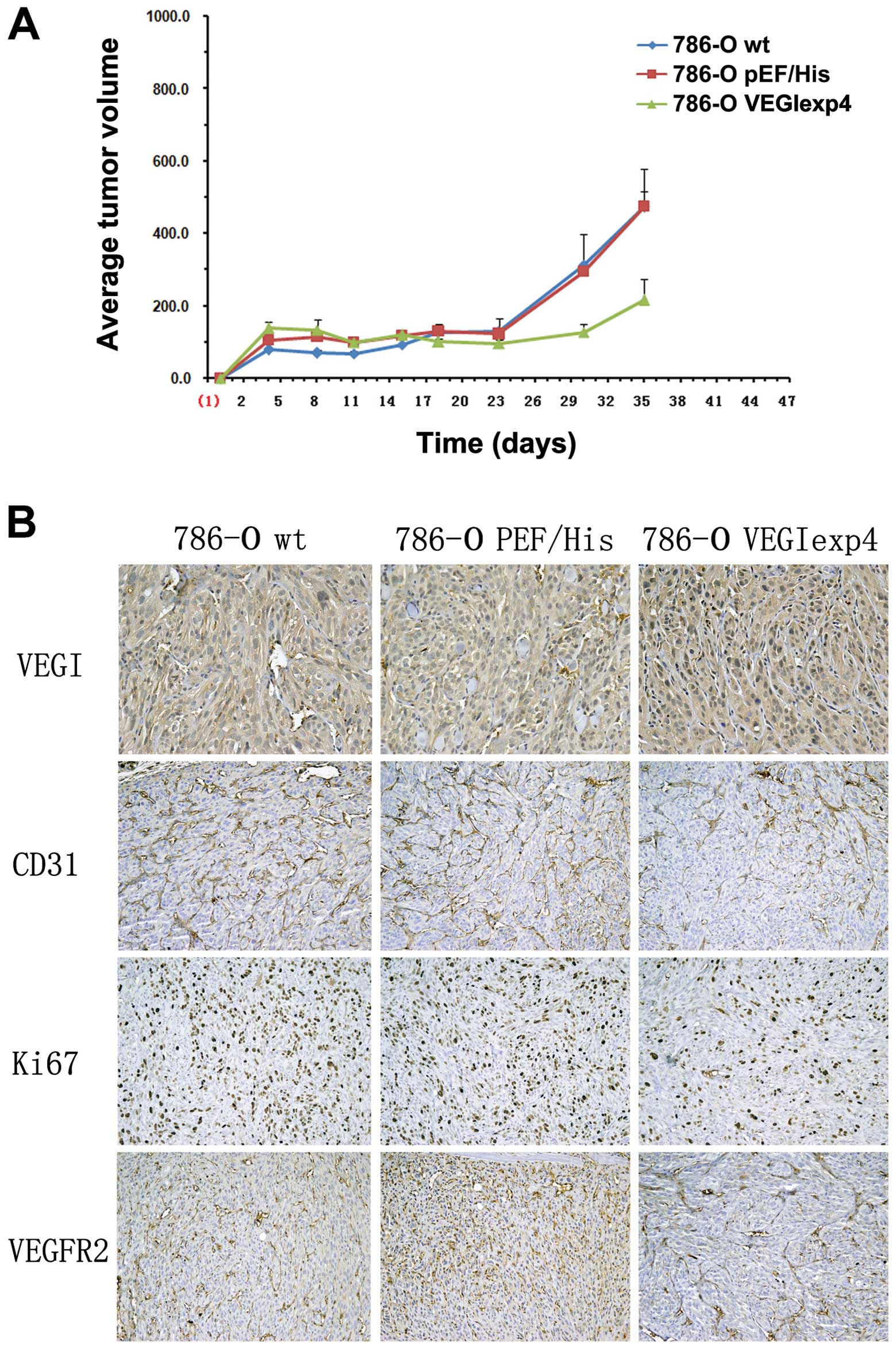 | Figure 5The effect of VEGI forced expression
on xenograft models and angiogenesis in xenograft models. (A) The
effect of forced expression of VEGI on xenograft models. Three
weeks after successful establishment of xenograft models, tumor
volume was found significantly reduced in the group of BALB/c nude
mice injected with 786-OVEGIexp4 cells, compared with
786-Owt, 786-OpEF/His cells (P<0.01). Error bars
represent the SD. (B) The effect of VEGI forced expression on
angiogenesis in xenograft models. In IHC, comparing with
786-Owt and 786-OpEF/His cells, VEGI staining
was increased in samples from xenograft tumors with
786-OVEGIexp4 cells (P<0.01). However, comparing with
786-Owt and 786-OpEF/His cells, Ki67 and
VEGFR2 expression was decreased, respectively, in tumors with
786-OVEGIexp4 cells (P<0.01). Significant reduction
in microvessel density, as indicated by reduced CD31 staining, was
observed in 786-OVEGIexp4 tumors compared to that in
786-Owt and 786-OpEF/His tumors
(P<0.01). |
Reduced angiogenesis in xenograft
models from 786-O cells with forced expression of VEGI
In immunohistochemical staining with samples from
xenograft tumor, comparing with 786-Owt and
786-OpEF/His cells, VEGI staining was increased in
tumors with 786-OVEGIexp4 cells (MOD was 0.45±0.22,
0.39±0.15 and 0.67±0.19, respectively, P<0.01) (Fig. 5B). Significant reduction in
microvessel density, as indicated by reduced CD31 staining, was
observed in 786-OVEGIexp4 tumors compared to that in
786-Owt and 786-OpEF/His tumors (87±21,
211±43 and 189±33 respectively, P<0.01) (Fig. 5B). Moreover, comparing with
786-Owt and 786-OpEF/His tumors, Ki67
(284±41, 331±29 and 119±37, P<0.01) and VEGFR2 (377±41, 321±29
and 144±24, P<0.01) expression were also decreased in
786-OVEGIexp4 tumors (Fig.
5B).
Discussion
VEGI, first identified from human umbilical vein
endothelial cells is known to exist abundantly in arterial
endothelial cells (21,22). Subsequently, it has been reported
that VEGI is also expressed in a wide variety of human cancer cell
lines, including breast, prostate, bladder, colorectal and liver
(24,35–38).
The role of VEGI in human cancer cells has been investigated.
According to the literature, it is able to induce apoptosis in
endothelial cells via an autocrine pathway (22–25).
Overexpression of VEGI has been shown to inhibit tumor
neovascularisation and progression in cellular and animal models.
Chew et al reported that overexpression of VEGI induced
apoptosis in endothelial cells and inhibited the growth of
xenograft tumors, together with a reduction in the microvessel
density (24). Zhai et al
found that the recombinant VEGI protein could markedly inhibit the
growth of breast and colon xenograft tumors and suggested that the
effect may be through an indirect inhibition on capillary-like
structures and cells growth (23).
Hou et al, using a Lewis lung cancer (LLC) murine tumor
model, demonstrated that systemic administration of VEGI gave rise
to a marked inhibition of tumor growth and to an increase in
survival time of the treated animals (29). Further investigation has shown that
the density of the endothelial cells exhibited an 88% decrease
within 1 week of treatment with recombinant human VEGI and a
further decrease within 3 weeks in Lewis lung cancer murine tumor
models. Parr et al reported that patients with breast tumors
expressing reduced levels of VEGI had a higher local recurrence,
shorter survival time and an overall poorer prognosis than those
patients expressing high levels of VEGI (39). Chen et al also reported that
purified rhVEGI-192 potently inhibited endothelial growth, induced
endothelial apoptosis and suppressed neovascularization in chicken
chorioallantoic membrane and demonstrated that VEGI-192 is capable
of forming polymeric structure, which is possibly required for its
anti-angiogenic activity (40).
Liang et al reported that VEGI inhibits bone marrow
(BM)-derived endothelial progenitor cells (EPC) mobilization and
prevents their incorporation into LLC tumors by inducing apoptosis
specifically of BM-derived cells, resulting in the inhibition of
EPC-supported tumor vasculogenesis and tumor growth (41). Overall, the above evidence
indicated that the antitumor activity of VEGI is more likely to be
the result of an interference with the development of tumor
associated vasculature. However, many reports have also suggested
that VEGI may inhibit the growth of some human tumor cell lines,
including human histiocytic lymphomas (U-937), human breast
carcinomas (MCF-7), human epithelial carcinoma (HeLa) and human
myeloid lymphomas ML-1a (28,42).
Our previous results also show that the overexpression of VEGI can
directly affect the motility and adhesion of urothelial tumor and
prostate cancer cells (26,27).
Given the dependence of RCC on angiogenesis and the
mechanisms of target therapy, it is speculated that VEGI may be an
effective target in treating RCC. The present study demonstrated
exciting evidence supporting our hypothesis.
First of all, VEGI mRNA was expressed in a wide
variety of human RCC cell lines, all the normal renal specimens and
a proportion of the RCC specimens. It is obviously that VEGI
transcript expression is at lower level in RCC tissues than that in
normal renal tissues. The immunohistochemical analysis clearly
indicates a similar trend. VEGI protein was found to be expressed
at lower level in RCC tissues than that in normal renal tissues and
was almost absent in tumors with high grade. The absence or
reduction of tumor VEGI expression suggests that there may be a
shift in the balance between pro- and anti-angiogenic stimuli. This
loss of balance may subsequently produce a microenvironment that is
conducive to tumor growth and survival.
Moreover, our results confirm that forced-expression
of VEGI was able to directly affect the motility and adhesion of
786-O cells. The number of adherent cells in VEGI forced-expression
cells was >50% decreased compared with the wild-type cells. The
average distance of VEGI expression cells was also decreased
dramatically compared with controls. Therefore, VEGI can directly
reduce aggressiveness of renal cancer cells, which is in line with
the decreased expression of VEGI in RCC specimens.
Our results demonstrated that force expression VEGI
suppressed renal cell carcinoma growth in vivo. Comparing
with controls, tumor volume was reduced >50% in the group of
BALB/c nude mice injected with 786-OVEGIexp4 cells.
However, manipulation of VEGI expression had no impact on growth,
or apoptosis of RCC cells in vitro. Combined with the result
of endothelial cell differentiation assay (tube formation on
Matrigel), when HUVECs with 786-OVEGIexp4 were
co-cultured, the tube formation decreased significantly in
comparison with controls. As indicated by reduced CD31 staining,
significant reduction in microvessel density was observed in
xenograft tumor samples. All the above suggested that the anti-RCC
growth in vivo of force expression VEGI is more likely to be
the result of an interference with the development of tumor
associated vasculature than that of a direct effect on tumor
cells.
The present study also shows that Ki67 and VEGFR2
protein level was decreased significantly in
786-OVEGIexp4 tumors. It is well known that the Ki-67
protein (also known as MKI67) is a cellular marker for
proliferation (43–45). Its high expression may be
independently associated with an increased risk of poor
disease-free survival for many tumors. Its decrease shows that
786-OVEGIexp4 cell proliferation was inhibited in
vivo. Probably, this kind of inhibition was also dependent on
suppressed vasculogenesis. The VEGF/VEGFR2 pathway plays a central
role in tumor vasculature. Blocking it may achieve inhibition of
the growth and metastasis of RCC (46,47).
VEGFR2 protein level decreased in 786-OVEGIexp4 tumors
indicating that VEGI suppressed angiogenesis through VEGF/VEGFR2
pathway except for DR3, SAPK/JNK and p38 MAPK and caspase pathway
(48,49). This is a new pathway of
VEGI to suppress angiogenesis not reported before. It is suggested
that reducing tumor neovascularisation and tumor cell proliferation
rate may be the reason for VEGI suppressing tumor growth in the
xenograft model.
In conclusion, the present study shows that VEGI
expression is decreased in renal cell carcinoma, particularly in
tumors with higher grade. VEGI, a potential cell migration and
adhesion regulating protein of TNFSF, suppressed tumor growth in
vivo. This is likely via its inhibitory role on
anti-angiogenesis, cell migration and adhesion. Our results suggest
that VEGI may be a putative tumor suppressor and a potential
therapeutic target for RCC.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China, grant no. 81072088 and Cancer Research
Wales (W.G.J.).
References
|
1.
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
2.
|
Bellmunt J and Guix M: The medical
management of metastatic renal cell carcinoma: integrating new
guidelines and recommendations. BJU Int. 103:572–577. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Chow WH, Devesa SS, Warren JL and Fraumeni
JF Jr: Rising incidence of renal cell cancer in the United States.
JAMA. 281:1628–1631. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Ferlay J, Autier P, Boniol M, Heanue M,
Colombet M and Boyle P: Estimates of the cancer incidence and
mortality in Europe in 2006. Ann Oncol. 18:581–592. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Bechtold RE and Zagoria RJ: Imaging
approach to staging of renal cell carcinoma. Urol Clin North Am.
24:507–522. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kane CJ, Mallin K, Ritchey J, Cooperberg
MR and Carroll PR: Renal cell cancer stage migration: analysis of
the National Cancer Data Base. Cancer. 113:78–83. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Bellmunt J, Eisen T, Fishman M and Quinn
D: Experience with sorafenib and adverse event management. Crit Rev
Oncol Hematol. 78:24–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Decastro GJ and McKiernan JM:
Epidemiology, clinical staging and presentation of renal cell
carcinoma. Urol Clin North Am. 35:581–592. vi:2008.PubMed/NCBI
|
|
9.
|
Wallen EM, Pruthi RS, Joyce GF and Wise M:
Kidney cancer. J Urol. 177:2006–2018. 2007. View Article : Google Scholar
|
|
10.
|
Hock LM, Lynch J and Balaji KC: Increasing
incidence of all stages of kidney cancer in the last 2 decades in
the United States: an analysis of surveillance, epidemiology and
end results program data. J Urol. 167:57–60. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Grandinetti CA and Goldspiel BR: Sorafenib
and sunitinib: novel targeted therapies for renal cell cancer.
Pharmacotherapy. 27:1125–1144. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Motzer RJ and Russo P: Systemic therapy
for renal cell carcinoma. J Urol. 163:408–417. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Vakkalanka BK and Rini BI: Targeted
therapy in renal cell carcinoma. Curr Opin Urol. 18:481–487. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Fisher RI, Rosenberg SA and Fyfe G:
Long-term survival update for high-dose recombinant interleukin-2
in patients with renal cell carcinoma. Cancer J Sci Am. 6(Suppl 1):
S55–S57. 2000.PubMed/NCBI
|
|
15.
|
Wilhelm SM, Adnane L, Newell P, Villanueva
A, Llovet JM and Lynch M: Preclinical overview of sorafenib, a
multikinase inhibitor that targets both Raf and VEGF and PDGF
receptor tyrosine kinase signaling. Mol Cancer Ther. 7:3129–3140.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Sonpavde G, Choueiri TK, Escudier B,
Ficarra V, Hutson TE, Mulders PF, Patard JJ, Rini BI, Staehler M,
Sternberg CN and Stief CG: Sequencing of agents for metastatic
renal cell carcinoma: can we customize therapy? Eur Urol.
61:307–316. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Porta C, Szczylik C and Escudier B:
Combination or sequencing strategies to improve the outcome of
metastatic renal cell carcinoma patients: a critical review. Crit
Rev Oncol Hematol. 82:323–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Paez-Ribes M, Allen E, Hudock J, Takeda T,
Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D and Casanovas O:
Antiangiogenic therapy elicits malignant progression of tumors to
increased local invasion and distant metastasis. Cancer Cell.
15:220–231. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Huang D, Ding Y, Zhou M, Rini BI, Petillo
D, Qian CN, Kahnoski R, Futreal PA, Furge KA and Teh BT:
Interleukin-8 mediates resistance to antiangiogenic agent sunitinib
in renal cell carcinoma. Cancer Res. 70:1063–1071. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Finke J, Ko J, Rini B, Rayman P, Ireland J
and Cohen P: MDSC as a mechanism of tumor escape from sunitinib
mediated anti-angiogenic therapy. Int Immunopharmacol. 11:856–861.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Tan KB, Harrop J, Reddy M, Young P,
Terrett J, Emery J, Moore G and Truneh A: Characterization of a
novel TNF-like ligand and recently described TNF ligand and TNF
receptor superfamily genes and their constitutive and inducible
expression in hematopoietic and non-hematopoietic cells. Gene.
204:35–46. 1997. View Article : Google Scholar
|
|
22.
|
Zhai Y, Yu J, Iruela-Arispe L, Huang WQ,
Wang Z, Hayes AJ, Lu J, Jiang G, Rojas L, Lippman ME, Ni J, Yu GL
and Li LY: Inhibition of angiogenesis and breast cancer xenograft
tumor growth by VEGI, a novel cytokine of the TNF superfamily. Int
J Cancer. 82:131–136. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Zhai Y, Ni J, Jiang GW, Lu J, Xing L,
Lincoln C, Carter KC, Janat F, Kozak D, Xu S, Rojas L, Aggarwal BB,
Ruben S, Li LY, Gentz R and Yu GL: VEGI, a novel cytokine of the
tumor necrosis factor family, is an angiogenesis inhibitor that
suppresses the growth of colon carcinomas in vivo. FASEB J.
13:181–189. 1999.PubMed/NCBI
|
|
24.
|
Chew LJ, Pan H, Yu J, Tian S, Huang WQ,
Zhang JY, Pang S and Li LY: A novel secreted splice variant of
vascular endothelial cell growth inhibitor. FASEB J. 16:742–744.
2002.PubMed/NCBI
|
|
25.
|
Yue TL, Ni J, Romanic AM, Gu JL, Keller P,
Wang C, Kumar S, Yu GL, Hart TK, Wang X, Xia Z, DeWolf WE Jr and
Feuerstein GZ: TL1, a novel tumor necrosis factor-like cytokine,
induces apoptosis in endothelial cells. Involvement of activation
of stress protein kinases (stress-activated protein kinase and p38
mitogen-activated protein kinase) and caspase-3-like protease. J
Biol Chem. 274:1479–1486. 1999. View Article : Google Scholar
|
|
26.
|
Zhang N, Sanders AJ, Ye L, Kynaston HG and
Jiang WG: Expression of vascular endothelial growth inhibitor
(VEGI) in human urothelial cancer of the bladder and its effects on
the adhesion and migration of bladder cancer cells in vitro.
Anticancer Res. 30:87–95. 2010.PubMed/NCBI
|
|
27.
|
Zhang N, Sanders AJ, Ye L, Kynaston HG and
Jiang WG: Vascular endothelial growth inhibitor, expression in
human prostate cancer tissue and the impact on adhesion and
migration of prostate cancer cells in vitro. Int J Oncol.
35:1473–1480. 2009.PubMed/NCBI
|
|
28.
|
Xiao Q, Hsu CY, Chen H, Ma X, Xu J and Lee
JM: Characterization of cis-regulatory elements of the vascular
endothelial growth inhibitor gene promoter. Biochem J. 388:913–920.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Hou W, Medynski D, Wu S, Lin X and Li LY:
VEGI-192, a new isoform of TNFSF15, specifically eliminates tumor
vascular endothelial cells and suppresses tumor growth. Clin Cancer
Res. 11:5595–5602. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Yao JJ, Zhang M, Miao XH, Zhao P, Zhu SY,
Ding H and Qi ZT: Isoform of vascular endothelial cell growth
inhibitor (VEGI72-251) increases interleukin-2 production by
activation of T lymphocytes. Acta Biochim Biophys Sin (Shanghai).
38:249–253. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Jin T, Kim S, Guo F, Howard A and Zhang
YZ: Purification and crystallization of recombinant human TNF-like
ligand TL1A. Cytokine. 40:115–122. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Park SS, Lillehoj HS, Hong YH and Lee SH:
Functional characterization of tumor necrosis factor superfamily 15
(TNFSF15) induced by lipopolysaccharides and Eimeria infection. Dev
Comp Immunol. 31:934–944. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Cai J, Jiang WG and Mansel RE: Inhibition
of the expression of VE-cadherin/catenin complex by gamma linolenic
acid in human vascular endothelial cells and its impact on
angiogenesis. Biochem Biophys Res Commun. 258:113–118. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Zhang N, Sanders AJ, Ye L and Jiang WG:
Vascular endothelial growth inhibitor in human cancer (Review). Int
J Mol Med. 24:3–8. 2009.PubMed/NCBI
|
|
35.
|
Migone TS, Zhang J, Luo X, Zhuang L, Chen
C, Hu B, Hong JS, Perry JW, Chen SF, Zhou JX, Cho YH, Ullrich S,
Kanakaraj P, Carrell J, Boyd E, Olsen HS, Hu G, Pukac L, Liu D, Ni
J, Kim S, Gentz R, Feng P, Moore PA, Ruben SM and Wei P: TL1A is a
TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell
costimulator. Immunity. 16:479–492. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Bamias G, Mishina M, Nyce M, Ross WG,
Kollias G, Rivera-Nieves J, Pizarro TT and Cominelli F: Role of
TL1A and its receptor DR3 in two models of chronic murine ileitis.
Proc Natl Acad Sci USA. 103:8441–8446. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Prehn JL, Thomas LS, Landers CJ, Yu QT,
Michelsen KS and Targan SR: The T cell costimulator TL1A is induced
by FcgammaR signaling in human monocytes and dendritic cells. J
Immunol. 178:4033–4038. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Parr C, Gan CH, Watkins G and Jiang WG:
Reduced vascular endothelial growth inhibitor (VEGI) expression is
associated with poor prognosis in breast cancer patients.
Angiogenesis. 9:73–81. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Chen X, Wu J, Liu H, He Z, Gu M, Wang N,
Ma J, Hu J, Xia L, He H, Yuan J, Li J, Li L, Li M and Zhu X:
Approaches to efficient production of recombinant angiogenesis
inhibitor rhVEGI-192 and characterization of its structure and
antiangiogenic function. Protein Sci. 19:449–457. 2010.PubMed/NCBI
|
|
40.
|
Liang PH, Tian F, Lu Y, Duan B, Stolz DB
and Li LY: Vascular endothelial growth inhibitor (VEGI; TNFSF15)
inhibits bone marrow-derived endothelial progenitor cell
incorporation into Lewis lung carcinoma tumors. Angiogenesis.
14:61–68. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Haridas V, Shrivastava A, Su J, Yu GL, Ni
J, Liu D, Chen SF, Ni Y, Ruben SM, Gentz R and Aggarwal BB: VEGI, a
new member of the TNF family activates nuclear factor-kappa B and
c-Jun N-terminal kinase and modulates cell growth. Oncogene.
18:6496–6504. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
McGuire BB and Fitzpatrick JM: Biomarkers
in renal cell carcinoma. Curr Opin Urol. 19:441–446. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Li XQ, Pei DS, Qian GW, Yin XX, Cheng Q,
Li LT, Li HZ and Zheng JN: The effect of methylated oligonucleotide
targeting Ki-67 gene in human 786-0 renal carcinoma cells. Tumour
Biol. 32:863–872. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Oka S, Uramoto H, Shimokawa H, Iwanami T
and Tanaka F: The expression of Ki-67, but not proliferating cell
nuclear antigen, predicts poor disease free survival in patients
with adenocarcinoma of the lung. Anticancer Res. 31:4277–4282.
2011.PubMed/NCBI
|
|
45.
|
Carmeliet P: VEGF as a key mediator of
angiogenesis in cancer. Oncology. 69(Suppl 3): 4–10. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Hicklin DJ and Ellis LM: Role of the
vascular endothelial growth factor pathway in tumor growth and
angiogenesis. J Clin Oncol. 23:1011–1027. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Yang CR, Hsieh SL, Teng CM, Ho FM, Su WL
and Lin WW: Soluble decoy receptor 3 induces angiogenesis by
neutralization of TL1A, a cytokine belonging to tumor necrosis
factor superfamily and exhibiting angiostatic action. Cancer Res.
64:1122–1129. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Cassatella MA, Pereira-da-Silva G, Tinazzi
I, Facchetti F, Scapini P, Calzetti F, Tamassia N, Wei P, Nardelli
B, Roschke V, Vecchi A, Mantovani A, Bambara LM, Edwards SW and
Carletto A: Soluble TNF-like cytokine (TL1A) production by immune
complexes stimulated monocytes in rheumatoid arthritis. J Immunol.
178:7325–7333. 2007. View Article : Google Scholar : PubMed/NCBI
|