Introduction
Systemic chemotherapy is an important breast cancer
treatment modality and its effectiveness has significantly improved
over the last decade (1).
Notwithstanding, the development of cancer cells that are resistant
to chemotherapeutic agents is a major clinical obstacle in the
successful treatment of breast cancer (2,3).
Understanding the structures underlying drug resistance development
and predisposition is critical to saving lives.
Overall, acquired drug resistance is a
multi-factorial phenomenon that involves multiple structures and
processes (2–5), including: a decreased uptake of drugs
(6), alterations in cell cycle and
signal transduction pathways (7,8),
increased repair of DNA damage (9), reduced apoptosis (7,10,11),
increased efflux of hydrophobic drugs (5,6,8,12,13)
and DNA damage tolerance (9).
Resistance to individual chemotherapeutic agents usually occurs
through alterations in the drug targets, but broad resistance can
also occur, affecting the utility of a variety of diverse and
unrelated antitumor drugs with different chemical structures and
different mechanisms of action (5,12,14–16).
Apoptosis avoidance is one of the key processes underlying multiple
drug resistance phenotypes (7,10,11,17).
Doxorubicin is an anthracycline drug frequently used
in the curative-intent, adjuvant therapy and palliative treatment
of metastatic breast cancer (18).
Although doxorubicin is among the most active agents in breast
cancer treatment, many patients will experience a relapse after the
drug therapy is completed. Furthermore, approximately half of
metastatic breast cancer patients will fail to respond to
doxorubicin entirely, and the majority of those showing initial
benefits will subsequently manifest acquired clinical resistance
demonstrated by tumor growth that will occur despite ongoing
anthracycline therapy (18).
It has also been reported that drug-resistant cancer
cells may fail to respond to cytotoxic radiotherapy and may develop
a multidrug-resistant phenotype (19–27).
However, the data on the radiation responses of chemoresistant
tumors is contradictory. For instance, some clinical studies
suggest significant benefits from a combination of chemo- and
radiotherapy for breast cancer management (28). On the other hand, there is proof
that chemotherapy used as an induction therapy before radiotherapy
has no significant additional antitumor effects (29). Breast tumors tend to resist and
reoccur after the aforementioned treatments (30). The exact nature and structure of
the radiation responses of chemoresistant tumor cells remain
unclear.
One of the key features of cancer cell resistance to
therapeutic agents is their associated resistance to apoptotic cell
death (7). Chemoresistant cells
and tumors have a strong capacity to withstand and avoid apoptosis
during chemotherapy treatment (7,31).
Ionizing radiation (IR) exposure is known to induce apoptosis in
exposed cells, yet little is known about the status of IR-induced
apoptosis in drug-resistant cell lines.
In this study, we analyzed the cellular and
molecular structures of radiation responses in MCF-7 breast
adenocarcinoma cells and their derivative line that is resistant to
doxorubicin (MCF-7/DOX). For the first time, we show that MCF-7/DOX
cells, while harboring an elevated potential to withstand
radiation-induced DNA damage, also have a significantly decreased
fidelity of DNA polymerases and a delayed radiation-induced
apoptosis.
Materials and methods
Cell lines and cell culture
conditions
MCF-7 and MCF-7/DOX multidrug-resistant human breast
adenocarcinoma cell lines were previously developed and described
elsewhere (17,32). Cells were grown and maintained in
Dulbecco’s modified Eagle’s medium (DMEM/F-12) with 2.5 mM
L-glutamine, without HEPES and Phenol Red (HyClone, Logan, UT),
supplemented with 10% heat-inactivated fetal bovine serum
(HyClone), in the presence of antibiotics 100 U/ml penicillin and
100 μg/ml streptomycin (Sigma-Aldrich Chemical Co., St.
Louis, MO), and in a 5% CO2 atmosphere at 37°C. Cells
were harvested for analyses by trypsinization (17,32).
Irradiation conditions
Cells were irradiated at a 60% confluency in DMEM.
Two radiation doses (0.5 and 5 Gy, 90 kVp, 5 mA) were applied to
check the cellular radiation responses. Unirradiated cells served
as the control. Cells were harvested 30 min, 24 and 48 h after
irradiation. All the cells were tested in triplicate. The
experiments were independently reproduced twice.
Whole-genome gene expression
profiling
RNA isolation
Total RNA was isolated using the Illustra RNAspin
mini kit (GE Healthcare Life Sciences, Buckinghamshire, UK).
Approximately 5×106 cultured cells were processed
following the manufacturer’s instructions. Samples were eluted in
Ultrapure DNase/RNase-free distilled water, which was provided in
the kit. RNA samples were quantified using ultraviolet spectroscopy
(NanoDrop, Wilmington, DE) and were further assessed for RNA
integrity (RIN) on the Agilent 2100 Bioanalyzer (Santa Clara, CA)
using the RNA Nano-chip Kit. RNA samples with RIN values of seven
or better were used for the further analysis.
Library preparation
CRNA was created using the Ambion’s Illumina
TotalPrep RNA Amplification Kit (Applied Biosystems, Carlsbad, CA)
with an input of 500 ng of total RNA per sample. Briefly, oligo-dT
primers were used to synthesize first strand cDNA containing a
phage T7 promoter sequence. Single-stranded cDNA was converted into
a double-stranded DNA template via DNA polymerase. RNase H
simultaneously acted to degrade the RNA. Samples of cDNA were
purified in filter cartridges to remove excess RNA, primers,
enzymes and salts. The recovered cDNA was subjected to in
vitro transcription using biotinylated UTPs. This step created,
labeled and amplified cRNA. A final purification step removed
unincorporated NTPs, salts, inorganic phosphates and enzymes, which
prepared the samples for hybridization.
Hybridization and detection
Illumina’s direct hybridization assay kit was used
to process samples according to the manufacturer’s protocol
(Illumina, San Diego, CA). Overnight, 750 ng from each cRNA sample
was hybridized into the Illumina HumanHT-12_v4 Whole Genome
Expression BeadChip arrays. Afterward, a 10-min incubation with a
supplied wash buffer at 55°C preceded a 5-min room temperature
wash. The arrays were incubated in 100% ethanol for 10 min. A
second room temperature wash lasted 2 min with gentle shaking,
which completed this high stringency wash step. The arrays were
blocked with a buffer for 10 min and washed before a 10-min
steptavidin-Cy3 (1:1,000) probing. After a 5-min wash at room
temperature, the BeadChips were dried and imaged. Six controls were
also built into the Whole-Genome Gene Expression Direct
Hybridization Assay system to cover aspects of the array
experiments, including controls for: the biological specimen (14
probes for housekeeping controls), 3 controls for hybridization (6
probes for Cy3-labeled hybridization, 4 probes for low stringency
hybridization, and 1 probe for high stringency hybridization),
signal generation (2 probes for biotin control), and approximately
800 probes for negative controls on an 8-sample BeadChip. The
arrays were scanned on the iScan platform (Illumina), and data were
normalized and scrutinized using Illumina BeadStudio Software.
BeadChip statistical analysis and data
processing
The false discovery rate (FDR) was controlled using
the Benjamini-Hochberg method. The Illumina Custom Model took the
FDR into account and was used to analyze the data. Differential
gene expression (at least a 1.5-fold change) from non-irradiated
cells was determined to be statistically significant if the p-value
after the Benjamini-Hochberg method adjustment was lower than 0.05.
The values were transformed to show a log2 scale.
Lists of regulated transcripts were inserted into
the web-based DAVID Bioinformatics Resources 6.7 (NIAID/NIH)
Functional Annotation Tool (33,34).
This program was used to group genes into functionally relevant
categories: metabolic processes, transport, response to
stimulus/stress, immune response, apoptosis and cell cycle
processes.
Quantitative real-time PCR
Quantitative real-time PCR was performed to confirm
the Whole-Genome Gene Expression results for the regulation
direction (either up or down) of select genes. Six genes (aurora B,
cyclin A, GADD45G, polymerases A, D and E) were selected
from the gene list of significantly differentially expressed
transcripts, representing a preliminary review of the acquired gene
expression data. 18SrRNA was used as a reference gene. All the
reactions were performed using cDNA synthesized from the same RNA
extraction as the BeadChip experiments, and 500 ng of the sample
was used for the Bio-Rad iScript Select cDNA Synthesis kit (Bio-Rad
Laboratories, Hercules, CA). Samples were stored at −20°C for
long-term storage and at 4°C until they were used for subsequent
qRT-PCR reactions.
Primers were designed using the NCBI database and
PrimerQuest (Integrated DNA Technologies Inc., Coralville, IA). The
following primers were designed: hAURKB forward primer
5′-TGA GGA GGA AGA CAA TGT GTG GCA-3′ and reverse primer 5′-AGG TCT
CGT TGT GTG ATG CAC TCT-3′; 18SrRNA reference gene primers 5′-GTC
AAG TTC GAC CGT CTT CT-3′ and 5′-AGC TTG CGT TGA TTA AGT CC-3′;
CCNA2 forward primer 5′-ATG AGC ATG TCA CCG TTC CTC CTT-3′
and reverse primer 5′-TCA GCT GGC TTC TTC TGA GCT TCT-3′;
hGADD45G forward primer 5′-TGC TGC GAG AAC GAC ATC GAC
ATA-3′ and reverse primer 5′-TCG AAA TGA GGA TGC AGT GCA GGT-3′;
hPOLA1 forward primer 5′-GGC AAT GGC TTT GAA ACC AGA CCT-3′
and reverse primer 5′-ATG CTG AAA GCC ATC ACG ACA AGC-3′;
hPOLD1 forward primer 5′-AAC CTG TGT TAC ACC ACG CTC CTT-3′
and reverse primer 5′-TCC GCA CTG AGG TCT TCA CAA ACT-3′;
hPOLE forward primer 5′-AGA TTG TGC AGA TCA GCG AGA CCA-3′
and reverse primer 5′-TTA CCT TGC GAT ACG AAG CAC CCT-3′. Reactions
were prepared using 1 μl of diluted cDNA, 10 pmol/μl
of each forward and reverse primer, and SsoFast EvaGreen Supermix
(Bio-Rad Laboratories) prepared according to the manufacturer’s
instructions. Samples were prepared in triplicate and were run on
the Bio-Rad C1000 Thermal Cycler equipped with the CFX96 Real-Time
System. The qRT-PCR protocol consisted of denaturation at 95°C for
2 min; 43 cycles of denaturation (95°C, 5 sec) and
annealing/extension (55°C, 5 sec); and a final extension at 65°C
for 5 sec. For every set of primers, annealing temperature
optimization, melting curve analysis and a gel analysis of the
amplicon were performed. To evaluate PCR efficiency, a standard
curve was established using a series of cDNA dilutions. Data were
captured and organized using Bio-Rad CFX Manager 2.1 software
(Bio-Rad Laboratories).
qRT-PCR statistical analysis
Quantification data from the Bio-Rad CFX Manager
software was analyzed using the Pfaffl method in Microsoft Excel
(35). Graphs showing a fold
change from the sham group were created, and transcript regulation
directions (up or downregulation) were matched to the Whole-Genome
Gene Expression results.
Western immunoblot analysis
Following radiation treatment, the cells were
harvested, washed in PBS, lysed and sonicated in 0.2 ml of 1%
sodium dodecyl sulphate (SDS). The lysates were cleared using
centrifugation. The protein content was determined using the
Bradford protein determination assay (Bio-Rad Laboratories). Equal
amounts of lysate protein were subsequently run on 10–12%
SDS-polyacrylamide gels and transferred to PVDF membranes (GE
Healthcare, Baie d’Urfé, QC, Canada).
Western immunoblot analysis was conducted using
well-established protocols (32,36).
The membranes were incubated with antibodies against goat
anti-polymerase ι, mouse anti-polymerase ε (1:1,000, Santa Cruz
Biotechnology Inc., Santa Cruz, CA ), mouse anti-polymerase β,
rabbit anti-polymerase δ (1:500 dilution, Abcam Inc., Cambridge,
MA), mouse anti-phospho-ATM (1:500, Cell Signaling Technology Inc.,
Danvers, MA), mouse anti-Ku-70 and mouse anti-Rad51 (1:1,000, Santa
Cruz Biotechnology Inc.). Antibody binding was revealed through
incubation with horseradish peroxidase-conjugated secondary
antibodies (GE Healthcare, Piscataway, NJ) and the ECL Plus
immunoblotting detection system (GE Healthcare). Chemiluminescence
was detected using BioMax MR film (Eastman Kodak, New Haven, CT).
Unaltered PVDF membranes were stained with Coomassie Blue (Bio-Rad
Laboratories) to prove equal protein loading.
Analysis of DNA polymerase fidelity in
MCF-7 and MCF-7/DOX cells
The DNA polymerase fidelity assay allows the
researcher to determine the activity of polymerases on damaged DNA
and the quality of the repair synthesis (37). The assay employs a FAM-labeled 15
bp primer as a component of the substrate. Its oligonucleotide can
be revealed on a gel. In the assay, different deoxyribonucleotides
were added to the reaction mixture to check the ability of
polymerases to incorporate the correct and incorrect dNTPs into the
template. Any increase in primer weight upon incorporation would
indicate higher DNA polymerase activity while a decrease is
associated with exonuclease activity. Misincorporation efficiency
is associated with changes in DNA polymerase fidelity.
Substrate (template/primer
complex)
In order to produce the substrate for the assay,
FAM-labeled 15bp primer was annealed using a 30 bp template (both
were PAGE purified). Template: AG030-PAGE
5′-TCATCGAGCATGATCACGTCGTGAC TGGGA-3′. Primer: AG031-PAGE
5′-FAM-TCCCAGTCACG ACGT-3′. The reaction was performed in 1 M
Tris-HCl (pH 8.0), β-mercaptoethanol, BSA (100X NEB), 100 μM
primer and 100 μM template, incubated at 95°C for 5 min and
slowly cooled at room temperature.
Cell extracts
MCF-7 and MCF-7/DOX control and irradiated
(harvested 24 h after a 5 Gy X-ray treatment) cells were harvested,
washed in 1X PBS, resuspended and sonicated in PBS, and centrifuged
at 4°C for 10 min at 14,000 × g. The total protein concentration in
the samples was determined using a Bradford Assay (Bio-Rad
Laboratories).
A DNA polymerase fidelity assay was carried out
according to Gening et al(37). The reaction was performed at 37°C
for 15 min, and it was quickly frozen afterward. The reaction
mixture contained: 50 mM Tris-HCl (pH 8.0), 5 mM MgCl2,
1 mM DTT, 70 μg of the tested lysate protein,
template/primer complex and 2 mM dNTP. When the reaction was
stopped, 5 μl of each sample was mixed with 10 μl of
a loading buffer (95% formamide, 50 mM EDTA, 0.05% bromophenol
blue), incubated at 95°C for 3 min and cooled on ice. The reaction
products were separated in 20% polyacrylamide gel in the presence
of an 8 M urea in a Tris-borate buffer at 750 V. PAGE gels were
scanned using a Typhoon 9410 imager (excitation 488 nm, emission
filter 520 BP 40, PMT 620 V, resolution 50 μm). The
intensity of the bands was measured using the ImageQuant 5.2
software program (Molecular Dynamics).
Annexin V assay
For the early detection of apoptosis, an Annexin
V-FITC Apoptosis Detection Kit I (BD Biosciences, San Jose, CA) was
used according to the manufacturer’s protocol. Cells were grown and
irradiated as previously described above in Irradiation conditions.
The analysis was performed 24 and 48 h after radiation exposure.
Cells were harvested, washed with PBS, resuspended in a 1X binding
buffer, stained with Annexin V and propidium iodide for 15 min at
25°C in the dark, and analyzed using flow cytometry within 1 h at
the Flow Cytometry Core Facility (University of Calgary, Calgary,
AB, Canada). The results were represented as a percentage of gated
Annexin V positive cells.
Alkaline comet assay
The alkaline comet assay protocol was based on Olive
and Bannath (38) and Tice and
Vasques (39) at cometassay.com. The cells that were grown in cultures
were trypsinised, collected in 15-ml tubes, and centrifuged for 3
min at 1,000 × g to form a pellet. Next, the pellet was washed
three times with ice cold phosphate-buffered saline (PBS) without
-Ca2+ and -Mg2+. Finally, the cells were
resuspended in their final concentration of 1,000 cells per 1
μl of cell suspension in ice-cold PBS. The cell suspension
was stored on ice during the course of the subsequent
procedures.
A total of 10 μl of cell suspension were
mixed with 75 μl of 1% low melting point (LMP) agarose
pre-heated to 40°C, mixed gently through pipetting up and down, and
applied to a fully frosted microscope slide (VWR) that was
pre-coated with normal melting point agarose. Agarose was overlaid
with a cover slip and allowed to solidify for 2 to 3 min on ice.
The removal of the cover slip was followed by an application of 85
μl of 1% LMP agarose pre-heated to 40°C in order to form a
protective layer on top of the layer containing the cell
suspension. The cover slip was re-positioned and the slides were
placed on ice to allow the agarose to solidify.
The cover slips were removed and the slides were
placed in a freshly prepared alkaline lysis solution [2.5 M NaCl,
100 mM Na2EDTA, 10 mM Tris-base, 1% Triton X-100 and
0.1% sodium lauroyl sarcosine (pH 10) adjusted to 4°C], left
overnight at 4°C and protected from light. Following the lysis
step, the slides were rinsed with a freshly prepared
electrophoresis solution [300 mM, 2 mM EDTA (pH >14)]. Next, the
slides were placed in an electrophoresis tank, covered with a thin
layer (1–2 mm) of electrophoresis buffer, and left for 30 min to
permit alkaline DNA unwinding. Electrophoresis was performed for 25
min at 0.7 V/cm. Each electrophoresis included slides that belonged
to the same experimental time-point.
After the completion of the electrophoresis, the
slides were washed three times for 5 min in a neutralization buffer
[0.4 M Tris (pH 7.5)]. The slides were stained with SYBR-Gold dye
(Invitrogen), comets were viewed under an epifluorescent microscope
(Zeiss), and the image information was collected using a Comet
Assay IV system (Perceptive Instruments).
Statistical analysis was performed for tail
intensity data using SPSS software (IBM). The data were collected
from three replicate Petri plates, at 2 slides per plate, and 50
cells were examined on each slide, avoiding those located near the
edges. A preliminary examination showed that the data were not
normally distributed and could not be normalized through
logarithmic transformation. Therefore, we applied non-parametric
methods for hypothesis testing. Kruskal-Wallis one-way analysis of
variance by ranks was used to compare the data distribution for
samples at a specific point in time. Following the Kruskall-Wallis
test, each of the treatment groups was compared to the control
group using a Mann-Whitney U test.
Immunofluorescence
For immunocytochemical analysis, the cells were
grown on Lab-Tek chambered 2-well slides (Nulge Nunc International
Corp., Naperville, IL) and irradiated. After irradiation, the cells
were fixed in 4% paraformaldehyde in PBS, permeabilized with 70%
ethanol and washed in PBS containing 0.1% Triton X-100. Blocking
was done in 8% BSA in PBS. For immunocytochemical detection, the
cells were incubated for 2 h at room temperature using the
following antibodies: anti-γH2AX (Ser 139) rabbit antibodies
(1:100, Cell Signaling Technology Inc.), anti-RAD51 rabbit
antibodies, anti-pATM and anti-KU70 mouse antibodies (1:100, Santa
Cruz Biotechnology Inc.). Afterward, the cells were rinsed and
incubated in 1:500 diluted secondary antibodies (goat anti-rabbit
IgG Alexa Fluor 488, goat anti-mouse IgG Alexa Fluor 546, and goat
anti-mouse IgG Alexa Fluor 488, Invitrogen Molecular Probes,
Eugene, OR). Cell nuclei were counterstained with 0.1 mg/ml
4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma-Aldrich
Chemical Co.). The slides were mounted with an anti-fade
fluorescence medium prepared from 1,4-diazabicyclo[2.2.2]octane
(DABCO), polyvinyl alcohol and glycerol, and analyzed using a Zeiss
epifluorescent microscope.
The number of γH2AX foci per cell was counted in at
least 400 cells from each cell group, as previously described
(40). The γH2AX levels are
presented as the mean ± SE; p≤0.05. The expression levels of pATM,
Ku70, and Rad51 were evaluated using the fluorescence intensity of
the corresponding antibody. The fluorescence intensity in each cell
was measured using CellProfiler cell image analysis software
(41,42).
The process used for the analysis is as follows: i)
load images; ii) measure image intensity; iii) identify primer
automatically; iv) measure object intensity; and v) export to
excel. The intensity was represented in intensity units or
arbitrary units as mean ± SD; p≤0.05.
Statistical analysis
Statistical analysis was performed using MS Excel
2007 and JMP5 software packages.
Results
Effect of radiation on whole genome
gene expression in MCF-7 and MCF-7/DOX cells
Isolated RNA from MCF-7 and MCF-7/DOX cell lines
(17,32) was used for gene expression
profiling. The background level of gene expression was extremely
different in the MCF-7 and MCF-7/DOX cells. In fact, most of the
reported housekeeping genes were expressed less in drug-resistant
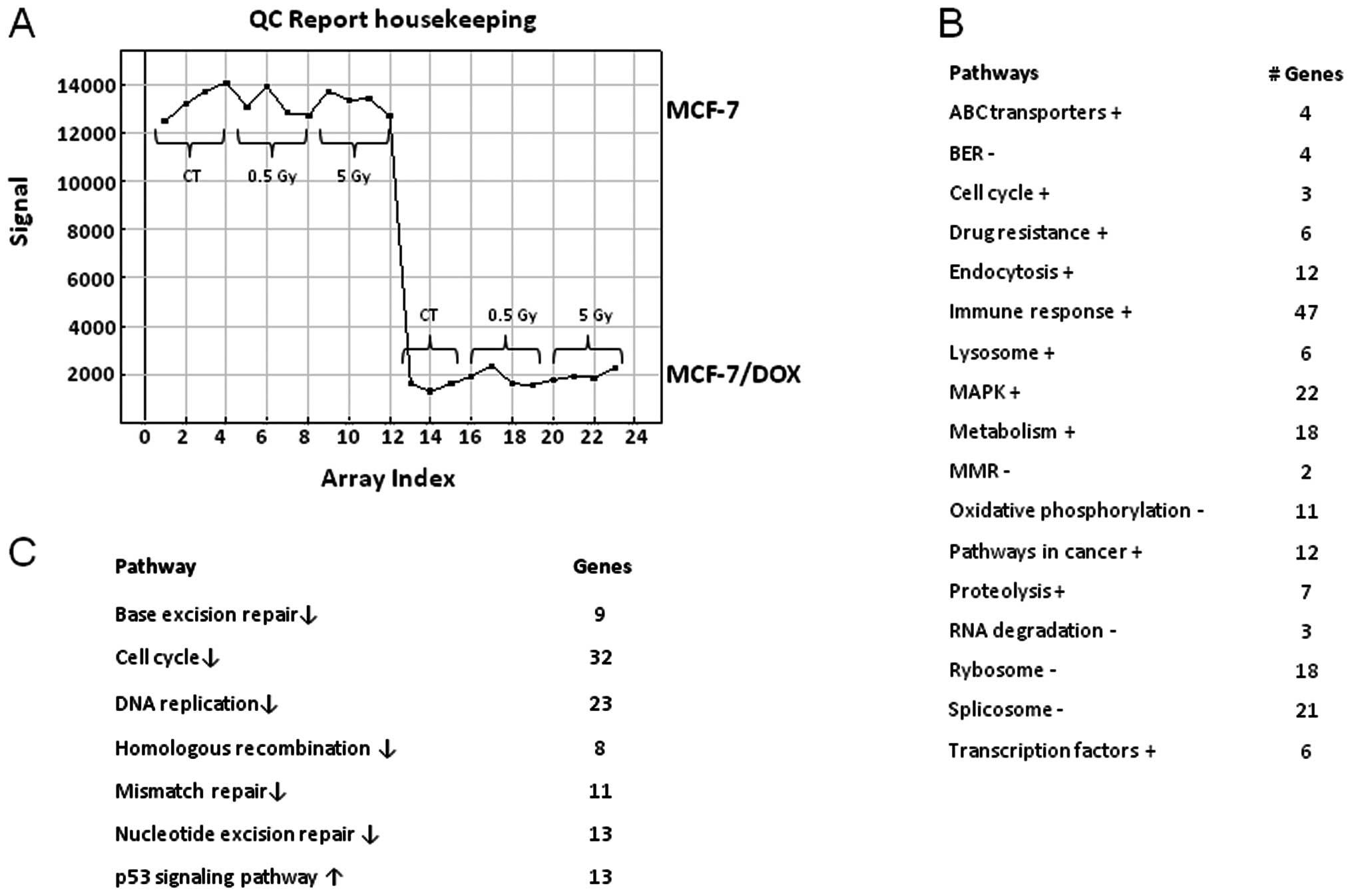
cells than in wild-type parental cells (Fig. 1A). With the help of the DAVID
functional annotation array analysis tools, we were able to
identify and group the evaluated genes according to their function
and possible role in certain pathways. Subsequently, the genes with
a similar or identical function were grouped together, and based on
their expression changes, the role of certain pathways was
evaluated and compared between the two cell lines (Fig. 1B). Fig. 1 demonstrates the identified
biological functions and their predominance or weakness in
MCF-7/DOX compared to MCF-7. MCF-7/DOX cells had a higher
expression of the ABC transporter genes, which when translated,
play a role in pumping the cytotoxic drugs out of the cells,
contributing to drug resistance. Similarly, a higher expression of
the genes corresponding to cell cycle progression, endocytosis,
lysosome, proteolysis, transcription factors, genes contributing to
the cancer pathways and drug resistance were found in
doxorubicin-resistant cells (Fig.
1B). The genetic profiling of MCF-7/DOX cells also showed an
increase in metabolism, immune response and some cell-signaling
pathways, such as the MAPK signaling pathway. The primary
downregulated processes in MCF-7/DOX cells in comparison to the
MCF-7 parental line were: oxidative phosphorylation, ribosome and
RNA degradation and splicing (Fig.
1B).
It is possible that the previously mentioned
difference in the genetic profiling of MCF-7/DOX cells could affect
the response of these cells to radiation treatment. Neither low
(0.5 Gy) nor high (5 Gy) X-ray doses caused any changes in the gene
expression of MCF-7/DOX. In contrast, MCF-7 cells showed an extreme
genetic response to the high (5 Gy) X-ray dose (Fig. 1C). Thirty-two cell cycle genes and
23 genes responsible for DNA replication were downregulated
(Fig. 1C). The primary repair
processes were shut down by the decreased expression of key genes.
Parental cells lost their MMR, NER, BER and HR due to the
downregulation of the 11, 13, 9 and 8 pathway genes, respectively
(Fig. 1C). These changes usually
lead to cell death. Moreover, the genes responsible for cell death
from the p53 signaling pathway were upregulated (Fig. 1C).
The validity of gene expression profiling was
confirmed by qRT-PCR for the genes with the most change and the
greatest radiation response. Therefore, the primary targets for
qRT-PCR were: DNA polymerases A, D and E, which are the key
components in DNA replication and DNA repair pathways, and cyclin
A, GADD45G, and aurora B, which play an important role in
cell cycle and p53 signaling pathways.
Aurora B is a protein kinase that functions through
the attachment of the mitotic spindle to the centromere and
provides equal chromosome movement and segregation during mitosis.
The level of AURKB transcripts gradually and significantly
decreased in the MCF-7 parental line after X-ray treatment
(Table I). There was no change in
AURKB expression found in MCF-7/DOX cells after irradiation
and the background expression level was significantly lower in the
drug-resistant cell line than the parental cells. Similar to
AURKB, cyclin A (CCNA) was downregulated in parental
cells after X-ray exposure (Table
I). Because cyclin A binds to S phase Cdk2 and is required for
the cell to progress through the S phase, the deficit of cyclin A
may contribute to cell cycle arrest. There was no change in cyclin
A expression found in MCF-7/DOX cells, and the background level of
the cyclin A expression was significantly lower in the cells
resistant to doxorubicin.
 | Table IFold change (corrected for internal
standard) in levels of gene transcripts of aurora B, cyclin A,
Gad45G and polymerases A, D and E detected by qRT-PCR. |
Table I
Fold change (corrected for internal
standard) in levels of gene transcripts of aurora B, cyclin A,
Gad45G and polymerases A, D and E detected by qRT-PCR.
| PT
| DOX
| CT
|
|---|
| Gene | CT | 0.5 | 5 | CT | 0.5 | 5 | PT | DOX |
|---|
| Aurora B | | | | | | | | |
| Relative fold
change | 1 | 0.9 | 0.8 | 1 | 1.1 | 1.1 | 1 | 0.2 |
| P-value | | 0.01 | 0.00 | | 0.09 | 0.10 | | 0.00 |
| Cyclin A | | | | | | | | |
| Relative fold
change | 1 | 1 | 0.9 | 1 | 0.9 | 1 | 1 | 0.2 |
| P-value | | 0.21 | 0.00 | | 0.09 | 0.28 | | 0.00 |
| Gad45G | | | | | | | | |
| Relative fold
change | 1 | 2.1 | 2 | 1 | 1.2 | 1.1 | 1 | 5.7 |
| P-value | | 0.00 | 0.02 | | 0.81 | 0.83 | | 0.00 |
| PolA | | | | | | | | |
| Relative fold
change | 1 | 0.8 | 0.3 | - | - | - | 1 | 0 |
| P-value | | 0.22 | 0.00 | | | | | |
| PolD | | | | | | | | |
| Relative fold
change | 1 | 1 | 0.9 | - | - | - | 1 | 0 |
| P-value | | 0.09 | 0.00 | | | | | |
| PolE | | | | | | | | |
| Relative fold
change | 1 | 1 | 0.8 | 1 | 1 | 1 | 1 | 0.3 |
| P-value | | 0.07 | 0.00 | | 0.71 | 0.48 | | 0.00 |
GADD45G is a growth arrest and DNA-damage-inducible
protein whose levels are increased following stressful growth
arrest conditions and treatment with DNA-damaging agents. The
protein encoded with GADD45G responds to environmental
stresses by mediating the activation of the p38/JNK pathway. Both
0.5 and 5 Gy X-rays caused an increase in GADD45G transcript
levels in MCF-7 cells, which is in contrast to levels in MCF-7/DOX
cells (Table I). Interestingly,
the background expression level of GADD45G was higher in
drug-resistant cells, which could be due to the genomic instability
in cells that acquired drug resistance. All three polymerases (A, D
and E) were significantly downregulated in response to a 5 Gy
radiation treatment in their parental cell lines (Table I), disabling the polymerization of
deoxyribonucleotides into a DNA strand. There were no changes in
the expression level of the three polymerases found in MCF-7/DOX
cells; moreover, the control expression level of polymerases was so
low that POLD and POLA could not be identified using
qRT-PCR.
Levels of DNA polymerase proteins in
MCF-7 and MCF-7/DOX cells
Taking the results into consideration, we wondered
how drug-resistant cells survived radiation, how they proliferated
and what DNA polymerases they used for replication and DNA repair.
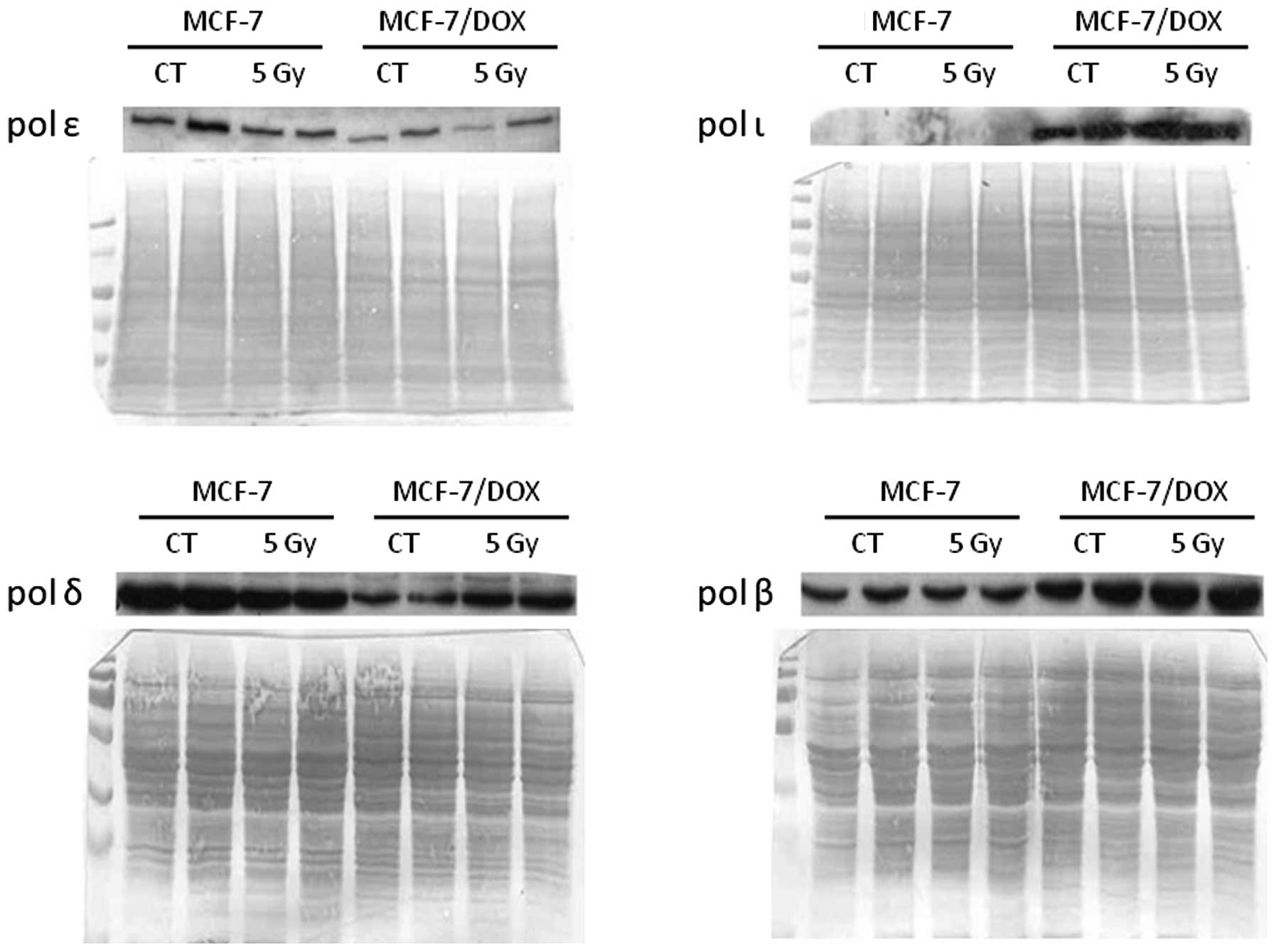
We, therefore, analyzed the protein levels of polymerases δ, ε, β,
and ι in the MCF-7 and MCF-7/DOX cells.
The expression level of both polymerases δ and ε
were found to be higher in MCF-7 cells, similar to gene expression
profiling analysis and qRT-PCR analysis (Fig. 2). Furthermore, the level of DNA
polymerase δ was slightly increased in doxorubicin-resistant
MCF-7/DOX cells after radiation exposure.
Two other polymerases (β and ι) were highly
expressed in MCF-7/DOX. While the polymerase β level was much lower
in MCF-7 cells than in MCF-7/DOX, polymerase ι was not detected in
the parental cells at all (Fig.
2). DNA polymerase ι was recently discovered as a polymerase
that catalyses error-prone DNA synthesis. It promotes the
replication of damaged DNA by misincorporating deoxynucleotides
opposite DNA lesions (43,44). We doubted whether the high
expression of polymerase ι provided a fast, yet inaccurate, DNA
repair in DOX cells following any DNA-damaging treatment, including
X-ray exposure.
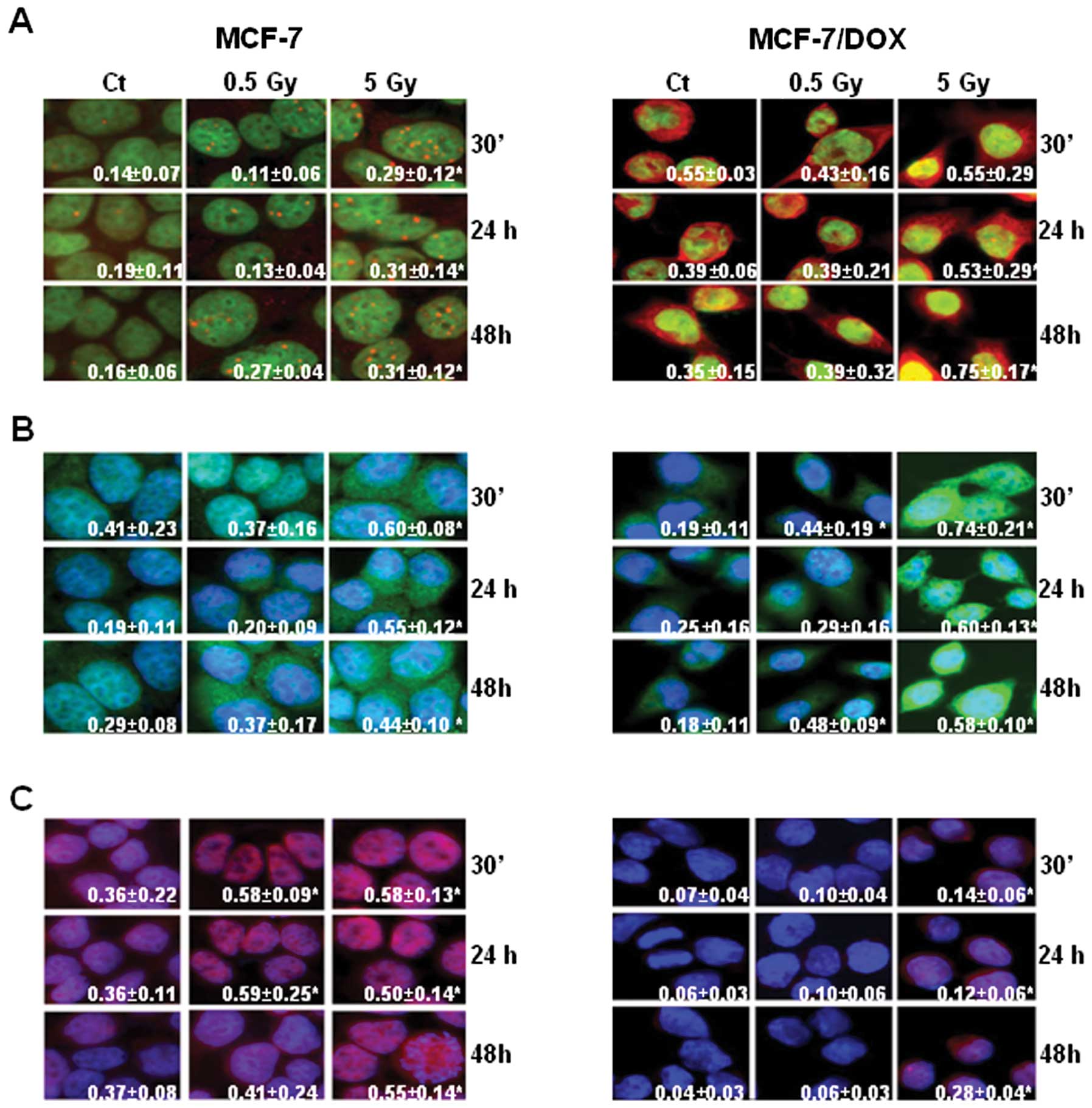
Analysis of the fidelity of DNA
polymerases in MCF-7 and MCF-7/DOX cells
All types of DNA repair involve the resynthesis of
DNA to replace damaged strands. To uncover any correlation between
the dynamics of the induction and repair of IR-induced DNA damage,
we studied the fidelity of the DNA polymerase pool in the cell
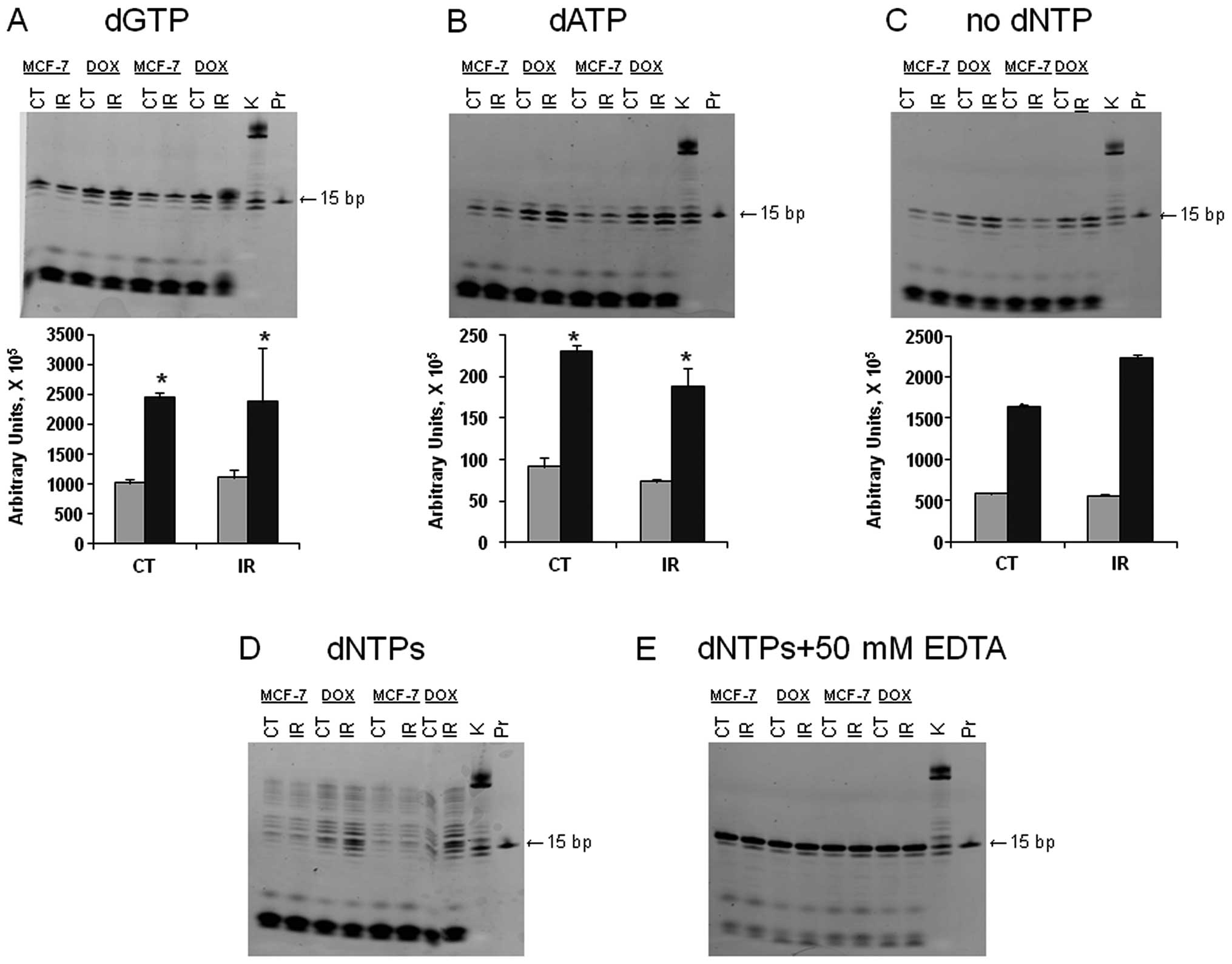
lysates from MCF-7 and MCF-7/DOX (Fig.
3). Because doxorubicin-resistant MCF-7/DOX cells managed to
survive DNA damage, we hypothesized that low fidelity DNA
polymerases may be more active in the resistant cells. Therefore,
we analyzed the DNA polymerase fidelity in MCF-7 and MCF-7/DOX
(37).
DNTPs were added to the mixture containing the
template and extracts of the unirradiated or irradiated MCF-7 or
MCF-7/DOX cells and the incorporation patterns were analyzed.
According to the template sequence, the next nucleotide to be
inserted was dGTP. When only dGTP was in the reaction mixture, we
obtained a 16-bp gel band with a higher intensity in MCF-7/DOX;
moreover, the band corresponding to the irradiated MCF-7/DOX had
the highest intensity (Fig. 3A).
The observed difference may be explained by higher DNA polymerase
activity or an increased amount of polymerases in resistant cells.
The latter idea would make sense only for certain polymerases, such
as polymerases β or ι; as polymerases α, ε and δ were previously
shown to be downregulated in MCF-7/DOX.
Furthermore, as shown in Fig. 3B, MCF-7/DOX had a higher level of
dATP misincorporation, which means that DNA polymerase specificity
or fidelity is lower in the drug-resistant cells. We did not
observe the incorporation of dTTP and dCTP. Therefore, we concluded
that ATP is the most common incorrect nucleotide to be inserted by
the low fidelity polymerases to continue synthesis in MCF-7/DOX
cells.
When adding both dGTPs and dATPs or all dNTPs to the
samples, we obtained 16 and 17 bp bands and completed synthesis,
respectively (Fig. 3D). In all the
cases, the activity of the polymerases was higher in the
MCF-7/DOX-resistant cell line. At the same time, we observed more
intense DNA cleavage in MCF-7 cells due to significant exonuclease
activity. The excision of incorrect nucleotides by exonucleases
reduces mismatches. The control sample did not contain any dNTPs,
and no band with a weight higher than 15 bp was observed (Fig. 3C). The negative control contained
all the dNTPs and EDTA (to inactivate all metal-using enzymes).
Under these conditions, the exonuclease activity was lower and the
intensity of all the bands was the same (Fig. 3E). Both controls indicated that
there were no endogenous oligonucleotides observed in the gels
(Fig. 3). In summary, we concluded
that irradiated and non-irradiated MCF-7/DOX cells exhibited
significantly higher processivity and significantly lower
polymerases fidelity.
Radiation-induced apoptosis in MCF-7
breast adenocarcinoma cells and their drug-resistant counterpart,
MCF-7/DOX cells
In this study, we characterized and compared the
responses of the MCF-7 breast adenocarcinoma line and its
doxorubicin-resistant variant (MCF-7/DOX) to ionizing radiation
(IR) in vitro. IR exposure is known to induce apoptotic cell
death in irradiated cells. Therefore, we analyzed the levels of
IR-induced apoptosis in MCF-7 and MCF-7/DOX cells. Early apoptosis
is characterized by various changes in the cellular plasma
membrane; the primary change is the translocation of
phosphatidylserine (PS) from the inner layer to the surface of the
membrane. Annexin V possesses a high affinity to PS, and this
allows for the early detection of apoptotic changes (45). Here, we analyzed IR-induced
apoptosis using the Annexin V assay.
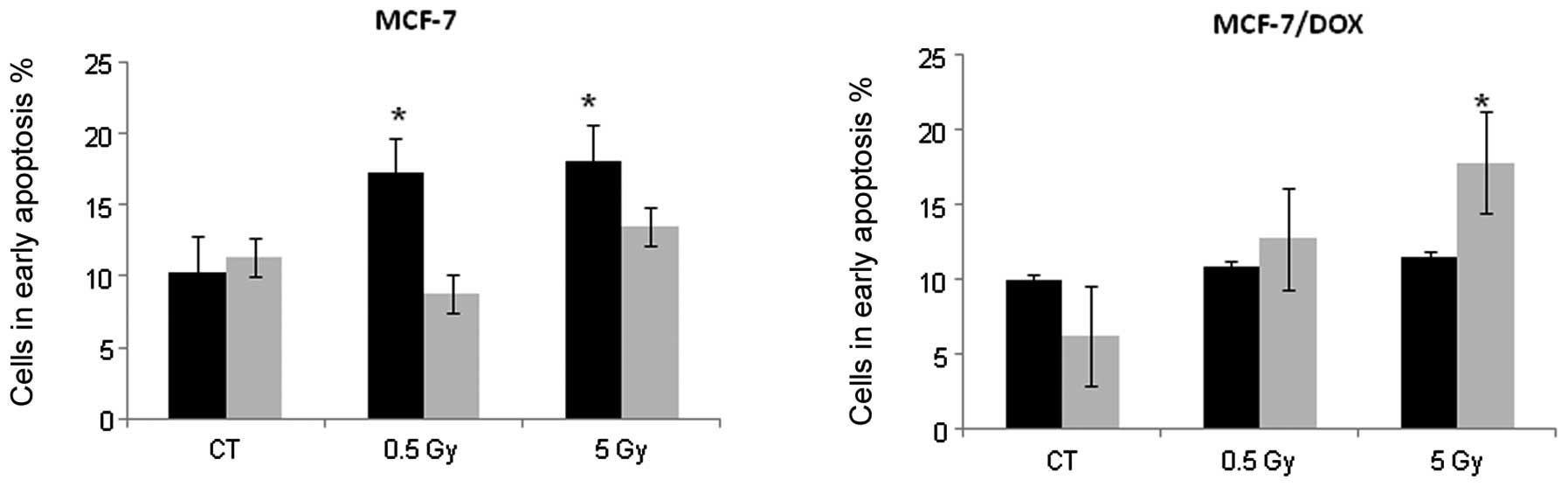
Fig. 4 shows that
MCF-7 cells began to undergo early apoptosis 24 h after
irradiation. We found a 1.67 and 1.75-fold increase in Annexin V
positive cells 24 h after exposure to 0.5 and 5 Gy X-rays,
respectively. The percentage of MCF-7 cells in early apoptosis
returned to the control level within 48 h (Fig. 4); however, the number of dead cells
increased at this time point. These changes may indicate that cells
that were undergoing early apoptosis 24 h after irradiation were
dead within 48 h. In contrast, MCF-7/DOX-resistant cells only
showed an apoptotic response 48 h after treatment with the high IR
dose (5 Gy). The 2.87-fold increase in Annexin V positive cells was
reached 48 h after the X-ray treatment of MCF-7/DOX cells (Fig. 4). Based on these data, we concluded
that MCF-7/DOX cells exhibit a significantly delayed apoptotic
response to ionizing radiation.
Radiation-induced DNA damage in MCF-7
and MCF-7/DOX cells
Next, we analyzed the structures associated with
such significant differences in IR-induced apoptotic responses in
the MCF-7 and MCF-7/DOX cells. IR is a potent DNA-damaging agent
capable of inducing cross linking, nucleotide base damage, and most
importantly, single and double strand breaks (DSBs), which are
well-known inducers of apoptosis (46,47).
Therefore, we analyzed and compared the levels of IR-induced DNA
damage in MCF-7 and MCF-7/DOX cells using the Comet assay and by
detecting γH2AX foci, a well accepted indicator of DNA
double-strand breaks (48).
In the comet assay, the super coiled duplex DNA
underwent unwinding and denaturation under strong alkaline
conditions (49). This led to DNA
fragment size reductions and the expression of alkali labile sites
as single-strand breaks, which are stretched out by
electrophoresis. A comet tail consisting of the damaged or broken
DNA fragments was analyzed through the intensity in both types of
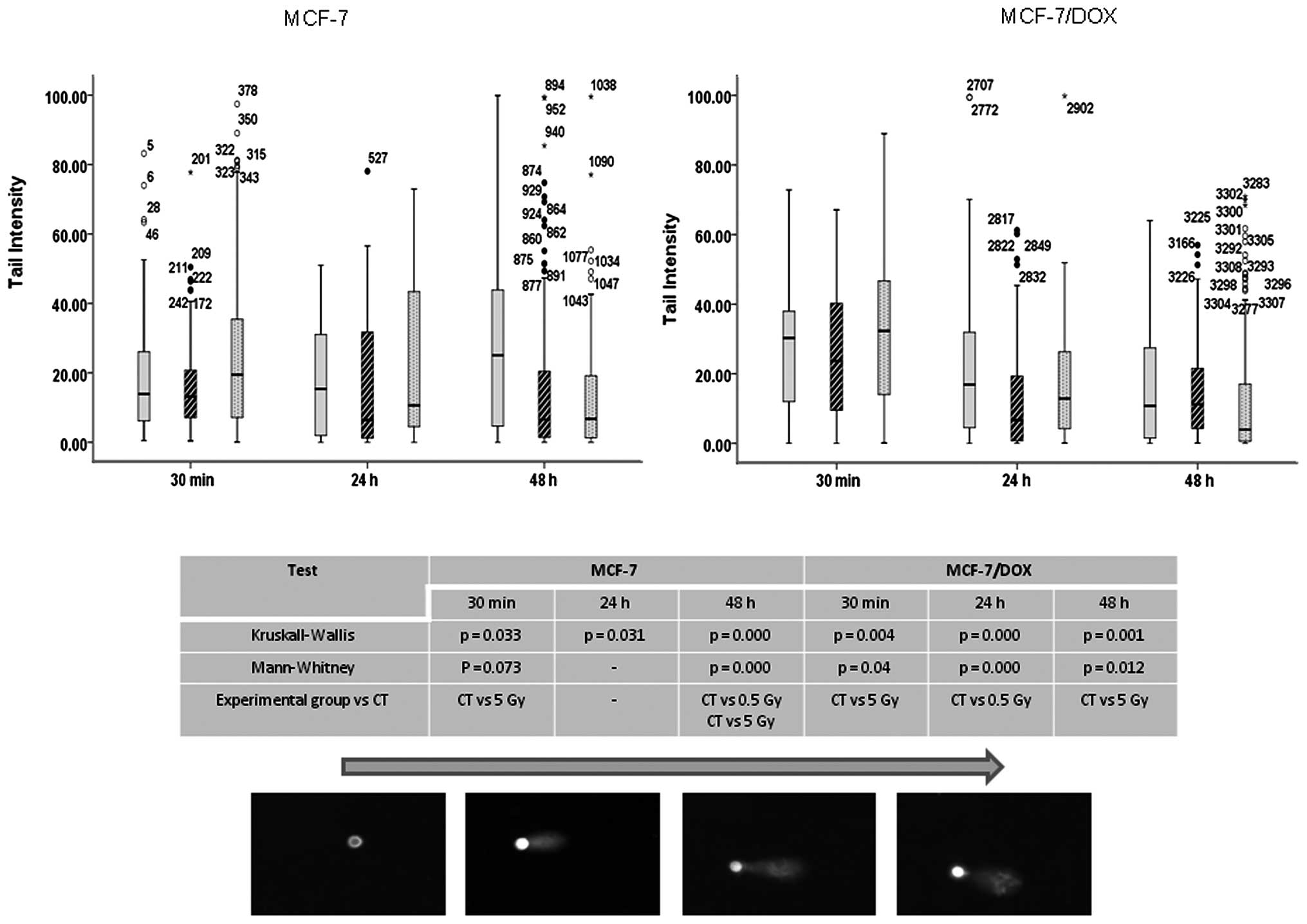
MCF-7 cells after radiation treatment (Fig. 5). A 5 Gy X-ray led to significant
damage in MCF-7 parental and drug-resistant cells immediately (30
min) after application. The damage is believed to represent DSBs,
SSBs, alkali labile sites and breaks from replication events. The
persistence of the damage was only observed for up to 24 h in the
parental line, and no significant damages were observed in the
drug-resistant line after 24–48 h (Fig. 5).
Similarly, both 0.5 and 5 Gy X-ray doses led to the
formation of γH2AX foci in MCF-7 and MCF-7/DOX cells. However,
MCF-7/DOX cells were much less sensitive to IR than MCF-7 cells
(Fig. 6). Specifically, the
irradiation of MCF-7 cells caused significant (2.6 and 8.5 times)
increases in the levels of γH2AX foci, from 3.14±0.22 foci per cell
in the control to 8.23±0.53 and 26.70±1.02 foci per cells 30 min
after 0.5 and 5 Gy treatments, respectively (Fig. 6A). The γH2AX foci induced by 0.5 Gy
X-rays disappeared 48 h after irradiation, indicating efficient DNA
repair. The application of 5 Gy X-rays led to the persistent
elevation of γH2AX foci, as detected 48 h after exposure.
In MCF-7/DOX cells, radiation exposure led to
significant (1.9 and 6.0 times) increases in the levels of γH2AX
foci, from 1.83±0.2 foci per cell in the control to 3.49±0.15 and
10.9±0.44 foci per cell after 0.5 Gy and 5 Gy treatments,
respectively (Fig. 6B). The levels
γH2AX foci significantly decreased 24 and 48 h after
irradiation.
Most importantly, the levels of γH2AX foci in all
cases in MCF-7 cells were significantly different from the levels
seen at the corresponding time-points in MCF-7/DOX cells (Fig. 6). MCF-7/DOX cells exhibited lower
levels of IR-induced DNA damage and faster repair of γH2AX compared
to the sensitive MCF-7 cells.
Analysis of the DNA repair machinery
in MCF-7 and MCF-7/DOX cells
The apparent differences in the levels of IR-induced
DNA damage between MCF-7 and MCF-7/DOX cells have led us to
question how the resistant cells repair the DNA lesions. In
mammalian cells, two processes exist to repair DSBs: homologous
recombination (HR) and non-homologous end-joining (NHEJ) (50–53).
The key component for both processes is the serine/threonine
specific protein kinase ATM. The phosphorylation of ATM is
necessary for DSB repair (48,54).
Therefore, we analyzed the level of phosphorylated ATM (pATM) in
MCF-7 and MCF-7/DOX cell lines after irradiation.
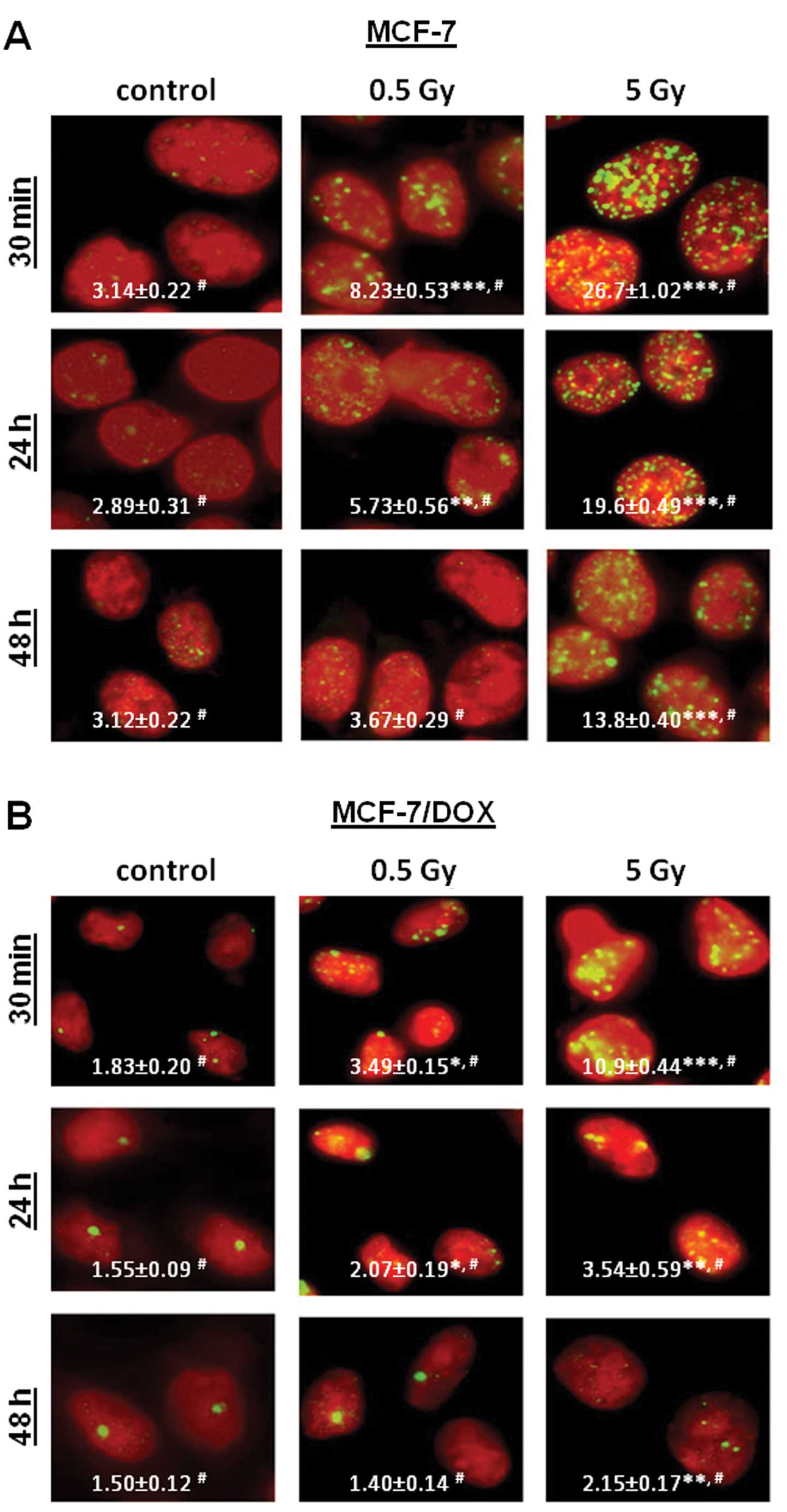
Overall, the level of pATM was higher in MCF-7/DOX
cells. Interestingly, the subcellular localization of the protein
was different in MCF-7 and MCF-7/DOX cell lines (Fig. 7A). For example, in MCF-7 cells,
pATM was detected as nuclear foci (Fig. 7A). The number of pATM nuclear foci
in MCF-7 cells increased after irradiation. The dynamics of pATM
expression were similar to that of γH2AX (Fig. 7A).
In MCF-7/DOX-resistant cells, slight pATM foci were
observed, and the protein was localized in both the nucleus and
cytoplasm. Yet, the general level of pATM in MCF-7/DOX cells
measured by fluorescent intensity was higher than that in the MCF-7
cells (Fig. 7A). With evidence of
different levels of γH2AX and pATM in MCF-7 and MCF-7/DOX cells, we
then asked if HR or NHEJ-related proteins were differentially
induced in these cell lines after irradiation. RAD51 is a key
protein essential for the repair of DSBs via HR in mammals
(55). KU70 is a key participant
in the NHEJ pathway that repairs DSBs (56,57).
Immunocytochemistry was performed to analyze the
levels of RAD51 and KU70 in MCF-7 and MCF-7/DOX cells after
irradiation. We found that the expression level of RAD51 increased
after irradiation in both cell lines (Fig. 7B), but the highest level was
observed in MCF-7/DOX cells after exposure to 5 Gy X-rays (Fig. 7B).
Interestingly, MCF-7 cells expressed relatively high
levels of KU70 prior to irradiation, and an abundant amount of the
protein was found after exposure (Fig.
7C). On the contrary, KU70 levels were almost undetectable in
non-irradiated MCF-7/DOX cells, and only exposure to 5 Gy X-rays
resulted in a noticeable upregulation of KU70 levels (Fig. 7C). Overall, we concluded that
MCF-7/DOX cells harbored higher DNA repair potential than sensitive
MCF-7 cells.
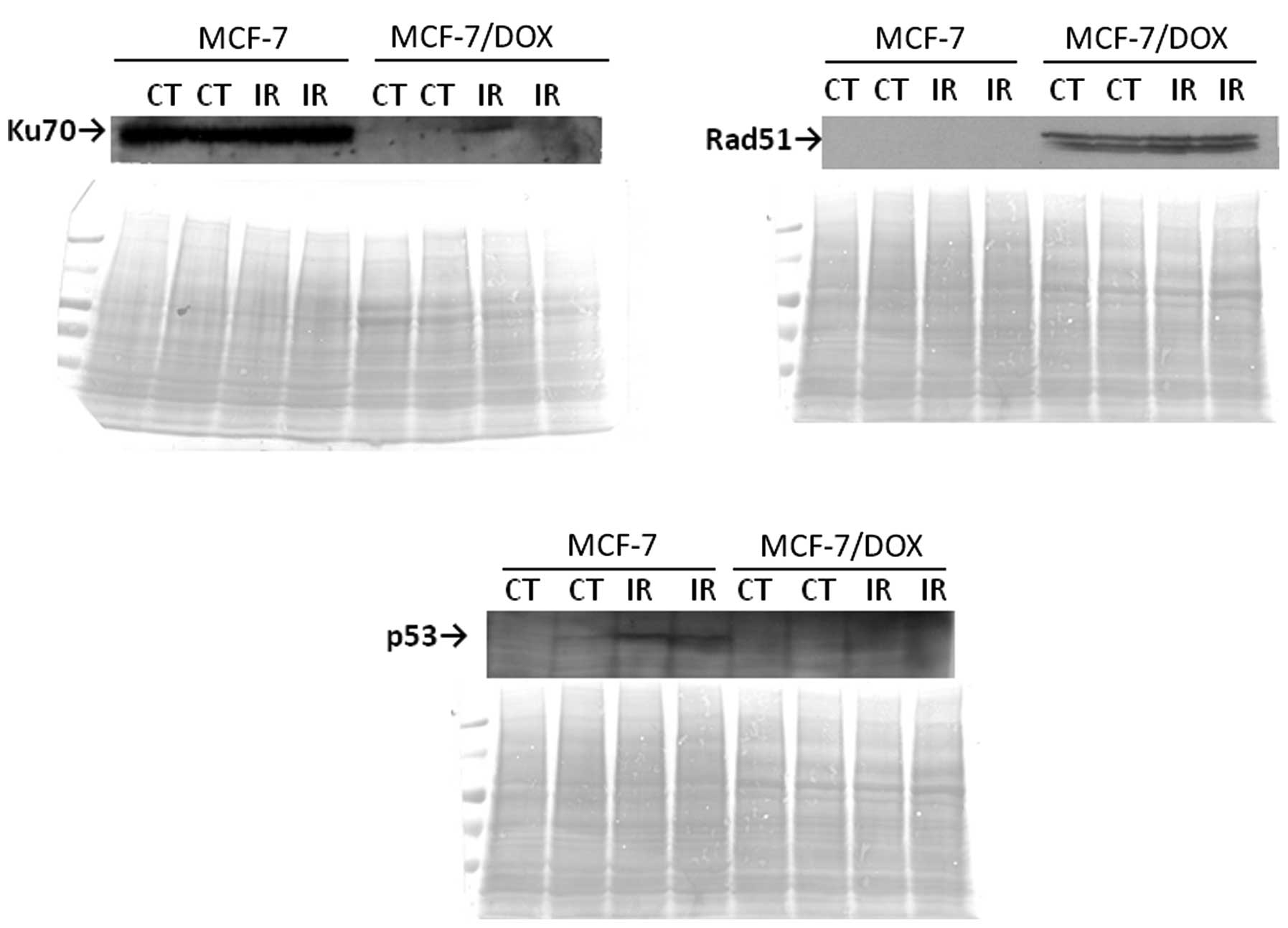
The immunocytochemistry results for RAD51 and KU70
repair proteins were confirmed using a western immunoblot assay
(Fig. 8). High expressions of KU70
in both the control and irradiated MCF-7 cells were observed, but
there was an absence of KU70-specific bands in MCF-7/DOX cells. In
contrast, RAD51 expression was only found in MCF-7/DOX cells
(Fig. 8). This difference in the
preference of the two cell lines to different types of DNA DSBs
repair may be due to the differences in the proliferative
potentials of these cell lines. The highly proliferative MCF-7/DOX
cells may use an available sister chromatid for the homology search
that is needed for HR.
The p53 protein is a well known DNA damage response
initiator that induces long-term checkpoint activation. As
expected, a 5 Gy X-ray treatment led to p53 elevation in MCF-7
cells. However, no change in the p53 protein level was observed in
the MCF-7/DOX line (Fig. 8).
Discussion
Relapse risk in breast cancer is largely dependent
on the combination of anticancer treatment modalities.
Anthracycline chemotherapy is increasingly used for treating
locally advanced breast cancer and hormone-resistant metastatic
breast tumors (1,15).
Unfortunately, resistance to chemotherapy occurs
frequently (15). Drug-resistant
tumors often become unresponsive to the use of other antitumor
therapies, acquire multidrug resistance, and fail to respond to
radiation therapy (8). Frequently,
the use of chemotherapy drugs as radiation sensitizers fails for
unknown reasons (28,29). Overall, data are scarce on the
radiation response of drug-resistant cells. Therefore, we set out
to dissect the mechanisms of radiation responses of cells resistant
to doxorubicin.
Doxorubicin is widely used in curative-intent
adjuvant breast cancer therapy (58). Mechanistically, doxorubicin, an
anthracycline antibiotic, intercalates DNA and inhibits the
progression of the enzyme topoisomerase 2α (Top2A) (58). Functionally, it stabilizes the
Top2A complex after it has broken the DNA chain, preventing DNA
resealing and, thereby, blocking replication (59). Therefore, because doxorubicin
treatment leads to the induction of strand breaks, we hypothesized
that cells exposed to doxorubicin for a prolonged period of time
could develop structures to effectively repair DSBs, thus avoiding
drug-induced apoptosis. Consequently, these structures may help
drug-resistant cells withstand the effects of other treatment
modalities that induce DNA strand breaks as the primary method of
their cell-killing action.
The purpose of this study was to investigate the
radiation-induced gene expression changes in the two cell lines of
breast adenocarcinoma: the parental MCF-7 and drug-resistant
MCF-7/DOX. Using microarray technology tools, we were able to
screen the differential gene expression between MCF-7 and
MCF-7/DOX. Here, we report the substantial variations in the
expression levels of most housekeeping genes between the
drug-sensitive and drug-resistant cells.
Housekeeping, or maintenance, genes control basic
metabolic functions, provide support through the cell cycle, and
are expected to retain an unchanged expression through various
cells and tissues during cell development, treatment or disease
anomalies (60). This makes
housekeeping genes a good reference for the normalization of gene
expression analysis following differential treatments or during
disease states. However, multiple studies found inconsistent
reliabilities in the housekeeping genes as the standard in cancer
experiments. Variability in housekeeping gene expression was
reported in colorectal, esophageal, gastric, hepatic, breast and
prostate cancers (60–63). In our study the expression level of
most housekeeping genes in MCF-7/DOX cells was at least six times
lower than that in MCF-7 cells (Fig.
1A). We believe that these differences not only reflect the
need for a cautious approach when studying differential gene
expression, but also can be involved in any cellular or tissue
changes in the morphology, physiology and sensitivity to treatment
modalities. MCF-7/DOX cells were previously characterized as larger
than initial MCF-7 cells with stronger adhesion, more complex
structural organization due to microtubule and microfilament
increases, and the existence of multivesicular bodies near the
plasma membrane that may be associated with increased drug efflux
(64).
Furthermore, the background differential gene
expression was evaluated in unexposed MCF-7/DOX cells, which
allowed us to identify possible changes in the biological processes
and pathways during the development of drug resistance. Four genes
encoding for the ATP-binding cassettes of MDR and MRP sub-families
of ABC transporters were upregulated in MCF-7/DOX cells compared to
the MCF-7cells (Fig. 1B). The
overexpression of ABC transporters is a well-characterized
structure of acquired drug resistance in cancer cells, particularly
in MCF-7/DOX (32,65).
In addition, 6 genes involved in drug metabolism
were upregulated in MCF-7/DOX cells. One of these genes was
microsomal glutathione S-transferase 3 (GST), a radical scavenger
that is involved in the metabolism of xenobiotics. It was
previously found that GST plays an important role in the
acquisition of DOX resistance through decreased intracellular drug
accumulation and the stimulation of the repair of drug-induced DNA
damage (66,67). Moreover, GST may be involved in the
resistance of cancer cells to radiation, and therefore, may be
considered one of the common structural indicators for chemoand
radio-resistance. An early study on human lung cancer cells showed
that the introduction of GST cDNA into cells modestly increased
resistance to ionizing radiation and adriamycin (68).
The gene profiling analysis showed higher rates of
the metabolism of sphingolipids, starch, sucrose, retinol,
riboflavin, amino-sugars, nucleo-sugars, androgen and estrogen. We
assume that high metabolic rates may also contribute to drug- and
radio-resistance and the overall survival ability of cancer cells.
It is important to analyze the cellular metabolism in relation to
mitochondrial functions.
Eleven genes that play a role in oxidative
phosphorylation were highly downregulated in MCF-7/DOX cells.
Amongst them are ATP synthases, proton-transporting mitochondrial
complexes, NADH dehydrogenases, and a cytochrome c oxidase
subunit. These data correlate highly with the previously formulated
parameters for drug-resistant cells: i) lower mitochondrial
membrane potential; ii) smaller proton gradient and proton leak;
iii) higher use of fat for fuel in mitochondria and higher rate of
glycolysis; and iv) lower levels of reactive oxygen and lower DNA
damage and susceptibility to apoptosis under stress (69). According to the authors, drug- and
radiation-resistant cancer cells switch their metabolism (Warburg
effect) from efficient respiration to highly inefficient
glycolysis, producing ATP to protect themselves from reactive
oxygen species. The combination of high glucose utilization, a
shift to fatty acids as a source of fuel, and low oxidative
phosphorylation is one structure of dual drug- and radio-resistance
(69,70).
With the help of David software, we were able to
reveal the activation of the MAPK pathway and at least 12 genes
from the pathway that contribute to cancer in MCF-7/DOX. These
results correlate with a recent study that found that the
inhibition of certain cell signaling pathways, including MAPK,
inhibited the invasive activities of MCF-7/DOX cells (71). The most upregulated genes were:
catenin (Wnt pathway), glycogen synthase kinase 3β (Wnt, Hedgehog,
ErbB pathways), peroxisome proliferator-activated receptor δ (Wnt,
PPAR pathways), son of sevenless (ErbB, Jak-STAT signaling
pathways), and a platelet-derived growth factor receptor (MAPK,
focal adhesion, gap junction). We believe that the difference
between MCF-7/DOX and its parental line may also contribute to the
observed higher invasiveness of drug-resistant cells.
Surprisingly, doxorubicin-resistant MCF-7 cells
showed an upregulated expression of at least 47 genes involved in
immune response (Table I). Most of
the genes are involved in hematopoietic cell lineage, B cell
receptor signaling pathways, natural killer cell mediated
cytotoxicity, antigen processing and presentation processes or
encode components of complement cascades and Ig-like receptors.
Anticancer drugs that induce immune responses are
considered to be very successful for cancer treatment. The
traditional view states that drug or radiation damage may cause
cell surface changes that are recognized by the immune system. The
ability of anticancer drugs to boost the host’s immune system
against tumor cells may have great therapeutic potential (72). Such immunomodulating effects were
shown for doxorubicin as well. The doxorubicin-cured mice had
memory T cell-dependent resistance to the reimplantation of the
tumor (73). In contrast,
therapy-resistant cells display certain molecular and metabolic
characteristics that mask them from the immune system. Based on our
results, we can speculate that doxorubicin-resistant cells express
modified cytokines and cell surface receptors that may recruit the
immune system to work for them. We assume that this ability may
defend resistant cells against harm due to repeated similar or
different treatments. The upregulation of endocytosis, lysosome and
proteolysis pathways in MCF-7/DOX may also confirm the unique
protective and metabolic characteristics of the drug-resistant
cells.
Two key mismatch repair genes, MLH1 and MSH3, along
with four base excision repair genes were highly downregulated in
MCF-7/DOX cells compared to MCF-7 cells. Meanwhile, six genes
encoded for transcription factors, including three members (TAF10,
TAF15 and TAF3) of basal transcription factor TAFIID and three cell
cycle components, were found to be upregulated in MCF-7/DOX
(Fig. 1B). We assume the
possibility that the inaccurate repair of nucleotide
misincorporation toward fast DNA replication and cell division may
result in MCF-7/DOX resistance. Interestingly, MCF-7/DOX cells
exhibited low expressions of ribosome subunits and splicing
components. The selective inhibition of RNA and protein synthesis
may be characteristic of drug-resistant cells, but we are unable to
explain this phenomenon in the present research; however, more
studies are required in this field.
The main purpose of this study was to examine and
compare the radiation responses of drug-sensitive MCF-7 cells and
doxorubicin-resistant MCF-7/DOX cells. Gene expression profiling
showed that the expression level of more than 500 genes was changed
in the sensitive cell line due to 5 Gy X-rays; however in MCF-7/DOX
cells, no changes in gene expression were observed. We believe that
the ability of the cells to retain their gene expression potential
on a constant level regardless of DNA-damaging insults may be due
to the features that cells acquired during drug resistance and are
shared in other forms of resistance, such as radio resistance.
MCF-7 cells exhibited the expected downregulation of
biological pathways, such as cell cycle, DNA replication, DNA
repair and the activation of the p53 pathway (Fig. 1C). Thirty-two cell cycle regulators
where downregulated, which led to cell cycle shut down. These genes
were encoded for cyclins (A2, B1, B2), cyclin-dependant kinases
(CDK2, CDK4), cell division cycle proteins (CDC20, CDC25A, CDC7),
E2F transcription factors (E2F2, E2F4), mitotic polo-like kinase
PLK1, checkpoint kinase CHEK1, mini-chromosome maintenance complex
components (MCM2, 3, 4, 5, 6, 7), and other cell cycle-associated
proteins.
The upregulation of the transforming growth factor-β
(TGF-β) and growth arrest and DNA damage-inducible factors (GADD45A
and GADD45G) also contributed to cell cycle deactivation.
Obviously, cell cycle deactivation paralleled inhibited DNA
replication. Twenty-three genes involved in replication were
downregulated: DNA polymerases [A1, A2, D1, D2, E, E2, E3 (except
of D4, which was upregulated)], replication factors (RFC2, 3, 4,
5), replication protein (RPA2), mini-chromosome maintenance complex
components (MCM2, 3, 4, 5, 6, 7), ligase 1, endonuclease FEN and
ribonucleaseH2 (RNASEH2A).
A specialized DNA damage response was initiated
through the activation of the p53 pathway due to the overexpression
of BCL2-associated X protein (BAX), damage-specific DNA-binding
protein (DDB2), sestrin1 (SESN1), and growth arrest and DNA
damage-inducible factors (GADD45A and GADD45G). DNA repair
processes were downregulated due primarily to the decrease in the
expression of specific repair polymerases and replication factors.
For instance, base excision repair downregulation was caused by a
low expression of polymerases (D1, D2, E, E2, E3), uracil-DNA
glycosylase (UNG), ligase 1 (LIG1) and endonuclease (FEN1); NER
deficiency was due to the same polymerases and ligase 1, and also
replication factors (RFC2, 3, 4, 5) and RPA2; MMR deactivation was
caused by a low level of MSH6, polymerases D1 and D2, LIG1, RPA2,
RFC2, 3, 4, 5, and exonuclease 1 (EXO1); and decreased homologous
recombination was caused by low expression levels of RAD54L, XRCC3,
polymerases D1 and D2, RPA2, Bloom syndrome, RecQ helicase-like
(BLM) and topoisomerase (TOP3A).
Gene expression profiling data were confirmed
through the qRT-PCR analysis of six genes that were changed in
MCF-7 cells after radiation treatment. Polymerases A, D and E were
involved in most of the biological processes that were affected in
MCF-7 cells after radiation exposure (Fig. 1C). Because GADD45G, cyclin A and
aurora B are involved in DNA damage responses, cell cycle and cell
division, their expression levels were of great interest to us as
well.
The members of the aurora kinase family have been
actively studied as mitotic progression targets in cancer studies.
Mutations associated with aurora gene amplification were reported
in human cancers (74). Tumor
development and progression due to aberrant chromosomal segregation
and aneuploidy is a common outcome of the misregulation of the
aurora B function (75).
Inhibition of aurora B during the fractionated
radiation treatment suppressed the repopulation of human cancer
cells (76). Similarly, 5 Gy
X-rays caused a significant down-regulation of aurora B in
drug-sensitive cell lines, which was correlated with slower mitotic
progression and the suppressed repopulation of the cells. Cyclin A
expression was also decreased, which may be associated with a lower
DNA replication status and suppressed cell cycle progression. In
addition, GADD45G, which is a member of growth arrest and
DNA-damage inducible genes, was overexpressed after both 0.5 and 5
Gy of irradiation. This indicates the existence of radiation stress
in the cells, which can result in cell cycle arrest, senescence and
apoptosis (77).
Significant downregulation of polymerases A, D and E
confirms the suppression of DNA replication and DNA repair
processes. Overall, gene expression profiling and qRT-PCR analysis
showed a strong response in MCF-7 cells to genotoxic agents, such
as ionizing radiation, allowing us to conclude that the parental
cells were radiation-sensitive. In contrast, the MCF-7/DOX cells
did not respond to X-rays on the gene expression level, which
signifies that they are radio-resistant. We assume that this
radio-resistance was gained in parallel with the acquired
resistance to doxorubicin.
All types of DNA repair involve the resynthesis of
DNA to replace the damaged strands. Therefore, DNA polymerases play
key roles not only in the DNA replication, but also in DNA repair
processes (53,56). Specifically, the high fidelity and
processivity of polymerases is crucial for faithful DNA replication
and the prevention of the accumulation of mutations. Indeed, the
efficient repair of DNA synthesis depends on the proper functioning
of DNA polymerases. Eukaryotic cells have 15 polymerases that
belong to several families (43,44).
Members of the B-family of polymerases include the major eukaryotic
DNA polymerases α, δ and ε (43,44,78).
Polymerases δ and ε harbor exonuclease activity (43). They take part in the replication
and processing of Okazaki fragments during replication processes
and are implicated in the repair of damaged DNA. As components of
recombination complexes, they are able to repair double-strand
breaks and participate in HR and NHEJ.
Some members of the X family of polymerases, such as
polymerase β, are required for base excision repair. Polymerase β
is not as accurate as replicative DNA polymerases because it lacks
proofreading capability. Polymerase β is a key player in base
excision repair; it is a mechanism that takes care of damaged bases
and single strand breaks (43,44,79).
In addition to replicative polymerases, there are a number of
translesion DNA polymerases, such as polymerase ι, which is another
member of X family of polymerases. These polymerases are involved
in bypassing DNA lesions that otherwise impede replication
polymerases (80).
A detailed analysis of DNA polymerases δ, ε, β and
ι demonstrated higher activity but lower fidelity of polymerases in
MCF-7/DOX-resistant cells in comparison to MCF-7 cells (Figs. 2 and 3). Low fidelity polymerases are thought
to be an evolutionary solution, allowing for the replication of
previously damaged DNA and avoiding apoptosis (81,82).
The ability to catalyze error-prone DNA synthesis belongs to DNA
polymerase ι, which was highly expressed in doxorubicin-resistant
cells and was not detected in parental MCF-7 cells (Figs. 2 and 3).
We also found higher exonuclease/proofreading
activity in MCF-7 cells than in MCF-7/DOX cells. In the current
study, we only analyzed four cellular DNA polymerases; therefore,
future analysis of other polymerases may shed more light on the
structure of chemo- and radiation-resistance. This study revealed
that drug-resistant MCF-7/DOX cells experienced more rapid DNA
repair, seemingly sacrificing the specificity and efficiency of
this process to gain higher survival potential. In the long run,
this may lead to an increased probability of the accumulation of
mutations and further the development of an even more pronounced
resistance phenotype.
In this study, we assessed the levels of IR-induced
apoptosis in MCF-7 and MCF-7/DOX cells. We noted that
drug-resistant cells were significantly less susceptible to
IR-induced apoptosis than their sensitive counterparts (Fig. 4). We assume that apoptosis delay in
MCF-7/DOX cells is a feature of resistance that could be developed
by cells after drug treatment. Indeed, it was previously suggested
that a drug-induced delay of apoptosis is considered a signifier
for pleiotropic drug resistance in tumor cells (83).
Seeking to explain this apparent discrepancy in the
levels of IR-induced apoptosis, we studied the formation and repair
of DNA damage, including DSBs, using a Comet assay and the
induction of γH2AX foci in MCF-7 and MCF-7/DOX cells after IR
exposure. Both methods showed a rapid increase in DNA damage 30 min
after radiation treatment in both cell types (Figs. 5 and 6). The level of damage was lowered 24 and
48 h after exposure, and increased efficiency was found in
MCF-7/DOX (Fig. 5). Importantly,
the background number of γH2AX foci in untreated MCF-7 cells
correlated with previous study data (40).
The γH2AX foci appear in the nuclei within 1 min of
irradiation and reach their maximum concentration by 30 min to 1 h.
Afterward, the number of γH2AX foci reduces due to the repair
processes (48,84). Our assay showed that non-resistant
MCF-7 cells are more radiosensitive (Fig. 6). MCF-7 cells were not able to
completely repair DNA damages after high dose (5 Gy) treatments,
and even after 48 h, the amount of residual foci was very high. In
contrast, drug-resistant MCF-7/DOX cells did not accumulate
substantial damage after low dose (0.5 Gy) treatments. The maximum
number of foci was observed 30 min after 5 Gy X-ray exposure and
was significantly lower than the number of foci detected in MCF-7
cells at this dose. Moreover, all DNA damage in the drug-resistant
MCF-7/DOX cells was repaired within 48 h (Fig. 6). Currently, it is thought that
γH2AX recruits proteins to repair DNA damage and γH2AX is
dephosporylated after the repair is complete (54). Therefore, we assume that the faster
foci disappear, the higher the DNA repair activity in the
cells.
DSBs can be repaired by two major processes:
homologous recombination (HR) and non-homologous end-joining (NHEJ)
(53,56,85).
HR allows cells to use the undamaged sister chromatid or the
homologous chromosome as a template for repair and is considered
error-free (50–53,56).
The error-free HR is controlled by the RAD51 protein (50,55,86,87).
RAD51 binds to single-stranded DNA and forms a nucleoprotein
filament that catalyses homology searching, strand pairing, and
strand exchange (86,88).
NHEJ is a fast, error-prone process of linking
broken DNA ends together without reference to accurate base pairing
(53,56). This DNA repair process is most
common in mammalian cells and requires a DNA-binding
component-heterodimer of KU70 and KU80 proteins (56,57).
A crucial signalling component for both pathways is the protein
kinase ATM. ATM coordinates DNA repair by phosphorylating the
downstream proteins involved in the actual repair (89). The ATM activity is increased 2.0 to
3.0-fold after exposure to IR (90). Such an increase in ATM activity is
thought to occur due to the autophosphorylation of serine 1981
(90).
Our study showed that MCF-7 and drug-resistant
MCF-7/DOX cell lines have different profiles of the aforementioned
DNA repair proteins. Although both cell lines exhibited elevated
levels of pATM, RAD51 and KU70 after exposure, the initial
pre-treatment levels of these proteins were different in MCF-7 and
MCF-7/DOX cells (Fig. 7). The
cytoplasmic localization of ATM in MCF-7/DOX cells was unusual, and
it is, at the moment, difficult to explain. The recent report that
ATM can be activated in cytoplasm by reactive oxygen species (ROS)
could possibly suggest that the cytoplasmic pATM in MCF-7/DOX cells
was activated by ROS generated during doxorubicin treatment
(91).
We found that MCF-7 cells express higher levels of
KU70, which is a key protein for NHEJ, while doxorubicin-resistant
MCF-7/DOX cell elevation of RAD51 could contribute to HR-mediated
DNA repair (Fig. 8). Why MCF-7 and
MCF-7/DOX cells display different preferences to error-free and
error-prone DSB repair strategies remains unknown, but it is
possible, that rapidly dividing MCF-7/DOX cells remain in S and M
phases more often, allowing them to use the present sister
chromatids for the repair of DNA damage. Nevertheless, we believe
that triggering certain steps of preferred repair pathways may
improve chemo- and radiotherapy responses. Our data are in
agreement with previous studies, showing higher DNA repair
potential in drug-resistant cells (20,21,23,27,92).
Further detailed studies are needed to determine
the cellular and molecular processes that are altered in resistant
cells that allow them to survive genotoxic treatments, such as
irradiation. This study may, therefore, provide a roadmap for the
analysis of the roles of DNA repair function and effectiveness, and
apoptosis in responses to radiation, chemotherapy and combinations
of both treatment modalities.
Abbreviations:
|
IR
|
ionizing radiation;
|
|
DSB
|
double strand DNA break;
|
|
HR
|
homologous recombination;
|
|
NHEJ
|
non-homologous end-joining
|
Acknowledgements
We are grateful to Rocio
Rodriguez-Juarez, Jody Filkowski and Dipankar Goyal for technical
assistance. This study was supported by Alberta Cancer Research
Institute grant to O. Kovalchuk. L. Luzhna is a recipient of the
Alberta Cancer Research Institute Graduate Scholarship.
References
|
1.
|
Guarneri V and Conte PF: The curability of
breast cancer and the treatment of advanced disease. Eur J Nucl Med
Mol Imaging. 31(Suppl 1): S149–S161. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Lehnert M: Clinical multidrug resistance
in cancer: a multifactorial problem. Eur J Cancer. 32A:912–920.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Szakacs G, Paterson JK, Ludwig JA,
Booth-Genthe C and Gottesman MM: Targeting multidrug resistance in
cancer. Nat Rev Drug Discov. 5:219–234. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Fojo T: Multiple paths to a drug
resistance phenotype: mutations, translocations, deletions and
amplification of coding genes or promoter regions, epigenetic
changes and microRNAs. Drug Resist Updat. 10:59–67. 2007.
View Article : Google Scholar
|
|
5.
|
O’Driscoll L and Clynes M: Molecular
markers of multiple drug resistance in breast cancer. Chemotherapy.
52:125–129. 2006.PubMed/NCBI
|
|
6.
|
Gottesman MM: Mechanisms of cancer drug
resistance. Annu Rev Med. 53:615–627. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Pommier Y, Sordet O, Antony S, Hayward RL
and Kohn KW: Apoptosis defects and chemotherapy resistance:
molecular interaction maps and networks. Oncogene. 23:2934–2949.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Stavrovskaya AA: Cellular mechanisms of
multidrug resistance of tumor cells. Biochemistry (Mosc).
65:95–106. 2000.PubMed/NCBI
|
|
9.
|
Karran P: Mechanisms of tolerance to DNA
damaging therapeutic drugs. Carcinogenesis. 22:1931–1937. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Rixe O and Fojo T: Is cell death a
critical end point for anti-cancer therapies or is cytostasis
sufficient? Clin Cancer Res. 13:7280–7287. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Hickman JA: Apoptosis and chemotherapy
resistance. Eur J Cancer. 32A:921–926. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Gottesman MM and Ling V: The molecular
basis of multidrug resistance in cancer: the early years of
P-glycoprotein research. FEBS Lett. 580:998–1009. 2006.PubMed/NCBI
|
|
13.
|
Modok S, Mellor HR and Callaghan R:
Modulation of multidrug resistance efflux pump activity to overcome
chemoresistance in cancer. Curr Opin Pharmacol. 6:350–354. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Coley HM: Mechanisms and strategies to
overcome chemotherapy resistance in metastatic breast cancer.
Cancer Treat Rev. 34:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Gonzalez-Angulo AM, Morales-Vasquez F and
Hortobagyi GN: Overview of resistance to systemic therapy in
patients with breast cancer. Adv Exp Med Biol. 608:1–22. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Petrelli A and Giordano S: From single- to
multi-target drugs in cancer therapy: when aspecificity becomes an
advantage. Curr Med Chem. 15:422–432. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Chekhun VF, Lukyanova NY, Kovalchuk O,
Tryndyak VP and Pogribny IP: Epigenetic profiling of
multidrug-resistant human MCF-7 breast adenocarcinoma cells reveals
novel hyper- and hypomethylated targets. Mol Cancer Ther.
6:1089–1098. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Dean-Colomb W and Esteva FJ: Emerging
agents in the treatment of anthracycline- and taxane-refractory
metastatic breast cancer. Semin Oncol. 35:S31–S40. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Ozols RF, Masuda H and Hamilton TC:
Mechanisms of cross-resistance between radiation and antineoplastic
drugs. NCI Monogr. 159–165. 1988.PubMed/NCBI
|
|
20.
|
Shimm DS, Olson S and Hill AB: Radiation
resistance in a multidrug resistant human T-cell leukemia line. Int
J Radiat Oncol Biol Phys. 15:931–936. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Belli JA: Interaction between radiation
and drug damage in mammalian cells. IV. Radiation response of
adriamycin-resistant V79 cells. Radiat Res. 119:88–100. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Lehnert S, Greene D and Batist G:
Radiation response of drug-resistant variants of a human breast
cancer cell line. Radiat Res. 118:568–580. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Lehnert S, Greene D and Batist G:
Radiation response of drug-resistant variants of a human breast
cancer cell line: the effect of glutathione depletion. Radiat Res.
124:208–215. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Alaoui-Jamali MA, Batist G and Lehnert S:
Radiation-induced damage to DNA in drug- and radiation-resistant
sublines of a human breast cancer cell line. Radiat Res. 129:37–42.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Miller PR, Hill AB, Slovak ML and Shimm
DS: Radiation resistance in a doxorubicin-resistant human
fibrosarcoma cell line. Am J Clin Oncol. 15:216–221. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Zhang Y, Sweet KM, Sognier MA and Belli
JA: Interaction between radiation and drug damage in mammalian
cells. VI. Radiation and doxorubicin age-response function of
doxorubicin-sensitive and -resistant Chinese hamster cells. Radiat
Res. 132:105–111. 1992. View Article : Google Scholar
|
|
27.
|
Lehnert S, Vestergaard J, Batist G and
Aloui-Jamali MA: Radiation resistance in a melphalan-resistant
subline of a rat mammary carcinoma. Radiat Res. 139:232–239. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Liang K, Lu Y, Jin W, Ang KK, Milas L and
Fan Z: Sensitization of breast cancer cells to radiation by
trastuzumab. Mol Cancer Ther. 2:1113–1120. 2003.PubMed/NCBI
|
|
29.
|
Koukourakis MI, Koukouraki S,
Giatromanolaki A, et al: Liposomal doxorubicin and conventionally
fractionated radiotherapy in the treatment of locally advanced
non-small-cell lung cancer and head and neck cancer. J Clin Oncol.
17:3512–3521. 1999.PubMed/NCBI
|
|
30.
|
Gewirtz DA: Growth arrest and cell death
in the breast tumor cell in response to ionizing radiation and
chemotherapeutic agents which induce DNA damage. Breast Cancer Res
Treat. 62:223–235. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Reinhold WC, Kouros-Mehr H, Kohn KW, et
al: Apoptotic susceptibility of cancer cells selected for
camptothecin resistance: gene expression profiling, functional
analysis, and molecular interaction mapping. Cancer Res.
63:1000–1011. 2003.
|
|
32.
|
Kovalchuk O, Filkowski J, Meservy J, et
al: Involvement of microRNA-451 in resistance of the MCF-7 breast
cancer cells to chemotherapeutic drug doxorubicin. Mol Cancer Ther.
7:2152–2159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009.PubMed/NCBI
|
|
34.
|
Huang da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009.PubMed/NCBI
|
|
35.
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Tryndyak VP, Kovalchuk O and Pogribny IP:
Loss of DNA methylation and histone H4 lysine 20 trimethylation in
human breast cancer cells is associated with aberrant expression of
DNA methyltransferase 1, Suv4-20h2 histone methyltransferase and
methyl-binding proteins. Cancer Biol Ther. 5:65–70. 2006.
View Article : Google Scholar
|
|
37.
|
Gening LV, Petrochenkov AN, Reshetnyak AB,
Andreeva LE and Tarantul VZ: DNA polymerase iota-like activity in
crude cell extracts of different mouse organs. Biochemistry (Mosc).
69:435–440. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Olive PL and Banath JP: The comet assay: a
method to measure DNA damage in individual cells. Nat Protoc.
1:23–29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Tice RR and Strauss GH: The single cell
gel electrophoresis/comet assay: a potential tool for detecting
radiation-induced DNA damage in humans. Stem Cells. 13(Suppl 1):
207–214. 1995.PubMed/NCBI
|
|
40.
|
Sedelnikova OA and Bonner WM: GammaH2AX in
cancer cells: a potential biomarker for cancer diagnostics,
prediction and recurrence. Cell Cycle. 5:2909–2913. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Carpenter AE, Jones TR, Lamprecht MR, et
al: CellProfiler: image analysis software for identifying and
quantifying cell phenotypes. Genome Biol. 7:R1002006. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Lamprecht MR, Sabatini DM and Carpenter
AE: CellProfiler: free, versatile software for automated biological
image analysis. Biotechniques. 42:71–75. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Bebenek K and Kunkel TA: Functions of DNA
polymerases. Adv Protein Chem. 69:137–165. 2004. View Article : Google Scholar
|
|
44.
|
Shcherbakova PV, Bebenek K and Kunkel TA:
Functions of eukaryotic DNA polymerases. Sci Aging Knowledge
Environ 2003. RE32003.PubMed/NCBI
|
|
45.
|
Vermes I, Haanen C, Steffens-Nakken H and
Reutelingsperger C: A novel assay for apoptosis. Flow cytometric
detection of phosphatidylserine expression on early apoptotic cells
using fluorescein labelled Annexin V. J Immunol Methods. 184:39–51.
1995. View Article : Google Scholar
|
|
46.
|
Little JB: Radiation carcinogenesis.
Carcinogenesis. 21:397–404. 2000. View Article : Google Scholar
|
|
47.
|
Huang L, Snyder AR and Morgan WF:
Radiation-induced genomic instability and its implications for
radiation carcinogenesis. Oncogene. 22:5848–5854. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Bonner WM, Redon CE, Dickey JS, et al:
GammaH2AX and cancer. Nat Rev Cancer. 8:957–967. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Olive PL: DNA damage and repair in
individual cells: applications of the comet assay in radiobiology.
Int J Radiat Biol. 75:395–405. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
West SC: Molecular views of recombination
proteins and their control. Nat Rev Mol Cell Biol. 4:435–445. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
McGlynn P and Lloyd RG: Recombinational
repair and restart of damaged replication forks. Nat Rev Mol Cell
Biol. 3:859–870. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Helleday T: Pathways for mitotic
homologous recombination in mammalian cells. Mutat Res.
532:103–115. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Hoeijmakers JH: DNA repair mechanisms.
Maturitas. 38:17–23. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Chowdhury D, Keogh MC, Ishii H, Peterson
CL, Buratowski S and Lieberman J: gamma-H2AX dephosphorylation by
protein phosphatase 2A facilitates DNA double-strand break repair.
Mol Cell. 20:801–809. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Lundin C, Schultz N, Arnaudeau C, Mohindra
A, Hansen LT and Helleday T: RAD51 is involved in repair of damage
associated with DNA replication in mammalian cells. J Mol Biol.
328:521–535. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Hoeijmakers JH: Genome maintenance
mechanisms for preventing cancer. Nature. 411:366–374. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Jin S and Weaver DT: Double-strand break
repair by Ku70 requires heterodimerization with Ku80 and DNA
binding functions. EMBO J. 16:6874–6885. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Fornari FA, Randolph JK, Yalowich JC,
Ritke MK and Gewirtz DA: Interference by doxorubicin with DNA
unwinding in MCF-7 breast tumor cells. Mol Pharmacol. 45:649–656.
1994.PubMed/NCBI
|
|
59.
|
Fortune JM and Osheroff N: Topoisomerase
II as a target for anticancer drugs: when enzymes stop being nice.
Prog Nucleic Acid Res Mol Biol. 64:221–253. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Khimani AH, Mhashilkar AM, Mikulskis A, et
al: Housekeeping genes in cancer: normalization of array data.
Biotechniques. 38:739–745. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Rubie C, Kempf K, Hans J, et al:
Housekeeping gene variability in normal and cancerous colorectal,
pancreatic, esophageal, gastric and hepatic tissues. Mol Cell
Probes. 19:101–109. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Barnard GF, Staniunas RJ, Bao S, et al:
Increased expression of human ribosomal phosphoprotein P0 messenger
RNA in hepatocellular carcinoma and colon carcinoma. Cancer Res.
52:3067–3072. 1992.PubMed/NCBI
|
|
63.
|
Henry JL, Coggin DL and King CR:
High-level expression of the ribosomal protein L19 in human breast
tumors that overexpress erbB-2. Cancer Res. 53:1403–1408.
1993.PubMed/NCBI
|
|
64.
|
Lukyanova NY, Rusetskya NV, Tregubova NA
and Chekhun VF: Molecular profile and cell cycle in MCF-7 cells
resistant to cisplatin and doxorubicin. Exp Oncol. 31:87–91.
2009.PubMed/NCBI
|
|
65.
|
Gyorffy B, Serra V, Jurchott K, et al:
Prediction of doxorubicin sensitivity in breast tumors based on
gene expression profiles of drug-resistant cell lines correlates
with patient survival. Oncogene. 24:7542–7551. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Huang J, Tan PH, Thiyagarajan J and Bay
BH: Prognostic significance of glutathione S-transferase-pi in
invasive breast cancer. Mod Pathol. 16:558–565. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
67.
|
Saleh EM, El-Awady RA, Abdel Alim MA and
Abdel Wahab AH: Altered expression of proliferation-inducing and
proliferation-inhibiting genes might contribute to acquired
doxorubicin resistance in breast cancer cells. Cell Biochem
Biophys. 55:95–105. 2009. View Article : Google Scholar
|
|
68.
|
Miyara H, Hida T, Nishida K, et al:
Modification of chemoradiosensitivity of a human lung cancer cell
line by introduction of the glutathione S-transferase pi gene. Jpn
J Clin Oncol. 26:1–5. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
69.
|
Harper ME, Antoniou A, Villalobos-Menuey
E, et al: Characterization of a novel metabolic strategy used by
drug-resistant tumor cells. FASEB J. 16:1550–1557. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
70.
|
Diehn M, Cho RW, Lobo NA, et al:
Association of reactive oxygen species levels and radioresistance
in cancer stem cells. Nature. 458:780–783. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71.
|
Kang JH, Song KH, Jeong KC, et al:
Involvement of Cox-2 in the metastatic potential of
chemotherapy-resistant breast cancer cells. BMC Cancer. 11:3342011.
View Article : Google Scholar : PubMed/NCBI
|
|
72.
|
Mihich E: On the immunomodulating effects
of anti-cancer drugs and their therapeutic exploitation. Jpn J Clin
Oncol. 30:469–471. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
73.
|
Ehrke MJ, Verstovsek S, Zaleskis G, et al:
Specific anti-EL4-lymphoma immunity in mice cured 2 years earlier
with doxorubicin and interleukin-2. Cancer Immunol Immunother.
42:221–230. 1996.PubMed/NCBI
|
|
74.
|
Cahill DP, Lengauer C, Yu J, et al:
Mutations of mitotic checkpoint genes in human cancers. Nature.
392:300–303. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
75.
|
Nguyen HG, Makitalo M, Yang D, Chinnappan
D, St Hilaire C and Ravid K: Deregulated Aurora-B induced
tetraploidy promotes tumorigenesis. FASEB J. 23:2741–2748. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
76.
|
Sak A, Stuschke M, Groneberg M, Kubler D,
Pottgen C and Eberhardt WE: Inhibiting the aurora B kinase potently
suppresses repopulation during fractionated irradiation of human
lung cancer cell lines. Int J Radiat Oncol Biol Phys. 84:492–499.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
77.
|
Liebermann DA, Tront JS, Sha X, Mukherjee
K, Mohamed-Hadley A and Hoffman B: Gadd45 stress sensors in
malignancy and leukemia. Crit Rev Oncog. 16:129–140. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
78.
|
Kunkel TA and Burgers PM: Dividing the
workload at a eukaryotic replication fork. Trends Cell Biol.
18:521–527. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79.
|
Matsuda T, van de Berg BJ, Bebenek K,
Osheroff WP, Wilson SH and Kunkel TA: The base substitution
fidelity of DNA polymerase beta-dependent single nucleotide base
excision repair. J Biol Chem. 278:25947–25951. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
80.
|
Kunkel TA, Pavlov YI and Bebenek K:
Functions of human DNA polymerases eta, kappa and iota suggested by
their properties, including fidelity with undamaged DNA templates.
DNA Repair (Amst). 2:135–149. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
81.
|
Beard WA, Shock DD, Vande Berg BJ and
Wilson SH: Efficiency of correct nucleotide insertion governs DNA
polymerase fidelity. J Biol Chem. 277:47393–47398. 2002. View Article : Google Scholar
|
|
82.
|
Goodman MF and Tippin B: Sloppier copier
DNA polymerases involved in genome repair. Curr Opin Genet Dev.
10:162–168. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
83.
|
Taylor ST, Hickman JA and Dive C: Survival
signals within the tumour microenvironment suppress drug-induced
apoptosis: lessons learned from B lymphomas. Endocr Relat Cancer.
6:21–23. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
84.
|
Banath JP, Macphail SH and Olive PL:
Radiation sensitivity, H2AX phosphorylation, and kinetics of repair
of DNA strand breaks in irradiated cervical cancer cell lines.
Cancer Res. 64:7144–7149. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
85.
|
Jeggo P and Lobrich M: Radiation-induced
DNA damage responses. Radiat Prot Dosimetry. 122:124–127. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
86.
|
Baumann P and West SC: Role of the human
RAD51 protein in homologous recombination and double-stranded-break
repair. Trends Biochem Sci. 23:247–251. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
87.
|
Dudas A and Chovanec M: DNA double-strand
break repair by homologous recombination. Mutat Res. 566:131–167.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
88.
|
Benson FE, Baumann P and West SC:
Synergistic actions of Rad51 and Rad52 in recombination and DNA
repair. Nature. 391:401–404. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
89.
|
Goodarzi AA, Block WD and Lees-Miller SP:
The role of ATM and ATR in DNA damage-induced cell cycle control.
Prog Cell Cycle Res. 5:393–411. 2003.PubMed/NCBI
|
|
90.
|
Goodarzi AA, Jonnalagadda JC, Douglas P,
et al: Auto-phosphorylation of ataxia-telangiectasia mutated is
regulated by protein phosphatase 2A. EMBO J. 23:4451–4461. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
91.
|
Alexander A, Cai SL, Kim J, et al: ATM
signals to TSC2 in the cytoplasm to regulate mTORC1 in response to
ROS. Proc Natl Acad Sci USA. 107:4153–4158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
92.
|
Harris AL: DNA repair: relationship to
drug and radiation resistance, metastasis and growth factors. Int J
Radiat Biol Relat Stud Phys Chem Med. 48:675–690. 1985. View Article : Google Scholar : PubMed/NCBI
|