Introduction
Osteosarcoma (OS) is the most common primary
malignant bone cancer in children and adolescents and is typically
observed in individuals between the ages of 10 and 25 years
(average age, 18 years) (1). In
most cases, OS originates from the metaphysis of long bones and is
mostly found in areas of rapid growth in children, i.e., the knees
and shoulders and the long bones of the arms and legs (2). OS is highly aggressive and primarily
metastasizes to the lungs (3). The
10-year disease-free survival rate is ∼60 and 30% in patients with
localized disease and patients with metastasis at diagnosis,
respectively, with the current use of adjuvant and neoadjuvant
chemotherapy involving doxorubicin, methotrexate, cisplatin and
vincristine (4,5). Therefore, new therapeutic strategies
need to be evaluated to improve OS survival, especially for
patients refractory to current chemotherapy regimens.
Various chemotherapeutic drugs are reported to
induce apoptosis in OS cells (6–8). Two
apoptotic pathways have been proposed: one pathway involves cell
death receptors (TNF-R or Fas) in the cell membrane, where binding
of ligands to the receptors activates the caspase-8 and, in turn,
activates downstream effector caspases (caspase-3 and -7) (9). The other pathway is associated with
mitochondrial alterations such as decrease in mitochondrial
membrane potential and release of cytochrome c from the
mitochondrial membrane, followed by activation of effector caspases
via the activation of caspase-9 (10). In both pathways, the activation of
effector caspases is known to be suppressed by X-linked
inhibitor-of-apoptosis protein (XIAP) (11). During the apoptotic process, the
apoptotic mechanism can be classified as a caspase-dependent or
caspase-independent pathway, depending on the involvement of
caspase activation (12). The
release of apoptosis-inducing factor (AIF) from the mitochondrial
membrane is believed to be a specific marker for the
caspase-independent pathway (13).
AIF contains a nuclear localization signal and translocates to the
nucleus where it participates in chromatin condensation and
large-scale DNA fragmentation (13).
During the last decade, autophagy has been gradually
recognized by another type of cell death machinery in several
cellular systems (14,15). Autophagy is a cellular process
whose primary function is to degrade long-lived proteins and
recycle cellular components (16).
Autophagy can be induced by various stimuli, including starvation
(17), cytokines (18), caspase inhibition (19) and chemical reagents such as
rapamycin (20). In mammalian
cells, autophagy has been implicated in cellular processes as
varied as cell survival (17),
death (19,20), pathogen clearance (18) and antigen presentation (21) and has also been associated with
pathological processes such as cancer progression and
neurodegenerative diseases (22,23).
Paclitaxel (Taxol®), a polyoxygenated
naturally occurring diterpenoid isolated from the bark of the
Pacific yew tree (Taxus brevifolia), has been considered one
of the most important cancer chemotherapeutic drugs (24). PCX has a potent ability to
stabilize microtubules. The anticancer activity of this drug is
ascribed to its unique mechanism of action, i.e., causing mitotic
arrest in cancer cells, which leads to apoptosis through inhibition
of microtubule depolymerization (25). However, the current protocols for
OS treatment do not incorporate this agent (26). The purpose of this study was to
evaluate the induction of cell death in OS cells after exposure to
PCX, to identify the cell death mechanism(s) activated by PCX and
to investigate whether autophagy is associated with PCX-induced
apoptosis.
Materials and methods
Reagents and antibodies
Paclitaxel (PCX) was obtained from Bristol-Myers
Squibb (New York, NY, USA). 3-methyladenine (3-MA),
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
propidium iodide (PI) and Rhodamine 123 and anti-actin antibody
were obtained from Sigma-Aldrich (St. Louis, MO, USA). Antibody for
microtubule associated protein 1 light chain 3 (LC3) was purchased
from Novus Biologicals (Littleton, CO, USA). Antibodies for
cytochrome c, Hsp60, XIAP and PARP were obtained from Santa Cruz
Biotech (Santa Cruz, CA, USA). Active-form specific antibody for
caspase-3 was from Cell Signaling Tech (Beverly, MA, USA). Rabbit
and mouse IgG-conjugated with horse-radish peroxidase were from
Amersham Pharmacia Biotech (Piscataway, NJ, USA).
Cell culture and drug treatment
Saos-2 cells (human osteosarcoma cell line; American
Type Culture Collection, Rockville, MD, USA) were cultured in
Dulbecco’s modified Eagle’s medium (DMEM) nutrient mixture F-12 HAM
(Sigma, St. Louis, MO, USA) containing 10% fetal bovine serum
(Invitrogen, Carlsbad, CA, USA) and 1.2 g/l sodium bicarbonate
supplemented with 10 μg/ml penicillin-streptomycin
(Invitrogen). The cells were incubated in a humidified incubator at
37°C with 5% CO2 and were exposed to PCX or 3-MC when
the confluency reached 30%.
MTT cell viability assay
Cells were seeded in 12-well plates at a density of
5×105 cells per well. After treatment at an appropriate
time (48, 72, or 96 h), the culture medium was removed and replaced
with a medium containing 0.5 mg of MTT dissolved in PBS (pH 7.2);
after 4 h, the formed crystals were dissolved with dissolved with
200 ml of DMSO. The intensity of the color in each well was
measured at a wavelength of 490 nm using a microplate reader
(BioTek EL-312e, VT, USA).
Cell cycle analysis
The cells were harvested, fixed with 95% ethanol for
24 h, incubated with 0.05 mg/ml PI and 1 μg/ml RNase A at
37°C for 30 min and analyzed by flow cytometry, using an Epics XL
and analysis software (EXPO32™; Beckman Coulter, MI, USA). The
cells belonging to the sub-G1 population were considered to be
apoptotic cells; the percentage of each phase of the cell cycle was
determined.
Annexin V cell death assay
The cells were stained using the AnnexinV-FITC
Apoptosis Detection kit (BD Biosciences, NJ, USA) according to the
manufacturer’s protocol. Stained cells were analyzed by flow
cytometry.
Western blot analysis
Whole-cell lysates were prepared by incubating cell
pellets in lysis buffer [30 mM NaCl, 0.5% Triton X-100, 50 mM
Tris-HCl (pH 7.4), 1 mM Na3VO4, 25 mM NaF, 10
mM Na4P2O7] for 30 min on ice.
After the insoluble fractions were removed by centrifugation at
14,000 rpm at 4°C for 30 min, the supernatants were collected and
protein concentration was determined with a BCA protein assay kit
(Pierce Biotechnology, Woburn, MA, USA). The same amounts of
proteins (∼30 μg) were subjected to SDS-PAGE and transferred
onto a nitrocellulose membrane. The membranes were incubated for 1
h at room temperature (RT) with a primary antibody in Tris-buffered
saline containing 0.05% Tween-20 [TBS-T (pH 7.4)] in the presence
of 5% non-fat dry milk. After the membranes were washed in TBS-T,
secondary antibody reactions were performed with an appropriate
source of antibody conjugated with horseradish peroxidase. The
signals were detected with an enhanced chemiluminescence detection
kit (Amersham Pharmacia Biotech) in the LAS-3000 detector
(Fujifilm, Japan). Immunoblotting for β-actin was performed in each
experiment as an internal control.
Preparation of mitochondrial
fractions
The cells (5×107) were washed in TD
buffer (135 mM NaCl, 5 mM KCl, 25 mM Tris-Cl, pH 7.6) and allowed
to swell for 15 min in ice-cold hypotonic CaRSB buffer [10 mM NaCl,
1.5 mM CaCl2, 10 mM Tris-Cl (pH 7.5) protease
inhibitors]. Cells were Dounce-homogenized with 30 strokes and
mitochondria stabilization buffer (210 mM mannitol, 70 mM sucrose,
5 mM EDTA, 5 mM Tris, pH 7.6) was added. After removing nuclear
contaminants (690 × g for 15 min), the supernatant was centrifuged
at 20,800 × g for 15 min. Finally, the pellet (mitochondria) was
directly diluted with lysis buffer [5 M NaCl, 1 M Tris-Cl (pH 7.6),
5% Triton X-100, protease inhibitors] and the mitochondria (pellet)
and supernatant (cytosol) were applied for protein analysis.
Caspase-3 activity assay
A fluorometric assay kit (Clontech, CA, USA), which
contains fluorogenic substrate specific for caspase-3 immobilized
in the wells, was used to evaluate enzyme activity. Ten micrograms
of the extracted proteins in homogenization buffer (50 mM Tris-HCl,
150 mM NaCl, 10% glycerin and 1% Triton X-100) were added to the
wells. The plate was incubated in the fluorescence plate reader at
37°C for 3 h and fluorescence was read every 10 min. The activity
was determined by fluorometric detection (excitation, 380 nm;
emission, 460 nm) and the negative control (blank, without sample)
was subtracted from all the samples. Results at 2 h were selected,
as the manufacturer suggested. Baseline values of negative controls
and samples with specific inhibitors did not increase during the
2-h interval.
Measurement of mitochondrial membrane
potential (MMP)
The cells (5×105) were incubated with 1
μM JC-1 dye at 37°C for 15 min, washed and resuspended with
PBS and then the fluorescence [red (585/590 nm); green (510/527
nm)] was measured by flow cytometer.
Immunocytochemistry
Harvested cells were attached on the slide glass by
cytospin centrifugation. The cells were fixed with 4% PFA, washed
with PBS and incubated with 0.2% Triton X-100. Then, the cells were
incubated with the appropriate primary antibody in 1% bovine serum
albumin at RT. For secondary antibody reaction, the cells were
incubated with an appropriate fluorescence-conjugated secondary
antibody at RT. For counterstaining of the nucleus, if required,
cells were incubated with PI (50 μg/ml) at RT. Finally,
cells were mounted and observed under a confocal microscope
(LSM510, Carl Zeiss, Germany).
Statistical analyses
Data were expressed as the mean ± SD of three or
four separate experiments. Where appropriate, data were subjected
analysis of variance (ANOVA) followed by Duncan’s post hoc
test. Means were considered significantly different at
p<0.05.
Results
PCX can induce cell death in osteosarcoma
Saos-2 cells
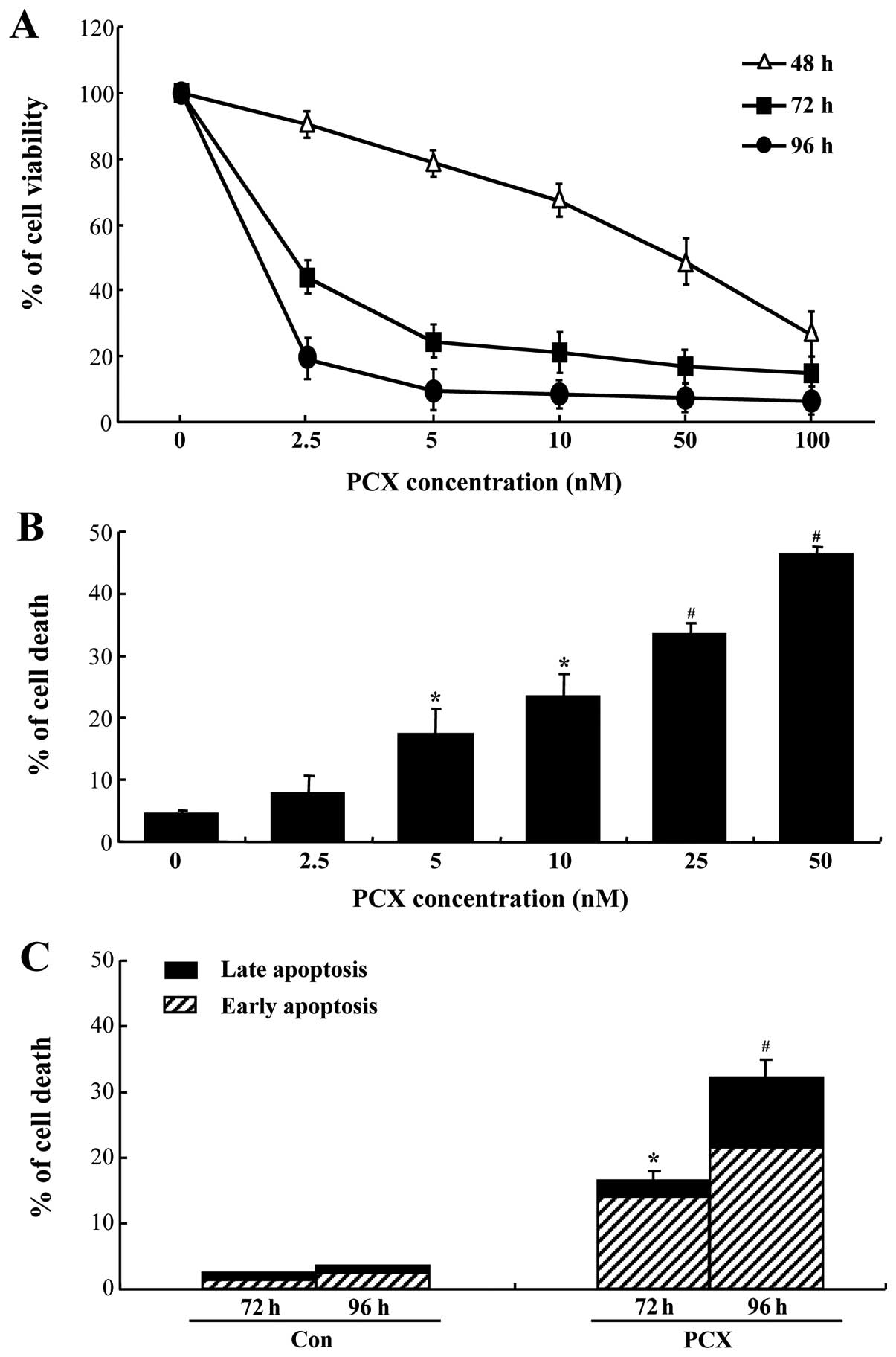
To investigate whether PCX has a cytotoxic effect on
osteosarcoma Saos-2 cells, the cells were exposed to various
concentrations of PCX (2.5–100 nM) for up to 96 h. Cell viability
decreased markedly by 2.5–5 nM PCX (Fig. 1A). The dose-response experiments
showed that 5 nM of PCX is considered to be optimal for the
time-course experiments in this study.
PCX-induced cell death exhibits apoptotic
characteristics in Saos-2 cells
PCX-induced Saos-2 cell death was characterized by
flow cytometry (Fig. 1B). Cell
death was increased significantly in a dose-response manner
(Fig. 3B), which confirmed that 5
nM of PCX is adequate for the time-course experiments. Annexin V
staining was used to identify the type of cell death evoked by PCX
(Fig. 1C). PCX significantly
increased proportion of apoptotic cells in Saos-2 cells (Fig. 1C).
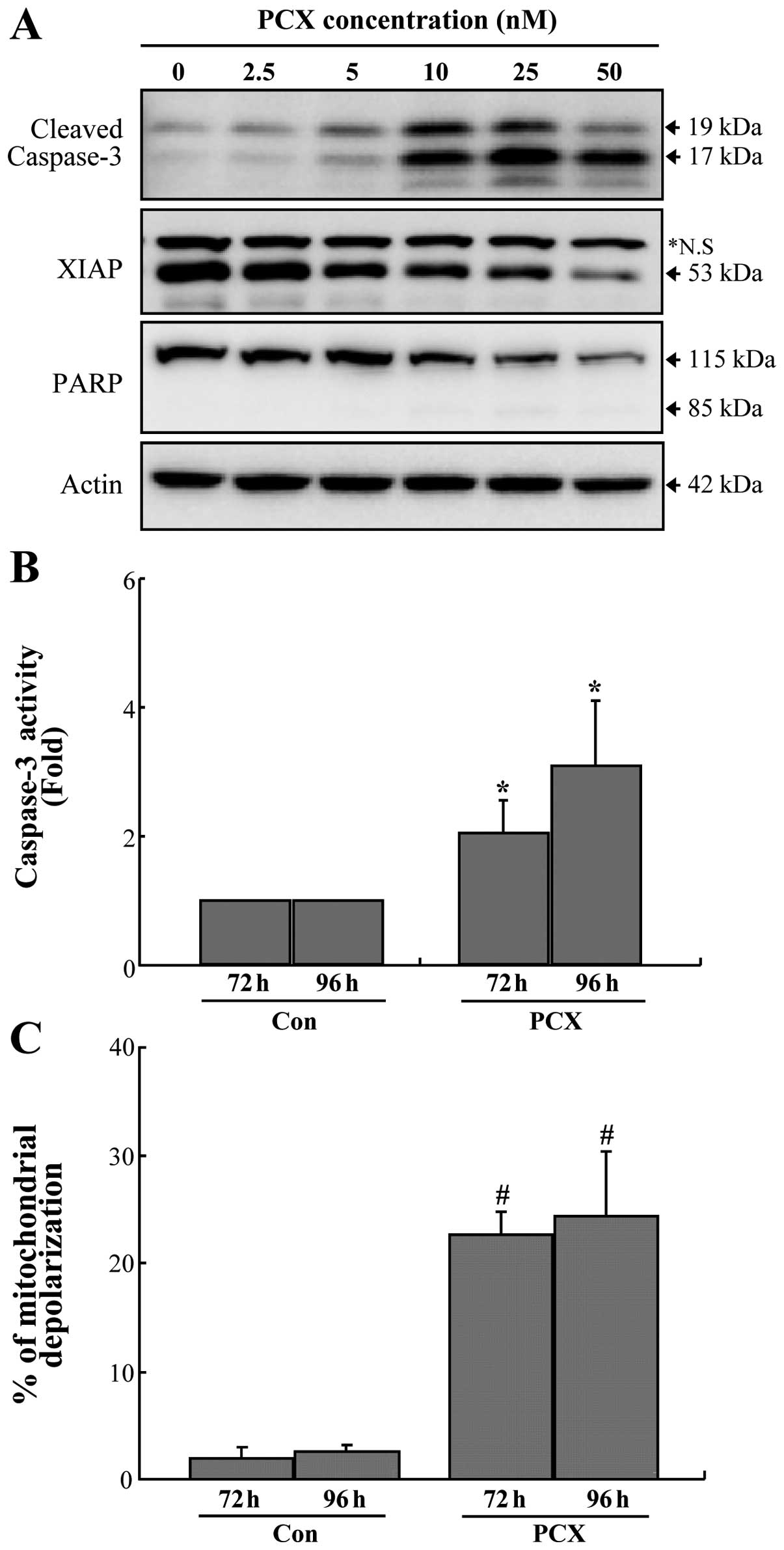
Caspase-3 activation and decrease of
mitochondrial membrane potential are involved in PCX-induced
apoptosis in Saos-2 cells
The involvement of caspase-3 activation in
PCX-induced apoptosis was examined in Saos-2 cells. Exposure to PCX
provoked an increase of caspase-3 cleavage (activation), which
reached a post-exposure maximum level with 10 nM concentration at
48 h (Fig. 2A). Caspase-3
activation was accompanied not only by decrease in XIAP protein
level but also by degradation of PARP protein (Fig. 2A). In addition, it has been
observed that caspase-3 activities gradually increase during the
duration of PCX treatment (Fig.
2B). In the present study, a decrease of MMP was observed in
PCX-treated cells. The rate of mitochondrial depolarization showed
a significant increase in PCX-treated cells compared to control
(Fig. 2C).
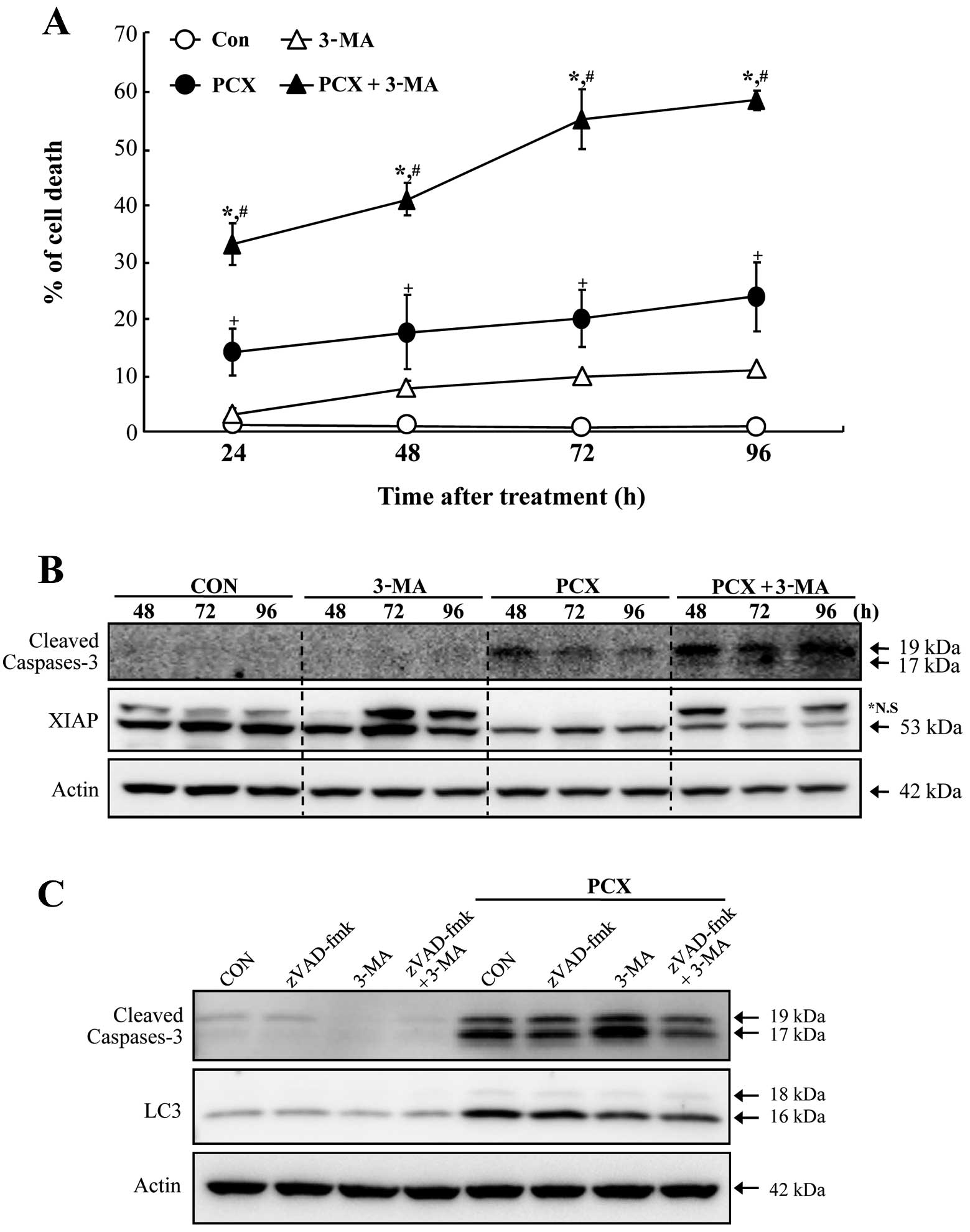
Enhancement of PCX-induced apoptosis by
the pretreatment of an autophagy inhibiting molecule
In order to investigate whether autophagic cell
death is associated with PCX-induced cell death in Saos-2 cells, a
specific inhibitor for autophagy (autophagosome formation;
3-methyladenine, 3-MA) was employed in the experiment of this
study. The pretreatment of 3-MA in Soas-2 cells resulted in a
significant enhancement of apoptosis provoked by exposure to PCX
(Fig. 3A). Although 3-MA alone
also induced cell death (≤10%), it appeared to be of minimal value
considering the capability of 3-MA in increasing cell death induced
by PCX. Augmentation of PCX-induced cell death by 3-MA revealed to
be involved with caspase-3 activation accompanying by decrease of
XIAP proteins (Fig. 3B). In
addition, pretreatment with 3-MA or z-VAD-fmk (a pancaspase
inhibitor) suppressed the expression of microtubule-associated
protein light chain 3 (LC3; specifically LC3-II), an early marker
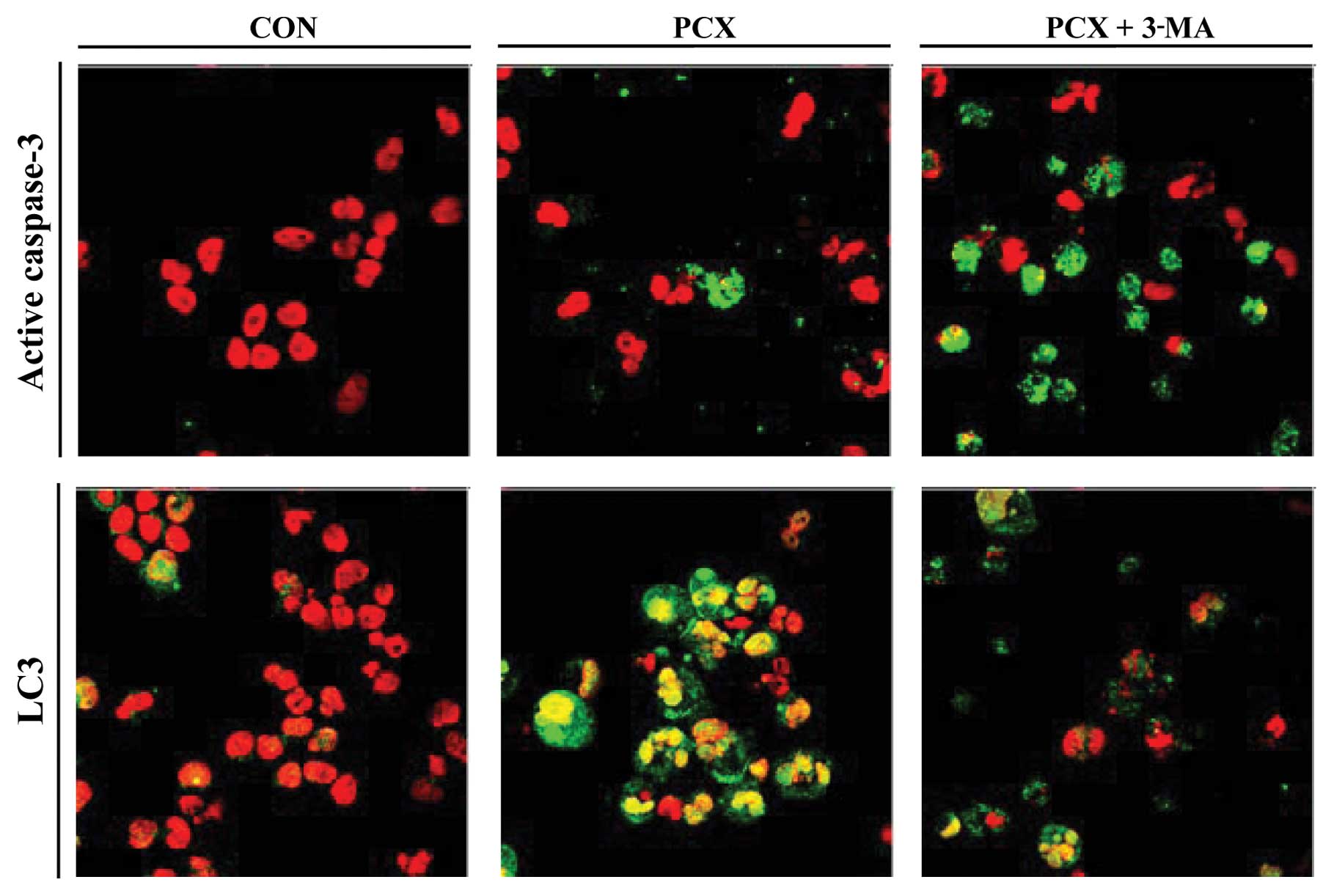
of autophagosome formation, in PCX-treated cells (Fig. 3C). Immunocytochemical staining of
LC3 confirmed that autophagic activity increased in the PCX-treated
cells, but was suppressed in the presence of 3-MA (Fig. 4). Concomitant with appearance of
LC3 proteins, immunoreactive cells for active caspase-3 protein
were observed in PCX-treated cells (Fig. 4). However, the pretreatment of 3-MA
abolished the LC3 immunoreactivity (Fig. 4) and increased caspase-3-positive
cells in PCX-treated cells (Fig.
4).
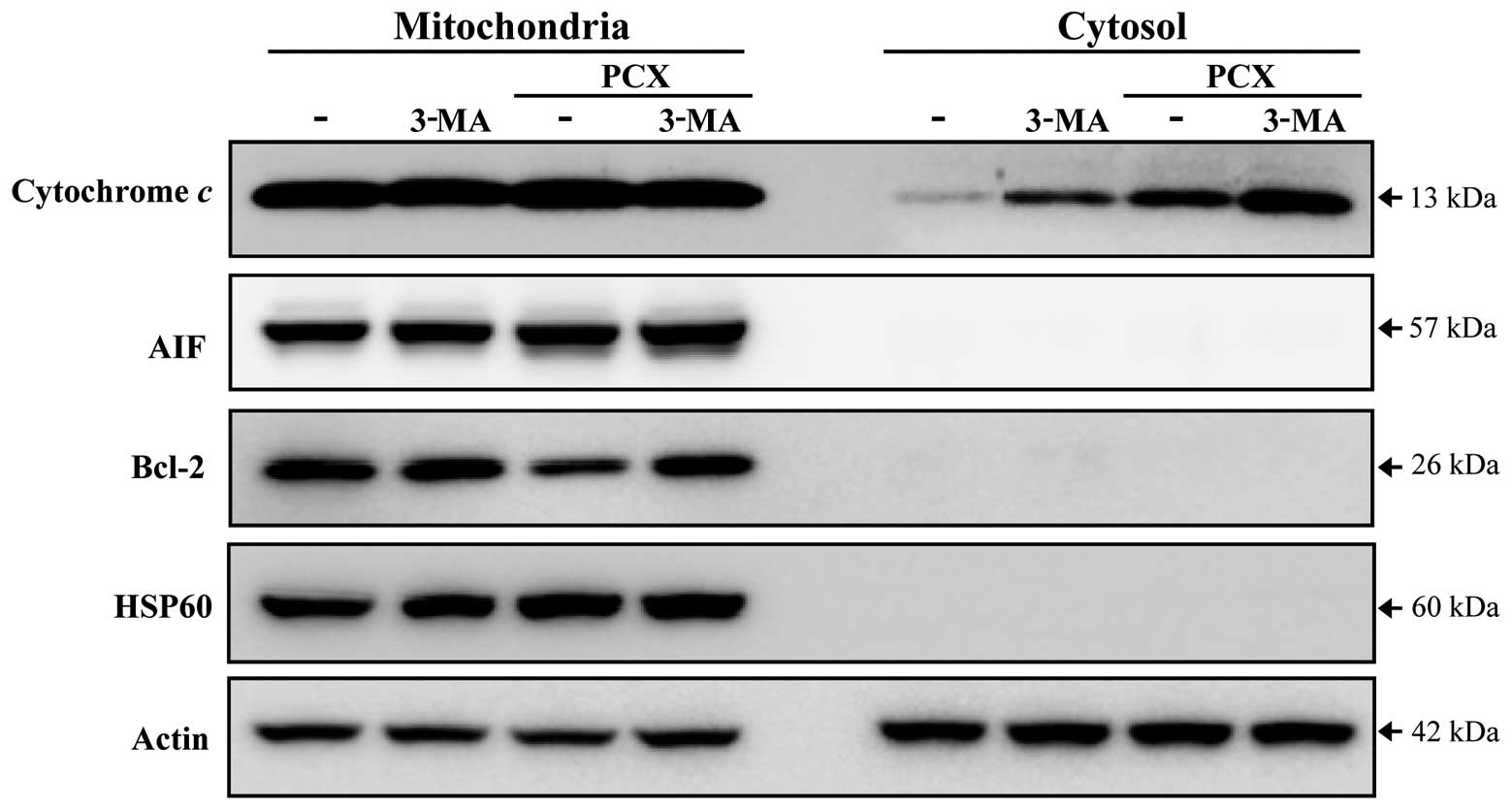
3-MA enhances cytochrome c release from
mitochondria in PCX-treated cells
In order to examine whether inhibition of
autophagosome formation correlates with the release of
mitochondrial apoptotic proteins, the levels of these proteins were
monitored in the cytosolic as well as mitochondrial fractions. We
showed that 3-MA enhanced PCX-induced release of cytochrome c from
the mitochondria (Fig. 5). In
contrast, AIF was not released from the mitochondria under these
conditions (Fig. 5). Furthermore,
the levels of Bcl-2, an anti-apoptotic mitochondrial protein,
appeared to be decreased after exposure to PCX, but were recovered
in the presence of 3-MA (Fig.
5).
Discussion
During the last few decades, taxanes have been
effectively used for the chemotherapeutic treatment of several
types of cancers, including breast (27), lung (28), prostate (29) and ovarian (30) cancers. In addition, many in
vitro experiments have shown that exposure to taxanes can
induce apoptotic cell death in various types of cancer cells
(31–33). PCX induces apoptosis in the human
osteogenic sarcoma cell lines Saos-2 (34) and U-2 OS (35). Insufficient information is
available on taxane-induced apoptosis in OS cells and therefore,
taxanes have not been considered for chemotherapeutic regimens for
treating OS (26).
Although previous studies have shown PCX-induced
apoptosis and related mechanisms in OS cells (34,35),
the concentrations of PCX (100–50,000 nM) used for inducing
apoptosis were relatively higher than that used in the present
study (5 nM). Because high-dose chemotherapy generally causes
serious cytotoxic adverse effects in patients, it is important to
develop lower-dose chemotherapeutic regimens. The results of the
present study confirmed that exposure to low PCX concentrations can
induce apoptotic cell death in Saos-2 cells; furthermore, caspase-3
activation, PARP degradation and XIAP downregulation were observed
in combination with PCX-induced apoptosis. Both degradation of PARP
and decrease in XIAP protein content appeared to be correlated with
increased caspase-3 activity. In addition, the potential
involvement of mitochondria (intrinsic apoptotic pathway) in
PCX-induced apoptosis of OS cells was confirmed by the alteration
(depolarization) of the mitochondrial membrane potential as well as
the release of cytochrome c from the mitochondria to the cytoplasm.
These results indicate that PCX-induced apoptosis in OS cells is
associated with the caspase-dependent pathway, which is consistent
with the previous findings for OS cells (34,35).
Similarly, involvement of caspase activation in
taxane-induced apoptosis has been shown in cancer cells originating
from the breasts (36), lungs
(37), ovaries (38) and prostate (33). In contrast, several studies have
shown that PCX-induced apoptosis is not related to caspase activity
in some types of cancer cells (39,40).
Currently, the precise reason for this inconsistency is not known,
but the inconsistency is presumed to be due to cellular differences
in tissue origin, status of differentiation, or cell cycle
checkpoint/regulatory proteins such as p53 (41). The XIAP protein is an endogenous
inhibitor of caspase-3 within cells (11) and has been suggested to be
associated with chemoresistance in cancer cells (42). Recently, the potential role of XIAP
in chemosensitivity or chemoresistance has been implicated in OS
cells (43). However, XIAP
downregulation in response to taxane therapy has not been shown in
OS cells before this study. Therefore, developing a taxane
chemotherapeutic regimen for XIAP downregulation could aid in
inducing caspase-dependent apoptosis in OS cells.
Autophagy is currently considered as cell death
machinery (programmed cell death type II) that differs from
apoptosis (programmed cell death type I) (14). Dysregulation or malfunction of
autophagy has been implicated in cancer (44), aging (45), diabetes (46), cardiovascular disease (47), inflammation (48) and neurodegenerative disease
(49). Therefore, the selectivity
of autophagy and its role in cell death and survival constitute an
important issue for understanding the wide spectrum of human health
and diseases (50). In the present
study, pretreatment with 3-methyladenine (3-MA), a specific
inhibitor of autophagy (51),
significantly increased PCX-induced apoptotic cell death in Soas-2
cells. The augmentation of PCX-induced apoptosis by 3-MA was
accompanied by increase in the cytochrome c release from the
mitochondria, caspase-3 activity and XIAP downregulation, which
suggests that inhibiting autophagy further stimulates the
PCX-induced mitochondrion-related (intrinsic) apoptotic pathway by
provoking caspase-3 activation.
Results similar to those obtained in this study have
recently been reported for A549 lung cancer cells (52). In these studies, the anti-apoptotic
function of autophagy was observed in PCX-induced apoptosis.
Collectively, these results indicate the cytoprotective role of
autophagy against apoptotic cell death in cancer cells. This
cytoprotection does not seem to be restricted to taxane treatment
in cancer cells. The cytoprotective function of autophagy has been
observed during 5-fluorouracil treatment in colon cancer cells
(53), sulforaphane treatment in
prostate cancer cells (54),
imatinib treatment in glioma cells (55) and suberoylanilide hydroxamic acid
treatment in leukemia cells (56).
Although the major cellular switch that determines cell destiny
(cell death vs. cell survival) by autophagy after exposure to
chemotherapeutic drugs remains unclear, the bcl-2 family proteins
(bcl-2 and bcl-xL) in the mitochondria are believed to play a
pivotal role in regulating autophagy (57).
Microtubule-associated protein light chain 3 (LC3)
is now widely used to monitor autophagy (58). LC3 is modified via a ubiquitin-like
system (59). LC3 exists in 2
forms: an 18-kDa cytosolic protein (LC3-I) and a processed 16-kDa
form (LC3-II) in cells engaged in autophagy. The LC3-II form is
mainly localized in autophagosome membranes and therefore, the
LC3-II level is a good early marker for autophagosome formation
(60). In the present study,
immunoreactivity for LC3 markedly increased in Saos-2 cells upon
PCX treatment but decreased in the presence of 3-MA. This obviously
indicates that PCX treatment increased autophagy. It can be
presumed that increased autophagy occurs during the rescue process
of cellular structures (i.e.,, organelles) damaged by PCX exposure.
Taken together, autophagy observed during PCX-induced apoptosis in
Saos-2 OS cells represents the role of cytoprotection in cellular
homeostatic processes.
In conclusion, the results of this study revealed
that PCX exposure effectively induces OS cell death by apoptosis
associated with the mitochondrion-mediated caspase-dependent
pathway. PCX can increase autophagic activity and suppressing
autophagy enhances PCX-induced apoptosis in OS cells. Therefore, it
is suggested that combination treatment involving low-dose PCX
therapy and autophagy inhibitor therapy could be an effective and
potent strategy for improved chemotherapy for OS in the near
future.
Acknowledgements
This study was supported by the Dong-A
University Research Fund.
References
|
1
|
Dorfman HD and Czerniak B: Bone cancers.
Cancer. 75:203–210. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rytting M, Pearson P, Raymond AK, Ayala A,
Murray J, Yasko AW, Johnson M and Jaffe N: Osteosarcoma in
preadolescent patients. Clin Orthop Relat Res. 373:39–50. 2000.
View Article : Google Scholar
|
|
3
|
Longhi A, Errani C, De Paolis M, Mercuri M
and Bacci G: Primary bone osteosarcoma in the pediatric age: state
of the art. Cancer Treat Rev. 32:423–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hudson M, Jaffe MR, Jaffe N, Ayala A,
Raymond AK, Carrasco H, Wallace S, Murray J and Robertson R:
Pediatric osteosarcoma: therapeutic strategies, results and
prognostic factors derived from a 10-year experience. J Clin Oncol.
8:1988–1997. 1990.PubMed/NCBI
|
|
5
|
Desandes E: Survival from adolescent
cancer. Cancer Treat Rev. 33:609–615. 2007. View Article : Google Scholar
|
|
6
|
Fellenberg J, Mau H, Nedel S, Ewerbeck V
and Debatin KM: Drug-induced apoptosis in osteosarcoma cell lines
is mediated by caspase activation independent of
CD95-receptor/ligand interaction. J Orthop Res. 18:10–17. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seki K, Yoshikawa H, Shiiki K, Hamada Y,
Akamatsu N and Tasaka K: Cisplatin (CDDP) specifically induces
apoptosis via sequential activation of caspase-8, -3 and -6 in
osteosarcoma. Cancer Chemother Pharmacol. 45:199–206. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fu W, Ma L, Chu B, Wang X, Bui MM, Gemmer
J, Altiok S and Pledger WJ: The cyclin-dependent kinase inhibitor
SCH 727965 (dinacliclib) induces the apoptosis of osteosarcoma
cells. Mol Cancer Ther. 10:1018–1027. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ashkenazi A and Dixit VM: Death receptors:
signaling and modulation. Science. 281:1305–1308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Deveraux QL, Takahashi R, Salvesen GS and
Reed JC: X-linked IAP is a direct inhibitor of cell-death
proteases. Nature. 388:300–304. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Broker LE, Kruyt FA and Giaccone G: Cell
death independent of caspases: a review. Clin Cancer Res.
11:3155–3162. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cande C, Cecconi F, Dessen P and Kroemer
G: Apoptosis-inducing factor (AIF): key to the conserved
caspase-independent pathways of cell death? J Cell Sci.
115:4727–4734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bursch W: The autophagosomal-lysosomal
compartment in programmed cell death. Cell Death Differ. 8:569–581.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tsujimoto Y and Shimizu S: Another way to
die: autophagic programmed cell death. Cell Death Differ.
12:1528–1534. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Grinde B: Autophagy and lysosomal
proteolysis in the liver. Experientia. 41:1089–1095. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lum JJ, Bauer DE, Kong M, Harris MH, Li C,
Lindsten T and Thompson CB: Growth factor regulation of autophagy
and cell survival in the absence of apoptosis. Cell. 120:237–248.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gutierrez MG, Master SS, Singh SB, Taylor
GA, Colombo MI and Deretic V: Autophagy is a defense mechanism
inhibiting BCG and Mycobacterium tuberculosis survival in infected
macrophages. Cell. 119:753–766. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu L, Alva A, Su H, Dutt P, Freundt E,
Welsh S, Baehrecke EH and Lenardo MJ: Regulation of an ATG7-beclin
1 program of autophagic cell death by caspase-8. Science.
304:1500–1502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shimizu S, Kanaseki T, Mizushima N, Mizuta
T, Arakawa-Kobayashi S, Thompson CB and Tsujimoto Y: Role of Bcl-2
family proteins in a non-apoptotic programmed cell death dependent
on autophagy genes. Nat Cell Biol. 6:1221–1228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Paludan C, Schmid D, Landthaler M,
Vockerodt M, Kube D, Tuschl T and Münz C: Endogenous MHC class II
processing of a viral nuclear antigen after autophagy. Science.
307:593–596. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Komatsu M, Waguri S, Chiba T, Murata S,
Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E and
Tanaka K: Loss of autophagy in the central nervous system causes
neurodegeneration in mice. Nature. 441:880–884. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar
|
|
24
|
Rowinsky EK, Onetto N, Canetta RM and
Arbuck SG: Taxol: the first of the taxanes, an important new class
of antitumor agents. Semin Oncol. 19:646–662. 1992.PubMed/NCBI
|
|
25
|
Ringel I and Horwitz SB: Effect of
alkaline pH on taxol-microtubule interactions. J Pharmacol Exp
Ther. 259:855–860. 1991.PubMed/NCBI
|
|
26
|
Anninga JK, Gelderblom H, Fiocco M, Kroep
JR, Taminiau AH, Hogendoorn PC and Egeler RM: Chemotherapeutic
adjuvant treatment for osteosarcoma: where do we stand? Eur J
Cancer. 47:2431–2445. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Saloustros E, Mavroudis D and Georgoulias
V: Paclitaxel and docetaxel in the treatment of breast cancer.
Expert Opin Pharmacother. 9:2603–2616. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chu Q, Vincent M, Logan D, Mackay JA and
Evans WK: Lung Cancer Disease Site Group of Cancer Care Ontario’s
Program in Evidence-based Care. Taxanes as first-line therapy for
advanced non-small cell lung cancer: a systematic review and
practice guideline. Lung Cancer. 50:355–374. 2005.
|
|
29
|
Obasaju C and Hudes GR: Paclitaxel and
docetaxel in prostate cancer. Hematol Oncol Clin North Am.
15:525–545. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Young M and Plosker GL: Paclitaxel: a
pharmacoeconomic review of its use in the treatment of ovarian
cancer. Pharmacoeconomics. 19:1227–1259. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Weigel TL, Lotze MT, Kim PK, Amoscato AA,
Luketich JD and Odoux C: Paclitaxel-induced apoptosis in non-small
cell lung cancer cell lines is associated with increased caspase-3
activity. J Thorac Cardiovasc Surg. 119:795–803. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Blajeski AL, Kottke TJ and Kaufmann SH: A
multistep model for paclitaxel-induced apoptosis in human breast
cancer cell lines. Exp Cell Res. 270:277–288. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim JY, Chung JY, Lee SG, Kim YJ, Park JE,
Yoo KS, Yoo YH, Park YC, Kim BG and Kim JM: Nuclear interaction of
Smac/DIABLO with Survivin at G2/M arrest prompts docetaxel-induced
apoptosis in DU145 prostate cancer cells. Biochem Biophys Res
Commun. 350:949–954. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pucci B, Bellincampi L, Tafani M,
Masciullo V, Melino G and Giordano A: Paclitaxel induces apoptosis
in Saos-2 cells with CD95L upregulation and Bcl-2 phosphorylation.
Exp Cell Res. 252:134–143. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lu KH, Lue KH, Chou MC and Chung JG:
Paclitaxel induces apoptosis via caspase-3 activation in human
osteogenic sarcoma cells (U-2 OS). J Orthop Res. 23:988–994. 2005.
View Article : Google Scholar
|
|
36
|
Kovar J, Ehrlichov M, Smejkalov B, Zanardi
I, Ojima I and Gut I: Comparison of cell death-inducing effect of
novel taxane SB-T-1216 and paclitaxel in breast cancer cells.
Anticancer Res. 29:2951–2960. 2009.PubMed/NCBI
|
|
37
|
Osaki S, Nakanishi Y, Takayama K, Pei XH,
Ueno H and Hara N: Transfer of IkappaBalpha gene increase the
sensitivity of paclitaxel mediated with caspase 3 activation in
human lung cancer cell. J Exp Clin Cancer Res. 22:69–75.
2003.PubMed/NCBI
|
|
38
|
Ueno NT, Bartholomeusz C, Herrmann JL and
Estrov Z: E1A-mediated paclitaxel sensitization in
HER-2/neu-overexpressing ovarian cancer SKOV3.ip1 through apoptosis
involving the caspase-3 pathway. Clin Cancer Res. 6:250–259.
2000.PubMed/NCBI
|
|
39
|
Huisman C, Ferreira CG, Broker LE,
Rodriguez JA, Smit EF, Postmus PE, Kruyt FA and Giaccone G:
Paclitaxel triggers cell death primarily via caspase-independent
routes in the non-small cell lung cancer cell line NCI-H460. Clin
Cancer Res. 8:596–606. 2002.PubMed/NCBI
|
|
40
|
Ofir R, Seidman R, Rabinski T, Krup M,
Yavelsky V, Weinstein Y and Wolfson M: Taxol-induced apoptosis in
human SKOV3 ovarian and MCF7 breast carcinoma cells is caspase-3
and caspase-9 independent. Cell Death Differ. 9:636–642. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vikhanskaya F, Vignati S, Beccaglia P,
Ottoboni C, Russo P, D’Incalci M and Broggini M: Inactivation of
p53 in a human ovarian cancer cell line increases the sensitivity
to paclitaxel by inducing G2/M arrest and apoptosis. Exp Cell Res.
241:96–101. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sasaki H, Sheng Y, Kotsuji F and Tsang BK:
Down-regulation of X-linked inhibitor of apoptosis protein induces
apoptosis in chemoresistant human ovarian cancer cells. Cancer Res.
60:5659–5666. 2000.PubMed/NCBI
|
|
43
|
Fellenberg J, Kunz P, Suhr H and Depeweg
D: Overexpression of inosine 5′-monophosphate dehydrogenase type II
mediates chemoresistance to human osteosarcoma cells. PLoS One.
5:e121792010.
|
|
44
|
Kimmelman AC: The dynamic nature of
autophagy in cancer. Genes Dev. 25:1999–2010. 2011. View Article : Google Scholar
|
|
45
|
Rubinsztein DC, Mariño G and Kroemer G:
Autophagy and aging. Cell. 146:682–695. 2011. View Article : Google Scholar
|
|
46
|
Jung HS and Lee MS: Role of autophagy in
diabetes and mitochondria. Ann NY Acad Sci. 1201:79–83. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nemchenko A, Chiong M, Turer A, Lavandero
S and Hill JA: Autophagy as a therapeutic target in cardiovascular
disease. J Mol Cell Cardiol. 51:584–593. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Levine B, Mizushima N and Virgin HW:
Autophagy in immunity and inflammation. Nature. 469:323–335. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Alirezaei M, Kemball CC and Whitton JL:
Autophagy, inflammation and neurodegenerative disease. Eur J
Neurosci. 33:197–204. 2011. View Article : Google Scholar
|
|
50
|
Yu L, Strandberg L and Lenardo MJ: The
selectivity of autophagy and its role in cell death and survival.
Autophagy. 4:567–573. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Seglen PO and Gordon PB: 3-Methyladenine:
specific inhibitor of autophagic/lysosomal protein degradation in
isolated rat hepatocytes. Proc Natl Acad Sci USA. 79:1889–1892.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xi G, Hu X, Wu B, Jiang H, Young CY, Pang
Y and Yuan H: Autophagy inhibition promotes paclitaxel-induced
apoptosis in cancer cells. Cancer Lett. 307:141–148. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li J, Hou N, Faried A, Tsutsumi S and
Kuwano H: Inhibition of autophagy augments 5-fluorouracil
chemotherapy in human colon cancer in vitro and in vivo model. Eur
J Cancer. 46:1900–1909. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Herman-Antosiewicz A, Johnson DE and Singh
SV: Sulforaphane causes autophagy to inhibit release of cytochrome
C and apoptosis in human prostate cancer cells. Cancer Res.
66:5828–5835. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shingu T, Fujiwara K, Bugler O, Akiyama Y,
Moritake K, Shinojima N, Tamada Y, Yokoyama T and Kondo S:
Inhibition of autophagy at a late stage enhances imatinib-induced
cytotoxicity in human malignant glioma cells. Int J Cancer.
124:1060–1071. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Carew JS, Nawrocki ST, Kahue CN, Zhang H,
Yang C, Chung L, Houghton JA, Huang P, Giles FJ and Cleveland JL:
Targeting autophagy augments the anticancer activity of the histone
deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug
resistance. Blood. 110:313–322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Levine B, Sinha S and Kroemer G: Bcl-2
family members: dual regulators of apoptosis and autophagy.
Autophagy. 4:600–606. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tanida I, Minematsu-Ikeguchi N, Ueno T and
Kominami E: Lysosomal turnover, but not a cellular level, of
endogenous LC3 is a marker for autophagy. Autophagy. 1:84–91. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ohsumi Y: Molecular dissection of
autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol.
2:211–216. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|