Introduction
Gallbladder carcinoma is the most common malignancy
of the biliary tract, the fifth or sixth common malignant neoplasm
of the digestive tract and the leading cause of cancer-related
deaths in West countries and China (1–5). The
low 5-year survival rate and poor prognosis of patients with
gallbladder carcinoma is related to diagnostic delay, low surgical
excision rate, high local recurrence and distant metastasis and
biological behavior of the tumor. Additionally, chemotherapy and
radiotherapy for the disease are disappointing (1,6–12).
Therefore, it is an urgent task to reveal the precise special
biological behavior of gallbladder carcinoma development and
provide a novel perspective for anticancer therapeutics.
The growth and metastasis of the tumor depend on an
effective microcirculation. The formation of a microcirculation can
occur via the traditionally recognized mechanisms of vasculogenesis
and angiogenesis and the recently found VM. VM, a newly-defined
pattern of tumor blood supply, provides a special passage without
endothelial cells and is conspicuously different from angiogenesis
and vasculogenesis (13) and is
associated with a poor prognosis for the patients with some
aggressive malignant tumors such as melanoma (13,14),
breast cancer (15), ovarian
carcinoma (16), hepatocellular
carcinoma (17,18), gastric adenocarcinomas (19), and colorectal cancer (20). However, the detailed mechanism of
the tumor cells that form VM remains to be further elucidated.
Currently, some signaling pathways involving factors which promote
cell migration, invasion and matrix remodeling are thought to
relative with the formation of tumor VM. These include PI3K, MMPs,
Ln-5γ2 chain (21–24), EphA2, FAK (25–29),
tissue factor (TF) and its pathway inhibitor (TFPI) (30) and vascular endothelial growth
factor α (VEGFα) (31), and others
(32,33). Therefore, understanding the key
molecular mechanisms that regulate VM would serve as an important
target for new cancer therapies.
We previously reported that VM existed in human
gall-bladder carcinomas and gallbladder carcinomas by both 3-D
matrices of highly aggressive GBC-SD cells in vitro and
GBC-SD nude mouse xenografts in vivo and correlated with the
patient’s poor prognosis and that poorly aggressive SGC-996 cells
did not form the vasculogenic-like networks when cultured under the
same conditions, but formed pattern, vasculogenic-like networks
when being cultured on a matrix preconditioned by the GBC-SD cells
(34–36). However, the exact mechanism
underlying VM in gallbladder carcinomas still needs to be
unraveled. In this study, we firstly present evidence that the
formation of VM in human gallbladder carcinomas through the
activation of the EphA2/FAK/Paxillin signaling pathway and the
PI3K/MMPs/Ln-5γ2 signaling pathway in the 3-D matrixes of GBC-SD
cells in vitro and GBC-SD nude mouse xenografts in
vivo and provide a potential target therapy for VM of
gallbladder carcinomas.
Materials and methods
Cell culture
Human gallbladder carcinoma (GBC-SD and SGC-996)
cell lines have been described previously (36) and were maintained in Dulbecco’s
modified Eagle’s media (DMEM, Gibco Co., USA) supplemented with 10%
fetal bovine serum (FBS, Hangzhou Sijiqing Bioproducts, China) and
105 U/ml penicillin and streptomycin (Shanghai
Pharmaceutical Works, China) in an incubator (Forma series II HEPA
Class 100, Thermo Co., USA) at 37°C with 5% carbon dioxide
(CO2).
Network formation assay in vitro
Matrigel and rat-tail collagen type I three
dimensional matrices were prepared as described previously
(36). Cells were allowed to
adhere to the matrix and untreated and treated with 100 nM TIMP-2
recombinant protein (Sigma Co., Germany) for 4 days. Phase contrast
microscopy (Olympus IX70, Japan) was used for analyzing the ability
of the cells to engage in VM. The images were taken digitally using
a Zeiss Televal inverted microscope (Carl Zeiss, Inc., Thornwood,
NY) and camera (Nikon, Japan) at the time indicated.
Tumor xenograft assay in vivo
Balb/c nu/nu mice (equal numbers of male and female
mice, 4-week old, ∼20 g) were provided by Shanghai Laboratory
Animal Center, Chinese Academy of Sciences and housed in specific
pathogen-free (SPF) conditions. All the procedures were performed
on nude mice according to the official recommendations of the
Chinese Community Guidelines. Tumor xenograft assay of GBC-SD and
SGC-996 cells in vivo was performed as described previously
(36). The mice, by 2 weeks when a
tumor xenograft was apparent in the axilback of all mice, were
randomly divided into a GBC-SD group (n=7), a SGC-996 group (n=7)
receiving intraperitoneal injections of 0.1 ml normal saline alone
twice each week and a GBC-SD+TIMP-2 group (n=6, each mouse with
GBC-SD xenograft receiving intratumoral injection of 100 nM TIMP-2
recombinant protein), twice each week for 6 weeks in all. The
maximum diameter (a) and minimum diameter (b) of the xenografts
were measured with calipers two times each week. The tumor volume
was calculated by the following formula: V (cm3) =
1/6πab2. Also, tumor growth curve of each group
was respectively evaluated.
Immunohistochemistry in vitro and in
vivo
Immunohistochemistry in vitro and in
vivo included H&E staining, PAS staining, CD31-PAS double
staining and the determination of MMP-2 or MT1-MMP protein for
sections and supernates from the cell culture tissues and sections
of tumor xenografts. i) H&E staining, PAS stainings and
CD31-PAS double staining were performed as described previously
(36). ii) MMP-2 and MT1-MMP
proteins from sections of 3-D culture samples and tumor xenografts
were determined by SABC method. The sections (4-μm) from
each group were dehydrated in xylene and graded ethanol series,
were added in order with primary antibody [MMP-2 (1: 200), MT1-MMP
(1:100); rabbit polyclonal antibody, Wuhan Boster Co., China)],
biotinylated secondary antibody, SABC reagents and DAB solution
(Wuhan Boster Co.), respectively. Then, sections were rinsed in
distilled water, dehydrated through alcohol and xylene and mounted
coverslip using a permanent mount medium and observed under an
optical microscope with ×10 and 40 objectives (Olympus CH-2,
Japan). For negative control, the slides were treated with PBS in
place of primary antibody. Ten sample slides in each group were
chosen by analysis. More than 10 visual fields were observed or
>500 cells were counted per slide. iii) MMP-2 and MT1-MMP
proteins from supernates of 3-D culture samples were determined by
ELISA. The supernates from each group and the diluted standard
solutions were added into 2 multiple wells, 2 zero adjusting wells
and a control TMB well. The former two wells were added with
biotinylated antibody (MMP-2, ELISA kits, Wuhan Boster Co.;
MT1-MMP, ELISA kits, DR, USA), ABC reagents and TMB solution (Wuhan
Boster Co.), respectively; the control TMB well did not include
reagents. Optical densities at 450 nm were measured using an ELISA
reader (Biorad model, Sigma).
Electron microscopy in vitro and in
vivo
For SEM and TEM, 3-D culture samples and fresh tumor
xenograft tissues (0.5 mm3) were fixed in cold 2.5%
glutaraldehyde in 0.1 mol/l of sodium cacodylate buffer and
postfixed in a solution of 1% osmium tetroxide, dehydrated and
embedded in a standard fashion. Specimens were subsequently
embedded, sectioned and stained by routine means for a Jeol-1230
TEM, or critically point-dried and sputter-coated with gold for a
Hitachi S-520 SEM.
Immunofluorescence detection in vivo
EphA2, FAK, PI3K, Ln-5γ2 and Paxillin-P protein
products from the xenografts of each group were determined by
indirect immunofluorescence method. The frozen sections (4
μm) of the xenografts from each group were pretreated with
99.5% acetone, methanol with 3% hydrogen peroxide and 20% normal
goat serum, were added with 50 μl (1:100) primary antibody
[EphA2 and FAK, rabbit anti-human polyclonal antibody, Santa Cruz,
USA; PI3K, mouse anti-human polyclonal antibody, Acris Antibodies
GmbH, USA; Ln-5γ2, mouse anti-human polyclonal antibody, Santa
Cruz; Paxillin (phosphor Y118), rabbit anti-human polyclonal
antibody, Abcam Plc, USA], biotinylated secondary antibody (1:100;
goat anti-rabbit IgG-FITC/GGHL-15F, or goat anti-mouse
IgG-FITC/GGHL-90F, Immunology Consultants Laboratory, USA),
respectively. Then, sections were rinsed in TBS solution and
distilled water, mounted with coverslip using buffer glycerine and
observed under a fluorescence microscope (Nikon, Japan). For
negative control, the slides were treated with PBS in place of
primary antibody. Ten sample slides in each group were chosen by
analysis. More than 10 visual fields were observed per slide.
Expression of each protein on slides of the xenografts showed a
fluorescent yellow-green stain. Fluorescent stain intensity was
classed into −, ±, +, ++, +++, ++++. Of these, − to +, negative
expression; ≥++, positive expression.
Western blot analysis in vivo
EphA2, FAK, PI3K, Ln-5γ2 and Paxillin-P proteins
from the xenografts of each group were determined by western blot
analysis. Cells were lysed with 200 ml of cell lysis buffer
(protein extraction kit, KangChen, KC-415, China) containing a
cocktail of protease inhibitors and the supernatant of the lysed
cells was recovered. BCA protein quantitative determination was
carried out with a protein quantitative kit (KangChen, KC-430;
China). Then, an aliquot of 20 mg of proteins was subjected to
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) under reducing condition and were subsequently
transferred to a PVDF membrane. An hour after being blocked with
PBS containing 5% non-fat milk, the membrane was incubated
overnight, then each primary antibody was added [EphA2, FAK, Ln-5γ2
(all from Santa Cruz); PI3K (P85-a, Acris Antibodies GmbH);
Paxillin (phosphor Y118, Abcam Plc): mouse anti-human antibody,
1:3,000; and GAPDH (mouse anti-human antibody, 1:10,000; Kangcheng
Bioengineering Co., Shanghai, China) diluted with PBST containing
5% non-fat milk at 4°C], an appropriate anti-mouse or anti-rabbit
HRP-labeled secondary antibody (1:5,000; Kangcheng Bioengineering
Co.). The target proteins were visualized by an enhanced
chemiluminescent (ECL) reagent (KC™ Chemiluminescent kit, KangChen,
KC-420, China), and imaged on the Bio-Rad chemiluminescence imager.
The gray value and gray coefficient ratio of each protein was
analyzed and calculated with Image J analysis software.
QRT-PCR analysis in vivo
Expression of MMP-2, MT1-MMP, EphA2, FAK, PI3K,
Ln-5γ2 and Paxillin-P mRNAs from the xenografts of each group was
respectively determined by qRT-PCR assay. QRT-PCR was performed as
described by the manufacturer. Total RNA from the xenograft cells
of each group was prepared using the TRizol reagent (Invitrogen,
USA). Concentration of RNA was determined by the absorption at
260–280. PCR amplifications were performed with gene-specific
primers (Table I) with annealing
temperature and number of amplification cycles optimized using cDNA
from the xenograft cells of each group. PCR amplification reactions
were performed as follows: 1 cycle of 94°C for 5 min; 35 cycles of
94°C for 10–22 sec, 57–60°C for 15–20 sec, 72°C for 20 sec, 82–86°C
(fluorescence collection) for 5–10 sec; 1 cycle of 72–99°C for 5
min. GAPDH primers were used as control for PCR amplification. PCR
products (10 μl) were placed onto 15 g/l agarose gel and
observed by ethidium bromide (EB, Huamei Bioengineering Co., China)
staining using the ABI PRISM 7300 SDS software.
 | Table IVM signaling-related markers. |
Table I
VM signaling-related markers.
| Gene | PCR primers
(forward-reverse) | Amplification size
(bp) | Cycle no. |
|---|
| MMP-2 |
5′-AAGAGCGTGAAGTTTGGAAGCA-3′
5′-TCTGAGGGTTGGTGGGATTGG-3′ | 290 | 35 |
| MT1-MMP |
5′-CAAAGGCAGAACAGCCAGAGG-3′
5′-ACAGGGACCAACAGGAGCAAG-3′ | 180 | 35 |
| EphA2 |
5′-TTAGGGAGAAGGATGGTGAGTT-3′
5′-GTTGCTGTTGACGAGGATGTT-3′ | 140 | 35 |
| FAK |
5′-CCCAGAAAGAAGGTGAACG-3′
5′-GGTCGAGGGCATGGTGTA-3′ | 152 | 35 |
| PI3K |
5′-TGTCGCAGCCCAGGTAGATT-3′
5′-CAGGAGGTGGTCGGGTCAAG-3′ | 269 | 35 |
| Ln-5γ2 |
5′-ACACGGGAGATTGCTACTCG-3′
5′-ACCCATTGTGACAGGGACAT-3′ | 123 | 35 |
| Paxillin-P |
5′-CTTCAAGGAGCAGAACGACAAA-3′
5′-TAGCAGGTGGTAGGGACGAGA-3′5 | 228 | 35 |
| GAPDH |
5′-CCTCTATGCCAACACAGTGC-3′
5′-GTACTCCTGCTTGCTGATCC-3′ | 211 | 35–40 |
Statistical analysis
The data are expressed as mean ± SD and performed
using SAS, 9.0 version software (SAS Institute Inc., Cary, NC,
USA). Statistical analyses to determine significance were tested
with the χ2, F or Student-Newman-Keuls t-tests.
P<0.05 was considered statistically significant.
Results
Vasculogenic-like network formation of
GBC-SD cells in vitro
As showed in Fig.
1, highly aggressive GBC-SD cells were able to form
vasculogenic-like network structures when cultured on Matrigel and
rat-tail collagen type I composed of the ECM gel in the absence of
endothelial cells and fibroblasts (Fig. 1Aa1–4). The tumor-formed
networks initiated formation within 48 h after seeding the cells
onto the matrix with optimal structure formation achieved by 2
weeks. However, poorly aggressive SGC-996 cells were unable to form
tubular-like structures with the same conditions (Fig. 1Ab1–4). SEM clearly
visualized channelized or hollowed vasculogenic-like networks in
GBC-SD cells (Fig.
1Ba1,2), with clear microvilli and tubular
structures surrounding a cluster of tumor cells. TEM showed some
microvilli outside the network, clear cellular organelle structures
and cell connection with an increased electron density (Fig. 1Bb1). The results were
concordant with our previous report (36). It is interesting that in the
process of vasculogenic-like structure formation, using TIMP-2
(Fig. 1Ac1–4) for 2
days, GBC-SD cells lost the capacity of network formation, with
visible cell aggregation, floating, nuclear fragmentation,
apoptosis and necrosis. Using TIMP-2 for 48 h after network
formation, the formed vasculogenic-like structures were destroyed,
with visible cell aggregation, floating, nuclear fragmentation and
apoptosis. It was shown that TIMP-2 inhibited and destroyed
formation of VM, and formed-VM from the 3-D culture of GBC-SD cells
in vitro.
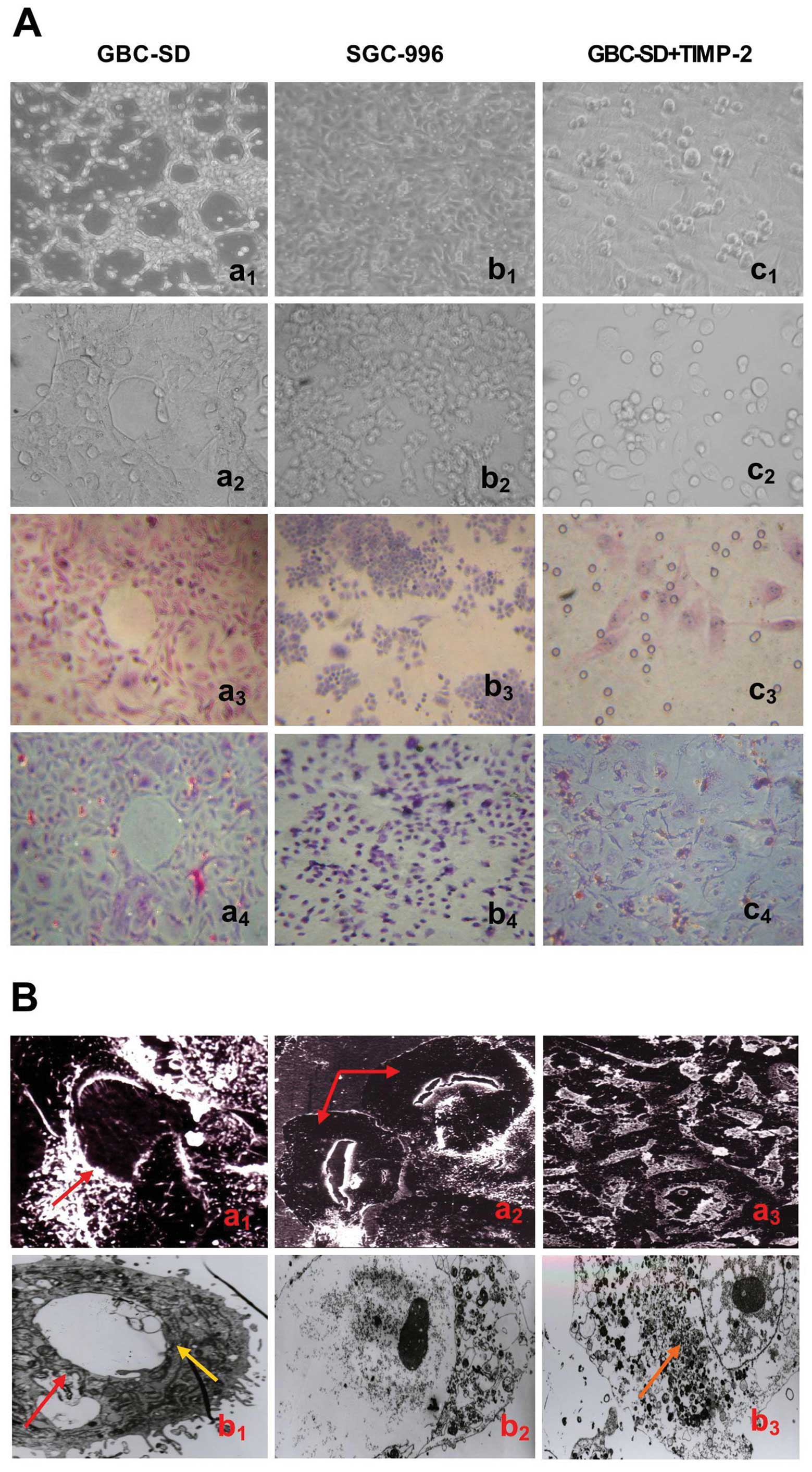 | Figure 1Phase contrast microscopy and
electron microscopy on 3-D cultures of GBC-SD and SGC-996 cells
in vitro. (A) Phase contrast microscopy of GBC-SD cells
cultured three dimensionally on Matrigel (a1,
b1 and c1, original magnification, ×200) and
rat-tail type I collagen matrix (a2–4, b2–4
and c2–4, original magnification, ×200) in vitro.
Highly aggressive GBC-SD cells formed patterned, vasculogenic-like
networks when cultured on Matrigel (a1) and rat-tail
type I collagen matrix (a2 and a3, H&E
staining) for 14 days. Similarly, the 3-D cultures of GBC-SD cells
when stained with PAS without hematoxylin counterstain showed the
vasculogenic-like structures; PAS-positive, cherry-red materials
were found in granules and patches in the cytoplasm of GBC-SD cells
appeared around the signal cell or cell clusters (a4).
However, poorly aggressive SGC-996 cells did not form these
networks when cultured under the same conditions (b1–4).
In the process of network formation, using TIMP-2 for 2 days,
GBC-SD cells lost the capacity of the vasculogenic-like network
formation, with visible cell aggregation, floating, nuclear
fragmentation, apoptosis and necrosis (c1–4). (B)
Vasculogenic-like microstructures on 3-D cultures of GBC-SD cells
by electron microscopy (Ba1–3, SEM ×500;
Bb1–3, TEM ×1200). SEM clearly visualized channelized or
hollowed vasculogenic-like networks formed in GBC-SD cells
(Ba1,2, red arrowhead), with clear microvillus
surrounding cluster of tumor cells. TEM shows some microvilli
outside the network, clear cellular organelle structures and cell
connection with an increased electron density (Bb1,
yellow arrowhead). After using TIMP-2 for 2 days, GBC-SD cells did
not grow along the collagen framework, were raised and deformed,
lost the capacity of network formation (Ba3), with
visible decreased microvillus, destroyed cellular organelles,
nuclear fragmentation, vacuolar degeneration and typical apoptotic
bodies (Bb2, 3, brown arrowhead). |
Tumor growth and VM formation of GBC-SD
xenografts in vivo
In the experiment, the tumor appeared gradually in
the subcutaneous area of right axilback of nude mice from the 6th
day after inoculation. As shown in Fig. 2A, xenograft formation rate in nude
mice after 2 weeks was 100% (7/7) for GBC-SD, 71.4% (5/7) for
SGC-996 and 33.3% (2/6) for GBC-SD+TIMP-2, with significant
difference between GBC-SD group and GBC-SD+TIMP-2 group
(P<0.01). In addition, the medium volume of nude mouse
xenografts at 6th weeks in GBC-SD+TIMP-2 group was smaller than
that of GBC-SD group (1.85+0.93 vs. 2.95+1.43 cm3,
P<0.001), but there was no significant difference between GBC-SD
group and SGC-996 group (P>0.05).
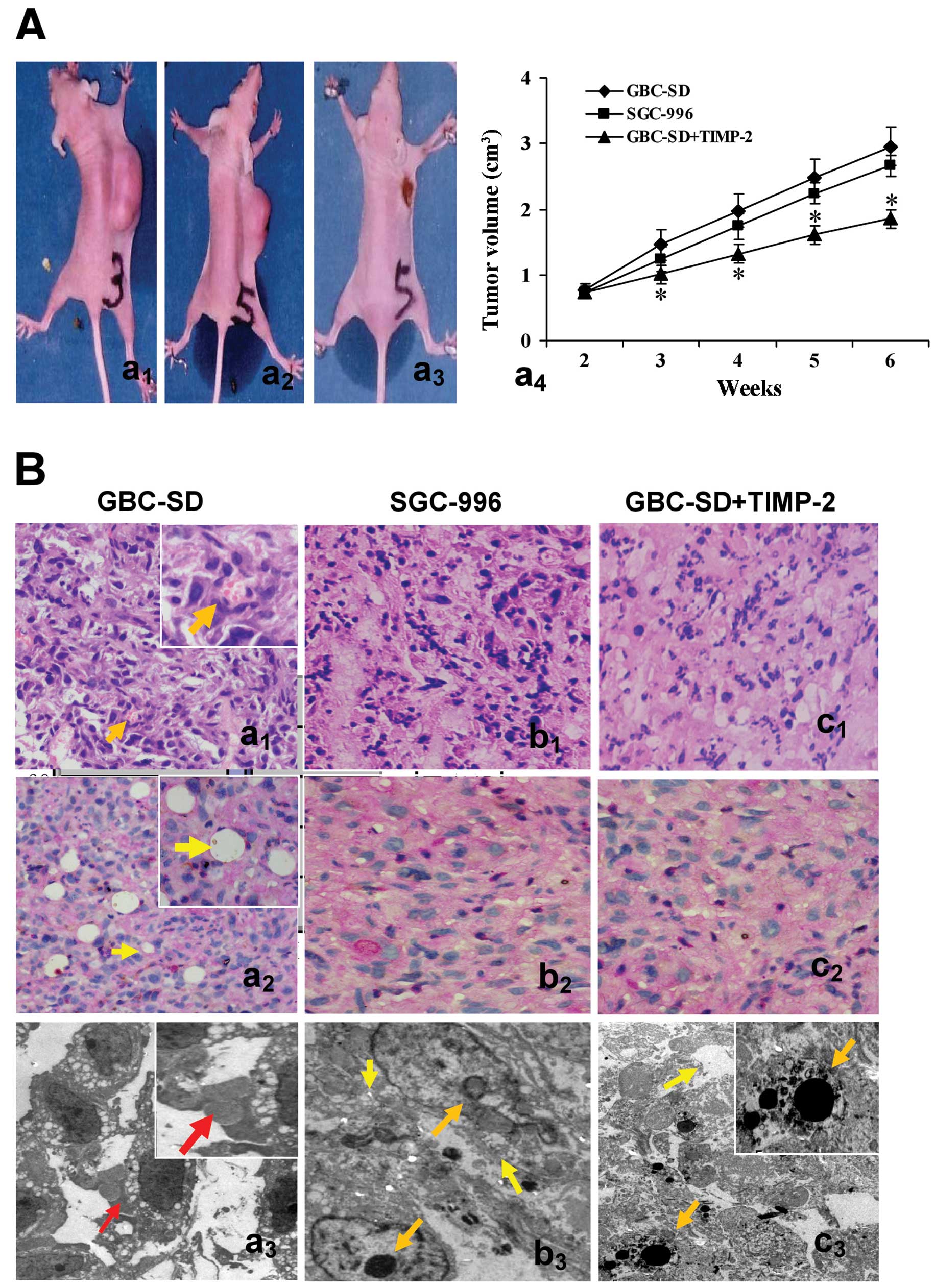 | Figure 2Growth and characteristic appearance
of GBC-SD and SGC-996 xenografts in vivo. (A) The xenografts
of GBC-SD (a1), SGC-996 (a2) and GBCSD+TIMP-2
(a3, the xenografts exhibited different degree of
necrosis, red arrowhead) groups and tumor growth curve in each
group (*P<0.001, vs. GBC-SD group or SGC-996 group
(a4). (B) Histomorphologic appearance of the xenografts
in GBC-SD (a1–3), SGC-996 (b1–3) and
GBC-SD+TIMP-2 (c1–3) groups. Using H&E (a1,
b1 and c1) and CD31-PAS double stain (a2,
b2 and c2, all original magnification, ×200),
sections of the xenografts in GBC-SD group showed tumor cell-lined
channels containing red blood cells (a1, orange
arrowhead) without any evidence of tumor necrosis; PAS-positive
substances line the channel-like structures; tumor cells form
vessel-like structure with single red blood cell inside
(a2, yellow arrowhead). TEM (original magnification,
×8,000) clearly visualized several red blood cells in the central
tumor nests in the xenografts of GBC-SD group (a3, red arrowhead).
However, similar phenomenon failed to occur in the xenografts of
SGC-996 group (b1–3) or GBC-SD+TIMP-2 (c1–3)
group, with destroyed cellular organelles, cell necrosis
(b3 and c3, yellow arrowhead), nuclear
pyknosis, fragmentation and apoptotic bodies (b3 and
c3, orange arrowhead). |
Morphology characteristics of xenografts were
observed via H&E staining and dual-staining with CD31-PAS under
optical microscopy and TEM. Microscopically, the xenografts in
GBC-SD group showed that tumor cells lined channels containing red
blood cells (Fig. 2Ba1)
without any evidence of tumor necrosis; the channel consisted of
tumor cells was negative for CD31 and positive for PAS; and tumor
cells formed vessel-like structures with single red blood cell
inside (Fig. 2Ba2). VM
positive rate was 85.7% (6/7) in GBC-SD group. Among 24 tissue
sections, 10 high-power fields in each section were counted to
estimate the proportion of vessels that were lined by tumor cells,
5.7% (17/300) channels were seen to contain red blood cells among
these tumor cell-lined vasculatures. In the central area of the
tumor, xenografts exhibited VM in the absence of ECs, central
necrosis or fibrosis (Fig.
2Ba2). For xenografts in GBC-SD group, TEM clearly
showed single, double and several red blood cells existed in the
centre of tumor nests (Fig.
2Ba3). There was no vascular structure between the
surrounding tumor cells and erythrocytes. Neither necrosis nor
fibrosis was observed in the tumor nests (Fig. 2Ba3). However, similar
phenomenon failed to occur in xenografts of SGC-996 group (Fig. 2Bb1–3) or GBC-SD+TIMP-2
group (Fig. 2Bc1–3)
with damaged cellular organelles, cell necrosis, nuclear pyknosis,
fragmentation and apoptotic bodies (Fig. 2Bb3 and c3).
These findings demonstrated VM in GBC-SD nude mouse xenografts, was
concordant with the results in vivo and in clinical report
by us (34,36). Additionally, TIMP-2 was able to
inhibit the VM formation of GBC-SD xenografts in nude mice in
vivo.
Expression of MMP-2, MT1-MMP
proteins/mRNAs in vitro and in vivo
Expression of MMP-2 and MT1-MMP proteins/mRNAs from
sections and supernates of 3-D culture samples in vitro and
from sections of tumor xenografts in vivo was shown in
Figs. 3 and 4. The positive expression site of MMP-2
and MT1-MMP proteins presented yellow-brown reactant in the
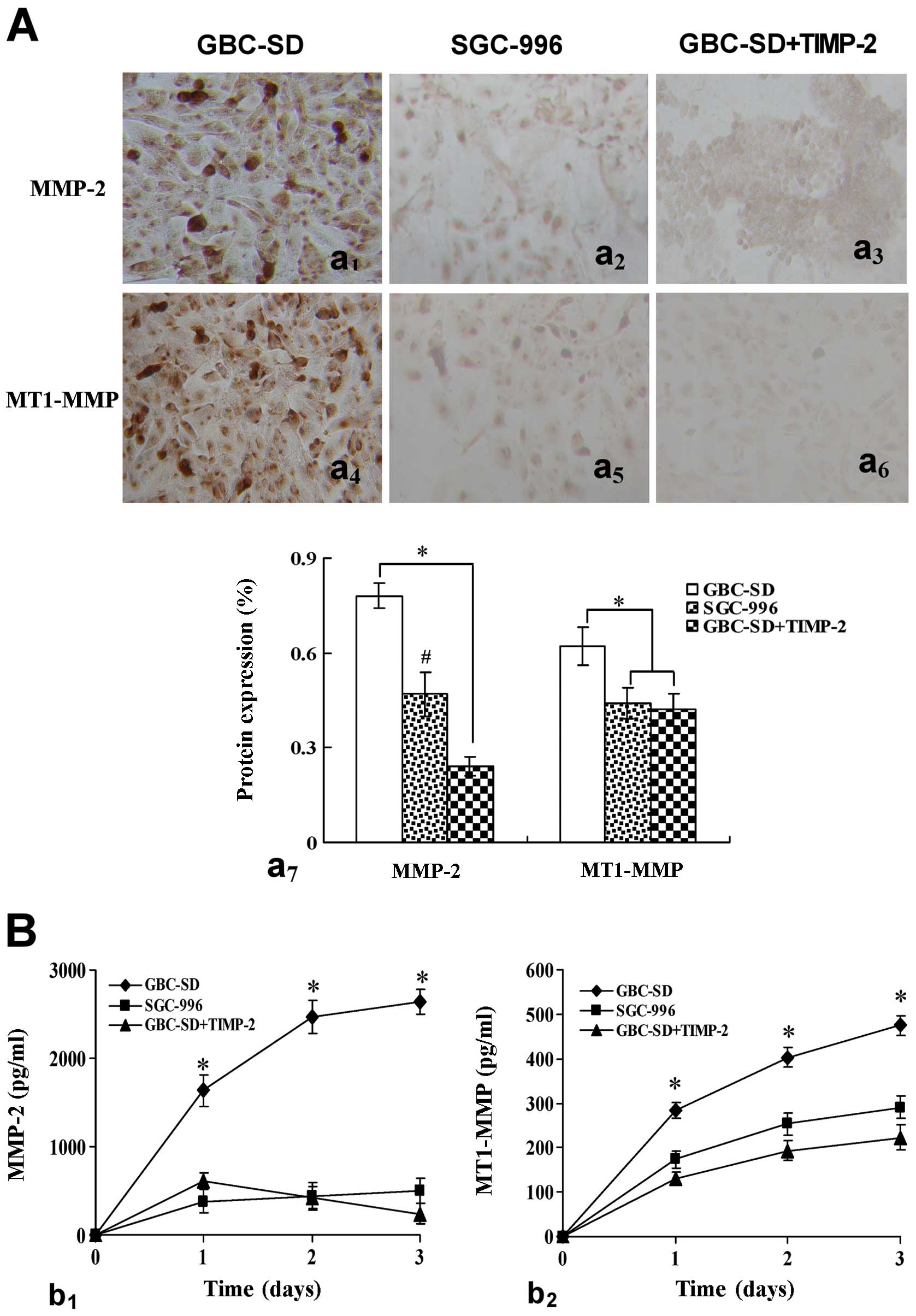
cytoplasm. Overexpression of MMP-2 (Fig. 3Aa1,7) and MT1-MMP
(Fig. 3Aa4,7) proteins
in GBC-SD group was observed in vitro. Expression of MMP-2
and MT1-MMP proteins in SGC-996 group (Fig. 3Aa2,5,7) and
GBC-SD+TIMP-2 (Fig.
3Aa3,6,7) group was significantly decreased
(*P<0.001, #P<0.01, vs. GBC-SD group).
Moreover, expression of MMP-2 (Fig.
3Bb1) and MT1-MMP (Fig.
3Bb2) proteins from supernates of 3-D culture
samples in vitro in GBC-SD group increased significantly
with time, when compared with SGC-996 group and GBC-SD+TIMP-2 group
(*P<0.001). Furthermore, overexpression of MMP-2
(Fig. 4Aa1,7 and B) and
MT1-MMP (Fig. 4Aa4,7 and
B) proteins or mRNAs from sections of tumor xenografts in
vivo in GBC-SD group was also observed; expression of MMP-2 and
MT1-MMP proteins or mRNAs in SGC-996 group (Fig. 4Aa2,5,7 and B) and
GBC-SD+TIMP-2 group (Fig.
4Aa3,6,7 and B) was significantly decreased
(*P<0.001, vs. GBC-SD group). The results showed that
highly aggressive GBC-SD cells formed in vitro and in
vivo VM networks overexpressing MMP-2 and MT1-MMP; however,
poorly aggressive SGC-996 cells or GBC-SD cells treated by TIMP-2,
which did not form these networks, markedly downregurated
expression of MMP-2 and MT1-MMP. Thus, TIMP-2 effectively inhibit
expression of these proteins, inhibiting VM of GBC-SD cells in
vitro and in vivo, as to disproof that highly aggressive
GBC-SD cells formed in vitro and in vivo VM through
the upreguration of MMP-2 and MT1-MMP expression.
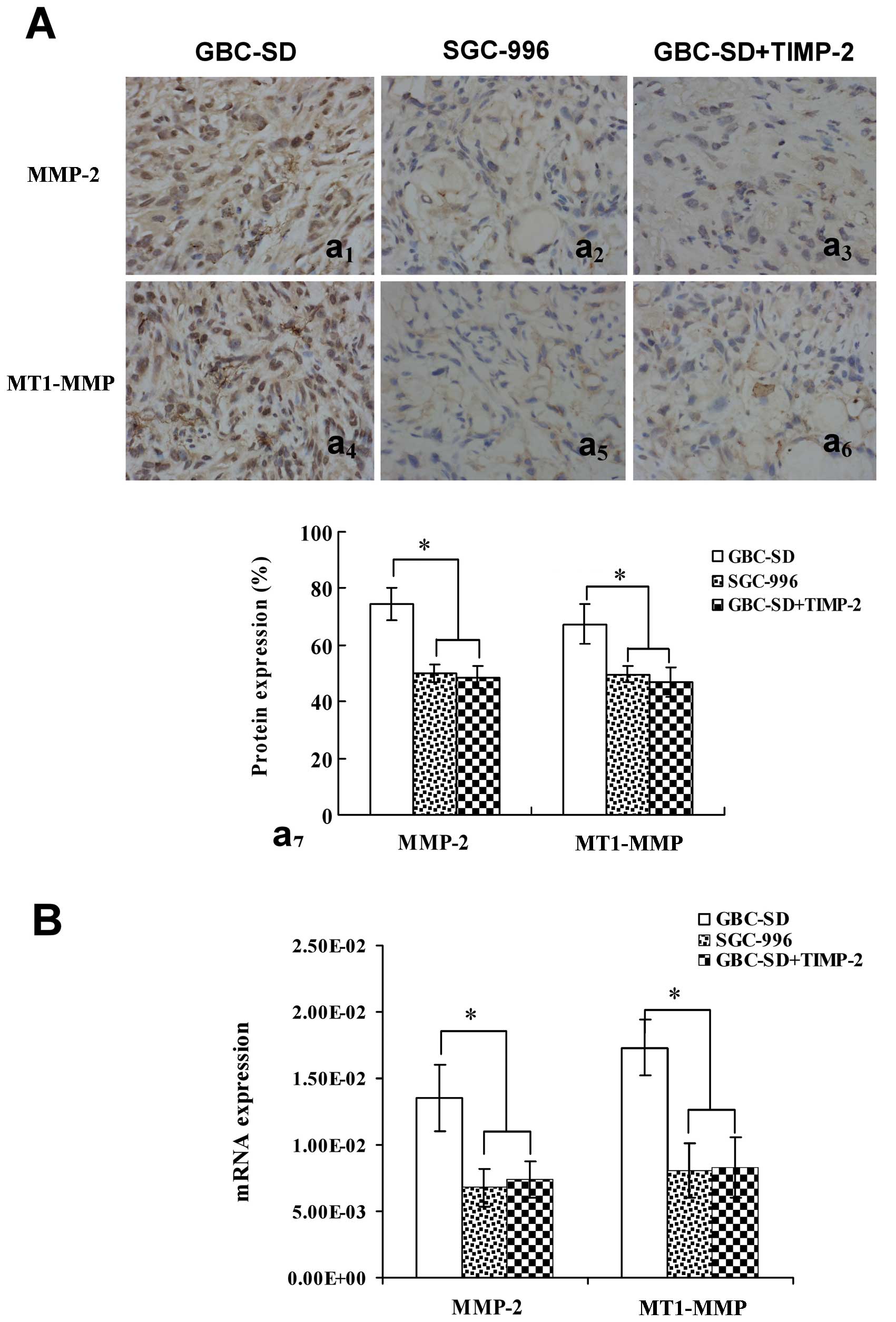 | Figure 4Expression of MMP-2, MT1-MMP
proteins/mRNAs from sections of tumor xenografts in vivo
[(A) SABC method, original magnification, ×200; (B) qRT-PCR)] in
GBC-SD, SGC-996 and GBC-SD+TIMP-2 groups. Overexpression of MMP-2
(Aa1,7) and MT1-MMP (Aa4,7) proteins or mRNA
(B) in GBC-SD group was observed in vivo. Expression of
MMP-2 and MT1-MMP proteins or mRNA in SGC-996 group
(Aa2,5,7 and B) and GBC-SD+TIMP-2 (Ac3,6,7
and B) group was significantly decreased (*P<0.001,
vs. GBC-SD group). |
Expression of EphA2, FAK, PI3K, Ln-5γ2
and Paxillin-P proteins/mRNAs of the tumor xenografts in vivo
Expression of EphA2, FAK, PI3K, Ln-5γ2 and
Paxillin-P proteins/mRNAs of the xenografts of each group in
vivo are shown in Figs. 5 and
6. Expression (bright yellow-green
fluorescent staining reactant in the cytoplasm, or western gray
value) of EphA2, FAK, PI3K, Ln-5γ2 and Paxillin-P proteins in
GBC-SD group (Figs. 5Aa1–5
and B and 6A and B) was all
upregulated markedly; however, expression of these VM
signal-related proteins in SGC-996 (Figs. 5Aa6–10 and B and
6A and B) and GBC-SD+TIMP-2
(Figs. 5Aa10–15 and B
and 6A and B) groups was
significantly downregulated (*P<0.001). Furthermore,
expression of EphA2, FAK, PI3K, Ln-5γ2 and Paxillin-P mRNAs in
GBC-SD group (Fig. 6C) was
increased significantly when compared with SGC-996 group and
GBC-SD+TIMP-2 group (*P<0.01). The results showed
that highly aggressive GBC-SD cells formed in vivo VM
networks overexpressing VM signal-related markers EphA2, FAK, PI3K,
Ln-5γ2 and Paxillin-P; poorly aggressive SGC-996 cells, which did
not form these networks, markedly downregurated expression of these
VM signal-related markers; TIMP-2 effectively inhibit expression of
these VM signal-related markers, then, as to disproof that highly
aggressive GBC-SD cells formed in vivo VM through
EphA2/FAK/Paxillin signaling and PI3K/MMPs/Ln-5γ2 signaling. Thus,
we deduced that EphA2/FAK/Paxillin and PI3K/MMPs/Ln-5γ2 signaling
pathways contributed to tumor growth and vasculogenic mimicry of
human gallbladder carcinoma GBC-SD cells in vitro and in
vivo.
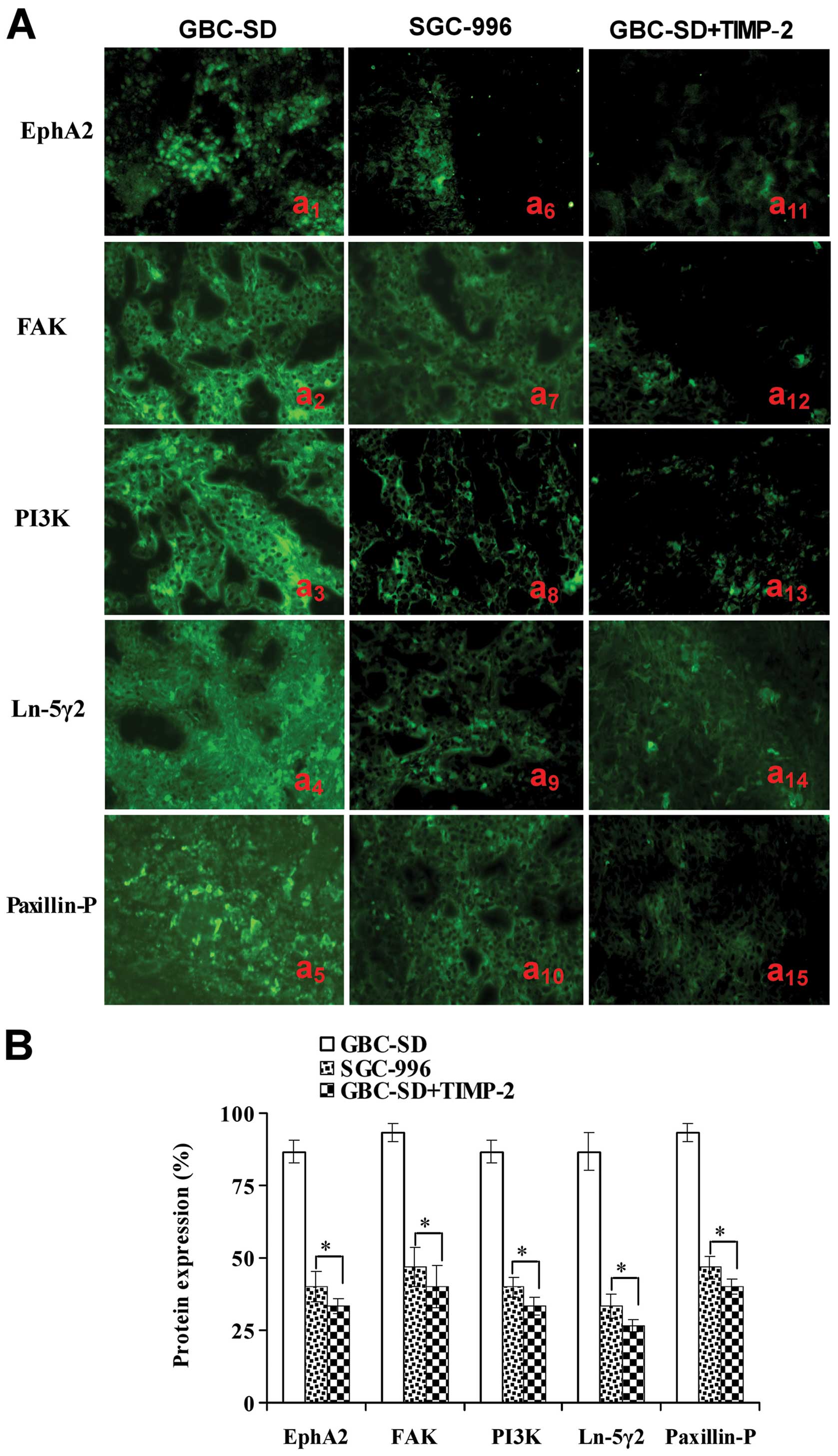 | Figure 5Expression of VM signal-related
proteins EphA2, FAK, PI3K, Ln-5γ2 and Paxillin-P of the xenografts
of each group in vivo (indirect immunofluorescence method,
original magnification, ×400). (A) Expression of EphA2, FAK, PI3K,
Ln-5γ2 and Paxillin-P proteins of the xenografts in GBC-SD
(a1–5), SGC-996 (a6–10) and GBC-SD+TIMP-2
(a11–15) groups. The positive expression site of these
proteins presented bright yellow-green fluorescent staining
reactant in the cytoplasm. Expression of these proteins in GBC-SD
group (Aa1–5 and B) was markedly upregulated. However,
expression of these proteins in SGC-996 (Aa6–10 and B)
and GBC-SD+TIMP-2 (Aa10–15 and B) groups was
significantly downregulated (*P<0.001, vs. GBC-SD
group). |
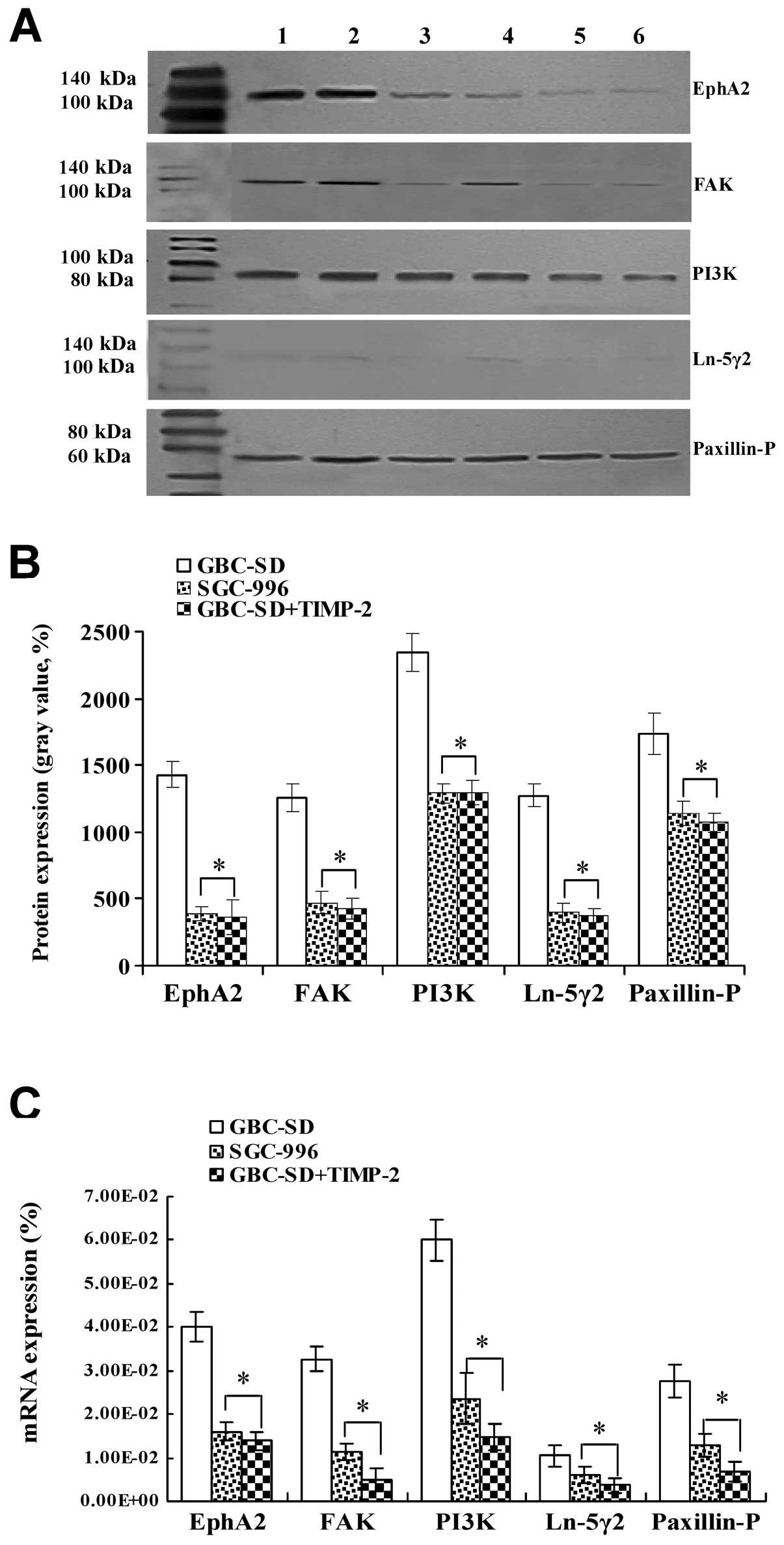 | Figure 6Expression of VM signal-related
proteins/mRNAs EphA2, FAK, PI3K, Ln-5γ2 and Paxillin-P of the
xenografts of each group in vivo [(A and B) western
blotting; lanes 1 and 2, GBC-SD group; lanes 3 and 4, SGC-996
group; lanes 5 and 6, GBC-SD+TIMP-2 group. (C) qRT-PCR)]. (A and B)
Overexpression of EphA2, FAK, PI3K, Ln-5γ2 and Paxillin-P proteins
of the xenografts in GBC-SD group was observed; but expression of
these proteins in SGC-996 or GBC-SD+TIMP-2 group was significantly
decreased (*P<0.001). (C) Expression of EphA2, FAK,
PI3K, Ln-5γ2 and Paxillin-P mRNAs of the xenografts in GBC-SD group
was increased significantly when compared with SGC-996 and
GBC-SD+TIMP-2 groups (*P<0.01). |
Discussion
VM is a novel paravascular tumor blood supply
pattern in some highly aggressive malignant tumors formed by tumor
cells instead of endothelial cells. VM describes the unique ability
of highly aggressive tumor cells to express endothelial
cell-associated genes and form ECM-rich, patterned tubular networks
when cultured on a three-dimensional matrix. We previously reported
that VM existed in human gallbladder carcinomas and correlated with
the patient’s poor prognosis (34,35).
In this study, we further investigated vasculogenic-like network
formation capability of human gallbladder carcinomas in
vitro and in vivo. The results shown that highly
aggressive GBC-SD cells were able to form vasculogenic-like network
structures when cultured on Matrigel and rat-tail collagen type I
and when injected subcutaneously into the right axilback of nu/nu
mice and then facilitated growth of tumor cells or xenografts; that
poorly aggressive SGC-996 cells were unable to form the
tubular-like structures with the same conditions; and TIMP-2 was
able to inhibit and destroy formation of VM from the 3-D culture of
GBC-SD cells in vitro and VM formation of GBC-SD xenografts
in nude mice in vivo, thus inhibiting tumor xenografts’
growth. The results were not only concordant with our previous
report (36), but also further
confirmed vasculogenic-like network formation capability of highly
aggressive GBC-SD cells in vitro and in vivo.
The molecular events underlying VM displayed by
highly aggressive malignant tumor cells, especially, aggressive
human gallbladder carcinomas remain poorly understood. Therefore,
understanding the key molecular mechanisms that regulate VM in
human gallbladder carcinomas would be an important event and
provide potential targets for new therapies of gallbladder
carcinomas. Recently, experimental evidence has shown the
importance of several key molecules or signaling pathways in the
formation of vasculogenic-like networks by aggressive malignant
tumor cells, including EphA2, FAK (25–29),
PI3K, MMPs, and Ln-5γ2 chain (21–24).
PI3K/MMPs/Ln-5γ2 signaling pathway is a key pathway
which regulate VM formation of aggressive malignant tumor cells.
PI3K, made up of four different 110-kDa catalytic subunits and a
smaller regulatory subunit, is a lipid kinase that phosphorylates
phosphatidylinositol or its derivatives on the 3-hydroxyl of the
inositol head group. The principle product of PI3K activity, PI
(3–5) -P3 acts as a binding site for many
intracellular proteins that include pleckstrin homology (PH)
domains with selectivity for this lipid. The PI3K signaling pathway
plays an integral role in many normal cellular processes, including
survival, proliferation, differentiation, metabolism and motility,
in a variety of cell types (37).
MMPs, divided into soluble MMPs and MT-MMP, is a broad family of
zinc-biding endopeptidases that participate in the ECM degradation
that accompanies cancer cell invasion, metastasis and angiogenesis
(38–41). Specifically, MT1-MMP and MMP-2 are
key mediators of invasion, metastasis, tumor angiogenesis and
recently tumor cell VM (22,42).
Numerous studies have indicated that MT1-MMP is important for
endothelial tubulogenesis in fibrin gels (43), in endothelial cell migration on 3-D
collagen gels (44) and both
MT1-MMP and MMP-2 are upregulated when endothelial cells are
cultured on a 3-D matrix (45).
Recent multiple studies have indicated that MMP-2 and MT1-MMP
expression was significantly related to VM formation in melanoma
and ovarian carcinoma cells in 3-D culture (21,24).
Microarray analysis revealed that MMPs (−1, −2, −9 and −14) were
all more highly expressed in aggressive melanoma with VM channels
compared with poorly aggressive melanoma with absence of VM
(46). The Ln-5γ2 chain, MMP-2 and
MT1-MMP act cooperatively and required highly aggressive melanoma
tumor cells to engage in VM when cultured on a three-dimensional
ECM (22). The Ln-5γ2 chain in the
ECM is able to remote VM formation (22,23).
Recent observation showed that highly aggressive melanoma tumor
cells can secrete the Ln-5γ2 chain and that the γ2 and γ2x chains,
antisense oligonucleotides to the Ln-5γ2 chain and antibodies to
MMP-2 or MT1-MMP may inhibit VM formation. Several recently
published reports have indicated that PI3K is an important adjustor
of directly affecting the cooperative interactions of MT1-MMP and
MMP-2 activity in highly aggressive melanoma tumor cells. PI3K
regulates MT1-MMP activity, which promotes the conversion of
pro-MMP into its active conformation through an interaction with
TIMP-2. Both enzymatically active MT1-MMP and MMP-2 may therefore
promote the cleavage of Ln-5γ2 chain into pro-migratory γ2 and γ2x
fragments. The deposition of these fragments into tumor
extracellular milieu may result in increased migration, invasion
and VM formation (22,23). Special inhibitors of PI3K may
impair VM formation and decrease MT1-MMP and MMP-2 activity.
Furthermore, inhibition of PI3K blocked the cleavage of Ln-5γ2
chain, resulting in decreased levels of the γ2 and γ2x promigratory
fragments (21). Similarly, in
aggressive ovarian tumor cells, MMP-2 or MT1-MMP seems to play an
important role in the VM channel. Human ovarian cancers with MMP
overexpression are more likely to have tumor cell-lined vasculature
(24). Thus, PI3K/MMPs/Ln-5γ2 may
represent the predominant targets for anti-VM of tumors and cancer
therapy. In this study, expression of MMP-2 and MT1-MMP
proteins/mRNAs from sections and supernates of 3-D culture samples
in vitro and from sections of tumor xenografts in
vivo in GBC-SD group was upregulated significantly
(P<0.001); however, expression of MMP-2 and MT1-MMP proteins/
mRNAs in SGC-996 group was significantly downregulated
(P<0.001). Furthermore, expression of PI3K and Ln-5γ2
proteins/mRNAs of the xenografts of GBC-SD group in vivo was
also upregulated markedly; however, expression of these VM
signal-related proteins in SGC-996 groups was significantly
downregulated (P<0.001). These results showed that highly
aggressive GBC-SD cells formed in vitro and in vivo
VM networks overexpressing VM signal-related markers PI3K, MMP-2,
MT1-MMP and Ln-5γ2; poorly aggressive SGC-996 cells, which did not
form these networks, markedly downregurated expression of these VM
signal-related markers. Thus, we deduced that highly aggressive
GBC-SD cells formed VM in vitro and in vivo through
the upregulation of PI3K/MMPs/Ln-5γ2 signaling, that
PI3K/MMPs/Ln-5γ2 signaling pathway contributed to tumor growth and
VM of human gallbladder carcinomas.
EphA2/FAK/Paxillin signaling pathway is another key
pathway which regulated VM formation of aggressive malignant tumor
cells. EphA2, a receptor tyrosine kinase and a member of the Eph
(ephrin receptor) family of protein tyrosine kinases (PTKs) which
could be pivotal factors of VM, has been found to play an important
role in angiogenesis and in the process of formation of VM
(24,28,47,48).
Microarray analyses revealed that EphA2 were dramatically
overexpressed in aggressive human cutaneous and uveal melanoma
cells, although not in poorly aggressive melanoma cells. Transient
knockout of EphA2 in vitro abrogated the ability of highly
aggressive melanoma cells to form the vasculogenic-like networks
(25,49). EphA2 upstream molecules regulate VM
formation. EphA2 and VEcad are colocalized at sites of cell-cell
adhesion. Knockdown of EphA2 expression does result in a
redistribution of EphA2 on the cell membrane and an inability of
the cells to form vasculogenic structures. When organized on the
cell membrane, EphA2 is capable of binding to its ligand EphA1,
resulting in the phosphorylation of EphA2. Phosphorylated EphA2
then forms an interaction with FAK, which leads to phosphorylation
and activation of FAK (49).
Additionally, EphA2 may converge to activate the PI3K (as effector
of EphA2 downstream) pathway leading to the activation of MMP-2 and
consequent cleavage of Ln-5γ2 (25–27,50,51).
Also, the localization of EphA2 in aggressive human melanoma
tissues is associated with areas containing patterns of
vasculogenic-like networks. FAK, non-receptor protein tyrosine
kinase, is a 125-kDa cytoplasmic tyrosine kinase associated with
focal adhesions and is the major protein to become tyrosine
phosphorylated after integrin activation. Recently, studies have
demonstrated FAK to be an important key mediator of the aggressive
melanoma phenotype, including VM (28,29).
FAK is phosphorylated on Tyr397 and Tyr576 in
aggressive human cutaneous and uveal melanoma cells cultured on a
3-D matrix in vitro, as well as in radial and vertical
growth phase melanomas in situ. Expression of FAK-related
non-kinase in melanoma cells, which acts to disrupt FAK signaling,
directly results in the inhibition of the aggressive phenotype, as
demonstrated by decreased invasion, migration and VM potential. FAK
signaling regulates invasion, migration and VM through two distinct
signaling pathways. Firstly, FAK signals through Erk1/2 increase
the levels of urokinase activity, thus regulating invasion of the
aggressive melanoma cells. Additionally, FAK seems to signal
through unknown downstream effectors to promote migration in
aggressive melanoma cells that may contribute to an increase of VM
potential. Secondly, Erk1/2 regulates MMP-2 and MT1-MMP activity,
thus promoting melanoma invasion and VM (28,29).
Collectively, these observations implicate FAK as a promoter of the
aggressive melanoma phenotype, thereby identifying it as a rational
target for therapeutic intervention of malignant melanoma. Paxillin
is a focal adhesion-associated, phosphotyrosine-containing protein
that may play a role in numerous signaling pathways. Paxillin
contains a number of motifs as docking sites that mediate
protein-protein interactions. Thus paxillin itself serves as a
docking protein to recruit signaling molecules to a specific
cellular compartment, the focal adhesions and/or to recruit
specific combinations of signaling molecules into a complex to
coordinate downstream signaling. The biological function of
paxillin coordinated signaling is likely to regulate cell spreading
and motility. Also, FAK plays an important role in tyrosine
phosphorylation of Paxillin (52).
In VM, activity of FAK, as bridging protein between EphA2 and
integrins, mediates Paxillin phosphorylation at local adhesion
sites, then regulating focal adhesion effect, increasing tumor cell
mobility, being conducive to the formation of VM (48). So, EphA2/FAK/Paxillin signaling
pathway may represent other predominant targets for anti-VM of
tumors and cancer therapy. In this study, expression (bright
yellow-green fluorescent staining reactant in cytoplast, or western
gray value) of EphA2, FAK and Paxillin-P proteins/ mRNAs of the
xenografts in GBC-SD group was upregulated markedly; however,
expression of these VM signal-related proteins in SGC-996 and
GBC-SD+TIMP-2 groups was significantly downregulated (all
P<0.001). The results showed that highly aggressive GBC-SD cells
formed in vivo VM networks overexpressing VM signal-related
markers EphA2, FAK and Paxillin-P; poorly aggressive SGC-996 cells,
which did not form these networks, significantly downregurated
expression of these VM signal-related markers. Thus, we deduced
that highly aggressive GBC-SD cells formed VM in vitro and
in vivo through the upregulation of EphA2/FAK/ Paxillin
signaling, and that EphA2/FAK/Paxillin signaling pathways also
contributed to tumor growth and VM of human gallbladder
carcinomas.
TIMP-2 is a 21-kDa protein which selectively forms a
complex with the latent proenzyme form of the 72-kDa type IV
collagenase. The secreted protein has 194 amino acid residues and
is not glycosylated. TIMP-2 inhibits at a 1:1 ratio the type IV
collagenolytic activity and the gelatinolytic activity associated
with the 72-kDa enzyme. Whereas the 72-kDa type IV collagenase is a
member of the collagenase enzyme family that has been closely
linked with the invasive phenotype of cancer cells. Both normal
cells and highly invasive tumor cells produce the 72-kDa type IV
procollagenase enzyme in a complexed form consisting of the
proenzyme and TIMP-2. The balance between activated enzyme and
available inhibitor is considered to be a critical determinant of
the matrix proteolysis associated with a variety of pathologic
processes, including tumor cell invasion. TIMP-2 is capable of
binding to both the latent and activated forms of the 72-kDa type
IV collagenase and will abolish the hydrolytic activity of all
members of the metalloproteinase family (53,54).
TIMP-2 is a potent inhibitor of cancer cell invasion through
reconstituted extracellular matrix (55,56).
TIMP-2 produced by the same tumor cells which make collagenase,
therefore, exists as a natural suppressor of invasion. Addition of
endogenous inhibitor TIMP-2 or antibodies to 72-kDa type IV
collagenase or specific antiserum against the 72-kDa type IV
collagenase achieved alteration of the type IV
collagenase-inhibitor balance, then inhibited HT-1080 cell invasion
(55). A significantly higher
concentration of TIMP-2 may effectively inhibit all of the
proteolytic activities associated with MMP-2 and/or MT1-MMP (plus
other MMPs in the culture that can bind TIMP-2). The inhibition of
either MMP-2 or MT1-MMP activity with antibodies is sufficient to
prevent formation of vasculogenic-like patterned networks (22). To determine whether MMPs,
especially MMP-2 or MT1-MMP are actively involved and required for
the vasculogenic process of 3-D culture matrices in vitro
and tumor xenografts in vivo, recombinant TIMP-2 was added
to the highly aggressive GBC-SD cells in 3-D culture matrices and
injected intratumorally into GBC-SD xenografts in vivo. The
results indicated that all of untreated GBC-SD cells and xenografts
formed patterned tubular networks within 2 weeks of seeding and
injecting and expression of MMP-2, MT1-MMP and EphA2, FAK, PI3K,
Ln-5γ2 proteins/mRNAs in these untreated GBC-SD cells and
xenografts was upregulaed to different degree; whereas TIMP-2
retarded the onset of the patterned network formation and markedly
downregurated expression of these proteins/mRNAs. Thus, we believed
that TIMP-2 inhibited VM formation of GBC-SD cells in vitro
and in vivo through two separate mechanisms. On one hand,
TIMP-2 inhibited PI3K/MMPs/Ln-5γ2 signaling pathway through
downregulation of MMP-2 and MT1-MMP expression. Inhibition of PI3K
not only reduced MT1-MMP and MMP-2 activity, but also blocked the
cleavage of Ln-5γ2 chain, resulting in decreased levels of the γ2
and γ2x promigratory fragments and impairment of VM formation. On
the other hand, TIMP-2 indirectly inhibited EphA2/FAK/Paxillin
signaling pathway through downregulation of EphA2 and FAK
expression. Inhibition of EphA2 and FAK through Erk1/2 not merely
decreased the levels of urokinase activity, thus regulating loss of
the invasive ability of aggressive GBC-SD cells, but also
downregulated MMP-2 and MT1-MMP activity, inhibiting tumor invasion
and VM (28,29). Additionally, inhibition of EphA2
did not converge to activate the PI3K pathway leading to the
activation of MMP-2 and consequently blocked cleavage of Ln-5γ2
(25–27,50,51).
Collectively, these results showed that TIMP-2 inhibited tumor
growth and VM formation of GBC-SD cells in vitro and in
vivo through diverse mechanisms; and served as to disproof that
highly aggressive GBC-SD cells formed in vitro and in
vivo VM through the upreguration of PI3K/ MMPs/Ln-5γ2
signaling, especially MMP-2 and MT1-MMP expression.
In conclusion, highly aggressive GBC-SD cells formed
VM in vitro and in vivo through the upregulation of
PI3K/ MMPs/Ln-5γ2 signaling and EphA2/FAK/Paxillin signaling.
PI3K/MMPs/Ln-5γ2 and EphA2/FAK/Paxillin signaling pathways
contributed to tumor growth and VM of human gall-bladder
carcinomas. PI3K/MMPs/Ln-5γ2 and EphA2/FAK/ Paxillin may act in a
coordinated manner as key signaling pathways in the process of
human gallbladder carcinoma VM and illustrate novel targets that
could be potentially exploited for therapeutic intervention.
Abbreviations:
|
VM
|
vasculogenic mimicry
|
|
EphA2
|
ephrin type a receptor 2
|
|
FAK
|
focal adhesion kinase
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
MMP
|
matrix metalloproteinase
|
|
MT1-MMP
|
membrane type 1-MMP
|
|
TIMP-2
|
tissue inhibitor of matrix
metalloproteinase-2
|
|
Ln-5
|
laminin 5
|
|
VE-cad
|
vascular endothelialcadherin
|
|
ECM
|
extracellular matrix
|
|
3-D culture
|
three-dimensional culture
|
|
PAS
|
periodic acid-Schiff
|
|
SABC
|
streptavidin-biotin complex
method
|
|
DAB
|
3,3-diaminob enzidine
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
TMB
|
tetramethylbenzidine
|
|
qRT-PCR
|
quantitative reverse
transcription-polymerase chain reaction
|
|
SEM
|
scanning electron microscopy
|
|
TEM
|
transmission electron microscopy
|
Acknowledgements
This study was supported by a grant
from the National Nature Science Foundation of China (no.
30672073).
References
|
1
|
Gourgiotis S, Kocher HM, Solaini L,
Yarollahi A, Tsiambas E and Salemis NS: Gallbladder cancer. Am J
Surg. 196:252–264. 2008. View Article : Google Scholar
|
|
2
|
Lazcano-Ponce EC, Miquel JF, Muñoz N,
Herrero R, Ferrecio C, Wistuba II, Alonso de Ruiz P, Aristi Urista
G and Nervi F: Epidemiology and molecular pathology of gallbladder
cancer. CA Cancer J Clin. 51:349–364. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reddy SK and Clary BM: Surgical management
of gallbladder cancer. Surg Oncol Clin North Am. 18:307–324. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li LD, Zhang SW, Lu FZ, Mu R, Sun XD and
Huangpu XM: Research on characteristics of mortality spectrum and
type composition of malignant tumors in China. Zhonghua Zhongliu
Zazhi. 19:323–328. 1997.PubMed/NCBI
|
|
5
|
Hsing AW, Gao YT, Devesa SS, Jin F and
Fraumeni JF Jr: Rising incidence of biliary tract cancers in
Shanghai, China. Int J Cancer. 75:368–370. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chakravarty KD, Yeh CN, Jan YY and Chen
MF: Factors influencing long-term survival in patients with T3
gallbladder adenocarcinoma. Digestion. 79:151–157. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Konstantinidis IT, Deshpande V, Genevay M,
Berger D, Fernandez-del Castillo C, Tanabe KK, Zheng H, Lauwers GY
and Ferrone CR: Trends in presentation and survival for gallbladder
cancer during a period of more than 4 decades: a single-institution
experience. Arch Surg. 144:441–447. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ishii H, Furuse J, Yonemoto N, Nagase M,
Yoshino M and Sato T: Chemotherapy in the treatment of advanced
gallbladder cancer. Oncology. 66:138–142. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morise Z, Sugioka A, Tanahashi Y, Okabe Y,
Ikeda M, Kagawa T and Takeura C: Treatment of patients with
unresectable advanced carcinoma of biliary tract chemotherapy and
surgical resection. Anticancer Res. 29:1783–1786. 2009.PubMed/NCBI
|
|
10
|
Mahantshetty UM, Palled SR, Engineer R,
Homkar G, Shrivastava SK and Shukla PJ: Adjuvant radiation therapy
in gallbladder cancers: 10 years experience at Tata Memorial
Hospital. J Cancer Res Ther. 2:52–56. 2006.PubMed/NCBI
|
|
11
|
Mojica P, Smith D and Ellenhorn J:
Adjuvant radiation therapy is associated with improved survival for
gallbladder carcinoma with regional metastatic disease. J Surg
Oncol. 96:8–13. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shukla PJ and Barreto SG: Gallbladder
cancer: we need to do better! Ann Surg Oncol. 16:2084–2085.
2009.
|
|
13
|
Maniotis AJ, Folberg R, Hess A, Seftor EA,
Gardner LM, Pe’er J, Trent JM, Meltzer PS and Hendrix MJ: Vascular
channel formation by human melanoma cells in vivo and in vitro:
vasculogenic mimicry. Am J Pathol. 155:739–752. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Warso MA, Maniotis AJ, Chen X, Majumdar D,
Patel MK, Shilkaitis A, Gupta TK and Folberg R: Prognostic
significance of periodic acid-Schiff-positive patterns in primary
cutaneous melanoma. Clin Cancer Res. 7:473–477. 2001.PubMed/NCBI
|
|
15
|
Shirakawa K, Wakasugi H, Heike Y, Watanabe
I, Yamada S, Saito K and Konishi F: Vasculogenic mimicry and
pseudo-comedo formation in breast cancer. Int J Cancer. 99:821–828.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sood AK, Fletcher MS, Zahn CM, Gruman LM,
Coffin JE, Seftor EA and Hendrix MJ: The clinical significance of
tumor cell-lined vasculature in ovarian carcinoma: implications for
anti-vasculogenic therapy. Cancer Biol Ther. 1:661–664. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun B, Zhang S, Zhang D, Du J, Guo H, Zhao
X, Zhang W and Hao X: Vasculogenic mimicry is associated with high
tumor grade, invasion and metastasis, and short survival in
patients with hepatocellular carcinoma. Oncol Rep. 16:693–698.
2006.PubMed/NCBI
|
|
18
|
Guzman G, Cotler SJ, Lin AY, Maniotis AJ
and Folberg R: A pilot study of vasculogenic mimicry
immunohistochemical expression in hepatocellular carcinoma. Arch
Pathol Lab Med. 131:1776–1781. 2007.PubMed/NCBI
|
|
19
|
Li M, Gu Y, Zhang Z, Zhang S, Zhang D,
Saleem AF, Zhao X and Sun B: Vasculogenic mimicry: a new prognostic
sign of gastric adenocarcinoma. Pathol Oncol Res. 16:259–266. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Baeten CI, Hillen F, Pauwels P, de Bruine
AP and Baeten CG: Prognostic role of vasculogenic mimicry in
colorectal cancer. Dis Colon Rectum. 52:2028–2035. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hess AR, Seftor EA, Seftor RE and Hendrix
MJ: Phosphoinositide 3-kinase regulates membrane Type 1-matrix
metalloproteinase (MMP) and MMP-2 activity during melanoma cell
vasculogenic mimicry. Cancer Res. 63:4757–4762. 2003.PubMed/NCBI
|
|
22
|
Seftor RE, Seftor EA, Koshikawa N, Meltzer
PS, Gardner LM, Bilban M, Stetler-Stevenson WG, Quaranta V and
Hendrix MJ: Cooperative interactions of laminin 5 gamma 2 chain,
matrix metalloproteinase-2, and membrane
type-1-matrix/metalloproteinase are required for mimicry of
embryonic vasculogenesis by aggressive melanoma. Cancer Res.
61:6322–6327. 2001.
|
|
23
|
Seftor RE, Seftor EA, Kirschmann DA and
Hendrix MJ: Targeting the tumor microenvironment with chemically
modified tetracyclines: inhibition of laminin 5 gamma2 chain
promigratory fragments and vasculogenic mimicry. Mol Cancer Ther.
1:1173–1179. 2002.
|
|
24
|
Sood AK, Fletcher MS, Coffin JE, Yang M,
Seftor EA, Gruman LM, Gershenson DM and Hendrix MJ: Functional role
of matrix metalloproteinases in ovarian tumor cell plasticity. Am J
Obstet Gynecol. 190:899–909. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hess AR, Seftor EA, Gardner LM,
Carles-Kinch K, Schneider GB, Seftor RE, Kinch MS and Hendrix MJ:
Molecular regulation of tumor cell vasculogenic mimicry by tyrosine
phosphorylation: role of epithelial cell kinase (Eck/EphA2). Cancer
Res. 61:3250–3255. 2001.PubMed/NCBI
|
|
26
|
Margaryan NV, Strizzi L, Abbott DE, Seftor
EA, Rao MS, Hendrix MJ and Hess AR: EphA2 as a promoter of melanoma
tumorigenicity. Cancer Biol Ther. 8:279–288. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hess AR, Margaryan NV, Seftor EA and
Hendrix MJ: Deciphering the signaling events that promote melanoma
tumor cell vasculogenic mimicry and their link to embryonic
vasculogenesis: role of the Eph receptors. Dev Dyn. 236:3283–3296.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hess AR and Hendrix MJ: Focal adhesion
kinase signaling and the aggressive melanoma phenotype. Cell Cycle.
5:478–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hess AR, Postovit LM, Margaryan NV, Seftor
EA, Schneider GB, Seftor RE, Nickoloff BJ and Hendrix MJ: Focal
adhesion kinase promotes the aggressive melanoma phenotype. Cancer
Res. 65:9851–9860. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ruf W, Seftor EA, Petrovan RJ, Weiss RM,
Gruman LM, Margaryan NV, Seftor RE, Miyagi Y and Hendrix MJ:
Differential role of tissue factor pathway inhibitors 1 and 2 in
melanoma vasculogenic mimicry. Cancer Res. 63:5381–5389.
2003.PubMed/NCBI
|
|
31
|
Wang JY, Sun T, Zhao XL, Zhang SW, Zhang
DF, Gu Q, Wang XH, Zhao N, Qie S and Sun BC: Functional
significance of VEGF-a in human ovarian carcinoma: role in
vasculogenic mimicry. Cancer Biol Ther. 7:758–766. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ge CY and Fan YZ: Vasculogenic mimicry and
its molecules signaling pathways. Chin Med Abstr (Surg).
15:344–350. 2006.
|
|
33
|
Fan YZ and Sun W: Molecular regulation of
vasculogenic mimicry in tumors and potential tumor-target therapy.
World J Gastrointest Surg. 2:117–127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fan YZ, Sun W, Zhang WZ and Ge CY:
Vasculogenic mimicry in human primary gallbladder carcinoma and
clinical significance thereof. Zhonghua Yi Xue Za Zhi. 87:145–149.
2007.PubMed/NCBI
|
|
35
|
Sun W, Shen ZY, Zhang H, Fan YZ, Zhang WZ,
Zhang JT, Lu XS and Ye C: Overexpression of HIF-1α in primary
gall-bladder carcinoma and its relation to vasculogenic mimicry and
unfavourable prognosis. Oncol Rep. 27:1990–2002. 2012.
|
|
36
|
Sun W, Fan YZ, Zhang WZ and Ge CY: A pilot
histomorphology and hemodynamic of vasculogenic mimicry in
gallbladder carcinomas in vivo and in vitro. J Exp Clin Cancer Res.
30:462011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Link W, Rosado A, Fominaya J, Thomas JE
and Carnero A: Membrane localization of all class I PI3-kinase
isoforms suppresses c-Myc-induced apoptosis in Rat1 fibroblasts via
Akt. J Cell Biochem. 95:979–989. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
McCawley LJ and Matrisian LM: Matrix
metalloproteinases: multifunctional contributors to tumor
progression. Mol Med Today. 6:149–156. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Noel A, Gilles C, Bajou K, Devy L, Kebers
F, Lewalle JM, Maquoi E, Munaut C, Remacle A and Foidart JM:
Emerging roles for proteinases in cancer. Invasion Metastasis.
17:221–239. 1997.PubMed/NCBI
|
|
40
|
Stetler-Stevenson WG: Matrix
metalloproteinases in angiogenesis: a moving target for therapeutic
intervention. J Clin Invest. 103:1237–1241. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Seiki M: Membrane-type matrix
metalloproteinases. APMIS. 107:137–143. 1999. View Article : Google Scholar
|
|
42
|
Chang C and Werb Z: The many faces of
metalloproteases: cell growth, invasion, angiogenesis and
metastasis. Trends Cell Biol. 11:S37–S43. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lafleur MA, Handsley MM, Knauper V, Murphy
G and Edwards DR: Endothelial tubulogenesis within fibrin gels
specifically requires the activity of membrane-type-matrix
metalloproteinases (MT-MMPs). J Cell Sci. 115:3427–3438.
2002.PubMed/NCBI
|
|
44
|
Koike T, Vernon RB, Hamner MA, Sadoun E
and Reed MJ: MT1-MMP, but not secreted MMPs, influences the
migration of human microvascular endothelial cells in 3-dimensional
collagen gels. J Cell Biochem. 86:748–758. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Haas TL, Davis SJ and Madri JA:
Three-dimensional type I collagen lattices induce coordinate
expression of matrix metalloproteinases MT1-MMP and MMP-2
inmicrovascular endothelial cells. J Biol Chem. 273:3604–3610.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hendrix MJ, Seftor EA, Kirschmann DA,
Quaranta V and Seftor RE: Remodeling of the microenvironment by
aggressive melanoma tumor cells. Ann NY Acad Sci. 995:151–161.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Brantley DM, Cheng N, Thompson EJ, Lin Q,
Brekken RA, Thorpe PE, Muraoka RS, Cerretti DP, Pozzi A, Jackson D,
Lin C and Chen J: Soluble Eph A receptors inhibit tumor
angiogenesis and progression in vivo. Oncogene. 21:7011–7026. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cheng N, Brantley DM, Liu H, Lin Q,
Enriquez M, Gale N, Yancopoulos G, Cerretti DP, Daniel TO and Chen
J: Blockade of EphA receptor tyrosine kinase activation inhibits
vascular endothelial cell growth factor-induced angiogenesis. Mol
Cancer Res. 1:2–11. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Miao H, Burnett E, Kinch M, Simon E and
Wang B: Activation of EphA2 kinase suppresses integrin function and
causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol.
2:62–69. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hendrix MJ, Seftor EA, Meltzer PS, Gardner
LM, Hess AR, Kirschmann DA, Schatteman GC and Seftor RE: Expression
and functional significance of VE-cadherin in aggressive human
melanoma cells: role in vasculogenic mimicry. Proc Natl Acad Sci
USA. 98:8018–8023. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hess AR, Seftor EA, Gruman LM, Kinch MS,
Seftor RE and Hendrix MJ: VE-cadherin regulates EphA2 in aggressive
melanoma cells through a novel signaling pathway: implications for
vasculogenic mimicry. Cancer Biol Ther. 5:228–233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Schaller MD: Paxillin: a focal
adhesion-associated adaptor protein. Oncogene. 20:6459–6472. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Stetler-Stevenson WG, Krutzsch HC and
Liotta LA: TIMP-2, a new member of the metalloproteinase inhibitor
family. J Biol Chem. 264:17374–17378. 1989.PubMed/NCBI
|
|
54
|
Goldberg GI, Marmer BL, Grant GA, Eisen
AZ, Wilhelm S and He C: Human 72-kDa type IV collagenase forms a
complex with a tissue inhibitor of metalloproteinase inhibitor.
Proc Nati Acad Sci USA. 86:8207–8211. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Albini A, Melchiori A, Santi L, Liotta LA,
Brown PD and Stetler-Stevenson WG: Tumor cell invasion inhibited by
TIMP-2. J Natl Cancer Inst. 83:775–779. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liotta LA and Stetler-Stevenson WG: Tumor
invasion and metastasis: an imbalance of positive and negative
regulation. Cancer Res. 51(Suppl): S5054–S5059. 1991.PubMed/NCBI
|