Introduction
Glioblastoma multiforme (GBM) is the most common and
the most aggressive brain tumor with a median survival of only 15
months (1,2). Despite conjugated surgery,
radiotherapy and chemotherapy most patients die within the first
year of diagnosis (3,4). The molecular mechanisms implicated in
the resistance of glioblastoma to chemotherapies and radiotherapies
overlap with those implicated in oncogenesis (5). Among those, the PI3K/AKT pathway
which is implicated in regulation of cell proliferation, cell
cycle, survival, apoptosis, migration and angiogenesis, is a major
one (6–16).
The activation of the AKT pathway promotes the
transition from anaplastic astrocytoma to glioblastoma (17), is correlated to histological
malignant evolution and is a negative prognosis factor (18,19).
Moreover, the intrinsic radioresistance of glioblastoma is
correlated with activation levels of AKT (15) and the activation of AKT confers
them radioresistance (7). During
carcinogenesis, the activation of the AKT pathway mainly occurs by
the gain of activity of upstream activators such as EGFR (12,20–23),
or by the loss of activity of an upstream inhibitor, PTEN (7,24,25).
PTEN dephosphorylates PIP3 into PIP2 via its lipid-phosphatase
activity and decreases the level of the phosphorylated active form
of AKT (24,26).
During gliomagenesis, the AKT pathway is also
frequently activated (27,28) and PTEN disrupted (29–31).
Consequently the inhibition of AKT by either PTEN re-expression or
PI3K inhibitors impairs DNA repair and radiosensitizes glioblastoma
(13,15,32,33).
Telomerase is a specific reverse transcriptase that
elongates the telomeres, enables unlimited proliferation of cancer
cells and is currently related to their radioresistance (34–36).
Consequently telomerase inhibition shortens telomeres and
radiosensitizes cells (37).
Telomerase is reactivated in 80–100% of glioblastomas (38) and its levels are correlated with
the pathological grade and the prognosis of the tumor (38–42).
This suggests that telomerase might also intervene in the
radioresistance of glioblastomas by either triggering telomere
maintenance and/or chromosome healing (43). Consequently telomere targeting or
telomerase inhibition radiosensitizes glioblastoma cell lines
(11,44–46).
The evidenced importance of telomerase activity in the biology and
the clinical outcomes of gliomas points out this enzyme as an
appropriate therapeutic target for the radiosensitization of
glioblastomas.
Interestingly, the telomerase activity is directly
regulated by AKT either by phosphorylation of the hTERT subunit
(47) or by both
post-translational and transcriptional mechanisms (48,49).
Furthermore, ionizing radiation increases the telomerase activity
in various cancer cell lines (35,50–53)
by a post-translational mechanism implicating PI3K/AKT pathway
(54). But still, the upregulation
of telomerase activity induced by ionizing radiation in
glioblastoma cells (46) remains
to be linked to PTEN/PI3-kinase/AKT pathway.
As both PI3K/AKT and telomerase appear to be
potential targets for cancer therapy and radio-sensitization of
brain cancers (5,11,15,16,43,45,55–57),
we decided to study the links between telomerase activity and AKT
pathway in human glioblastomas in order to challenge the idea of a
‘killing two birds with one stone’ radio-sensitizing strategy.
Therefore, we evaluated the effects of a specific
PI3K inhibitor (Ly-294002) (58)
in the radioresponse of two telomerase positive high-grade glioma
cell lines: CB193 (grade III WHO) a PTEN null one (59,60)
and a T98G (grade IV WHO) a PTEN harbouring one (61,62).
Materials and methods
Cell culture
Human malignant glioma cell lines CB193
(astrocytoma, grade III) (59) and
T98G (glioblastoma multiforme, grade IV) (61,62)
were kindly provided by Dr G. Gras (CEA, France). Cultures
(5×105 cells/flask) were maintained in DMEM medium (Life
Technologies, Grand Island, NY, USA) supplemented with 10% fetal
bovine serum (Life Technologies), 2 mM glutamine (Sigma, St. Louis,
MO, USA) and antibiotics (penicillin, 100 U/ml and streptomycin,
100 μg/ml; Sigma), in a 5% CO2 atmosphere at
37°C. Cells were collected by trypsin treatment and counted using
trypan blue. Ly-294002 (Ly, Biomol) a potent inhibitor of
phosphoinositol 3-kinase (PI3K) was dissolved in DMSO (Sigma) and
stored at −20°C. This solution was diluted in culture medium 24 h
after seeding to treat cultures during exponential asynchronous
growth to a final concentration of 50 μM. Control cells were
treated with the corresponding concentration of DMSO (0.2%). Cells
were γ-irradiated during exponential asynchronous growth at 2 or 5
Gy (IBL637, CisBio International). When cells were treated with
PI3K inhibitor and γ-irradiated, 50 μM of Ly-294002 was
added to culture medium 1 h before irradiation.
Western blot analysis
Cells were lysed in ice-cold CHAPS lysis buffer. The
protein concentration was estimated in the supernatant using the
Bio-Rad protein assay according to the manufacturer’s protocol.
Lysates (30 μg for CB193 and 25 μg for T98G) were
separated by SDS-PAGE under reducing conditions before transfer
onto nitrocellulose membranes (Life Technologies). Equal protein
loading was confirmed by Ponceau staining. Blots were blocked in
TBS buffer containing 5% non-fat dried milk for 1 h at room
temperature. The membranes were incubated for 1 h at room
temperature or overnight at 4°C with the primary antibodies: rabbit
anti-γ-AKT Ser473 clone 193H12 (Cell Signaling Technology, Danvers,
MA, USA), mouse anti-AKT (Cell Signaling), mouse anti-PTEN clone
A2B1 (Becton-Dickinson, Franklin Lakes, NJ, USA) or mouse
anti-β-actin (Sigma). Membranes were then washed and incubated with
the secondary antibody (GE Healthcare, Velizy, France) for 1 h at
room temperature before washes. Detection of antibody binding was
performed by enhanced chemiluminescence according to the
manufacturer’s instructions (ECL Super Signal Western blotting
detection kit, GE Healthcare).
Colony-forming unit (CFU) assay
For CFU assay, CB193 and T98G (5×105
cells/T25 flask) were cultured for 24 h at 37°C then treated with
Ly-294002 or the corresponding concentration of DMSO (Sigma) and
γ-irradiated as described above. Cultures were incubated at 37°C
for another 24 h. Cultures were then trypsinized and counted using
Trypan blue. A fixed number of experimentally determined living
cells (600 cells for T98G, 800 cells for CB193) were re-seeded in
6-well plates in fresh culture medium without PI3K-inhibitor and
CFU (>50 cells) were stained with methylene blue and counted
after 14–20 days in culture.
Apoptosis assay
Apoptotic cells were quantified by the detection of
cleaved capsase-3 by immunostaining. Briefly, cells were grown in
8-well Lab-Tek chamber slides and fixed in 4% paraformaldehyde and
permeabilized using 0.1% Triton X-100 and 0.1% sodium citrate.
After a blocking step (7.5% goat serum and 7.5% fetal calf serum in
PBS, 1 h at room temperature), cells were incubated with a 1:200
dilution of rabbit antibody specific for the cleaved form of
caspase-3 (cleaved caspase-3 (Asp175) antibody, Cell Signaling) for
1 h at room temperature. After washings, cells were incubated with
1:125 dilution of Texas-Red-conjugated anti-rabbit IgG for 50 min
at room temperature and then counterstained with DAPI before
observation under a fluorescence microscope (Olympus BX51).
Cell cycle analysis
Cells were collected by trypsin, washed with PBS,
fixed in 80% ethanol and kept at −20°C for ≥24 h. They were then
washed in PBS and resuspended in 50 μg/ml propidium iodide
and RNase-DNase free (10 μg/ml). The cell suspension was
incubated for 30 min at room temperature and cell cycle
distribution was determined by flow cytometry (FACSCalibur, BD,
Franklin Lakes, NJ, USA), with CellQuest software analysis and
quantification using Win-MDI software.
Immunostaining
Cells were grown in 8-well Lab-Tek chamber slides
and fixed in 4% paraformaldehyde and permeabilized using 0.1%
Triton X-100 and 0.1% sodium citrate. After a blocking step (7.5%
goat serum and 7.5% fetal calf serum in PBS, 1 h at room
temperature), cells were incubated with the primary antibody: mouse
anti-γ-H2AX clone JBW301 (Merck Millipore, MA, USA), diluted in
blocking buffer (1:200) for 1 h at room temperature. Then, cells
were washed and incubated with Alexa-594 anti-mouse antibody (Life
Technologies) diluted in blocking buffer (1:400) for 50 min at room
temperature. After washing, cells were then counterstained with
DAPI before observation under a fluorescence microscope (Olympus
BX51).
Telomerase activity assay
Telomerase activity was assessed with the TRAPeze
ELISA Telomerase Detection kit (S7750, Merck Millipore) according
to the manufacturer’s instructions. Briefly, the cells were seeded
(2×106 cells/T75 flask) for 24 h at 37°C then treated
with Ly-294002 or the corresponding concentration of DMSO and
γ-irradiated as described above. Cultures were transferred to an
incubator at 37°C for another 24 h. Then the cells were collected
by trypsin treatment in cold PBS and counted in triplicate using
trypan blue. Cells were lysed in ice-cold CHAPS lysis buffer. After
incubation at 4°C for 30 min and a centrifugation at 16,000 g for
25 min at 4°C, cell extracts were kept frozen at −80°C. Telomerase
activity was then measured on proteins corresponding to an
experimentally fixed number of cells (234 cells CB193 and 166 cells
for T98G) in a 50-μl reaction mixture containing 10
μl of 5X TRAP reaction mix and 2 U of Taq DNA
polymerase (GE Healthcare). The reaction mixture was incubated for
30 min at 30°C. The extended products were amplified by a
polymerase chain reaction (PCR, 32 cycles at 94°C for 30 sec and at
59°C for 30 sec) on a PTC-200 thermocycler (MJ Research). The
amplification products were immobilized onto streptavidin-coated
microtitre plates and detected by an anti-DNP antibody conjugated
to horseradish peroxidase (HRP). After addition of the peroxidase
substrate (3,3′, 5, 5′-tetramethylbenzidine), the amount of TRAP
products was determined by measuring the absorbance at 450 and 690
nm. Telomerase activity was semi-quantified using an internal
standard curve.
Statistical analysis
All statistical analyses were performed using the
StatView software (Abcus Concepts) and Student’s t-test was used to
evaluate the statistical significance of mean values between
conditions. In each figure error bars represent standard error of
the mean and statistical significance levels are noted as follows:
*P<0.05, **P<0.01,
***P<0.001.
Results
Ly-294002 radiosensitizes glioma cell
lines
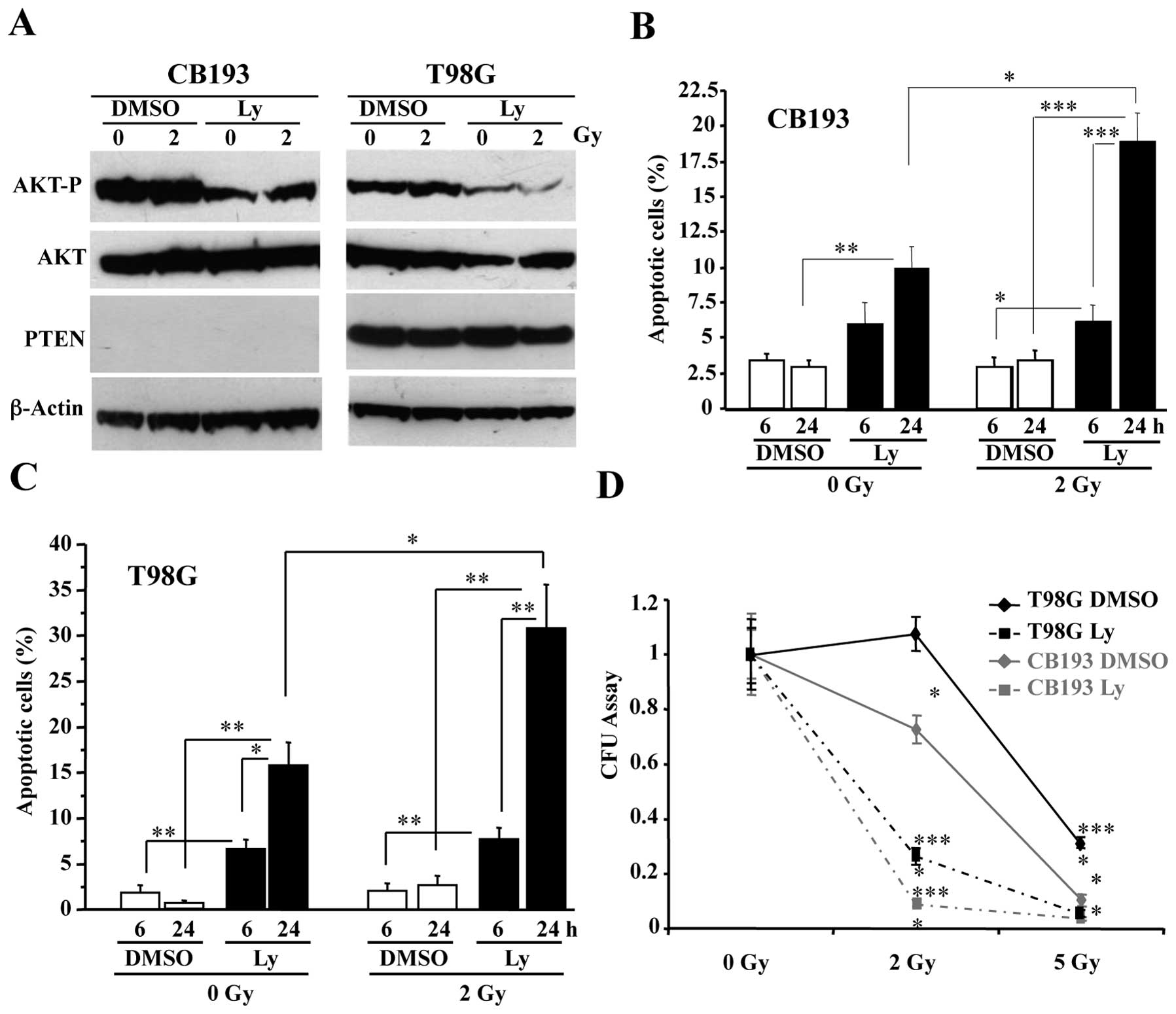
As shown in Fig.
1A, treatment with 50 μM Ly-294002 resulted in a
significant dephosphorylation of AKT in both CB193 and T98G glioma
cell lines, but 2-Gy radiation had no detectable effect on AKT
phosphorylation. Consistent with the importance of AKT
phosphorylation for cell survival, immuno-detection of
cleaved-caspase-3 showed that apoptosis increased in
Ly-294002-treated cultures (Fig. 1B
and C). Moreover, 2-Gy radiation did not significantly induce
apoptosis in DMSO-treated glioma cell lines, but nearly doubled
apoptosis levels in Ly-294002-treated cells 24 h after irradiation
(PI) (30.9±4.6 vs 15.7±2.6% in T98G cells and 18.9±2.0 vs. 9.2±1.5%
in CB193 cells), showing that Ly-294002 radiosensitizes glioma cell
lines.
This was further confirmed by determining the
capacity of irradiated glioma cells to form colonies after a 24 h
treatment with 50 μM Ly-294002 or with DMSO in a CFU assay
(Fig. 1D). Ly-294002 strongly
decreased the clonogenicity of 2-Gy-irradiated CB193 and T98G
cells, whereas 2-Gy radiation alone had no (T98G) or only a
moderate (CB193) effect on DMSO-treated glioma cell clonogenicity.
Radiosensitization by Ly-294002 was also observed in T98G cells
after 5 Gy, a dose that was sufficient to abolish CB193
clonogenicity.
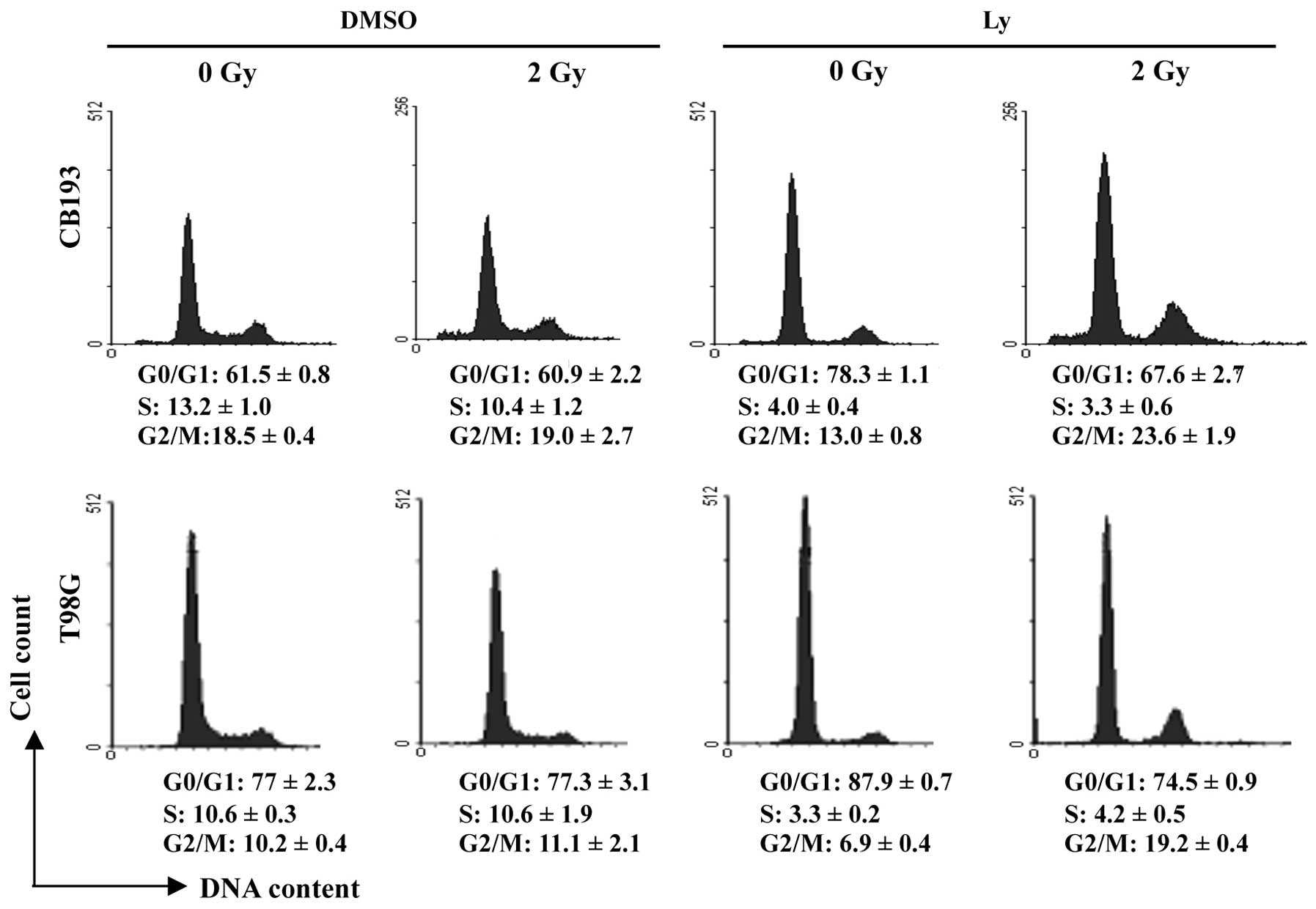
Radiation-induced G2/M arrest in
Ly-294002-treated glioma cells
The PI3K/AKT pathway plays multiple roles in cell
cycle progression (63). Measuring
DNA content by flow cytometry showed that Ly-294002 induced a G1
arrest in glioma cells, consistently with the requirement of
PI3K/AKT pathway for G1/S transition that has been previously
reported in many cell types (63).
Consistent with the little or absent effect of 2-Gy
radiation on glioma cell viability, as shown above (Fig. 1D), the cell cycle progression was
not altered in irradiated DMSO-treated cells (Fig. 2). Besides, a significant decrease
in S phase cells showed that Ly-294002 blocked G1/S transition in
irradiated cultures similarly to the non-irradiated ones. Moreover,
irradiation induced an increase in G2/M cells in Ly-294002-treated
cultures, which was more pronounced in T98G than in CB193 cells.
These data revealed that, besides its effects at the G1/S
transition, Ly-294002 also inhibited cell cycle progression at the
G2/M transition after radiation-induced DNA damage.
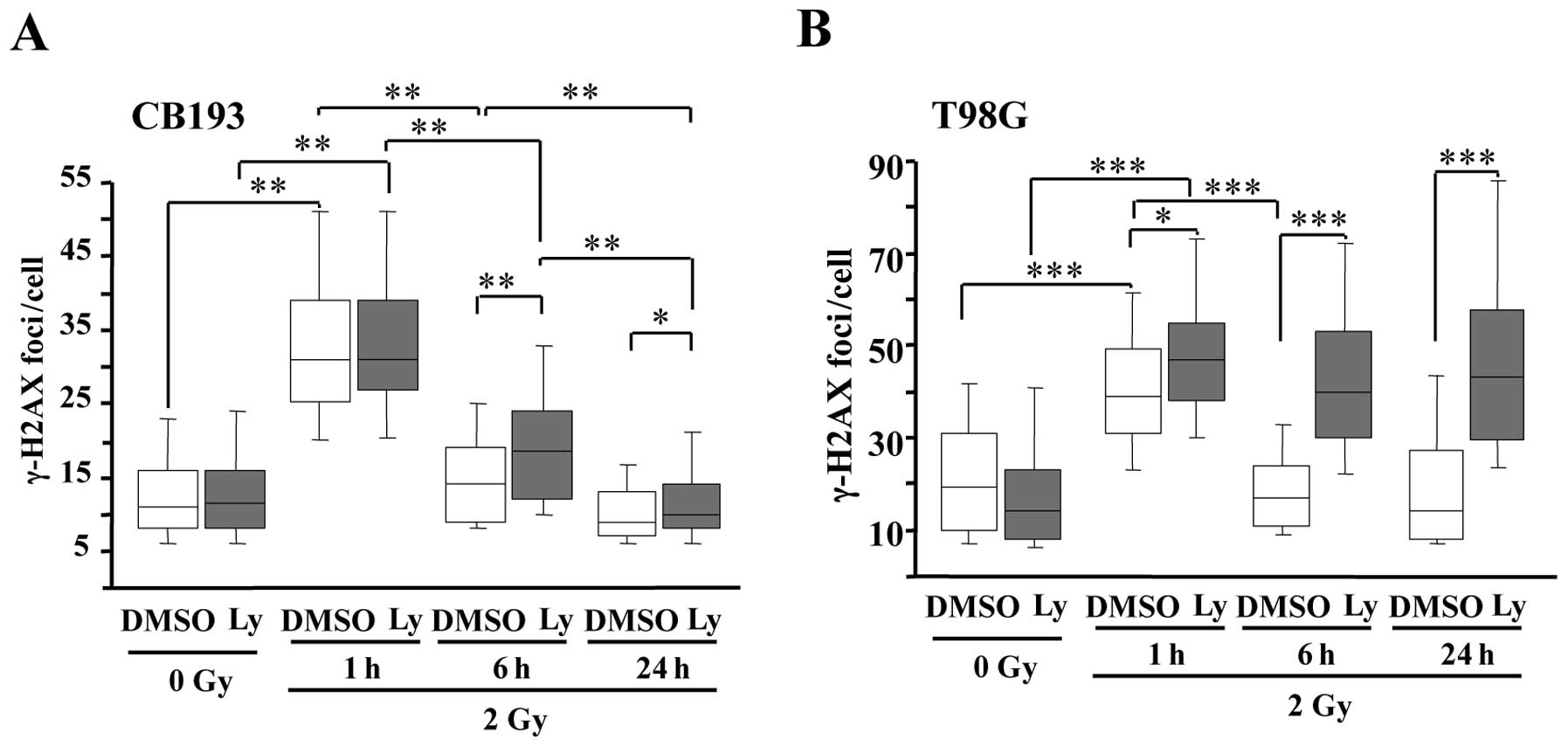
Ly-294002 delays DNA double strand break
(DSB) repair
DNA damage and repair can be evaluated by
quantifying γ-H2AX nuclear foci (64,65).
H2AX is a member of the nucleosome core histone H2A family, which
is recruited and phosphorylated on serine 139 in chromatin
surrounding the site of double strand breaks (DSBs) by kinases of
the PI-3K family, ATM, DNA-PKcs or ATR (66,67).
In both CB193 and T98G cells, 2-Gy irradiation induced a
significant increase in γ-H2AX foci at 1 h PI, which returned to
basal levels at 6 h PI, revealing no difference in the kinetics of
DNA repair between the two glioma cell lines. Ly-294002 did not
modify the number of γ-H2AX foci at 1 h PI in irradiated cells
(Fig. 3). This confirms that PI3K
inhibition does not prevent DSB signaling at the concentration we
used in agreement with previous studies (13,68).
By contrast, Ly-294002 inhibited the decrease in γ-H2AX foci in
irradiated T98G cells at 6 and 24 h PI, suggesting that PI3K
inhibition suppressed DSB repair. Ly-294002 had smaller effects on
CB193 since the number of foci was only slightly increased at 6 h
PI in Ly-294002-treated cells compared with DMSO treated controls
and recovered its basal level at 24 h PI. Altogether these data
evidenced difference in the effects of Ly-294002 on DNA repair
between the two cell lines. As we have shown above, the compound
had similar effects on apoptosis induction and clonogenicity of the
two glioma stem cells after irradiation, thus our data suggest that
the radiosensitization by Ly-294002 is not strictly related to its
effects on DNA repair.
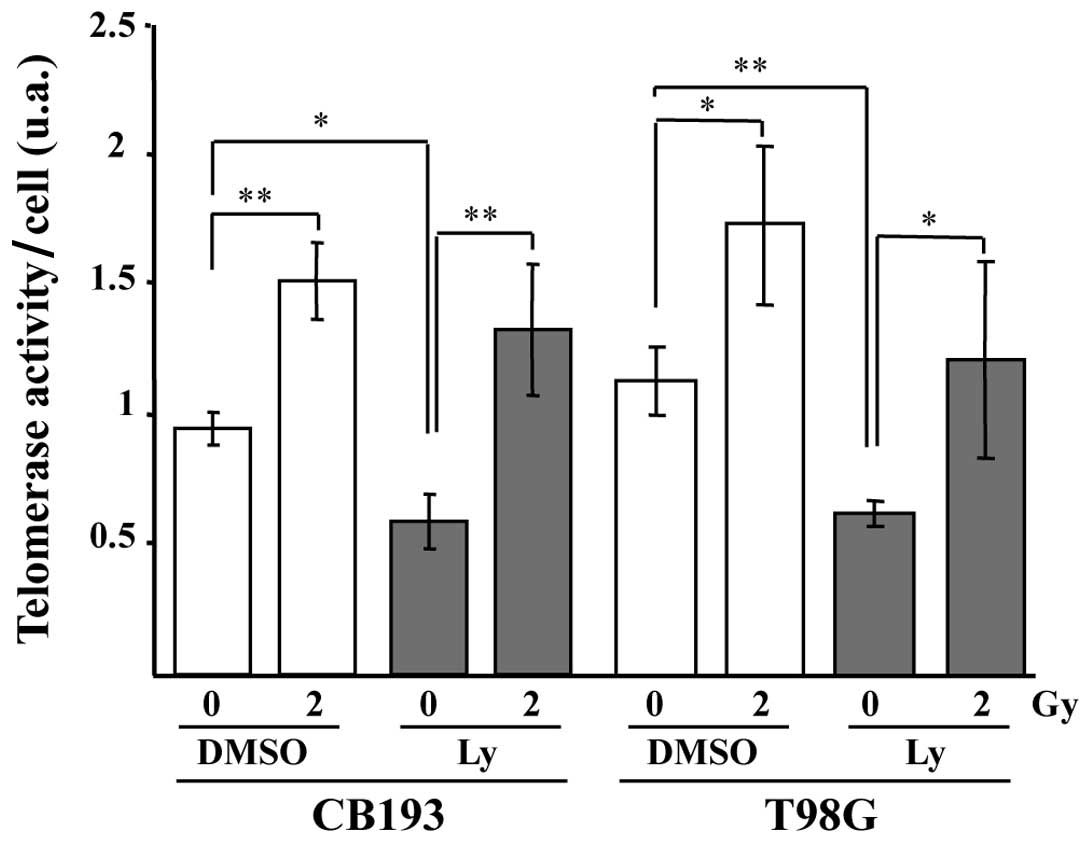
Ly-294002 does not prevent
radiation-induced upregulation of telomerase activity
PI3K inhibition induced by Ly-294002 decreases the
telomerase activity (Fig. 4) and
dephosphorylates AKT in both sham-irradiated CB193 and T98G,
suggesting that telomerase activity could be regulated by PI3K and
AKT phosphorylation in glioblastomas, as in many cell types
(47,49). Therefore, PI3K/AKT seems to
regulate at least partly basal telomerase activity in our
model.
We also found that radiation significantly increased
telomerase activity in both CB193 and T98G at 24 h PI (Fig. 4). However, whereas Ly-294002
significantly decreased telomerase activity in unirradiated glioma
cells, it failed to prevent the radiation-induced increase in
telomerase activity in irradiated cells, ruling out a role of the
PI3K/AKT pathway in the radiation-induced upregulation of
telomerase activity in our model.
Discussion
The PI3-kinase/AKT pathway is more and more regarded
as an interesting therapeutic target for the radiosensitization of
glioblastoma, but the mechanisms of radiosensitization resulting
from the inhibition of the PI3K/AKT pathway remain still unclear.
Its inhibition has been reported to impair DNA repair in
glioblastoma cells following ionizing radiation, thereby blocking
cell cycle progression and cell death (13). In this study, we have shown that
the radiosensitization of two glioma cell lines by the PI3K
inhibitor, Ly-294002, correlated with the induction of G1 and G2/M
arrests, but was inconsistently linked to a delayed DSBs repair.
The PI3K/AKT pathway has been also shown to activate
radioprotective factors such as telomerase, which inhibition may
contribute to radiosensitization (11,44–46).
However, we have shown that radiation upregulated telomerase
activity in Ly-294002-treated glioma cells as well as in untreated
controls, regardless of their PTEN status, evidencing a PI3K/AKT
independent pathway of telomerase activation. High-grade gliomas
are known for their inter- and intra-patient heterogeneity. They
express diversely telomerase activity and telomerase sub-units, but
this expression is strongly correlated to their progression in
malignancy and a poor clinical outcome (38,39,42,69–71).
Our study tends to indicate that the strategy of radiosensitization
of high-grade gliomas should combine different approaches and
should be adapted to the individual characteristics of the tumor
especially regarding their telomerase status.
Numerous previous reports have shown that inhibition
of the PI3K/AKT pathways radiosensitize gliomas (13,15,32,33),
consistently with the activation of PI3K/AKT conferring
radioresistance (7). Ionizing
radiation has been shown to increase Akt phosphorylation in various
cell lines including gliomas (32,72).
However, we did not find any radiation-increase of AKT
phosphorylation in our two glioma cells, consistently with the
study by Li et al(32)
showing that AKT phosphorylation occurred only in a subset of
glioblastoma cells.
Ly-294002 induced a G1 arrest in both CB193 and T98G
cells in accordance with the importance of the PI3K/AKT signaling
for G1/S transition (73–75). Moreover, as previously reported in
other cell lines (76,77), inhibition of the PI3K/AKT pathway
resulted in an accumulation in G2/M phase, but only after
irradiation. Inhibition of the PI3K pathway has been shown to
impair DNA repair after ionizing radiation, suggesting that the
blocking at the G2/M transition and subsequent cell death may
result from an inhibition of DSB repair (13,78).
However, this is not fully sustained by our present study showing
that the G2/M arrest was correlated with a delay in DSBs repair
only in T98G but not in CB193 cells, after the treatment with
Ly-294002. Activation of AKT has been also shown to promote G2/M
transition through the activation of downstream molecules such as
cyclin B associated kinase, NF-Y, Chk1 and FOXO3A (79–81).
Our data suggest that beside possible inhibition of DNA repair
depending on the cellular context, Ly-294002 inhibits the signaling
pathway required to pass the G2/M checkpoint independently of DNA
repair completion in irradiated cells.
Irradiation has been shown to upregulate telomerase
activity in various cell lines (35,50–53)
including a glioblastoma cell line (46). AKT is able to phosphorylate hTERT,
the catalytic subunit of telomerase and activate telomerase
activity (47). Recently, AKT has
been also shown to facilitate nuclear import of hTERT (82). Moreover, ionizing radiation has
been reported to upregulate telomerase activity in cancer cell
lines by post-translational mechanism via the PI3K/AKT pathway
(54). While Ly-294002 decreased
telomerase activity in unirradiated CB193 and T98G cells,
concomitantly with AKT dephosphorylation and G1 arrest, we have
shown that it did not prevent the radiation-induced increase of
telomerase activity, which was not correlated with an increase of
AKT phosphorylation in these cell lines. These results rule out a
predominant role of the PI3K/AKT pathway in the radiation-induced
upregulation of telomerase activity in our glioma cells lines
suggesting that an alternative pathway is involved which remains to
be determined. Such AKT/PKB independent upregulation of telomerase
activity after irradiation have been already observed in other cell
lines (83) but related to delayed
DSB repair. Complementary studies of DSB repair-related molecules
are needed in our model.
Telomerase is thought to increase the radiation
resistance of cancer cells by either protecting telomeres from
fusion or by its anti-apoptotic functions or by promoting DNA
repair through its actions on the chromatin structure (11,34–36,84–87).
A telomerase antagonist, imetelstat in combination with radiation
and temozolomide had a dramatic effect on cell survival of primary
human glioblastoma tumor-initiating cells (45). Telomere targeting with a
G-quadruplex ligand, has been recently reported to enhance
radiation-induced killing of human glioblastoma cells (44).
The personalization of glioblastoma medicine around
telomere profiling in radiation therapy is already under study
(88), and could be extended to
telomerase activity. Our results showing that telomerase
upregulation was not abolished by the PI3K/AKT pathway inhibition,
suggests that personalized combined therapies associating PI3K and
telomerase inhibitors or telomere G-quadruplex ligands should be
considered to improve the radiosensitization in telomerase
expressing high-grade gliomas.
References
|
1.
|
Stupp R, Mason WP, van den Bent MJ, et al:
Radiotherapy plus concomitant and adjuvant temozolomide for
glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Stupp R, Hegi ME, Mason WP, et al: Effects
of radiotherapy with concomitant and adjuvant temozolomide versus
radiotherapy alone on survival in glioblastoma in a randomised
phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet
Oncol. 10:459–466. 2009.
|
|
4.
|
Koukourakis GV, Kouloulias V, Zacharias G,
et al: Temozolomide with radiation therapy in high grade brain
gliomas: pharmaceuticals considerations and efficacy; a review
article. Molecules. 14:1561–1577. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Kondo Y, Hollingsworth EF and Kondo S:
Molecular targeting for malignant gliomas (Review). Int J Oncol.
24:1101–1109. 2004.PubMed/NCBI
|
|
6.
|
Tanaka H, Yoshida M, Tanimura H, et al:
The selective class I PI3K inhibitor CH5132799 targets human
cancers harboring oncogenic PIK3CA mutations. Clin Cancer Res.
17:3272–3281. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Jiang Z, Pore N, Cerniglia GJ, et al:
Phosphatase and tensin homologue deficiency in glioblastoma confers
resistance to radiation and temozolomide that is reversed by the
protease inhibitor nelfinavir. Cancer Res. 67:4467–4473. 2007.
View Article : Google Scholar
|
|
8.
|
Mason WP, Belanger K, Nicholas G, et al: A
phase II study of the Ras-MAPK signaling pathway inhibitor TLN-4601
in patients with glioblastoma at first progression. J Neurooncol.
107:343–349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Fassl A, Tagscherer KE, Richter J, et al:
Notch1 signaling promotes survival of glioblastoma cells via
EGFR-mediated induction of anti-apoptotic Mcl-1. Oncogene.
31:4698–4708. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Stockhausen MT, Broholm H, Villingshoj M,
et al: Maintenance of EGFR and EGFRvIII expressions in an in vivo
and in vitro model of human glioblastoma multiforme. Exp Cell Res.
317:1513–1526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Ji XM, Xie CH, Fang MH, et al: Efficient
inhibition of human telomerase activity by antisense
oligonucleotides sensitizes cancer cells to radiotherapy. Acta
Pharmacol Sin. 27:1185–1191. 2006. View Article : Google Scholar
|
|
12.
|
Chakravarti A, Zhai G, Suzuki Y, et al:
The prognostic significance of phosphatidylinositol 3-kinase
pathway activation in human gliomas. J Clin Oncol. 22:1926–1933.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Kao GD, Jiang Z, Fernandes AM, Gupta AK
and Maity A: Inhibition of phosphatidylinositol-3-OH kinase/Akt
signaling impairs DNA repair in glioblastoma cells following
ionizing radiation. J Biol Chem. 282:21206–21212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Gallia GL, Tyler BM, Hann CL, et al:
Inhibition of Akt inhibits growth of glioblastoma and glioblastoma
stem-like cells. Mol Cancer Ther. 8:386–393. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Chautard E, Loubeau G, Tchirkov A, et al:
Akt signaling pathway: a target for radiosensitizing human
malignant glioma. Neurooncology. 12:434–443. 2010.PubMed/NCBI
|
|
16.
|
Carnero A, Blanco-Aparicio C, Renner O,
Link W and Leal JF: The PTEN/PI3K/AKT signalling pathway in cancer,
therapeutic implications. Curr Cancer Drug Targets. 8:187–198.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Sonoda Y, Ozawa T, Aldape KD, Deen DF,
Berger MS and Pieper RO: Akt pathway activation converts anaplastic
astrocytoma to glioblastoma multiforme in a human astrocyte model
of glioma. Cancer Res. 61:6674–6678. 2001.PubMed/NCBI
|
|
18.
|
Suzuki Y, Shirai K, Oka K, et al: Higher
pAkt expression predicts a significant worse prognosis in
glioblastomas. J Radiat Res. 51:343–348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Ermoian RP, Furniss CS, Lamborn KR, et al:
Dysregulation of PTEN and protein kinase B is associated with
glioma histology and patient survival. Clin Cancer Res.
8:1100–1106. 2002.PubMed/NCBI
|
|
20.
|
Choe G, Horvath S, Cloughesy TF, et al:
Analysis of the phosphatidylinositol 3′-kinase signaling pathway in
glioblastoma patients in vivo. Cancer Res. 63:2742–2746. 2003.
|
|
21.
|
Hayashi Y, Ueki K, Waha A, Wiestler OD,
Louis DN and von Deimling A: Association of EGFR gene amplification
and CDKN2 (p16/MTS1) gene deletion in glioblastoma multiforme.
Brain Pathol. 7:871–875. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Sugawa N, Ekstrand AJ, James CD and
Collins VP: Identical splicing of aberrant epidermal growth factor
receptor transcripts from amplified rearranged genes in human
glioblastomas. Proc Natl Acad Sci USA. 87:8602–8606. 1990.
View Article : Google Scholar
|
|
23.
|
Huang HS, Nagane M, Klingbeil CK, et al:
The enhanced tumorigenic activity of a mutant epidermal growth
factor receptor common in human cancers is mediated by threshold
levels of constitutive tyrosine phosphorylation and unattenuated
signaling. J Biol Chem. 272:2927–2935. 1997. View Article : Google Scholar
|
|
24.
|
Haas-Kogan D, Shalev N, Wong M, Mills G,
Yount G and Stokoe D: Protein kinase B (PKB/Akt) activity is
elevated in glioblastoma cells due to mutation of the tumor
suppressor PTEN/MMAC. Curr Biol. 8:1195–1198. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Li J, Yen C, Liaw D, et al: PTEN, a
putative protein tyrosine phosphatase gene mutated in human brain,
breast and prostate cancer. Science. 275:1943–1947. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Myers MP, Pass I, Batty IH, et al: The
lipid phosphatase activity of PTEN is critical for its tumor
supressor function. Proc Natl Acad Sci USA. 95:13513–13518. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Holland EC, Celestino J, Dai C, Schaefer
L, Sawaya RE and Fuller GN: Combined activation of Ras and Akt in
neural progenitors induces glioblastoma formation in mice. Nat
Genet. 25:55–57. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Rajasekhar VK, Viale A, Socci ND, Wiedmann
M, Hu X and Holland EC: Oncogenic Ras and Akt signaling contribute
to glioblastoma formation by differential recruitment of existing
mRNAs to polysomes. Mol Cell. 12:889–901. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Smith JS, Tachibana I, Passe SM, et al:
PTEN mutation, EGFR amplification and outcome in patients with
anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer
Inst. 93:1246–1256. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Knobbe CB, Merlo A and Reifenberger G:
Pten signaling in gliomas. Neurooncology. 4:196–211.
2002.PubMed/NCBI
|
|
31.
|
Koul D: PTEN signaling pathways in
glioblastoma. Cancer Biol Ther. 7:1321–1325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Li HF, Kim JS and Waldman T:
Radiation-induced Akt activation modulates radioresistance in human
glioblastoma cells. Radiat Oncol. 4:432009. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Nakamura JL, Karlsson A, Arvold ND, et al:
PKB/Akt mediates radiosensitization by the signaling inhibitor
LY294002 in human malignant gliomas. J Neurooncol. 71:215–222.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Turriziani M, Di Giacomo AM, Cardillo A,
et al: Residual telomerase activity: a marker of cell survival
after exposure to gamma radiation in vitro. Anticancer Res.
23:4561–4569. 2003.PubMed/NCBI
|
|
35.
|
Wang X, Liu Y, Chow LS, et al: Regulation
of telomerase activity by gamma-radiation in nasopharyngeal
carcinoma cells. Anticancer Res. 20:433–437. 2000.PubMed/NCBI
|
|
36.
|
Perez Mdel R, Dubner D, Michelin S,
Leteurtre F, Carosella ED and Gisone PA: Radiation-induced
upregulation of telomerase in KG1a cells is influenced by dose-rate
and radiation quality. Int J Radiat Biol. 78:1175–1183.
2002.PubMed/NCBI
|
|
37.
|
Wong KK, Chang S, Weiler SR, et al:
Telomere dysfunction impairs DNA repair and enhances sensitivity to
ionizing radiation. Nat Genet. 26:85–88. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Falchetti ML, Pallini R, D’Ambrosio E, et
al: In situ detection of telomerase catalytic subunit mRNA in
glioblastoma multiforme. Int J Cancer. 88:895–901. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Huang F, Kanno H, Yamamoto I, Lin Y and
Kubota Y: Correlation of clinical features and telomerase activity
in human gliomas. J Neurooncol. 43:137–142. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Harada K, Kurisu K, Tahara H, Tahara E and
Ide T: Telomerase activity in primary and secondary glioblastomas
multiforme as a novel molecular tumor marker. J Neurosurg.
93:618–625. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Fukushima T, Yoshino A, Katayama Y,
Watanabe T, Kusama K and Moro I: Prediction of clinical course of
diffusely infiltrating astrocytomas from telomerase expression and
quantitated activity level. Cancer Lett. 187:191–198. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Boldrini L, Pistolesi S, Gisfredi S, et
al: Telomerase activity and hTERT mRNA expression in glial tumors.
Int J Oncol. 28:1555–1560. 2006.PubMed/NCBI
|
|
43.
|
Wesbuer S, Lanvers-Kaminsky C,
Duran-Seuberth I, et al: Association of telomerase activity with
radio- and chemosensitivity of neuroblastomas. Radiat Oncol.
5:662010. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Merle P, Evrard B, Petitjean A, et al:
Telomere targeting with a new G4 ligand enhances radiation-induced
killing of human glioblastoma cells. Mol Cancer Ther. 10:1784–1795.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Marian CO, Cho SK, McEllin BM, et al: The
telomerase antagonist, imetelstat, efficiently targets glioblastoma
tumor-initiating cells leading to decreased proliferation and tumor
growth. Clin Cancer Res. 16:154–163. 2010. View Article : Google Scholar
|
|
46.
|
Zhou FX, Liao ZK, Dai J, et al:
Radiosensitization effect of zidovudine on human malignant glioma
cells. Biochem Biophys Res Commun. 354:351–356. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Kang SS, Kwon T, Kwon DY and Do SI: Akt
protein kinase enhances human telomerase activity through
phosphorylation of telomerase reverse transcriptase subunit. J Biol
Chem. 274:13085–13090. 1999. View Article : Google Scholar
|
|
48.
|
Zhou C, Bae-Jump VL, Whang YE, Gehrig PA
and Boggess JF: The PTEN tumor suppressor inhibits telomerase
activity in endometrial cancer cells by decreasing hTERT mRNA
levels. Gynecol Oncol. 101:305–310. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Uziel O, Fenig E, Nordenberg J, et al:
Imatinib mesylate (Gleevec) downregulates telomerase activity and
inhibits proliferation in telomerase-expressing cell lines. Br J
Cancer. 92:1881–1891. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Leteurtre F, Li X, Gluckman E and
Carosella ED: Telomerase activity during the cell cycle and in
gamma-irradiated hematopoietic cells. Leukemia. 11:1681–1689. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Finnon P, Silver AR and Bouffler SD:
Upregulation of telomerase activity by X-irradiation in mouse
leukaemia cells is independent of Tert, Terc, Tnks and Myc
transcription. Carcinogenesis. 21:573–578. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Hande MP, Balajee AS and Natarajan AT:
Induction of telomerase activity by UV-irradiation in Chinese
hamster cells. Oncogene. 15:1747–1752. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Hyeon Joo O, Hande MP, Lansdorp PM and
Natarajan AT: Induction of telomerase activity and chromosome
aberrations in human tumour cell lines following X-irradiation.
Mutat Res. 401:121–131. 1998.PubMed/NCBI
|
|
54.
|
Ram R, Uziel O, Eldan O, et al: Ionizing
radiation upregulates telomerase activity in cancer cell lines by
post-translational mechanism via ras/phosphatidylinositol
3-kinase/Akt pathway. Clin Cancer Res. 15:914–923. 2009. View Article : Google Scholar
|
|
55.
|
Lefranc F, Rynkowski M, DeWitte O and Kiss
R: Present and potential future adjuvant issues in high-grade
astrocytic glioma treatment. Adv Tech Stand Neurosurg. 34:3–35.
2009.PubMed/NCBI
|
|
56.
|
Cully M, You H, Levine AJ and Mak TW:
Beyond PTEN mutations: the PI3K pathway as an integrator of
multiple inputs during tumorigenesis. Nat Rev Cancer. 6:184–192.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Hennessy BT, Smith DL, Ram PT, Lu Y and
Mills GB: Exploiting the PI3K/AKT pathway for cancer drug
discovery. Nat Rev Drug Discov. 4:988–1004. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Vlahos CJ, Matter WF, Hui KY and Brown RF:
A specific inhibitor of phosphatidylinositol 3-kinase,
2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol
Chem. 269:5241–5248. 1994.PubMed/NCBI
|
|
59.
|
Menet A, Speth C, Larcher C, et al:
Epstein-Barr virus infection of human astrocyte cell lines. J
Virol. 73:7722–7733. 1999.PubMed/NCBI
|
|
60.
|
Pennarun G, Granotier C, Gauthier LR, et
al: Apoptosis related to telomere instability and cell cycle
alterations in human glioma cells treated by new highly selective
G-quadruplex ligands. Oncogene. 24:2917–2928. 2005. View Article : Google Scholar
|
|
61.
|
Walker SM, Leslie NR, Perera NM, Batty IH
and Downes CP: The tumour-suppressor function of PTEN requires an
N-terminal lipid-binding motif. Biochem J. 379:301–307. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Sano T, Asai A, Mishima K, Fujimaki T and
Kirino T: Telomerase activity in 144 brain tumours. Br J Cancer.
77:1633–1637. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Liang J and Slingerland JM: Multiple roles
of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell
Cycle. 2:339–345. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Olive PL: Detection of DNA damage in
individual cells by analysis of histone H2AX phosphorylation.
Methods Cell Biol. 75:355–373. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Nowak E, Etienne O, Millet P, et al:
Radiation-induced H2AX phosphorylation and neural precursor
apoptosis in the developing brain of mice. Radiat Res. 165:155–164.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Rogakou EP, Pilch DR, Orr AH, Ivanova VS
and Bonner WM: DNA double-stranded breaks induce histone H2AX
phosphorylation on serine 139. J Biol Chem. 273:5858–5868. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
67.
|
Fernandez-Capetillo O, Lee A, Nussenzweig
M and Nussenzweig A: H2AX: the histone guardian of the genome. DNA
Repair. 3:959–967. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68.
|
Stiff T, O’Driscoll M, Rief N, Iwabuchi K,
Lobrich M and Jeggo PA: ATM and DNA-PK function redundantly to
phosphorylate H2AX after exposure to ionizing radiation. Cancer
Res. 64:2390–2396. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
69.
|
Hiraga S, Ohnishi T, Izumoto S, et al:
Telomerase activity and alterations in telomere length in human
brain tumors. Cancer Res. 58:2117–2125. 1998.PubMed/NCBI
|
|
70.
|
Tchirkov A, Rolhion C, Kemeny JL, et al:
Clinical implications of quantitative real-time RT-PCR analysis of
hTERT gene expression in human gliomas. Br J Cancer. 88:516–520.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
71.
|
Shervington A, Patel R, Lu C, et al:
Telomerase subunits expression variation between biopsy samples and
cell lines derived from malignant glioma. Brain Res. 1134:45–52.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
72.
|
Hirao T, Urata Y, Kageyama K, et al:
Dehydroepiandrosterone augments sensitivity to gamma-ray
irradiation in human H4 neuroglioma cells through down-regulation
of Akt signaling. Free Radic Res. 42:957–965. 2008. View Article : Google Scholar
|
|
73.
|
Ramaswamy S, Nakamura N, Vazquez F, et al:
Regulation of G1 progression by the PTEN tumor suppressor protein
is linked to inhibition of the phosphatidylinositol 3-kinase/Akt
pathway. Proc Natl Acad Sci USA. 96:2110–2115. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
74.
|
Gottschalk AR, Basila D, Wong M, et al:
p27Kip1is required for PTEN-induced G1 growth arrest.
Cancer Res. 61:2105–2111. 2001.PubMed/NCBI
|
|
75.
|
Liang J, Zubovitz J, Petrocelli T, et al:
PKB/Akt phosphorylates p27, impairs nuclear import of p27 and
opposes p27-mediated G1 arrest. Nat Med. 8:1153–1160. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
76.
|
Rosenzweig KE, Youmell MB, Palayoor ST and
Price BD: Radiosensitization of human tumor cells by the
phosphatidylinositol3-kinase inhibitors wortmannin and LY294002
correlates with inhibition of DNA-dependent protein kinase and
prolonged G2-M delay. Clin Cancer Res. 3:1149–1156. 1997.
|
|
77.
|
Park JK, Jung HY, Park SH, et al:
Combination of PTEN and gamma-ionizing radiation enhances cell
death and G(2)/M arrest through regulation of AKT activity and p21
induction in non-small-cell lung cancer cells. Int J Radiat Oncol
Biol Phys. 70:1552–1560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
78.
|
Toulany M, Kehlbach R, Florczak U, et al:
Targeting of AKT1 enhances radiation toxicity of human tumor cells
by inhibiting DNA-PKcs-dependent DNA double-strand break repair.
Mol Cancer Ther. 7:1772–1781. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79.
|
Shtivelman E, Sussman J and Stokoe D: A
role for PI 3-kinase and PKB activity in the G2/M phase of the cell
cycle. Curr Biol. 12:919–924. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
80.
|
Lee SR, Park JH, Park EK, Chung CH, Kang
SS and Bang OS: Akt-induced promotion of cell-cycle progression at
G2/M phase involves upregulation of NF-Y binding activity in PC12
cells. J Cell Physiol. 205:270–277. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
81.
|
He L, Yang X, Cao X, Liu F, Quan M and Cao
J: Casticin induces growth suppression and cell cycle arrest
through activation of FOXO3a in hepatocellular carcinoma. Oncol
Rep. 29:103–108. 2013.PubMed/NCBI
|
|
82.
|
Chung J, Khadka P and Chung IK: Nuclear
import of hTERT requires a bipartite nuclear localization signal
and Akt-mediated phosphorylation. J Cell Sci. 125:2684–2697. 2012.
View Article : Google Scholar
|
|
83.
|
Neuhof D, Zwicker F, Kuepper JH, Debus J
and Weber KJ: Activation of telomerase by ionizing radiation:
differential response to the inhibition of DNA double-strand break
repair by abrogation of poly (ADP-ribosyl)ation, by LY294002, or by
Wortmannin. Int J Radiat Oncol Biol Phys. 69:887–894. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
84.
|
Fu W, Begley JG, Killen MW and Mattson MP:
Anti-apoptotic role of telomerase in pheochromocytoma cells. J Biol
Chem. 274:7264–7271. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
85.
|
Blackburn EH: Switching and signaling at
the telomere. Cell. 106:661–673. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
86.
|
Akiyama M, Yamada O, Kanda N, et al:
Telomerase overexpression in K562 leukemia cells protects against
apoptosis by serum deprivation and double-stranded DNA break
inducing agents, but not against DNA synthesis inhibitors. Cancer
Lett. 178:187–197. 2002. View Article : Google Scholar
|
|
87.
|
Masutomi K, Possemato R, Wong JM, et al:
The telomerase reverse transcriptase regulates chromatin state and
DNA damage responses. Proc Natl Acad Sci USA. 102:8222–8227. 2005.
View Article : Google Scholar
|
|
88.
|
Ferrandon S, Saultier P, Carras J, et al:
Telomere profiling: toward glioblastoma personalized medicine. Mol
Neurobiol. 47:64–76. 2013. View Article : Google Scholar : PubMed/NCBI
|