Introduction
Tripterygium wilfordii Hook F. has been used
for centuries in traditional Chinese medicine to treat rheumatoid
arthritis, an autoimmune disease associated with the increased
production of the pro-inflammatory cytokine, tumor necrosis factor
(TNF)-α. Triptolide, a compound originally purified from T.
wilfordii Hook F., which has potent anti-inflammatory and
immunosuppressant activities (1,2), has
been reported to exert anti-neoplastic activity mainly by inducing
apoptosis in various cancer cells (3–6). The
exact mechanism responsible for the anti-neoplastic and
anti-inflammatory effects of triptolide is not clearly
understood.
Apoptosis is mediated through at least 3 major
pathways, which are regulated by the death receptors, mitochondria
and the endoplasmic reticulum. Activation of apoptosis pathways is
a key mechanism by which cytotoxic drugs kill tumor cells and
defects in apoptosis signaling contribute to tumor cell drug
resistance (7,8). Among the important regulators of
apoptosis are the members of the Bcl-2 family. This family of
proteins includes both anti-apoptotic molecules, such as Bcl-2 and
pro-apoptotic molecules, such as Bax (9). Triptolide induces apoptosis in T-cell
hybridomas and peripheral T cells (10) and can sensitize TNF-α-resistant
tumor cell lines to TNF-α-induced apoptosis (5).
Nuclear factor-κB (NF-κB) is active in most tumor
types and regulates a series of pivotal events in the tumor
progress, including cell survival, cell proliferation, angiogenesis
and invasion (11). For cell
survival, NF-κB mainly suppresses apoptosis by increasing the
transcription of genes encoding anti-apoptotic proteins, for
instance Bcl-2 and Bcl-XL(12). Thus, suppression of the NF-κB
pathway should be effective in inducing apoptosis of tumor
cells.
Moreover, mitogen-activated protein kinases (MAPKs)
play a critical role in the regulation of cell growth and
differentiation in the control of cellular responses to stress and
cytokines. MAPK signaling pathways have also been shown to play
critical roles in tumorigenesis. The activities of MAPKs are
negatively regulated via dephosphorylation of certain conserved
tyrosine and threonine residues by a family of MAPK phosphatases
(MKPs) (13). Triptolide has been
reported to suppress the expression of MKP-1, which inactivates
extracellular signal-regulated kinase (ERK), p38 MAPK and c-Jun
N-terminal kinase (JNK), to exert its anti-proliferative and
pro-apoptotic activities (14).
In the present study, we used a human monocytic
leukemia cell line, THP-1, which had been differentiated into
macrophage-like cells by treatment with phorbol myristate acetate
(PMA). We postulated that triptolide mediated its effects through
multiple mechanisms, including activating cell cycle arrest and
caspase-dependent pathways, as well as blocking NF-κB activation
and potently activating the MAPK pathway.
Materials and methods
Cell culture
Human monocytic leukemia THP-1 cells were supplied
by the Korean Cell Line Bank. Cells were cultured in RPMI-1640
medium (Gibco, Grand Island, NY, USA) containing 10% fetal bovine
serum, 100 U/ml penicillin and 100 μg/ml streptomycin. Cells
were incubated at 37°C in a humidified atmosphere of 5%
CO2 in 95% air. THP-1 cells were treated with 100 nM of
phorbol myristate acetate (PMA, Sigma-Aldrich Co., St, Louis, MO,
USA) for 24 h to induce differentiation of the cells into
macrophages. After differentiation, non-attached cells were removed
by aspiration and the adherent macrophages were washed with
RPMI-1640 medium 3 times and then incubated in cell culture medium
at 37°C.
MTT assay
Cell proliferation was measured with CellTiter 96
Aqueous One Solution (Promega, Madison, WI, USA). Cells were seeded
at 1×104 cells per well in 96-well plates and incubated
with different concentrations of triptolide (Sigma-Aldrich) at 37°C
for 24, 48 and 72 h. Cell viability was determined using a
colorimetric assay with PMS/MTS solution. The absorbance was
determined at 490 nm, with background subtraction at 650 nm.
Cell cycle analysis
Cells (5×105) were incubated with 5, 10
and 25 nM of triptolide for 48 h. After incubation, the cells were
harvested and washed with PBS. Cells were fixed with 70% ethanol
for 1 h, treated with RNase A (20 μg/ml) at 37°C for 1 h,
before being stained with propidium iodide (50 μg/ml). DNA
content at each cell cycle stage was analyzed using a FACSCalibur
with CellQuest software (Becton-Dickinson, USA).
Apoptosis assay
For determining apoptosis in THP-1 cells, apoptotic
cells were quantified using a cell death detection
ELISAplus kit (Roche Molecular Biochemicals, Mannheim,
Germany). Cells (1×104) were incubated with 5, 10 and 25
nM of triptolide for 48 h. Cells were lysed with cell lysis buffer
(200 μl). Cell lysates were assayed for DNA fragments using
the cell death ELISAplus kit according to the
manufacturer’s protocol. DNA fragmentation was measured at 405 nm
against an untreated control. To measure the enzymatic activity of
caspase proteases, a caspase colorimetric assay kit (R&D
Systems, Inc. Minneapolis, MN, USA) was used. THP-1 cells
(2×106) were treated with 5, 10 and 25 nM of triptolide
for 48 h. Cells were harvested and cell pellets were lysed in 50
μl of lysis buffer on ice for 10 min. The proteins
concentration in the supernatant (cytosolic extract) was measured
by BCA assay. The activities of caspase-3-, -8 and -9-like
proteases were measured by proteolytic cleavage of substrates,
including DEVD-pNA (caspase-3 substrate), IETD-pNA
(caspase-8 substrate) and LEHD-pNA (caspase-9 substrate)
respectively. These colorimetry substrates were solubilized in an
assay buffer. After incubation with the substrates at 37°C for 1 h
in the dark, color production in the lysates was measured with a
microplate reader at 405 nm. Caspase-3, -8 and -9 activities were
determined by direct comparison to the level of the uninduced
control. To assess the effect of caspase inhibitor treatment, THP-1
cells (1×104 cells) were pretreated with a pan-caspase
inhibitor, Z-VAD-FMK, or a caspase-3-specific inhibitor, Z-DEVD-FMK
(R&D Systems), for 2 h, followed by addition of 50 nM
triptolide. After 48 h, cell viability was determined by a
colorimetric assay with PMS/MTS solution. The absorbance was
determined at 492 nm with background subtraction at 650 nm.
Nuclear staining with Hoechst 33258
Cells (1×105) were treated with 5, 10 and
25 nM of triptolide for 48 h and then washed with ice-cold PBS. The
cells were fixed with 4% paraformaldehyde for 10 min, permeabilized
with 0.05% Triton X-100 for 5 min and stained with 50 ng/ml of
Hoechst 33258 (Sigma-Aldrich). The nuclear areas were observed and
photographed with a fluorescent microscope and calculated with an
Olympus 13×51 fluorescence microscope equipped with a Nuance 2.1
Multispectral Imaging System (Cambridge Research &
Instrumentation, Inc., MA, USA).
RNA extraction and real-time PCR
procedures
Total RNA was purified from cultured cells using the
RNA-Bee solution kit (Tel-test, Friendswood, TX, USA), following
the manufacturer’s protocol. First-strand cDNA synthesis was then
made using 1 μg of RNA using a reverse transcriptase system
(Promega). Reverse transcription was primed using random hexamers.
The primer sequences and product sizes were as follows: cyclin D1
forward 5′-CCGTCCATGCGGAAGATC-3′, reverse 5′-ATGGCCAGCGGGAAGAC-3′,
86 bp; p21 forward 5′-CAGACCAGCATGACAGATTTC-3′, reverse 5′-TTAGGGC
TTCCTCTTGGAGA-3′, 66 bp; p27 forward 5′-CCGGCTAA CTCTGAGGACAC-3′,
reverse 5′-AGAAGAATCGTCGGT TGCAG-3′, 120 bp; Bcl-2 forward
5′-GATTGATGGGATCGT TGCCTTA, reverse 5′-CCTTGGCATGAGATGCAGGA-3′, 200
bp; Bax forward 5′-GGATGCGTCCACCAAGAAG-3′, reverse
5′-GCCTTGAGCACCAGTTTGC-3′, 216 bp; survivin forward
5′-GGCCCAGTGTTTCTTCTGCTT-3′, reverse 5′-GCAACCGGACGAATGCTTT-3′, 91
bp; β-actin forward 5′-GCGAGAAGATGACCCAGATC-3′, reverse 5′-GGATAGC
ACAGCCTGGATAG-3′, 77 bp. Real-time PCR was performed on a
StepOneplus real-time PCR system (Applied Biosystems, Foster, CA,
USA) with Power SYBR Green PCR Master Mix. PCRs were performed with
1 μl cDNA in 20-μl reaction mixtures that consisted
of 10 μl Power SYBR Green PCR Master Mix, 2 μl
primers and 7 μl PCR-grade water. The reactions were
performed with a denaturation step at 95°C for 10 min, followed by
40 cycles each consisting of 95°C for 15 sec and 60°C for 1 min.
The crossing point of target genes with β-actin was calculated
using the formula 2−(target gene − β-actin) and the
relative amounts were quantified.
Immunoblot analysis
Cells (2×106) were treated with 5, 10 and
25 nM of triptolide for 48 h. After treatment, cells were washed
with cold PBS and lysed using lysis buffer [20 mM Tris-HCl (pH
7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton,
2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM
Na3VO4, 1 μg/ml leupeptin] containing
1 mM PMSF. The protein concentration was determined by means of a
BCA protein assay according to the manufacturer’s protocol. Thirty
micrograms of protein was fractionated on 12% SDS-PAGE and
transferred by electrophoresis onto a nitrocellulose membrane. The
membranes were blocked with 5% non-fat dry milk for 1 h at room
temperature and incubated with anti-NF-κB p65, anti-p38,
anti-phosphop38, anti-MEK1/2, anti-phospho-MEK1/2, anti-ERK1,
anti-phospho-ERK1/2 (Cell Signaling Technology, Danvers, MA, USA)
and β-actin antibodies (Sigma-Aldrich) at a 1:1,000 dilution with
Tris-buffered saline containing 0.05% Tween-20 (TBS-T) at 4°C for
18 h. After washing with TBS-T for 1 h, the membranes were treated
with horseradish peroxidaseconjugated secondary antibody, diluted
1:2,500 with TBS-T, for 1 h at room temperature. After washing the
membranes with TBS-T for 1 h, proteins were detected using an
Enhanced Chemiluminescence kit (Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA). Protein expression levels were analyzed using
a Chemiluminescence Imaging system (Davinch-Chemi™, Seoul,
Korea).
Statistical analysis
Values are expressed as the mean ± SD. Student’s
t-test was used to evaluate differences between the control samples
and triptolide-treated samples. Inhibition of apoptosis was
estimated by the differences between the triptolide-treated sample
and samples treated with a combination of caspase inhibitor and
triptolide. *P<0.05 and **P<0.01 were
considered statistically significant.
Results
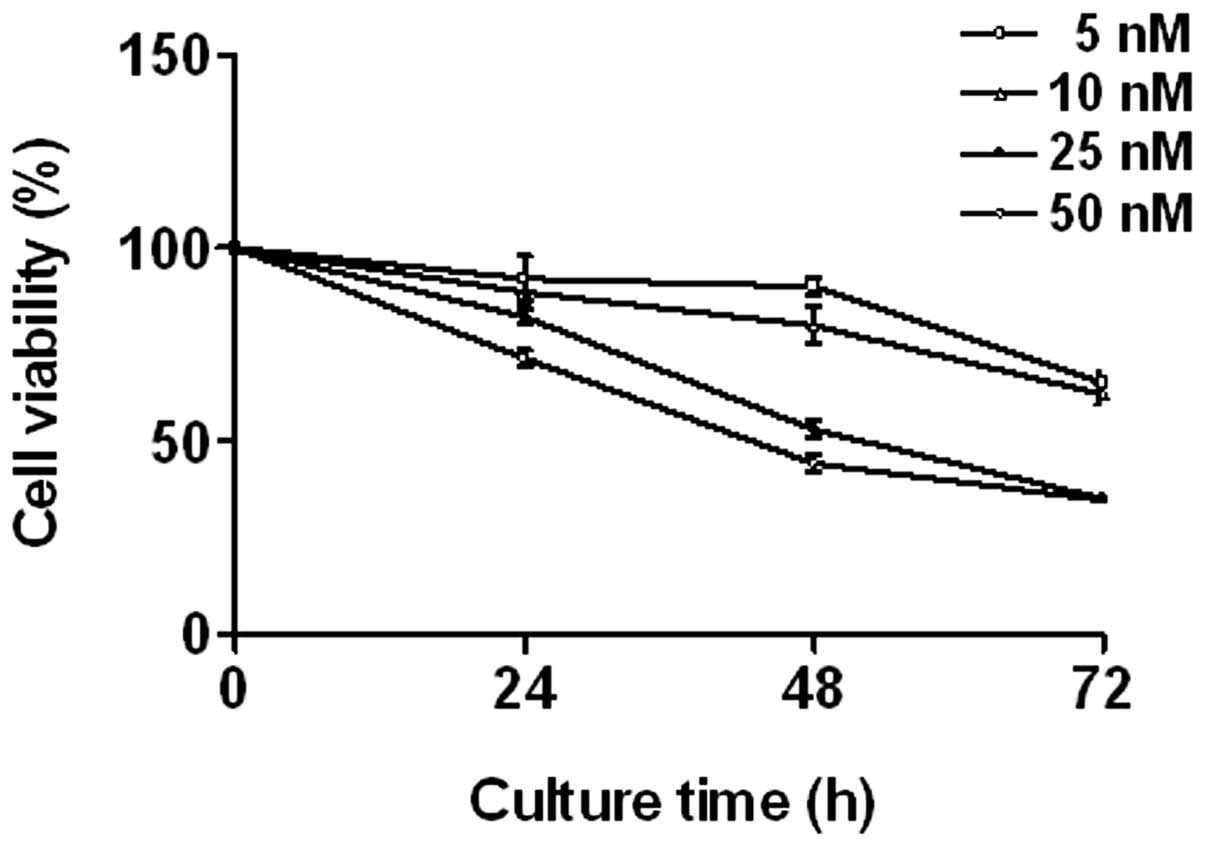
Triptolide inhibits cell proliferation in
THP-1 cells
THP-1 cells were treated with various concentrations
of triptolide (0–50 nM) for 24, 48 and 72 h. The effect of
triptolide on cell proliferation was measured using an MTT assay.
Triptolide induced a significant decrease in THP-1 cell
proliferation in a dose- and time-dependent manner (Fig. 1).
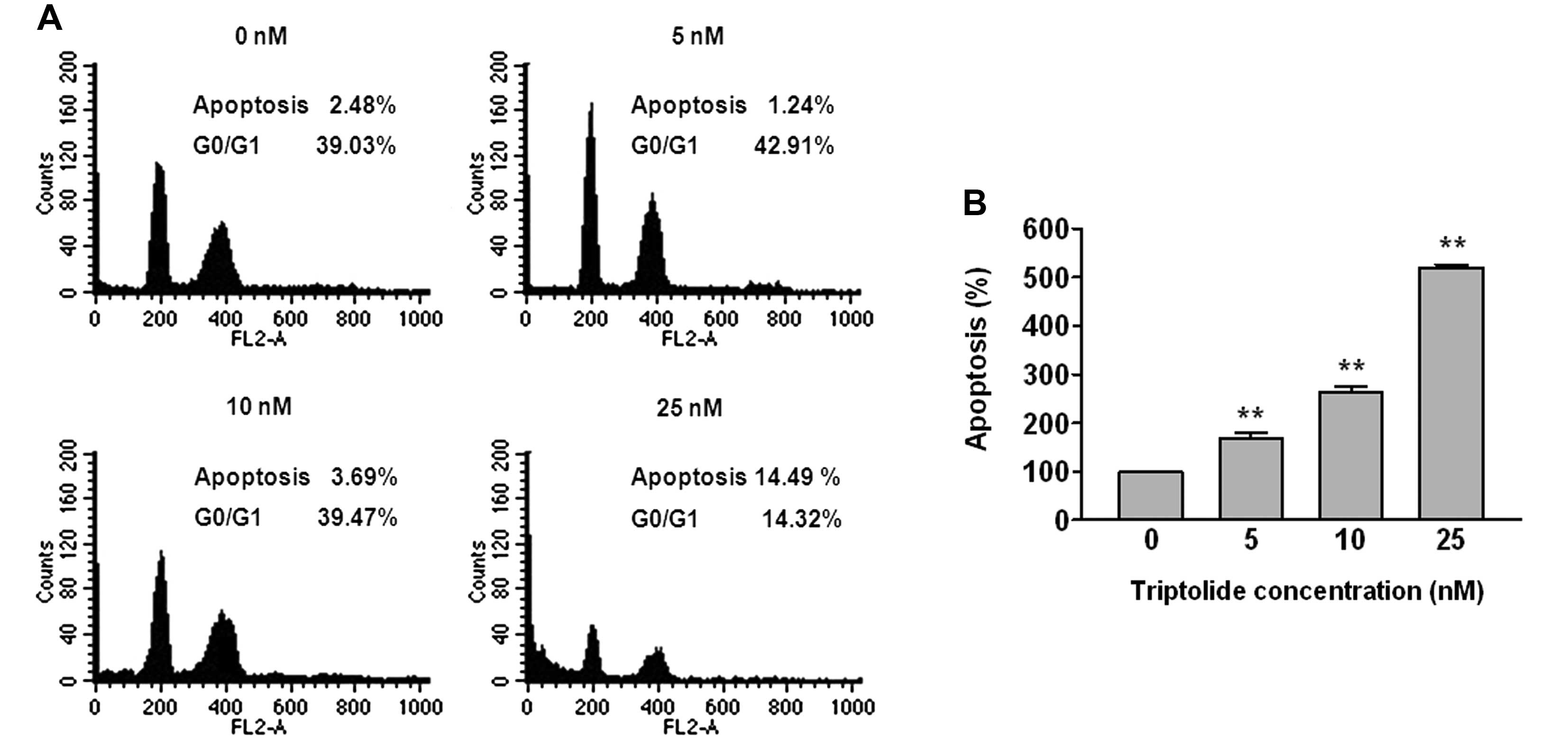
Triptolide induces cell populations
THP-1 cells were treated with 5, 10 and 25 nM of
triptolide for 48 h, after which flow cytometric analyses were
performed (Fig. 2A). The apoptosis
fraction increased by 2.48, 1.24, 3.69 and 14.49%, whereas the
G0/G1 phase decreased by 39.03, 42.91, 39.47 and 14.32%,
respectively, in THP-1 cells.
Triptolide induces apoptosis
THP-1 cells were treated with 5, 10 and 25 nM of
triptolide for 48 h and apoptotic cells were quantified using a
cell death detection ELISA (Fig.
2B). The number of apoptotic cells increased in a
dose-dependent manner. Hoechst 33258 staining was used to observe
the morphology of cell apoptosis. When THP-1 cells were exposed to
triptolide for 48 h, apoptotic cells characterized by morphological
alteration such as condensed nuclei and cell shrinkage were
observed and the apoptotic cell number increased with the dose
(Fig. 2C). Caspases are
cysteine-aspartate proteases that play critical roles during the
initiation and execution of apoptosis. To further elucidate the
mechanism involved in the observed apoptosis, intracellular
caspase-3, -8 and -9 activities were measured in THP-1 cells
treated with various concentrations of triptolide (Fig. 2D), using a colorimetric ELISA.
Caspase-3 and -9 activities were increased in response to
triptolide treatment in a dose-dependent manner at 48 h. To confirm
whether the activation of caspases is involved in
triptolide-induced apoptosis, cell growth of THP-1 cells by
triptolide was determined by MTT assay in the presence of the
pan-caspase inhibitor Z-VAD-FMK and the caspase-3-specific
inhibitor Z-DEVD-FMK. As shown in Fig.
2E, treatment with 25 nM triptolide resulted in an increase in
proliferation of THP-1 cells after pretreatment with 10–50
μM Z-VAD-FMK as well as by 10–50 μM Z-DEVD-FMK.
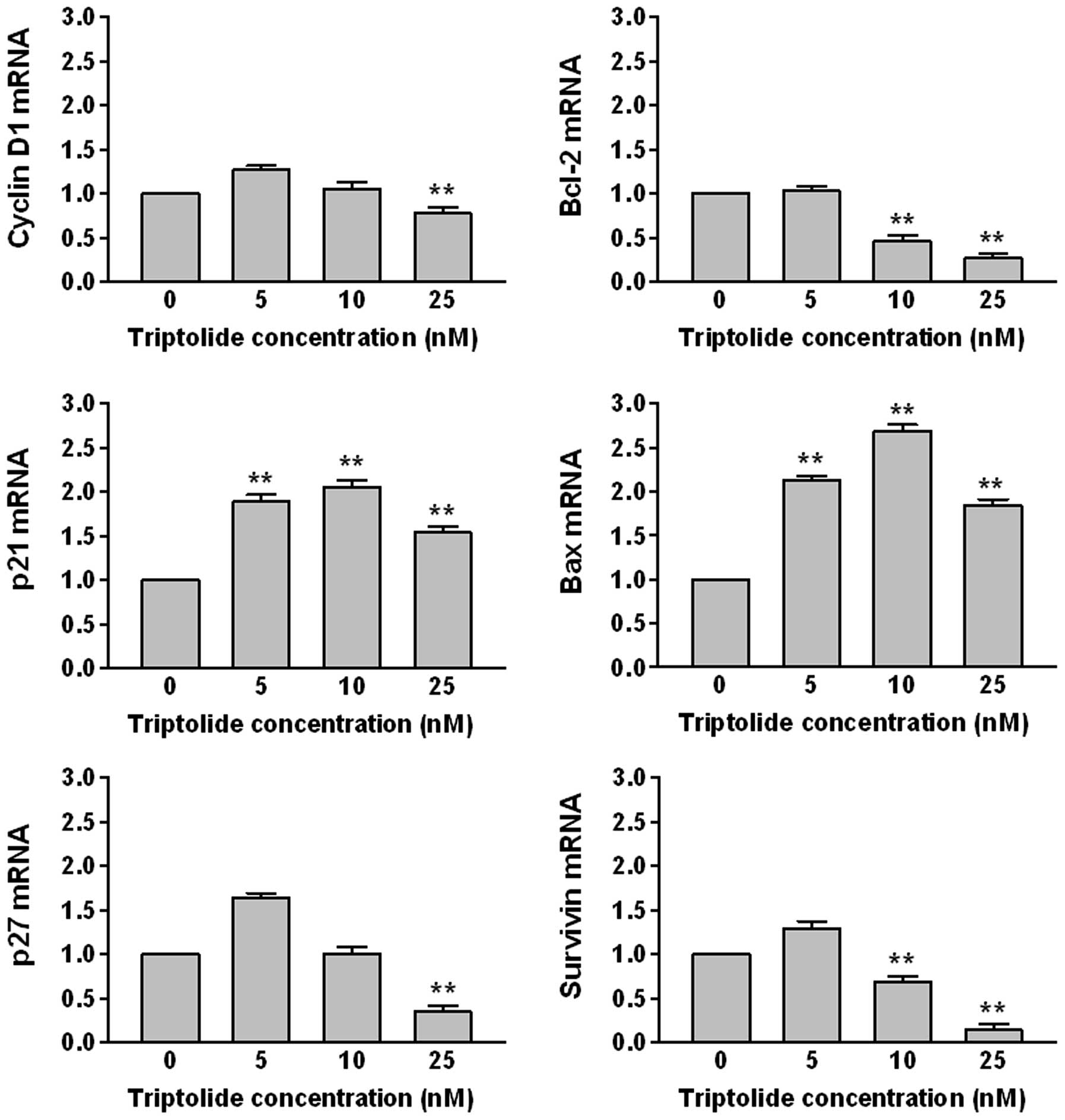
Triptolide mediates gene expression
The level of mRNAs transcribed from cell
cycle-related genes (cyclin D1, p21, p27) and apoptosis-related
genes (Bcl-2, Bax, survivin) were examined by real-time PCR
(Fig. 3). THP-1 cells were treated
with 5, 10 and 25 nM of triptolide for 48 h. The levels of Bcl-2,
cycline D1, p27 and survivin mRNA were decreased, whereas those of
Bax and p21 mRNA were increased in a dose-dependent manner.
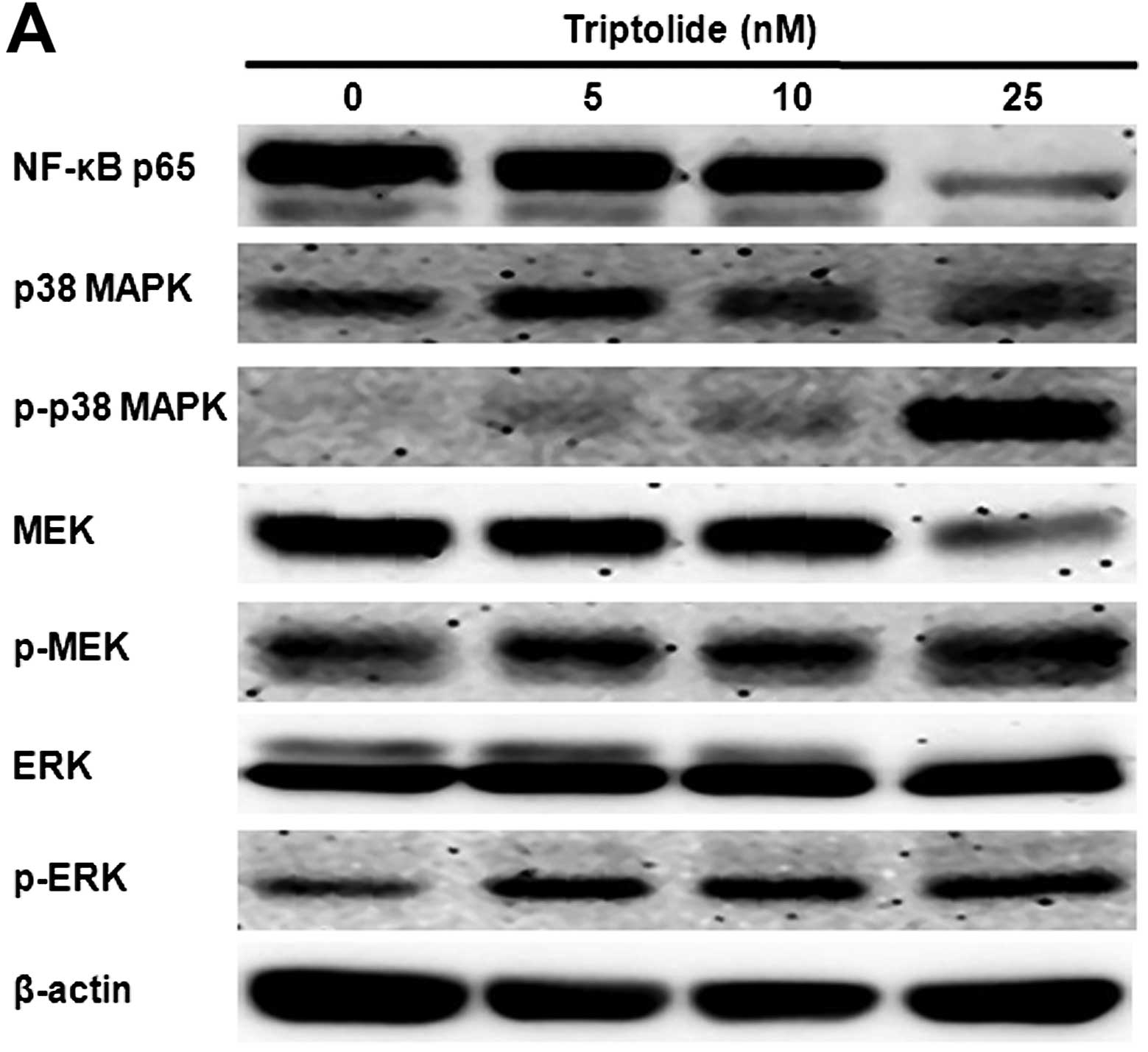
Triptolide inhibits NF-κB activation and
enhances p38 MAPK and MEK/ERK phosphorylation
THP-1 cells were incubated with triptolide at 5, 10
and 25 nM for 48 h. After triptolide treatment, protein expression
levels were measured by western blot analysis (Fig. 4). The levels of NF-κB p65
significantly declined after cells were exposed to triptolide.
Triptolide induced a marked increase in the levels of
phosphorylated p38 MAPK, MEK and ERK1/2.
Discussion
Triptolide is known to affect many cellular events,
to have anti-neoplastic activity and induce apoptosis. Triptolide
inhibits cell growth in variety of tumor cells (15,16).
In this study, human monocytic leukemia THP-1 cells that had been
PMA-differentiated showed inhibition of cell proliferation after
triptolide treatment, in a dose-and time-dependent manner. To
elucidate whether triptolide decreased cell viability by inducing
apoptosis, we investigated its effect on the expression of
apoptosis-related factors and found that triptolide increased the
apoptosis fraction at cell cycle and the number of apoptotic THP-1
cells in a dose-dependent manner. These results suggested that
triptolide not only inhibits THP-1 cell growth and blocks cell
cycle progression at the G1 phase, but also induces apoptosis.
Triptolide has also been reported to induce significant apoptosis
and minor accumulation in the S phase in THP-1 cells (17). However, treatment with triptolide
was shown to reduce the viability of HeLa and Caski cells in a
concentration-dependent manner and to increase accumulation of
sub-G1 phase cells and apoptotic bodies (16).
Triptolide not only regulates cell growth, but also
induces programmed cell death in several types of cells. It appears
that triptolide induces apoptosis by activating caspases, the
proteases responsible for cell death in multiple myeloma cells and
leukemic cells (18,19). Caspases play important roles in the
apoptotic process (20,21). When investigating the molecular
mechanism underlying apoptosis of THP-1 cells in response to
triptolide, we found that treatment of THP-1 cells with triptolide
produced increases in intracellular caspase-3 activity. This
finding was confirmed by experiments using the pancaspase inhibitor
Z-VAD-FMK and the caspase-3-specific inhibitor Z-DEVD-FMK, which
enhanced cell growth in triptolide-treated cells. These results
suggest that the apoptotic effect of triptolide in THP-1 cells may
result from the regulation of the caspase pathways. Caspase-3 and
-9 activity play important roles in apoptosis through a
mitochondria-dependent pathway. It has been shown that the
induction of apoptosis in cervical cancer cells by triptolide is
associated with activation of caspases (16). Moreover, it was reported that
triptolide may induce apoptosis through a mitochondria-mediated
apoptotic pathway in a caspase-dependent way in human melanoma A375
cells (22).
Bcl-2 gene-family members have been widely
considered to be regulators of cell death (23). In this study, our results revealed
that triptolide treatment of THP-1 cells upregulated the mRNA
expression of genes encoding Bax and p21 and downregulated those
encoding Bcl-2, cycline D1, p27 and survivin. These results
suggested that triptolide regulated transcription factors of cell
cycle-related genes and apoptosis-related genes. It has previously
been shown that triptolide treatment leads to increased Bax
expression and decreased Bcl-2 expression (24). In contrast, Bax expression was
significantly up-regulated in SW1990 cells treated with triptolide,
while Bcl-2 mRNA was not (25).
Triptolide has also been shown to decrease
anti-apoptotic mechanisms through NF-κB inhibition (26,27).
We investigated whether this regulatory mechanism is also involved
in the process of apoptosis in THP-1 cells; our results indicated
that triptolide suppressed NF-κB activation, suggesting that
triptolide induced apoptosis through inhibition of NF-κB
activation. Recent reports demonstrated that the NF-κB signal is
clearly downregulated during apoptosis induced by triptolide
(15). Bcl-2 and Bcl-XL
are members of the Bcl-2 family whose expression is regulated by
NF-κB. Triptolide induces apoptosis by means of inhibiting NF-κB
through downregulating the expression of the genes encoding Bcl-2
and Bcl-XL(22). Our
study supports that the NF-κB pathways are involved in the process
of THP-1 cell apoptosis induced by triptolide.
The MAPK pathway is a key signaling mechanism that
regulates many cellular functions, such as cell growth,
transformation and apoptosis (28). MAPKs can mediate apoptotic
signaling induced by antineoplastic agents (29). ERK, JNK and p38 MAPK constitute 3
major subfamilies of MAPKs that appear to mediate cellular
responses, including proliferation, differentiation and apoptosis
(30). Gleditsia sinensis
thorns (WEGS) induced phosphorylation of ERK1/2, p38 MAPK and JNK
in human colon cancer cells (31).
Danshensu increased phosphorylation of Akt and ERK1/2 in H9c2
cardiomyocytes (32). Induction of
cell death can be mediated by activating ERK1/2 (33,34).
We found that triptolide enhanced the level of p38 MAPK and MEK/ERK
phosphorylation in THP-1 cells. These results suggest that
triptolide mediates cell growth and apoptosis through the
activation of the MEK/ERK pathway.
In conclusion, we demonstrated that triptolide
inhibits the growth of THP-1 cells by inducing apoptosis through
the activation of caspases, as well as inhibiting NF-κB and
activating MAPK pathways. However, studies are needed to explore
further details of the mechanisms underlying the effect of
triptolide on human monocytic leukemia cells.
Acknowledgements
This study was supported by
Bio-industry Technology Development Program, Ministry for Food,
Agriculture, Forestry and Fisheries, Republic of Korea (grant no.
311059-4) and partially supported by the Rural Development
Administration, Republic of Korea (grant no. PJ008475022012). We
thank Waterborne Virus Bank for the technical support of real-time
PCR work.
References
|
1.
|
Chen BJ: Triptolide, a novel
immunosuppressive and anti-inflammatory agent purified from a
Chinese herb Tripterygium wilfordii Hook F. Leuk Lymphoma.
42:253–265. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Qiu D and Kao PN: Immunosuppressive and
anti-inflammatory mechanisms of triptolide, the principal active
diterpenoid from the Chinese medicinal herb Tripterygium
wilfordii Hook. f. Drugs R D. 4:1–18. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Shamon LA, Pezzuto JM, Graves JM, et al:
Evaluation of the mutagenic, cytotoxic and antitumor potential of
triptolide, a highly oxygenated diterpene isolated from
Tripterygium wilfordii. Cancer Lett. 112:113–117. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Tengchaisri T, Chawengkirttikul R,
Rachaphaew N, Reutrakul V, Sangsuwan R and Sirisinha S: Antitumor
activity of triptolide against cholangiocarcinoma growth in vitro
and in hamsters. Cancer Lett. 133:169–175. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Lee KY, Chang W, Qiu D, Kao PN and Rosen
GD: PG490 (triptolide) cooperates with tumor necrosis factor-α to
induce apoptosis in tumor cells. J Biol Chem. 274:13451–13455.
1999.PubMed/NCBI
|
|
6.
|
Chan EW, Cheng SC, Sin FW and Xie Y:
Triptolide induced cytotoxic effects on human promyelocytic
leukemia, T cell lymphoma and human hepatocellular carcinoma cell
lines. Toxicol Lett. 122:81–87. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Tsuruo T, Naito M, Tomida A, et al:
Molecular targeting therapy of cancer: drug resistance, apoptosis
and survival signal. Cancer Sci. 94:15–21. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Debatin KM: Apoptosis pathways in cancer
and cancer therapy. Cancer Immunol Immunother. 53:153–159. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Levine B, Sinha S and Kroemer G: Bcl-2
family members: dual regulators of apoptosis and autophagy.
Autophagy. 4:600–606. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Yang Y, Liu Z, Tolosa E, Yang J and Li L:
Triptolide induces apoptotic death of T lymphocyte.
Immunopharmacology. 40:139–149. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Aggarwal BB: Nuclear factor-κB: the enemy
within. Cancer Cell. 6:203–208. 2004.
|
|
12.
|
Karin M: Nuclear factor-κB in cancer
development and progression. Nature. 441:431–436. 2006.
|
|
13.
|
Keyse SM: Protein phosphatases and the
regulation of mitogen-activated protein kinase signaling. Curr Opin
Cell Biol. 12:186–192. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Liu Q: Triptolide and its expanding
multiple pharmacological functions. Int Immunopharmacol.
11:377–383. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Zhu W, Hu H, Qiu P and Yan G: Triptolide
induces apoptosis in human anaplastic thyroid carcinoma cells by a
p53-independent but NF-κB-related mechanism. Oncol Rep.
22:1397–1401. 2009.PubMed/NCBI
|
|
16.
|
Kim MJ, Lee TH, Kim SH, Choi YJ, Heo J and
Kim YH: Triptolide inactivates Akt and induces caspase-dependent
death in cervical cancer cells via the mitochondrial pathway. Int J
Oncol. 37:1177–1185. 2010.PubMed/NCBI
|
|
17.
|
Pigneux A, Mahon FX, Uhalde M, et al:
Triptolide cooperates with chemotherapy to induce apoptosis in
acute myeloid leukemia cells. Exp Hematol. 36:1648–1659. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Yinjun L, Jie J and Yungui W: Triptolide
inhibits transcription factor NF-kappaB and induces apoptosis of
multiple myeloma cells. Leuk Res. 29:99–105. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Carter BZ, Mak DH, Schober WD, et al:
Triptolide induces caspase-dependent cell death mediated via the
mitochondrial pathway in leukemic cells. Blood. 108:630–637. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Kim R, Emi M and Tanabe K:
Caspase-dependent and -independent cell death pathways after DNA
damage (Review). Oncol Rep. 14:595–599. 2005.PubMed/NCBI
|
|
21.
|
Denault JB and Salvesen GS: Caspases: keys
in the ignition of cell death. Chem Rev. 102:4489–4500. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Tao Y, Zhang ML, Ma PC, et al: Triptolide
inhibits proliferation and induces apoptosis of human melanoma A375
cells. Asian Pac J Cancer Prev. 13:1611–1615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Chao DT and Korsmeyer SJ: BCL-2 family:
regulators of cell death. Annu Rev Immunol. 16:395–419. 1998.
View Article : Google Scholar
|
|
24.
|
Li J, Zhu W, Leng T, et al:
Triptolide-induced cell cycle arrest and apoptosis in human renal
cell carcinoma cells. Oncol Rep. 25:979–987. 2011.PubMed/NCBI
|
|
25.
|
Zhou GX, Ding XL, Huang JF, et al:
Apoptosis of human pancreatic cancer cells induced by Triptolide.
World J Gastroenterol. 14:1504–1509. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Lee KY, Park JS, Jee YK and Rosen GD:
Triptolide sensitizes lung cancer cells to TNF-related
apoptosis-inducing ligand (TRAIL)-induced apoptosis by inhibition
of NF-κB activation. Exp Mol Med. 34:462–468. 2002.PubMed/NCBI
|
|
27.
|
Frese S, Pirnia F, Miescher D, et al:
PG490-mediated sensitization of lung cancer cells to
Apo2L/TRAIL-induced apoptosis requires activation of ERK2.
Oncogene. 22:5427–5435. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Rubinfeld H and Seger R: The ERK cascade:
a prototype of MAPK signaling. Mol Biotechnol. 31:151–174. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Fan M and Chambers TC: Role of
mitogen-activated protein kinases in the response of tumor cells to
chemotherapy. Drug Resist Updat. 4:253–267. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Davis RJ: The mitogen-activated protein
kinase signal transduction pathway. J Biol Chem. 268:14553–14556.
1993.PubMed/NCBI
|
|
31.
|
Lee SJ, Park K, Ha SD, Kim WJ and Moon SK:
Gleditsia sinensis thorn extract inhibits human colon cancer cells:
the role of ERK1/2, G2/M-phase cell cycle arrest and p53
expression. Phytother Res. 24:1870–1876. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Yin Y, Guan Y, Duan J, et al:
Cardioprotective effect of Danshensu against myocardial
ischemia/reperfusion injury and inhibits apoptosis of H9c2
cardiomyocytes via Akt and ERK1/2 phosphorylation. Eur J Pharmacol.
699:219–226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Stanciu M, Wang Y, Kentor R, et al:
Persistent activation of ERK contributes to glutamate-induced
oxidative toxicity in a neuronal cell line and primary cortical
neuron cultures. J Biol Chem. 275:12200–12206. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Wang X, Martindale JL and Holbrook NJ:
Requirement for ERK activation in cisplatin-induced apoptosis. J
Biol Chem. 275:39435–39443. 2000. View Article : Google Scholar
|