Introduction
Glioblastoma is the most common type of aggressive
brain tumor in adults, with a poor prognosis. Chemotherapy is often
performed in addition to radiation therapy as an adjuvant therapy
for glioblastoma. However, a major obstacle for successful
treatment is that it is difficult for chemotherapeutic agents to
cross the blood-brain barrier to achieve sufficient concentrations
(1).
Most chemically synthesized phenoxazines, which are
the reductive form, are hardly soluble in water and exert little
anticancer effect. However, some oxidative forms of phenoxazines
[e.g., 2-aminophenoxazine-3-one (Phx-3)], which is synthesized by
the biological reactions of o-aminophenol with bovine
hemoglobin solution or bovine erythrocytes (2,3),
exhibit anticancer activity in vitro and in vivo
(4–10). Phx-3 inhibits cell growth and
induces apoptosis in various cancer cell lines in vitro
(5,6,8).
Phx-3 also suppresses the growth of mouse malignant melanoma B16
cells transplanted in mice (7).
Azuine et al reported that phenoxazine derivatives can pass
through the blood-brain barrier (11). Because of this feature, Phx-3 is a
potential anticancer agent for glioblastoma; however, its effect on
glioblastoma has not been well characterized.
Regarding cellular fate in response to cell damage,
commitment to apoptosis or evasion of apoptosis is determined by
the balance of the complex survival-and-death signaling network.
Various cell growth factors [e.g., epidermal growth factor receptor
(EGFR), platelet-derived growth factor (PDGF), basic fibroblast
growth factor (bFGF), transforming growth factor (TGF)-β and
insulin-like growth factor (IGF)-1)] are overexpressed and
activated in glioblastoma cells and thus are advantageous for
oncogenesis (12). In the growth
factor receptor signaling transduction, the most important pathways
involved in oncogenesis are the phosphatidylinositide 3-kinases
(PI3K)/Akt and Ras/MEK/ERK pathways (13). Akt, the central node in the complex
cascade of PI3K signaling, is activated in various tumors,
including glioblastoma and is involved in cell survival and
proliferation as well as the anti-apoptotic effect (14). Extracellular signal-regulated
kinase (ERK) activation is generally associated with cell survival,
proliferation and death. Some studies suggest that the balance
between the intensity and the duration of pro- versus
anti-apoptotic signals transmitted by ERK determines whether the
cell undergoes proliferation or apoptosis (15). In addition, c-jun N-terminal kinase
(JNK), another subgroup of the mitogen-activated protein kinase
(MAPK) family, has been implicated in pro-apoptotic signaling and
is activated by many types of stress (e.g., UV irradiation,
inhibition of protein synthesis and exposure to anticancer
reagents) (16).
In the present study, we demonstrated the potent
apoptosis-inducing effect of Phx-3 on glioblastoma cell line LN229.
We further investigated the effect of Phx-3 on ERK-Akt signaling
and JNK signaling in order to clarify the molecular mechanism of
its apoptosis induction.
Materials and methods
Materials
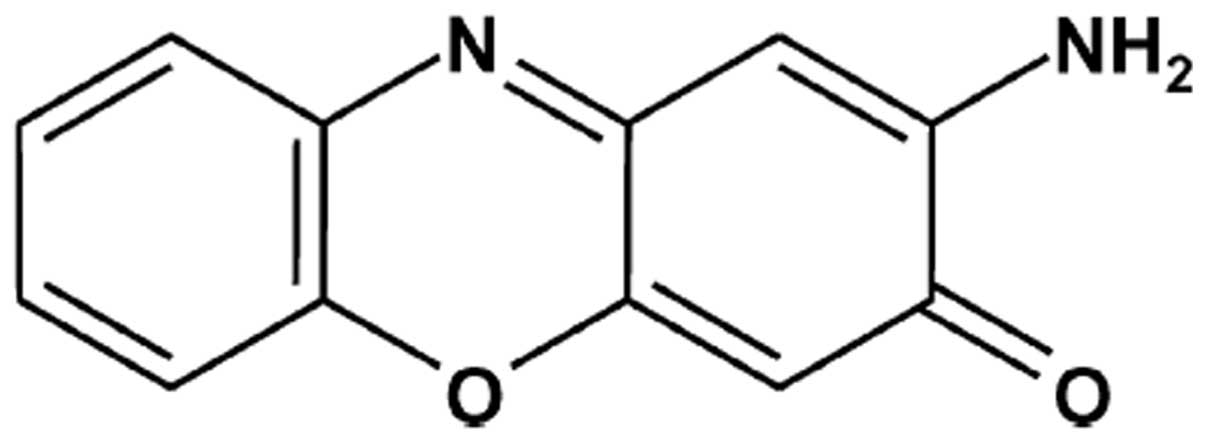
Phx-3 (Fig. 1) was
prepared by reacting o-aminophenol with bovine erythrocyte
suspension, following the method of Nakachi et al (17). Phx-3 was dissolved in a mixture of
dimethylsulfoxide and ethyl alcohol (3:1) as a vehicle to make a
20-mM solution and was diluted appropriately with an isotonic
saline for the experiments. Primary antibodies of phospho (p)-Akt,
Akt, p-JNK, p-mTOR, mTOR and XIAP were purchased from Cell
Signaling Technology (Beverly, MA, USA). Antibodies against p-ERK,
ERK, JNK, PARP and GAPDH were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-LC-3 monoclonal
antibody (mAb) was obtained from Novus Biologicals (Littleton, CO,
USA). A polyclonal antibody (Ab) against survivin, MP001, was
prepared in our laboratory. Melatonin was purchased from Nakariai
Tesque, Inc. (Kyoto, Japan) and N-acetyl-L-cysteine (NAC) was
obtained from Sigma (St. Louis, MO, USA). U0126 and SP600125 were
obtained from Merck KgaA (Darmstadt, Germany) and
5-(and-6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate
(CM-H2DCFDA) was obtained from Invitrogen (Carlsbad, CA,
USA).
Cell culture and viability assay
Human glioblastoma cell line LN229 was cultured in
Dulbecco’s modified Eagle’s medium (Sigma) supplemented with 10%
heat-inactivated fetal calf serum (Equitech-Bio, Kerrville, TX,
USA) at 37°C in a 5% CO2 humidified atmosphere. LN229
cells (3,000 cells/well) in 100 μl of culture medium were
incubated with or without various concentrations of Phx-3 for 24–96
h in 96-well plates. The number of viable cells was assessed using
a CellTiter-Blue Cell Viability Assay kit (Promega Co., Ltd.,
Madison, WI, USA), with fluorescence measurements at 570 nm for
excitation and 590 nm for emission using a Powerscan HT
Multidetection Microplate Reader (Dainippon Pharmaceutical, Osaka,
Japan).
Cell morphology
The LN229 cells were plated in 24-well Ezview Glass
Bottom Culture Plates (Iwaki, Asahi Techno Glass, Tokyo, Japan) at
a density of 3×105 cells/well and cultured for 24 h to
completely attach them to the surface of the plates. The cells were
treated with or without different concentrations of Phx-3 (2, 5 and
10 μM) in the presence or absence of 10 μM U0126, 10
μM SP600125, or 0.5 mM melatonin for 6 and 20 h.
Morphological changes were examined by digital microscopy using
BZ-8000 (Keyence Co., Osaka, Japan).
Apoptosis detection
Apoptotic cells were quantitatively evaluated by
flow cytometry using an Annexin V-Fluorescein Staining kit (Wako
Pure Chemical, Inc., Osaka, Japan). LN229 cells were treated with
various concentrations of Phx-3 for the indicated time in the
presence or absence of 10 μM U0126, 10 μM SP600125,
or 0.5 mM melatonin. The cells were then harvested and stained with
Annexin V and propidium iodide (PI). The levels of fluorescent
staining of the cells were analyzed using a flow cytometer (Partec
PAS; Partec, Münster, Germany) to estimate the population of
apoptotic cells gated in Annexin V+/PI−,
Annexin V+/PI+ and Annexin
V−/PI+.
Immunoblotting
Immunoblotting was performed as previously described
(18). Briefly, cells were lysed
with RIPA lysis buffer (Nacalai Tesque, Inc., Kyoto, Japan)
containing 1 mM PMSF, 0.15 U/ml aprotinin, 10 mM EDTA, 10 mg/ml
sodium fluoride and 2 mM sodium orthovanadate. Cellular proteins
were quantified using a Protein Assay Bicinchoninate kit (Nakalai
Tesque, Inc.). Lysates containing 40 μg proteins were
subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and
then transferred to an Immobilon-P membrane (Millipore, Bedford,
MA). The membrane was incubated with primary Ab (dilution of
1:1,000) overnight at 4°C. Immunoreactive proteins were detected
with a horseradish peroxidase-conjugated secondary Ab and enhanced
chemiluminescence reagent (ECL) (Millipore). Densitometry was
performed using a Molecular Imager ChemiDoc XRS System
(Bio-Rad).
Detection of intracellular ROS
Reactive oxygen species (ROS) production was
determined using CM-H2DCFDA, which diffuses through the
cell membrane and is hydrolyzed by esterases to non-fluorescent
dichlorofluorescein (DCFH) in cells. In the presence of ROS, this
compound is oxidized to highly fluorescent dichlorofluorescein
(DCF) in cells. For these experiments, LN229 cells were treated
with or without Phx-3 (2, 5, or 10 μM) for 2.5 h at 37°C in
the presence or absence of 0.5 mM melatonin or 5 mM NAC. The cells
were then incubated with 10 μM H2DCFDA for an
additional 30 min at 37°C and trypsinized and washed with ice-cold
phosphate buffered saline (PBS). Fluorescence was quantified using
a flow cytometer Partec PAS (Partec).
Detection of mitochondrial membrane
potential
Reduced mitochondrial membrane potential was
monitored using a sensitive fluorescent dye,
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetra-ethylbenzimidazolylcarbocyanine
iodide (JC-1) (Molecular Probes, Eugene, OR, USA) as previous
described (10). Red fluorescence
is attributed to potential-dependent aggregation in the
mitochondria; green fluorescence, reflecting the monomeric form of
JC-1, represents the dye in the cytosol after mitochondrial
membrane depolarization. LN229 cells were treated with Phx-3 in the
presence or absence of 10 μM of U0126 or SP600125, or 0.5 mM
melatonin for 3 and 20 h. The cells were then stained with DMEM
medium containing 5 μM JC-1 for 30 min at 37°C. Relative
fluorescence intensities were monitored by flow cytometry and
instantly assessed for red and green fluorescence using a flow
cytometer (Partec PAS; Partec).
Statistical analysis
All the quantitative data were expressed as mean ±
standard deviation (SD). Student’s t-test was used to compare the
values for the population of apoptotic cells. A p-value of <0.05
was considered to be statistically significant.
Results
Phx-3 induces apoptosis in human
glioblastoma cell line LN229
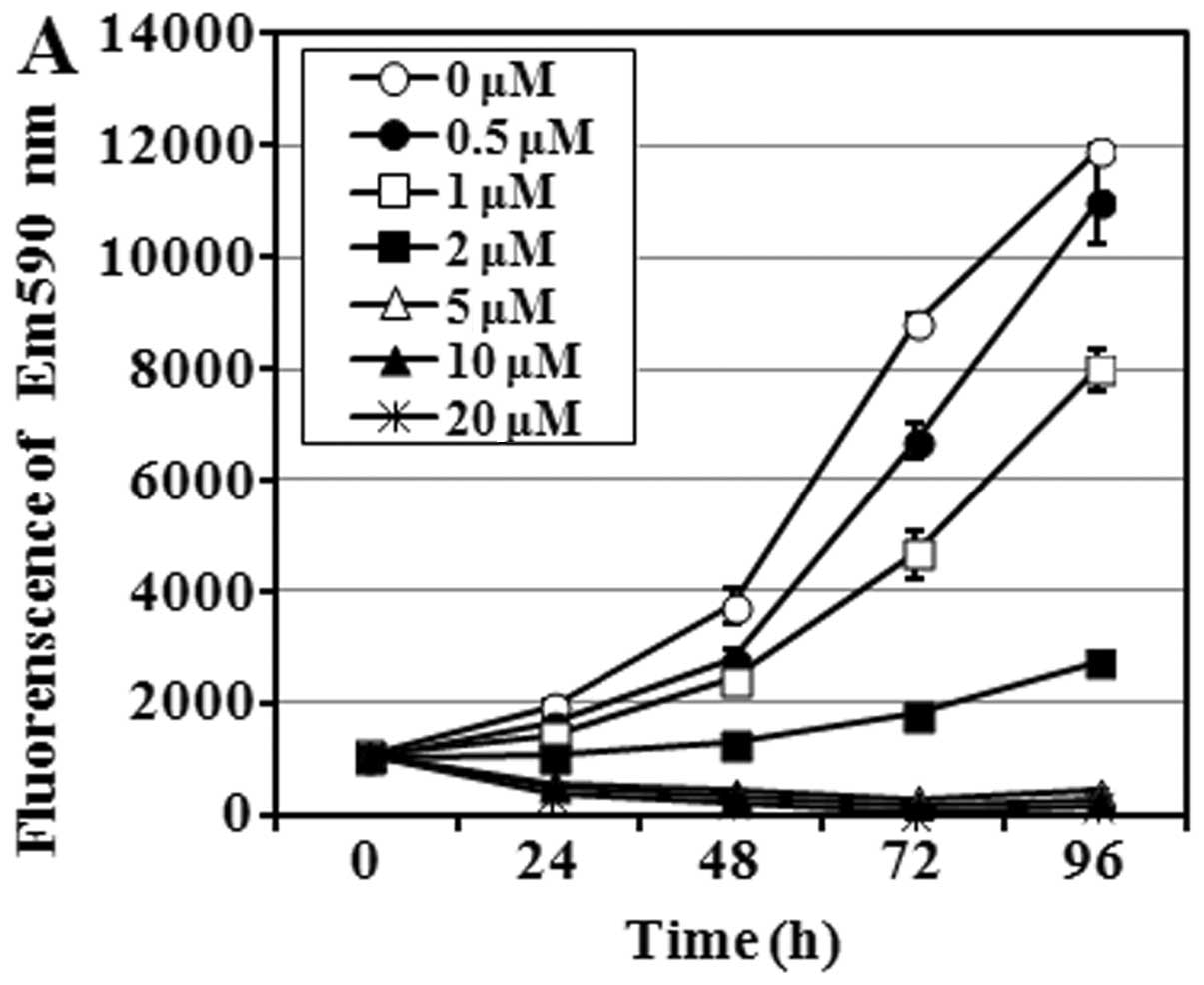
The growth of LN229 cells was significantly
inhibited at 1 μM Phx-3 and was almost completely inhibited
at >2 μM Phx-3 (Fig.
2A). The IC50 values (50% inhibition of cell growth)
of Phx-3 on LN229 cells were 2.602±0.087 μM for 24 h and
1.655±0.093 μM for 48 h. These results indicated that Phx-3
exhibits a more potent cell growth inhibitory effect at a lower
concentration in glioblastoma cell line LN229 than in other cell
lines in our previous studies (8).
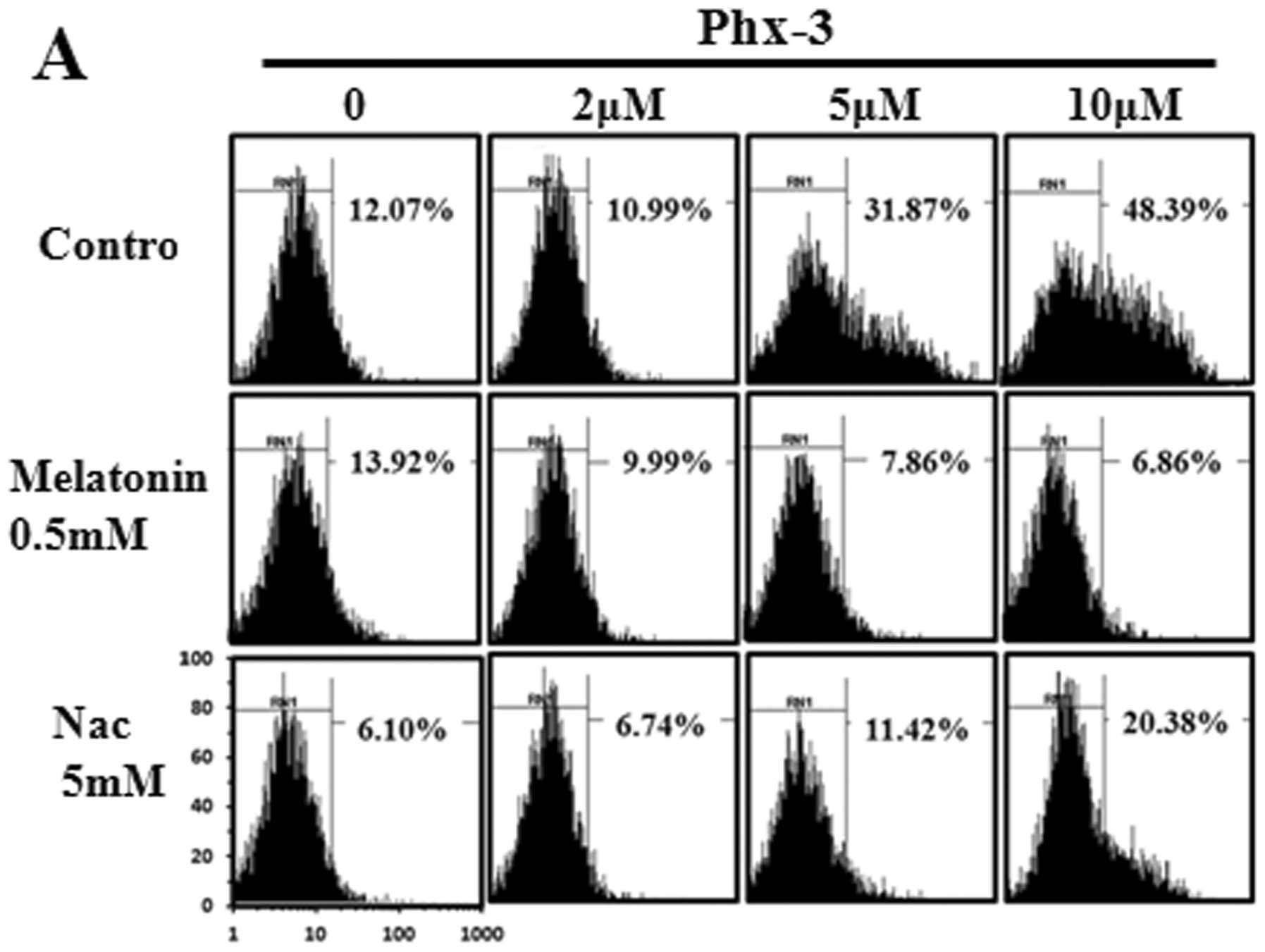
We next examined apoptosis induction in LN229 cells treated with
the indicated concentration of Phx-3 for 6 and 20 h (Fig. 2B). The population of apoptotic
cells [Annexin V(+)/PI(−) + Annexin V(+)/PI(+) + Annexin
V(−)/PI(+)] induced by Phx-3 increased in a dose- and a
time-dependent manner. The characteristics of apoptotic morphology
(e.g., cytoplasm shrinking, plasma membrane blebbing and nuclear
condensation) were observed (Fig.
2C).
We next investigated the expression profiles of
survivin, XIAP and poly (ADP-ribose) polymerase (PARP). Survivin
and XIAP, members of the inhibition of apoptosis (IAP) family,
suppress cell death by inhibiting the caspase family. PARP, which
is the substrate of caspase-3, is cleaved during drug-induced
apoptosis. Expressions of survivin and XIAP decreased after
treatment with Phx-3 in a time- and dose-dependent manner (Fig. 2D). Although cleaved PARP was not
clear, the 114 KDa of PARP expressions significantly decreased
after treatment with Phx-3.
JNK activation is the main pathway of
Phx-3-induced apoptosis
Both the suppression of survival signaling and the
activation of apoptotic signaling could induce apoptosis. The Akt
and ERK signaling pathway plays a key role in cell proliferation,
survival and differentiation (14,15),
whereas the activation of JNK pathways is involved in pro-apoptotic
signaling in response to various chemical and environmental
stresses (16). In order to
clarify the molecular mechanisms of Phx-3-induced apoptosis in
LN229 cells, we focus on the ERK-Akt and the JNK signaling
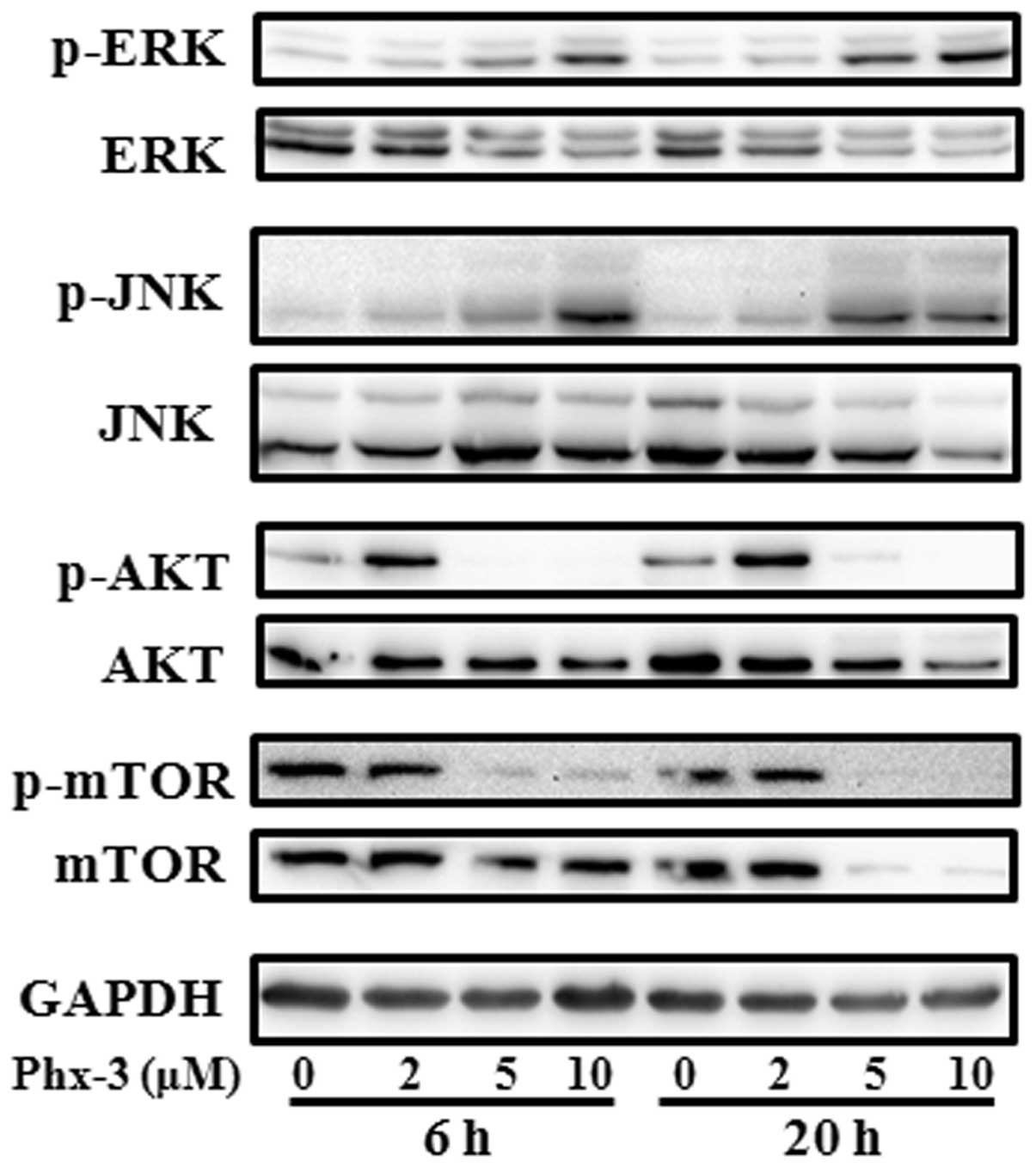
pathways. The level of p-ERK and p-JNK significantly increased
after treatment with Phx-3 for 6 h and was sustained for ≥20 h
(Fig. 3). Conversely, the
phosphorylation levels of Akt and mTOR, a downstream target of Akt,
were suppressed after treatment with Phx-3. These results suggest
that activation of ERK and JNK, as well as suppression of Akt and
mTOR, might contribute to apoptosis induction by Phx-3.
To investigate whether ERK plays an important role
in apoptosis induction by Phx-3, we treated the cells with Phx-3 in
the presence or absence of ERK inhibitor U0126 for 6 and 20 h; and
apoptosis induction was assessed by flow cytometry. Although the
phosphorylation of ERK induced by Phx-3 was almost completely
suppressed in the presence of U0126 (Fig. 4A), Phx-3-induced apoptosis was only
partially blocked (Fig. 4B and C).
This result indicated that the ERK-Akt axis does not appear to be
the major pathway for apoptosis induction. In addition, the
suppression of Akt, but not mTOR was almost completely restored by
U0126, which suggesting that Akt acts on downstream ERK
signaling.
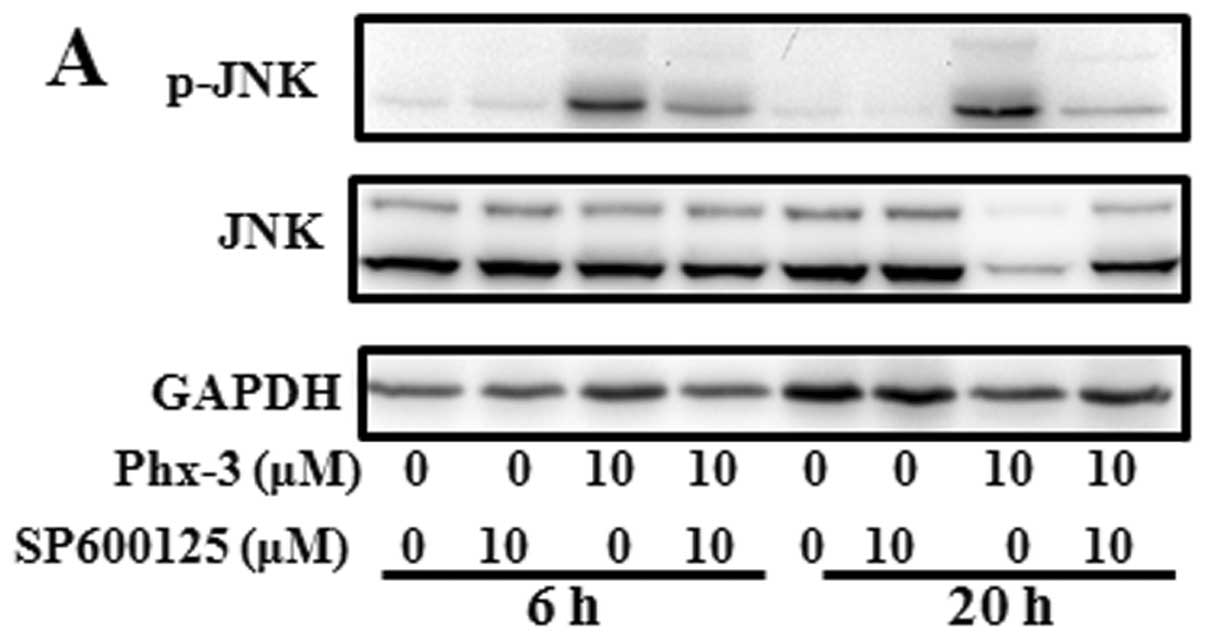
We next investigated the role of JNK activation.
Activation of JNK in response to Phx-3 was repressed in the
presence of 10 μM SP600125, a specific JNK inhibitor
(Fig. 5A). It was noteworthy that,
unlike U0126, the apoptosis induced by Phx-3 was almost completely
blocked in the presence of SP600125 (Fig. 5B and C). Furthermore, the
activation of ERK and the suppression of Akt were almost completely
restored, while repression of survivin and XIAP and suppression of
mTOR phosphorylation were still detectable (Fig. 5D and E). These results indicated
that activation of JNK plays a critical role in apoptosis induction
by Phx-3 in LN229 cells. In addition, JNK appears to localize
upstream of ERK/Akt signaling.
JNK activation depends on Phx-3-induced
ROS generation
The next question was how JNK was activated after
treatment with Phx-3. In our previous study, we found that
treatment of the lung adenocarcinoma cell line A549 with Phx-3
resulted in localization of Phx-3 in mitochondria along with ROS
generation (10). Other studies
indicated that suppression of JNK significantly protected against
apoptosis induction by ROS (19).
We therefore postulated that ROS generation might be involved in
the activation of JNK in response to Phx-3. We first examined ROS
generation in LN229 cells by measuring the fluorescence of
H2DCFDA. Fluorescence increased after 3-h exposure to
Phx-3 in a dose-dependent manner (Fig.
6A). ROS production was significantly inhibited in the presence
of 0.5 mM melatonin or 5 mM NAC, both of which are ROS scavengers.
Melatonin inhibits ROS more strongly than NAC, so we used melatonin
for the following studies. Notably, the presence of 0.5 mM
melatonin at 6 and 20 h almost completely prevented apoptosis
induction by Phx-3 (Fig. 6B and
C). These results indicated that ROS generation in response to
Phx-3 was strongly involved in apoptosis induction in LN229 cells.
After elimination of ROS with melatonin treatment, Phx-3-induced
JNK activation was also inhibited (Fig. 6E). As well as SP600125 treatment
(Fig. 5E), melatonin also
suppressed ERK phosphorylation and restored the phosphorylation
state of Akt without any effect on the repression of survivin and
XIAP, as well as suppression of mTOR phosphorylation (Fig. 6D and E). These results suggest that
ROS generated by Phx-3 plays a pivotal role in apoptosis induction
and JNK activation in LN229 cells.
To confirm that ROS is the direct activator of JNK,
we treated LN229 cells with various concentrations of
H2O2 for 1–20 h (Fig. 7A). The results indicated that JNK
phosphorylation increased transiently after treatment with 100
μM H2O2. Also, glucose oxidase
catalyzes the oxidation of glucose to hydrogen peroxide and
D-glucono-δ-lactone. With the addition of glucose oxidase to LN229
cell culture, the glucose in the medium should be continuously
metabolized to hydrogen peroxide. The level of phospho-JNK
increased in the presence of glucose oxidase (Fig. 7B). Therefore, ROS is the direct
activator of JNK in LN229 cells.
JNK activation by ROS is involved in
depolarization of the mitochondrial membrane
Mitochondria are considered the main source of ROS
(20). Conversely, continuous ROS
generation by mitochondria also causes the dysfunction of
mitochondria (21). In addition,
JNK translocates into the mitochondria, inactivates Bcl-2 and
Bcl-xL and decreases the mitochondrial membrane potential, which
leads to the activation of caspase cascade. We examined
mitochondrial depolarization by JC-1 staining to evaluate the
mitochondrial membrane integrity after treatment with Phx-3 in the
presence or absence of U0126, SP600125 and melatonin. In intact
cells, JC-1 accumulates in mitochondria and emits primarily red
fluorescence; in the mitochondrial depolarized cells, JC-1 emits
green fluorescence when leaking from mitochondria to cytoplasm.
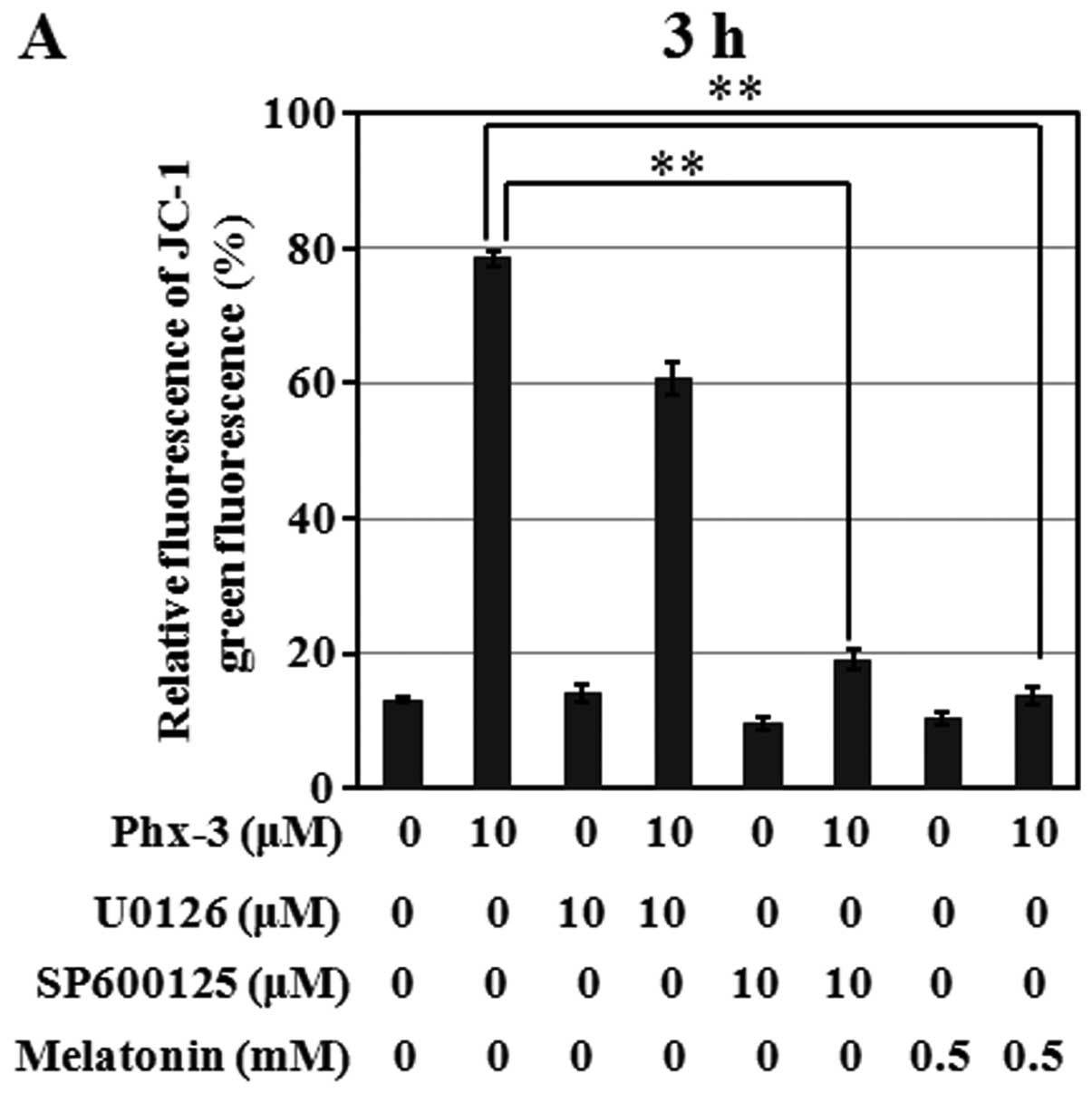
Relative green fluorescence significantly increased in the cells
treated with 10 μM Phx-3 for 3 h (Fig. 8A). This depolarization was blocked
in the presence of either SP600125 or melatonin, but not in the
presence of U0126. However, with exposure time extended to 20 h
(Fig. 8B), mitochondrial
depolarization was only slightly blocked even in the presence of
SP600125 or melatonin. Therefore, sustained mitochondrial
dysfunction by some alternative mechanism(s) may cause apoptosis
even if the cells are treated with JNK inhibitor or ROS
scavenger.
All the data above suggest that for short-time
exposure to Phx-3, ROS generation, followed by JNK activation is
the main axis for apoptosis induction in response to Phx-3.
However, for longer exposure to Phx-3, mitochondrial dysfunction
may cause other apoptotic machinery in LN229 cells.
Discussion
In the present study, we found that the oxidative
form of Phx-3 potently induced apoptosis in the glioblastoma cell
line LN229 (Fig. 2). In addition,
we confirmed that the ROS/JNK axis is the pivotal pathway for
apoptosis induction. This conclusion is supported by the following
results (1). Phx-3 induced
concomitant activation of both JNK and ERK pathways; however, JNK
inhibitor SP600125 completely blocked apoptosis induction by Phx-3,
but ERK inhibitor U0126 did not (Figs.
4 and 5) (2). Phx-3 treatment induced ROS generation
within 3 h (Fig. 6A); however, the
ROS scavenger melatonin almost completely blocked Phx-3-induced
apoptosis and JNK activation (Fig. 6B,
C and E) (3). Exogenous ROS
generation with the addition of H2O2 or
glucose oxidase in culture medium resulted in JNK activation
(Fig. 7).
Cellular endogenous ROS is implicated in
genotoxicity, tumor initiation and progression and a high level of
ROS induces cell death via apoptotic and/or necrotic mechanisms
(22). In addition to Phx-3, the
anticancer agents that are generally used (e.g., cisplatin,
vinblastine and doxorubicin) exert their pharmacological effects
through ROS-mediated cell death induction (21). Mitochondria are the main source of
endogenous ROS and antioxidant systems such as superoxide dismutase
(SOD) and glutathione peroxidase (GPx) eliminate over-generated
ROS, keeping the cells in homeostasis (23). Therefore, either exogenous ROS
production or inhibition of the antioxidant systems induces an
accumulation of cellular ROS. In this study, significant increase
of ROS generation (Fig. 6A) and
dramatic decrease of mitochondrial membrane potential (Fig. 8A) were detected after 3-h treatment
with Phx-3. In our previous study, microscopic examination
indicated that Phx-3 localized mainly in the mitochondria within
1-h exposure in A549 cells, a lung adenocarcinoma cell line and ROS
generation induced by Phx-3 contributed to apoptosis induction
(10). Therefore, mitochondria
appear to be the direct target of Phx-3 and overwhelming ROS
generation by mitochondria appears to be involved in apoptotic
induction.
Several signaling pathways (e.g., ERK, JNK and p38
kinases) are activated in response to ROS generation (24–26).
In response to DNA damage by ROS, sustained activation of both JNK
and ERK mediates cell death (24,25).
As well as these previous reports, JNK and ERK were activated after
treatment with Phx-3 in LN229 cells (Fig. 3) and their activation was
concomitantly blocked in the presence of melatonin, a ROS
scavenger, along with inhibition of Phx-3-induced apoptosis
(Fig. 6E). We analyzed which
signal is critical for Phx-3-induced apoptosis and found that
apoptosis induction by Phx-3 was completely blocked by SP600125, a
specific inhibitor of JNK. However, U0126, an inhibitor of ERK,
only partially suppressed apoptosis induction (Figs. 4C and 5C). This result indicates that activation
of JNK mediated through ROS generation is the main pathway of
Phx-3-induced apoptosis. Additionally, treatment with SP600125
inhibited ERK activation (Fig.
5E), which suggests that ERK is located on one of the
downstream pathways of JNK.
The molecular mechanism of JNK activation in
response to ROS is still unclear in our system. It has been
reported that signaling pathways such as ASK1 (27,28),
Src kinase (29) and glutathione
S-transferase Pi (GSTπ) (30,31)
are involved in ROS-mediated JNK activation. In the presence of
ROS, oxidized thioredoxin and/or glutaredoxin, which is dissociated
from ASK1, resulted in activation of ASK1 followed by subsequent
JNK phosphorylation (27,28). Src kinase is also reported to be an
important redox-sensitive pathway for JNK activation after
treatment with H2O2(29). Additionally, the monomeric form of
GSTπ suppresses JNK activity (30), while the oligomeric form of GSTπ
induced by H2O2 treatment activates JNK
(31). Further studies are
required to clarify the pathway from ROS to JNK activation after
Phx-3 treatment.
It is noteworthy that SP600125 and melatonin did not
suppress the depolarization of mitochondria after 20-h treatment
with Phx-3, although these reagents blocked Phx-3-induced apoptosis
(Fig. 8B). When Phx-3 exposure
time was extended to 72 h, apoptosis in LN229 cells was detectable
even in the presence of SP600125 or melatonin (data not shown).
These results suggest that the ROS/JNK pathway is indeed the main
axis for apoptosis induction during short-time exposure to Phx-3,
however, with longer exposure, some alternative mechanism(s)
causing irreversible mitochondrial demoralization may be involved
in apoptosis induction. Phx-3-induced suppression of mTOR activity
and downregulation of survivin and XIAP may also be involved in
these alternative mechanisms. These molecules were not restored in
the presence of U0126, SP600125, or melatonin (Figs. 4D and E, 5D and E and 6D and E), so they are considered to be
independent of the ROS/JNK axis. However, Phx-3 efficiently
inhibited Akt activation and this inhibition was completely
restored in the presence of U0126, as well as SP600125 and
melatonin. This result suggests that Akt is located downstream of
ERK. Sinha et al reported that withdrawal of survival
factors leads to activation of ERK and suppression of Akt activity,
along with apoptosis induction in mouse renal proximal tubular
cells (32). This apoptosis was
inhibited by U0126 or PD98059, indicating that ERK plays a role in
suppressing survival signaling through decreasing Akt activity
(32). Similarly, U0126 partially
blocked the apoptosis induced by Phx-3 (Fig. 5B and C), suggesting that the
suppression of Akt might partially promote cell death in our system
during exposure to Phx-3. However, the Phx-3-induced inhibition of
mTOR, which is a key molecule of Akt signaling, was not restored
when Akt activation was restored in the presence of melatonin,
SP600125, or U0126 (Figs. 4E,
5E and 6E). Therefore, the inhibition of mTOR by
Phx-3 may be mediated through the Akt-independent pathway.
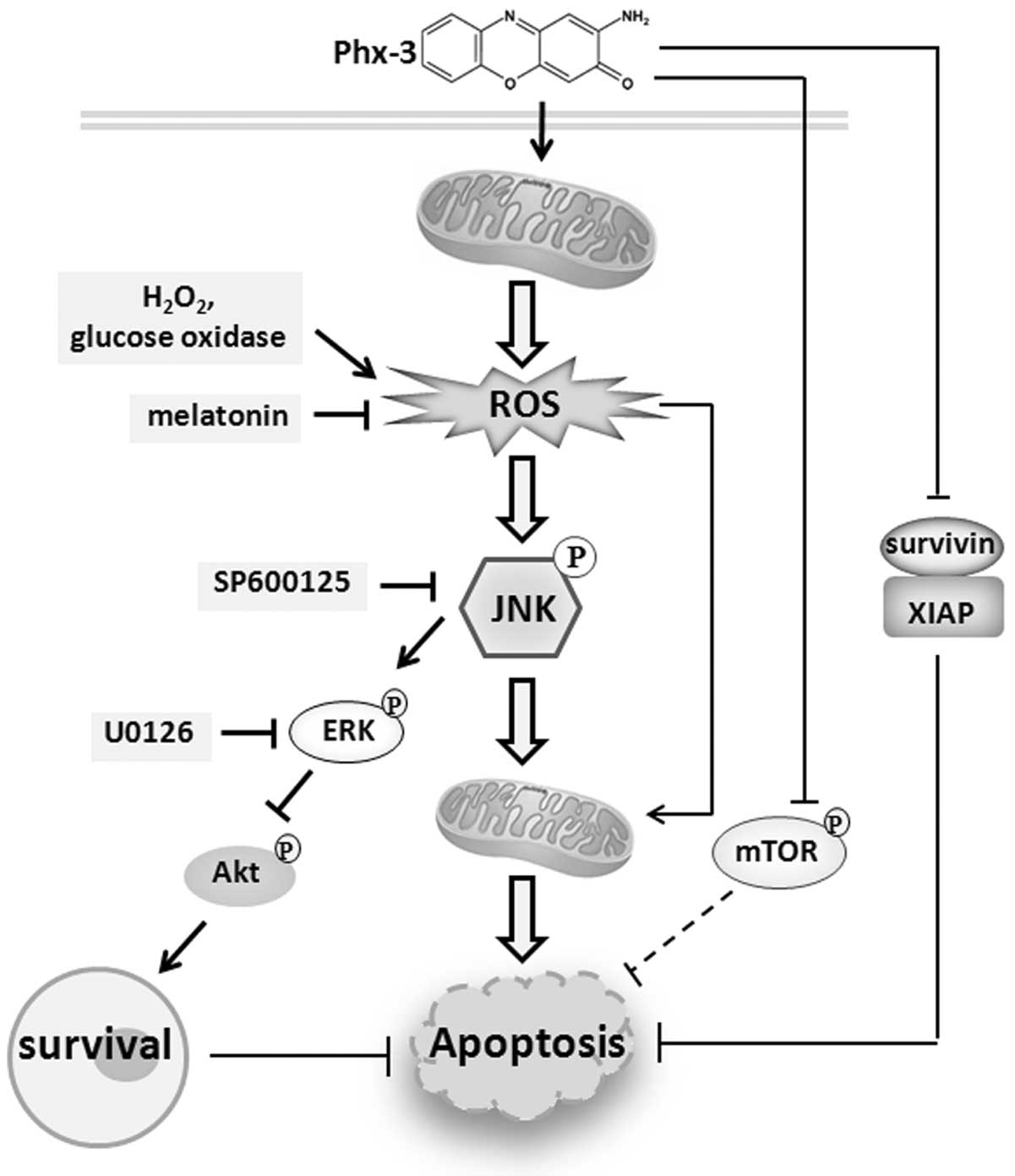
Collectively, Phx-3 appears to induce apoptosis
through multiple pathways (Fig.
9). In 20-h exposure to Phx-3, ROS/JNK signaling is the pivotal
pathway for apoptosis induction. However, with longer exposure,
some alternative mechanism(s) causing irreversible mitochondrial
demoralization may also be involved in apoptosis induction.
Therefore, if the cells have any functional defects in the pathway
from ROS to JNK, some alternate mechanisms (e.g., downregulation of
survivin and XIAP, or inhibition of mTOR activity) may be involved
in apoptosis induction. Phenoxazine derivatives are able to pass
through the blood-brain barrier (11), so our findings suggest that Phx-3
has high potential as an anticancer reagent for glioblastoma
therapy.
Acknowledgements
This study was supported by funds from
the Private University Strategic Research-Based Support Project
(Molecular Information-based Intractable Disease Research Project)
from the Ministry of Education, Culture, Sports, Science and
Technology of Japan to A.T. and K.M. (2008–2012), and a
Grant-in-Aid from the Tokyo Medical University for Cancer Research
to X.C., Japan.
References
|
1.
|
Bidros DS and Vogelbaum MA: Novel drug
delivery strategies in neuro-oncology. Neurotherapeutics.
6:539–546. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Tomoda A, Arai S, Ishida R, Shimamoto T
and Ohyashiki K: An improved method for the rapid preparation of
2-amino-4,4·-di-hydro-4,7-dimethyl-3Hphenoxazine-3-one, a novel
antitumor agent. Bioorg Med Chem Lett. 11:1057–1058.
2001.PubMed/NCBI
|
|
3.
|
Shimizu S, Suzuki M, Tomoda A, Arai S,
Taguchi H, Hanawa T and Kamiya S: Phenoxazine compounds produced by
the reactions with bovine hemoglobin show antimicrobial activity
against non-tuberculosis mycobacteria. Tohoku J Exp Med. 203:47–52.
2004. View Article : Google Scholar
|
|
4.
|
Shimamoto T, Tomoda A, Ishida R and
Ohyashiki K: Antitumor effects of a novel phenoxazine derivative on
human leukemia cell lines in vitro and in vivo. Clin Cancer Res.
7:704–708. 2001.PubMed/NCBI
|
|
5.
|
Shirato K, Imaizumi K, Miyazawa K,
Takasaki A, Mizuguchi J, Che XF, Akiyama S and Tomoda A: Apoptosis
induction preceded by mitochondrial depolarization in multiple
myeloma cell line U266 by 2-aminophenoxazine-3-one. Biol Pharm
Bull. 31:62–67. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Shirato K, Imaizumi K, Abe A and Tomoda A:
Phenoxazine derivatives induce caspase-independent cell death in
human glioblastoma cell lines, A-172 and U-251MG. Oncol Rep.
17:201–208. 2007.PubMed/NCBI
|
|
7.
|
Miyano-Kurosaki N, Kurosaki K, Hayaashi M,
Takaku H, Hayafune M, Shrato K, Kasuga T, Endo T and Tomoda A:
2-Aminophenoxazine-3-one suppresses the growth of mouse malignant
melanoma B16 cells transplanted into C57BL/6Cr Slc mice. Biol Pharm
Bull. 29:2197–2201. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Che XF, Zheng CL, Akiyama S and Tomoda A:
2-Amino phenoxazine-3-one and
2-amino-4,4α-dihydro-4α,7-dimethyl-3H-phenoxazine-3-one cause
cellular apoptosis by reducing higher intracellular pH in cancer
cells. Proc Jpn Acad Ser B Phys Biol Sci. 87:199–213. 2011.
|
|
9.
|
Takasaki A, Hanyu H, Iwamoto T, Shirato K,
Izumi R, Toyota H, Izuguchi J, Miyazawa K and Tomoda A:
Mitochondrial depolarization and apoptosis associated with
sustained activation of c-jun-N-terminal kinasein the human
multiple myeloma cell line U266 induced by
2-aminophenoxazine-3-one. Mol Med Rep. 2:199–203. 2009.PubMed/NCBI
|
|
10.
|
Zheng CL, Che XF, Akiyama S, Miyazawa K
and Tomoda A: 2-Aminophenoxazine-3-one induces cellular apoptosis
by causing rapid intracellular acidification and generating
reactive oxygen species in human lung adenocarcinoma cells. Int J
Oncol. 36:641–650. 2010.
|
|
11.
|
Azuine MA, Tokuda H, Takayasu J, Enjyo F,
Mukainaka T, Konoshima T, Nishino H and Kapadia GJ: Cancer
chemopreventive effect of phenothiazines and related
tri-heterocyclic analogues in the
12-O-tetradecanoylphorbol-13-acetate promoted Epstein-Barr virus
early antigen activation and the mouse skin two-stage
carcinogenesis models. Pharmacol Res. 49:161–169. 2004. View Article : Google Scholar
|
|
12.
|
Nakada M, Kita D, Watanabe T, Hayashi Y,
Teng L, Pyko IV and Hamada J: Aberrant signaling pathways in
glioma. Cancers. 3:3242–3278. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Soni D, King JA, Kaye AH and Hovens CM:
Genetics of glioblastoma multiforme: mitogenic signaling and cell
cycle pathways converge. J Clin Neurosci. 12:1–5. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Los M, Maddika S, Erb B and
Schulze-Osthoff K: Switching Akt: from survival signaling to deadly
response. Bioessays. 31:492–495. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Pearson G, Robinson F, Beers Gibson T, Xu
BE, Karandikar M, Berman K and Cobb MH: Mitogen-activated protein
(MAP) kinase pathways: regulation and physiological functions.
Endocr Rev. 22:153–183. 2001.PubMed/NCBI
|
|
16.
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Nakachi T, Tabuchi T, Takasaki A, Arai S,
Miyazawa K and Tomoda A: Anticancer activity of phenoxazines
produced by bovine erythrocytes on colon cancer cells. Oncol Rep.
23:1517–1522. 2010.PubMed/NCBI
|
|
18.
|
Kawaguchi T, Miyazawa K, Moriya S, Ohtomo
T, Che XF, Naito M, Itoh M and Tomoda A: Combined treatment with
bortezomib plus bafilomycin A1 enhances the cytocidal effect and
induces endoplasmic reticulum stress in U266 myeloma cells:
crosstalk among proteasome, autophagy-lysosome and ER stress. Int J
Oncol. 38:643–654. 2011.
|
|
19.
|
Nohl H, Gille L and Staniek K:
Intracellular generation of reactive oxygen species by
mitochondria. Biochem Pharmacol. 69:719–723. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Gogvadze V, Orrenius S and Zhivotovsky B:
Mitochondria as targets for cancer chemotherapy. Semin Cancer Biol.
19:57–66. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Maundrell K, Antonsson B, Magnenat E,
Camps M, Muda M, Chabert C, Gillieron C, Boschert U, Vial-Knecht E,
Martinou JC and Arkinstall S: Bcl-2 undergoes phosphorylation by
c-Jun N-terminal kinase/stress-activated protein kinases in the
presence of the constitutively active GTP-binding protein Rac1. J
Biol Chem. 272:25238–25242. 1997. View Article : Google Scholar
|
|
22.
|
Basu A and Haldar S: Identification of a
novel Bcl-xL phospho rylation site regulating the sensitivity of
taxol- or 2-methoxyestradiol-induced apoptosis. FEBS Lett.
538:41–47. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Fulda S, Gorman AM, Hori O and Samali A:
Cellular stress responses: cell survival and cell death. Int J Cell
Biol. 2010:2140742010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Trachootham D, Lu W, Ogasawara MA, Valle
NR and Huang P: Redox regulation of cell survival. Antioxid Redox
Signal. 10:1343–1374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Feligioni M, Brambilla E, Camassa A, Sclip
A, Arnaboldi A, Morelli F, Antoniou X and Borsello T: Crosstalk
between JNK and SUMO signaling pathways: deSUMOylation is
protective against H2O2-induced cell injury.
PLoS One. 6:e281852011. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Conde de la Rosa L, Schoemaker MH, Vrenken
TE, Buist-Homan M, Havinga R, Jansen PL and Moshage H: Superoxide
anions and hydrogen peroxide induce hepatocyte death by different
mechanisms: involvement of JNK and ERK MAP kinases. J Hepatol.
44:918–929. 2006.PubMed/NCBI
|
|
27.
|
Matos TJ, Duarte CB, Goncalo M and Lopes
MC: Role of oxidative stress in ERK and p38 MAPK activation induced
by the chemical sensitizer DNFB in a fetal skin dendritic cell
line. Immunol Cell Biol. 83:607–614. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Tobiume K, Matsuzawa A, Takahashi T,
Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T and
Ichijo H: ASK1 is required for sustained activations of JNK/p38 MAP
kinases and apoptosis. EMBO Rep. 2:222–228. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Song JJ, Rhee JG, Suntharalingam M, Walsh
SA, Spitz DR and Lee YJ: Role of glutaredoxin in metabolic
oxidative stress: glutaredoxin as a sensor of oxidative stress
mediated by H2O2. J Biol Chem.
277:46566–46575. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Yoshizumi M, Abe J, Haendeler J, Huang Q
and Berk BC: Src and Cas mediate JNK activation but not ERK1/2 and
p38 kinases by reactive oxygen species. J Biol Chem.
275:11706–11712. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Wang T, Arifoglu P, Ronai Z and Tew KD:
Glutathione S-transferase P1-1 (GSTP1-1) inhibits c-Jun N-terminal
kinase (JNK1) signaling through interaction with the C terminus. J
Biol Chem. 276:20999–21003. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Sinha D, Bannergee S, Schwartz JH,
Lieberthal W and Levine JS: Inhibition of ligand-independent ERK1/2
activity in kidney proximal tubular cells deprived of soluble
survival factors upregulates Akt and prevents apoptosis. J Biol
Chem. 279:10962–10972. 2004. View Article : Google Scholar
|