Introduction
Chemotherapeutic drugs used in cancer therapy induce
apoptosis to tumor cells (1,2).
Apoptosis is a physiological form of cell death that plays an
important role in normal development, tissue homeostasis and
pathological situation (3,4). Two major pathways of apoptosis have
been widely recognized, i.e. extrinsic (death receptor; DR) pathway
(5,6), and intrinsic (mitochondria mediated)
pathway (7,8). More recently, endoplasmic reticulum
(ER) stress has drawn attention as the third pathway of apoptosis
(9), and has an impact on
alternative cell death pathways as potential new targets for cancer
therapy.
ER is a multifunctional organelle, and plays roles
as an intracellular reservoir of Ca2+, and in synthesis
of lipid and cholesterol, and synthesis and controlling of the
quality of membrane proteins or secreted proteins. A polypeptide
translated from mRNA needs to be formatted into a proper
higher-order structure to be functional, and this process is called
‘folding’. Folding in ER includes not only formation of a
higher-order structure but also glycosylation and formation of
disulfide bonds, namely the unique reactions which cannot be seen
in folding in the cytoplasm. Correctly folded proteins are
transported to Golgi apparatus through vesicular trafficking, where
post-translational modifications take place, forming mature
proteins. Some proteins that have been misfolded in this process
are refolded by ER chaperons such as calnexin and immunoglobulin
heavy chain binding protein/glucose regulated protein 78
(Bip/GRP78), or degraded by endoplasmic reticulum-associated
degradation (ERAD). Collapse of ER homeostasis induces ER stress
derived from the unfolded protein response (UPR), a sequential,
pro-survival process for restoring ER functions. UPR is accompanied
by the augmentation of folding capacity through increase of
molecular chaperon expression and suppression of protein synthesis
at transcription or translation level, therefore causing relief of
the ER stress by unloading the folding in ER (10–12).
The UPR is initiated by activation of three sensors, inositol
requiring enzyme 1 (IRE1), activating transcription factor 6
(ATF6), and PKR-like endoplasmic reticulum kinase (PERK). These
proteins are transmembrane proteins that monitor accumulation of
misfolded proteins in ER lumen, and function as signal transducers
of ER stress to cytosol (13).
However, in the case of severe prolonged ER stress when UPR and
ERAD are not sufficient for the evading the stress, the misfolded
proteins are eliminated with whole cells by ER-stress mediated
apoptosis. It has been reported that ER stress-dependent apoptosis
is mediated mainly by the pro-apoptotic transcription factor CHOP,
proapoptotic members of the Bcl-2 family and direct calcium
transfer from ER to mitochondria (14,15).
Association of ER stress-mediated apoptosis to some pathology such
as Alzheimer (16), Parkinson
(17), and diabetes (18) has been reported as well as the
involvement of the cytotoxic mechanism in the medicinal drugs such
as Bortezomib (19) and Nelfinavir
(20). It is suggested that
targeting ER stress and UPR is a promising strategy for cancer
treatment (21).
Sialic acid-binding lectin (SBL) isolated from
bullfrog (Rana catesbeiana) oocytes was found as a lectin,
because SBL agglutinates various kinds of tumor cells and the
agglutination was inhibited by sialoglycoprotein or ganglioside
(22–24). Agglutination induced by SBL was
observed only in tumor cells but not in normal red blood cells and
fibroblasts (24). Amino acid
sequence of SBL shows that it has homology to the member of RNase A
superfamily, and it has been revealed that SBL has pyrimidine
base-specific ribonuclease activity (25–28).
The antitumor effect of SBL was reported using p388 and L1210
murine leukemia cells in vitro and sarcoma 180, Ehrlich and
Mep 2 ascites cells in vivo (29–31).
We have recently reported that SBL shows cytotoxity for various
human leukemia cells including MDR cells, and that cytotoxity is
induced through caspase-dependent apoptosis in which mitochondrial
perturbation occurs as upstream events (32). However, the detail of molecular
mechanisms implicated in SBL-induced apoptosis is still unknown. In
this study, we investigated the involvement of ER stress in
apoptosis triggered by SBL.
Materials and methods
Materials
SBL was isolated in sequential chromatography on
Sephadex G-75, DEAE-cellulose, hydroxyapatite and SP-Sepharose as
described previously (24).
Thapsigargin (TG) was purchased from Calbiochem (Darmstadt,
Germany). Caspase inhibitors (z-LEVD-fmk, z-VAD-fmk, z-IETD-fmk and
z-LEHD-fmk), anti-caspase-4 antibody and anti-caspase-9 antibody
were purchased from Medical and Biological Laboratories Co., Ltd
(Nagoya, Japan). Anti-caspase-8 antibody and anti-caspase-3
antibody were from Cell Signaling Technology (Beverly, MA, USA).
Anti-Bip/GRP78 antibody was from Becton-Dickinson (Franklin Lakes,
NJ, USA). Horseradish peroxidase (HRP)-conjugated anti-mouse IgG
actibody and HRP-conjugated anti-rabbit IgG andibody were from
Zymed (South San Francisco, CA, USA) and Cedarlane Laboratories Ltd
(Hornby, ON, Canada), respectively.
Cell culture
Human leukemia Jurkat T-cells, were obtained from
the Cell Resource Center of the Biomedical Research, Institute of
Development, Ageing and Cancer, Tohoku University (Sendai, Japan).
Cells were routinely kept in RPMI-1640 medium (Nissui
Pharmaceutical Co. Ltd., Tokyo, Japan) supplemented with 10% fetal
calf serum (FCS), penicillin (100 U/ml) and streptomycin (100
μg/ml) at 37°C in a 95% air and 5% CO2
atmosphere.
Detection of sub G1 population
SBL- and TG-treated cells were harvested, washed and
re-suspended in PBS. Then, equal amount of PBS containing Triton
X-100 (0.2%), EDTA (4 mM, pH 8.0), RNase A (20 μg/ml),
propidium iodide (PI; 40 μg/ml) was added. DNA contents of
cells were determined by FACSCalibur (Becton-Dickinson), and the
cell population that indicated low DNA contents was counted as sub
G1 population.
Western blot analysis
Whole cell lysate was prepared by lysing the cells
with extraction buffer [10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1%
Triton X-100, 5 mM EDTA (pH 8.0), 1 mM phenylmethylsulfonyl
fluoride (PMSF) and 1 tablet/10 ml protease inhibitor cocktail
(Roche Applied Science, Indianapolis, IN, USA)]. Soluble proteins
were collected and concentrations were measured by DC protein assay
kit (Bio-Rad, Richmond, CA, USA) in accordance with instructions.
Proteins were separated by SDS-PAGE, and transferred to
polyviniliden difluoride (PVDF) membrane (GE Healthcare, Little
Chalfont, UK). The membrane was blocked by 5% fat-free skim milk
for 1 h. After the membrane was washed with TBST [20 mM Tris-HCl
(pH 7.6), 137 mM NaCl, 0.05% Tween-20], primary and secondary
antibodies were added to the membrane, respectively. The proteins
on membrane were detected using ECL western blotting detection
reagents (GE Healthcare).
Detection of x-box binding protein 1
(XBP-1) splicing
Total cellular RNA was isolated from cells using a
TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Reverse
transcription (RT) was performed using ReverTra Ace (Toyobo, Osaka,
Japan) with total RNA (1 μg) and oligo (dT)12–18
primers. Splicing of XBP-1 was detected by following the methods of
Nakamura et al (33). The
RT reaction mixture (1 μl) was subjected to PCR for 23
cycles in a final volume of 50 μl of Taq DNA polymerase
(1.25 units) (ABgene, Epsom, UK), gene specific forward primer
(5′-ACCACAGTCCATGCCATCAC-3′) and reverse primer
(5′-TCCACCACCCTGTTGCTG-3′). After initial denaturation at 94°C for
2 min, each of the cycles comprised: at 94°C for 30 sec, at 58°C
for 30 sec and at 72°C for 30 sec. To confirm the total expression
of XBP-1, PCR products were separated on 1.5% agarose gel, and
bands were visualized with ethidium bromide (EtBr) staining. GAPDH
expression was also detected as internal control using gene
specific forward primer (5′-ACCACAGTCCATGCCATCAC-3′) and reverse
primer (5′-TCCACCACCCTGTTGCTGTA-3′). To detect splicing of XBP-1,
PCR products were digested with ApaLI (10 units) at 37°C for
90 min. Digested sample were separated on 2.5% agarose gel, and
bands were visualized with EtBr.
Treatment of caspase inhibitors
The role of caspase activation in the process of
SBL-induce apoptosis was studied by the addition of z-VAD-fmk
(pan-caspase inhibitor), z-LEVD-fmk (caspase-4 specific inhibitor),
z-IETD-fmk (caspase-8 specific inhibitor) and z-LEHD-fmk (caspase-9
specific inhibitor). Each of the caspase inhibitors [z-LEVD-fmk (2,
10, 30 μM for DNA fragmentation assay, and 30 μM for
other assays), z-VAD-fmk, z-IETD-fmk and z-LEHD-fmk (50 μM
for all assays)] was added to culture medium 30 min before the
addition of SBL or TG.
Detection of DNA fragmentation
The cells (2×105/ml) were cultured in 100
μl in 96-well plates. After treatment with SBL, the cells
were collected by centrifugation, washed with PBS, then lysed with
cell lysis buffer [50 mM Tris-HCl (pH 6.8), 10 mM EDTA, 0.5w/v%
sodium-N-lauroylsarcosinate]. The samples were incubated for 30 min
with RNase A (final concentration: 500 μg/ml) at 50°C,
before being digested for 30 min with proteinase K (final
concentration: 500 μg/ml) at 50°C. After the samples were
electrophoresed on 1.8% agarose gel, DNA bands were visualized by
EtBr staining.
Observation of nuclear morphology
The cells (2×105/ml) were cultured in 5
ml in 6-well plates. After treatment with SBL, the cells were
collected by centrifugation and washed with PBS. Then the cells
were fixed with 1% paraformaldehyde (100 μl) for 15 min at
4°C, and stained with Hoechst 33258 (50 μl, 1 mg/ml) for 15
min at 4°C. After three washes with PBS, the cells were mounted on
slide glass using Prolong gold antifade reagent (Molecular Probes).
The fluorescence was visualized with a fluorescence microscope,
IX71 microscope (Olympus Corporation, Tokyo, Japan).
Flow cytometric analysis of Annexin V
binding and PI incorporation
Annexin V binding and PI incorporation were detected
with a MEBCYTO apoptosis kit (Medical and Biological Laboratories)
according to manufacturer’s directions. The cells
(2×105/ml) were cultured in 1 ml in 24-well plates.
Fluorescence intensity of fluorescein isothiocyanate (FITC)-Annexin
V and PI was determined using a FACSCalibur flow cytometer
(Becton-Dickinson).
Reduction of mitochondrial membrane
potential (MMP)
MMP was assessed using a fluorescent probe 5, 50, 6,
60-tetra-chloro-1, 10, 3, 30-tetraethylbenzamidazolocarbocyanin
iodide (JC-1, AnaSpec, Fremont, CA, USA). Red emission from the dye
is attributed to the potential of aggregation of JC-1 in the
mitochondria. Green fluorescence reflects the monomeric form of
JC-1, appearing in the cytoplasm after mitochondrial membrane
depolarization. Cells were cultured in condition of each
experiment, and then incubated with JC-1 (2 μM) dye diluted
in culture medium at 37°C for 15 min. The cells were washed three
times with PBS, and analyzed immediately using FACSCalibur
(Becton-Dickinson).
Statistical analysis
Results were collected from three independent
experiments, each performed in triplicate, and data are expressed
as mean ± SD. Statistical analysis was performed using GraphPad
Prism 3.0 and comparisons were made using one-way or two-way
analysis of variance (ANOVA), followed by Bonferroni’s post hoc
tests.
Results
Time course of apoptotic events in SBL-
and TG-treated Jurkat cells
We have recently shown that SBL possess anti-tumor
effect for various leukemia cells including multidrug resistant
cells, because SBL executes caspase-dependent apoptosis in which
mitochondrial perturbation occurs as upstream events (32). To analyze the detail of the
signaling pathway of SBL-induced apoptosis, we first observed the
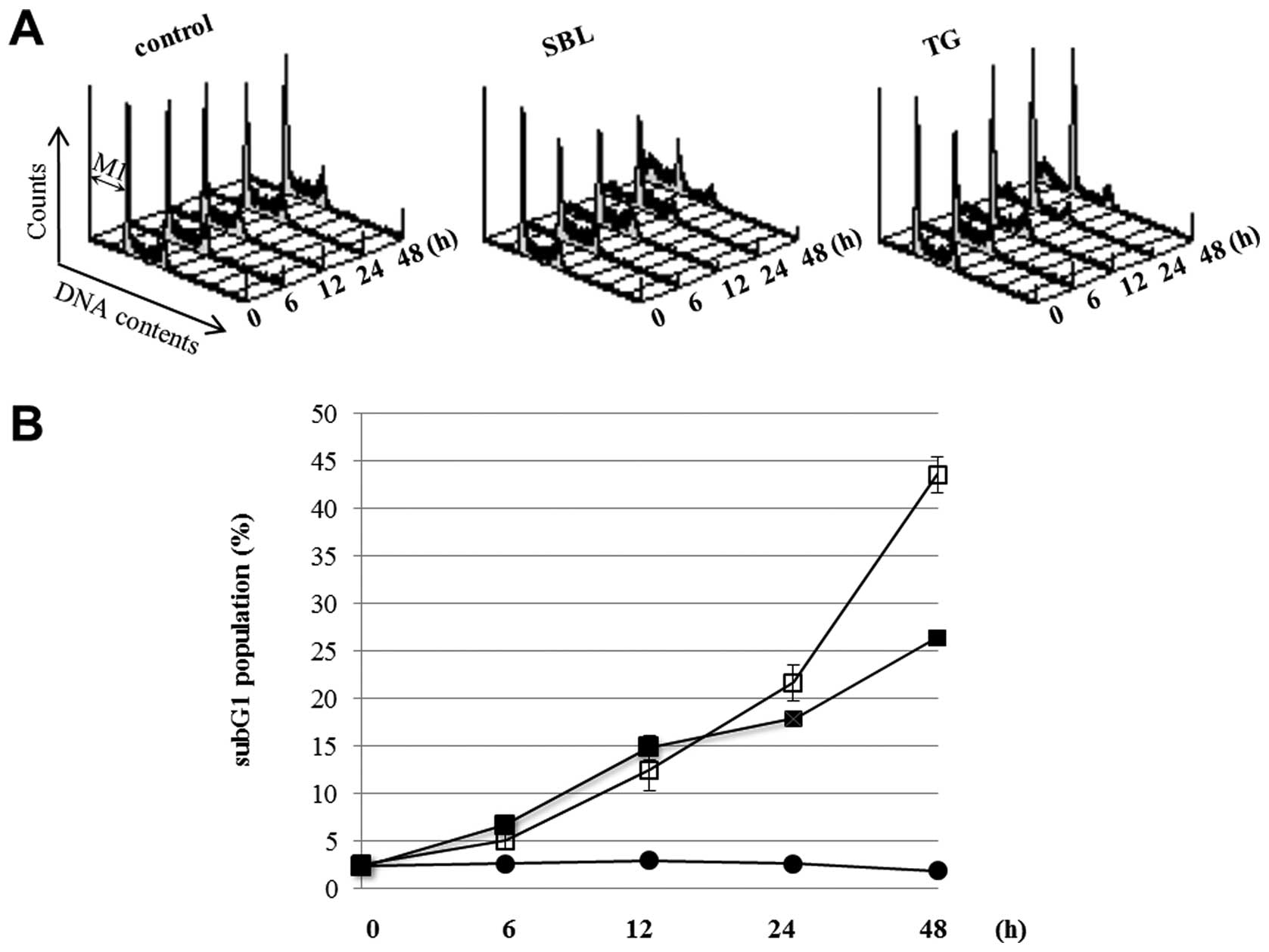
time course of apoptotic events caused by SBL treatment. During
apoptosis, it was observed that the cell population indicated low
DNA contents resulting from the fragmentation of nucleus and
chromatin, and the formation of apoptotic bodies. The sub G1
population described above is considered as an indicator of
execution phase of apoptosis. We compared SBL with TG, an
endoplasmic reticulum Ca2+-ATPase inhibitor, using ER
stress inducer as a control. DNA contents of SBL- and TG-treated
Jurkat cells were analyzed by flow cytometry, and the sub G1
populations in the cells treating with SBL or TG for 24 and 48 h
were 22 and 44% or 18 and 26%, respectively (Fig. 1). Time course of activation of
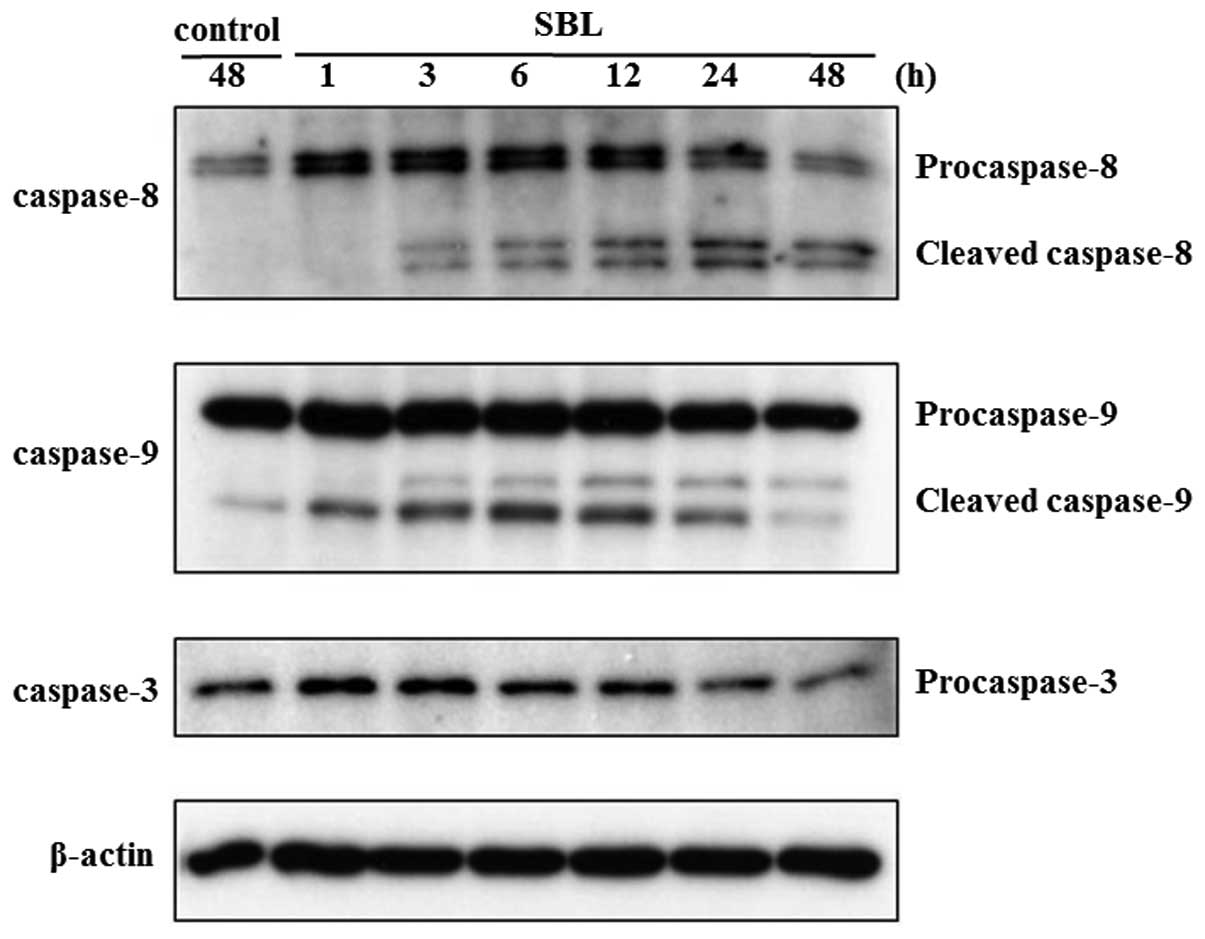
caspases, key proteases in apoptotic process, is also assessed by
western blot analysis. As shown in Fig. 2, SBL-induced cleavage of
procaspases 9, 8 and 3 was detected from 1, 3 and 24-h treatment,
respectively. These results indicate that SBL-induced apoptotic
signal is detected from 1-h treatment, as we observed initiator
caspase-9 activation.
Activation of ER stress signaling in
SBL-treated Jukat cells
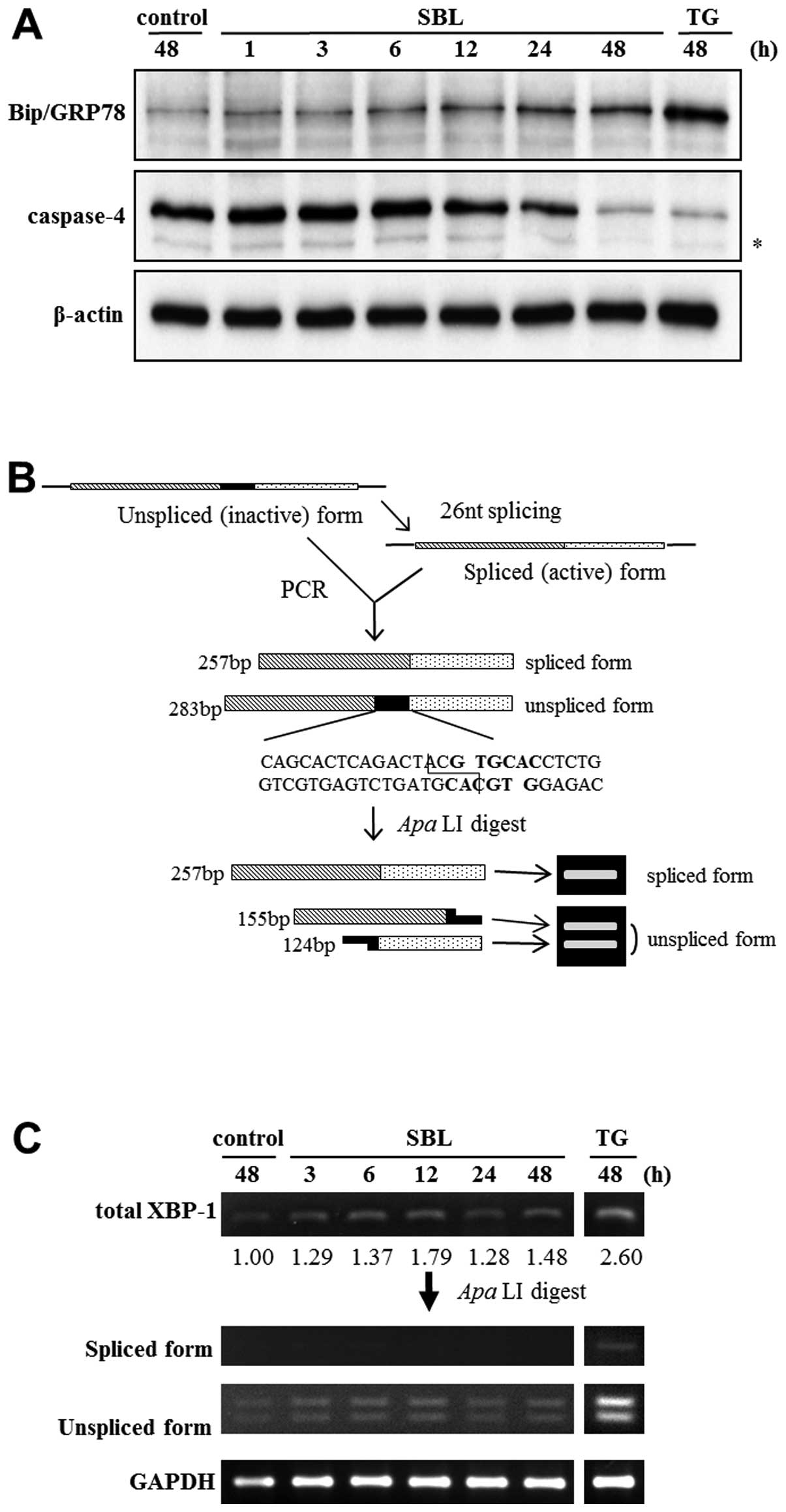
To investigate whether SBL induces unfolded protein
response (UPR) and ER stress-mediated apoptosis, we assessed the
expression of Bip/GRP78 and activation of caspase-4 by western blot
analysis, and the elevation of specific splicing of XBP-1 mRNA by
RT-PCR followed by subsequent restriction enzyme digestion. Results
showed that expression of Bip/GRP78 was elevated after 6 to 48-h of
SBL treatment, and that degradation of procaspase-4, namely
activation of caspase-4, was detected after 24 to 48-h treatment
with SBL (Fig. 3A). It is known
that once ER stress was induced, XBP-1 is spliced specifically by
IRE1. Because there is an ApaLI digestion site on the 27 nt
domain on the unspliced form of XBP-1, ApaLI digests only
unspliced form of XBP-1, and results in smaller two fragments.
Digestion with ApaLI makes it easier to discern the
expression of spliced and unspliced form of XBP-1 (Fig. 3B, upper panel). We found that the
spliced and unspliced form of XBP-1 mRNA were detected by RT-PCR
followed by subsequent ApaLI digestion, and that the total
expression of XBP-1 was increased by SBL treatment, but the spliced
form of XBP-1 was not increased (Fig.
3B, lower panel).
Participation of ER stress to SBL-induced
apoptosis in Jurkat cells
To assess the participation of ER stress signaling
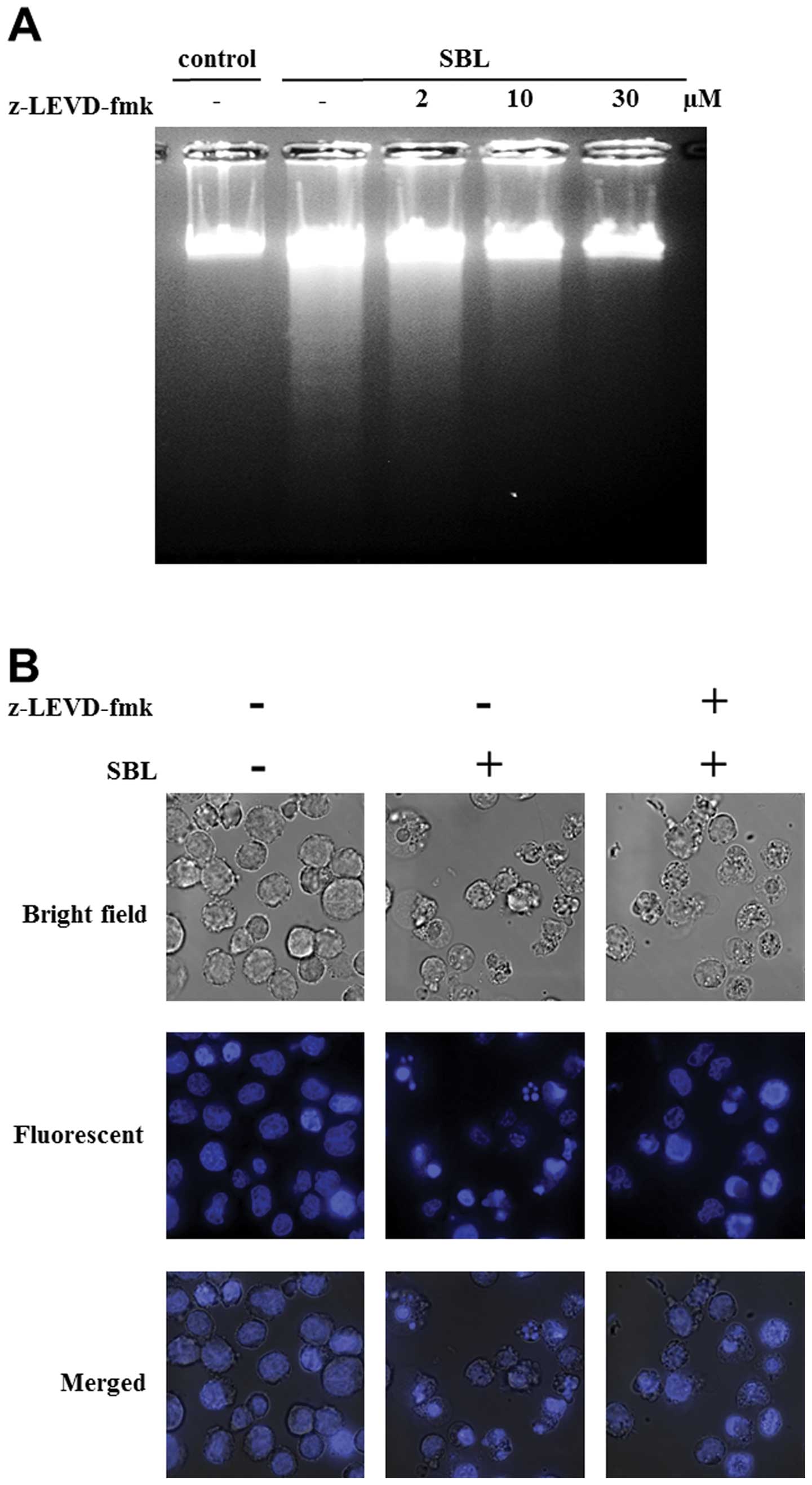
to apoptosis induced by SBL, we performed experiments using
caspase-4 inhibitor, z-LEVD-fmk. DNA fragmentation induced by SBL
was inhibited by z-LEVD-fmk (from 10 μM, in a
concentration-dependent manner), and nuclear fragmentation was
almost completely inhibited by z-LEVD-fmk at 30 μM (Fig. 4A and B). Staining with Annexin V-PI
showed that 54% cells were Annexin V positive in SBL-treated cells,
but z-LEVD-fmk-pretreated cells resulted in 20% reduced percentage
of Annexin V positive cells (Fig.
4C). These results indicate that caspase-4 activation is
involved in SBL-induced apoptosis.
Comparison of the effects of each caspase
inhibitor
Three apoptotic signaling pathways: i) death
receptor pathway; ii) mitochondria pathway; and iii) ER stress
mediated pathway are well known, and caspase-8, -9 and -4 are
considered as the initiator caspase of each pathway, respectively.
In SBL-induced apoptotic signal, we detected the activation of
caspase-8, -9 and -3 (32), and
studied which caspase was activated upstream of apoptotic signal
and which caspase is the most important in SBL-induced apoptosis
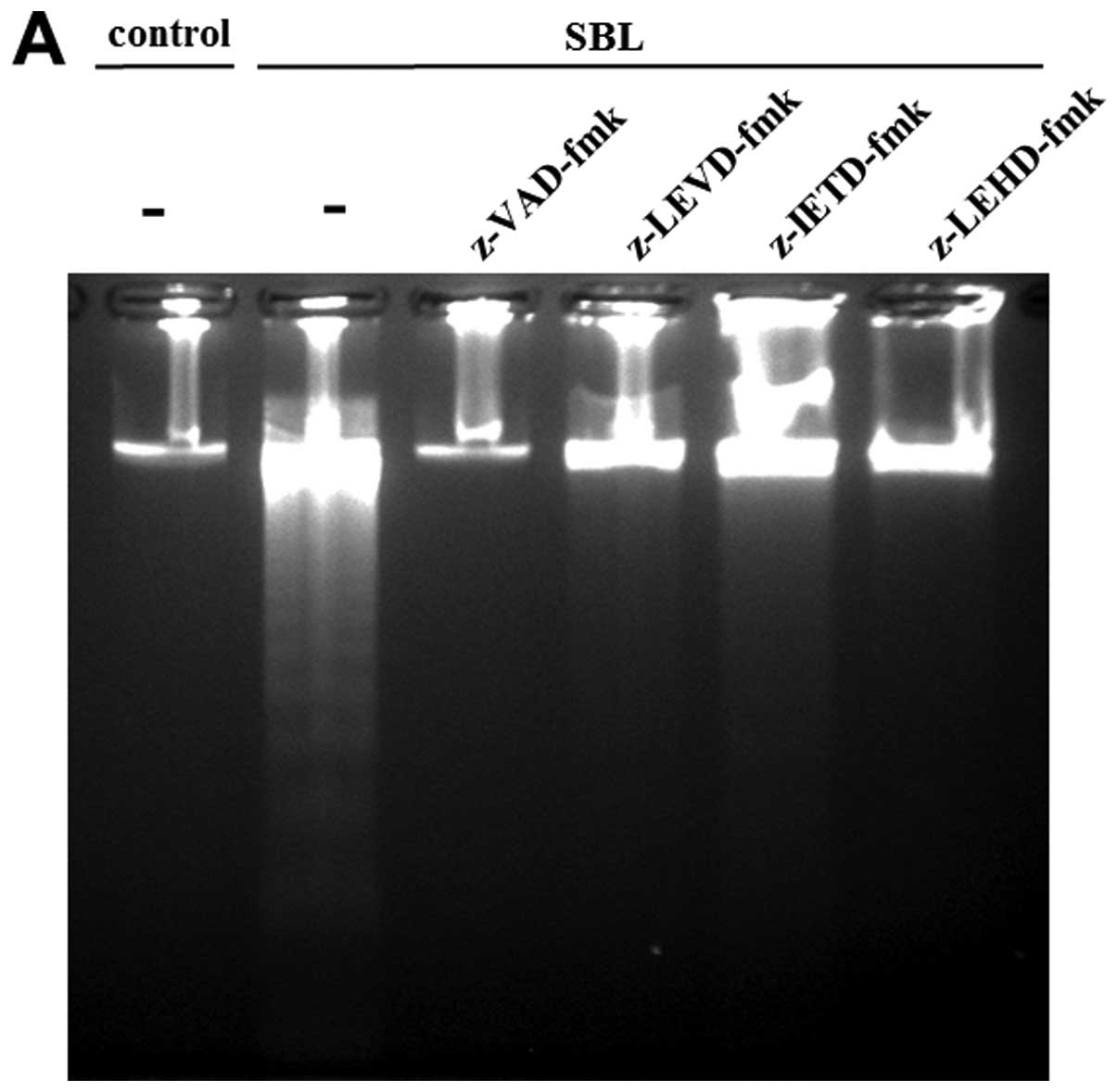
using specific caspase inhibitors. As a result, DNA fragmentation
caused by SBL was inhibited completely by pan-caspase inhibitor:
z-VAD-fmk; and caspase-4 inhibitor: z-LEVD-fmk. Pretreatment with
caspase-9 inhibitor: z-LEHD-fmk also inhibited the DNA
fragmentation, but caspase-8 inhibitor, z-IETD-fmk shows relatively
low inhibition (Fig. 5A). The
effect on induction of apoptosis was also assessed by Annexin V-PI
staining, and 36, 19, 9 and 12% inhibition were detected by
pretreatment with z-VAD-fmk, z-LEVD-fmk, z-IETD-fmk and z-LEHD-fmk,
respectively (Fig. 5B).
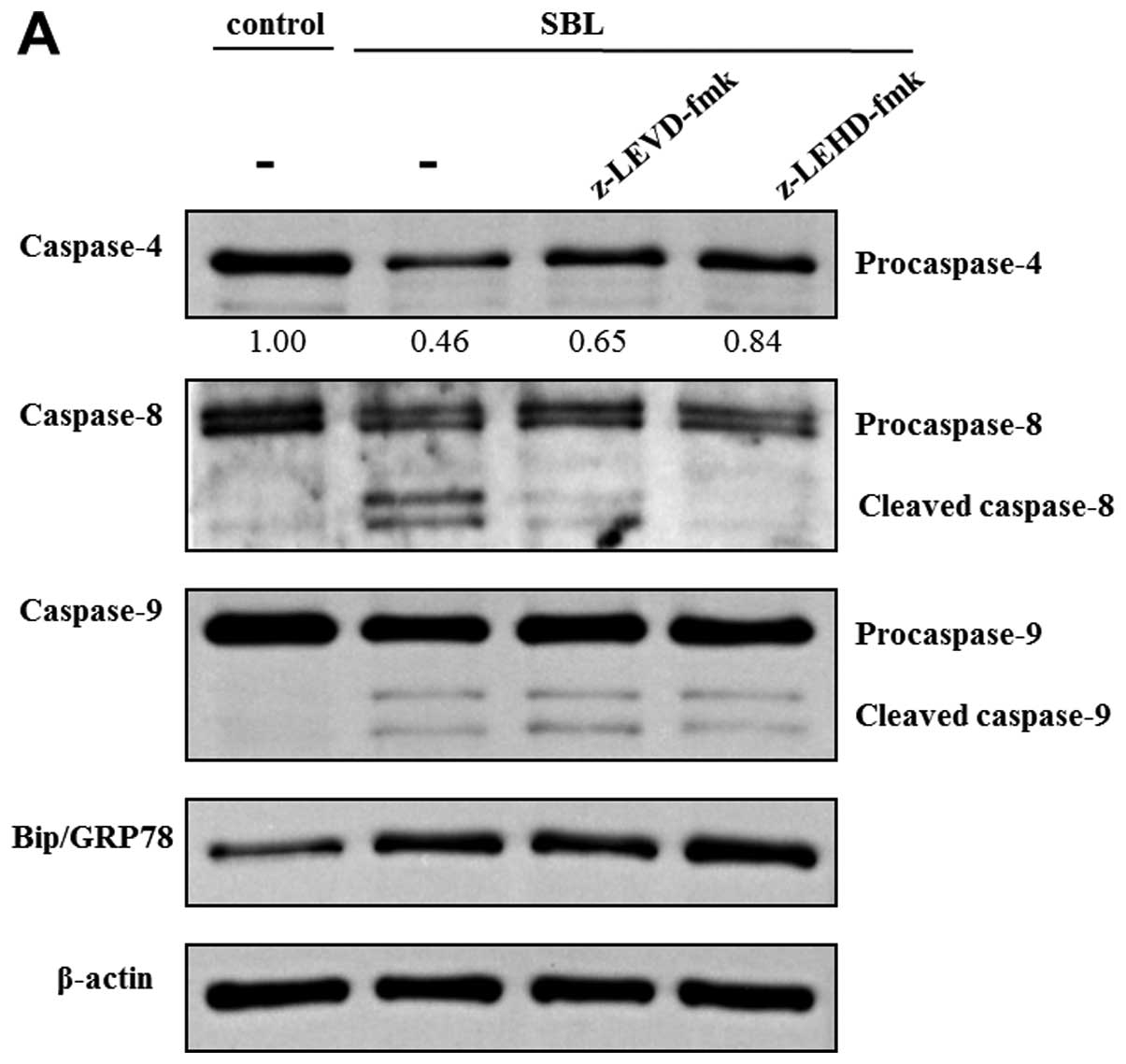
Because z-LEVD-fmk and z-LEHD-fmk inhibited
SBL-induced DNA fragmentation, we assessed the effect of these
specific caspase inhibitors on other caspase activation and
expression of Bip/FRP78 induced by SBL. In z-LEVD-fmk-pretreated
cells, activation of caspase-8 was inhibited, but there was no
effect on activation of caspase-9 (Fig. 6A). On the other hand, in
z-LEHD-fmk-pretreated cells, activation of caspase-4 as well as
that of caspase-8 was diminished. The elevation of Bip/GRP78
expression was not inhibited either by pretreatment with z-LEVD-fmk
or z-LEHD-fmk. Previously, we reported that SBL caused rigid
mitochondrial perturbation. In the present study, we assessed the
effect of caspase-4 and -9 inhibitors on the mitochondrial
perturbation induced by SBL. These inhibitors, however, did not
inhibit the reduction of MMP triggered by SBL, indicating that
mitochondrial perturbation caused by SBL may occur upstream of
caspase activation and other events which could be inhibited by
caspase inhibitors (Fig. 6B).
Discussion
We demonstrated that ER stress participated in
SBL-induced apoptosis. Disruption of the balance between newly
synthesis and quality control mechanism of proteins leads to
accumulation of abnormal proteins, i.e. ER stress. The cells try to
suppress the stress by elevating the folding clearance though UPR
and ERAD. The signal of UPR in eukaryotes can start from each of
three transmembrane proteins IRE1, ATF6 and PERK. IRE1 is a type I
transmembrane protein activated by dimerization and
phosphorylation. Cytosolic domain of IRE1 possesses RNase activity,
and activate IRE1 splices XBP-1 mRNA by its RNase activity
independently of spliceosome. XBP-1 translated from spliced form of
XBP-1 mRNA works as transcription factor, and activates the
expression of chaperons such as Bip/GRP78 and factors of ERAD
(34–36). ATF6 is a type II transmembrane
protein. Once ATF6 detects the accumulation of misfolded proteins,
it translocates from ER to golgi through vesicle transport, and is
activated by specific cleavage. N-terminal fragment of the cleavage
product itself works as a transcription factor, and induces
transcription of XBP-1 and molecular chaperones similarly to IRE1
(13). PERK is a type I
transmembrane protein which has homology to IRE1, and has kinase
activity in its cytosolic domain. PERK is activated by
phosphorylation, and the kinase activity causes phosphorylation of
the eukaryotic initiation factor 2α (eIF2α), resulting in
suppression of global gene expression and unloading protein folding
in ER (12). Furthermore,
phosphorylation of eIF2α elevates the translation of ATF4, and it
is known that ATF4 increases the expression of CHOP and ATF3
(37–40). In the UPR associated factors above,
we assessed the expression of Bip/GRP78 and XBP-1 in SBL-treated
Jurkat cells to analyze if SBL causes induction of UPR. The results
showed that SBL induced elevation of Bip/GRP78 expression, in a
time-dependent manner (Fig. 3A).
Also, elevated expression of XBP-1 mRNA itself was observed in 48-h
treatment with SBL, but the elevation of the active (spliced) form
was not observed (Fig. 3C). The
elevation of expression of Bip/GRP78 and XBP-1 mRNA suggested that
SBL caused the induction of UPR by ER stress, while the fact that
there was no elevation of spliced XBP-1 mRNA suggested that the
signal transduction may not be mediated by IRE1.
Because the induction of UPR attributed to ER stress
was observed in SBL-treated Jurkat cells, we next analyzed whether
SBL induces ER stress-mediated apoptosis or not. Caspase-4, a human
homolog of mouse caspase-12 is known as an initiator caspase of ER
stress-mediated apoptosis (41,42).
We revealed that the cleavage of procaspase-4, that is, activation
of caspase-4 occurred in SBL-treated Jurkat cells, suggesting that
ER stress-mediated apoptosis is involving in SBL-induced apoptosis
(Fig. 3).
To assess the participation of ER stress in
SBL-induced apoptosis, we performed experiments using caspase-4
specific inhibitor, z-LEVD-fmk. Pretreatment with z-LEVD-fmk
diminished SBL-induced DNA fragmentation, dose-dependently
(Fig. 4A and B). The percentage of
apoptotic cells detected by Annexin V binding assay was also
decreased in the cells pretreated with z-LEVD-fmk. Therefore, it is
suggested that caspase-4 may play an important role in SBL-induced
apopotosis. Caspase-8 and -9, are known as initiator caspases of DR
and mitochondrial pathway, respectively, as mentioned above. To
examine the contribution of initiator caspases to SBL-induced
apoptosis, comparative study was performed with specific caspase
inhibitors. We showed that caspase-4 and -9 were prominently
involved, because z-LEVD-fmk and z-LEHD-fmk inhibited SBL-induced
apoptosis. However, the caspase-8 inhibitor z-IETD was less
effective.
Cephalostatin 1 derived from Cephalodiscus
gilchristi induces ER stress-dependent apoptosis to Jukat
cells, and activates caspase-4 causing activation of caspase-9
independently of apoptosome formation. It is reported that
caspase-4 is activated upstream of caspase-9 activation in ER
stress-dependent apoptosis induced by TG or tunicamycin. Because
the clear involvement of caspase-4 and -9 was clarified in
SBL-induced apoptosis, we investigated whether SBL-induced
apoptotic signal is transduced similarly to the ER stress inducers,
by focusing on caspase activation in specific caspase
inhibitor-pretreated cells. The results showed the activation of
caspase-9 was not affected by the caspase-4 inhibitor z-LEVD-fmk,
while activation of caspase-4 was partially diminished by
pretreatment with thecaspase-9 inhibitor z-LEHD-fmk (Fig. 6A), indicating that activation of
caspase-4 occurs not upstream of caspase-9 activation, but
partially dependent on caspase-9 activation. We have recently
reported that caspase-8 is activated at downstream of caspase-9
activation in SBL-treated Jurkat cells (32). Taken together, it is suggested that
in caspase cascade activated by SBL, caspase-9 is activated as
initiator caspase, and it escalates activation of caspase-4, then
caspase-8 is activated at downstream of these caspases.
Furthermore, activation of caspase-8 is depending on caspase-9 and
-4 activation. It is suggested that it participates in the
amplification of the apoptotic signal mediated by Bid cleavage. On
the other hand, elevated expression of Bip/GRP78 was not affected
by z-LEHD-fmk (Fig. 6A),
indicating that activation of caspase-9 is not implicating to ER
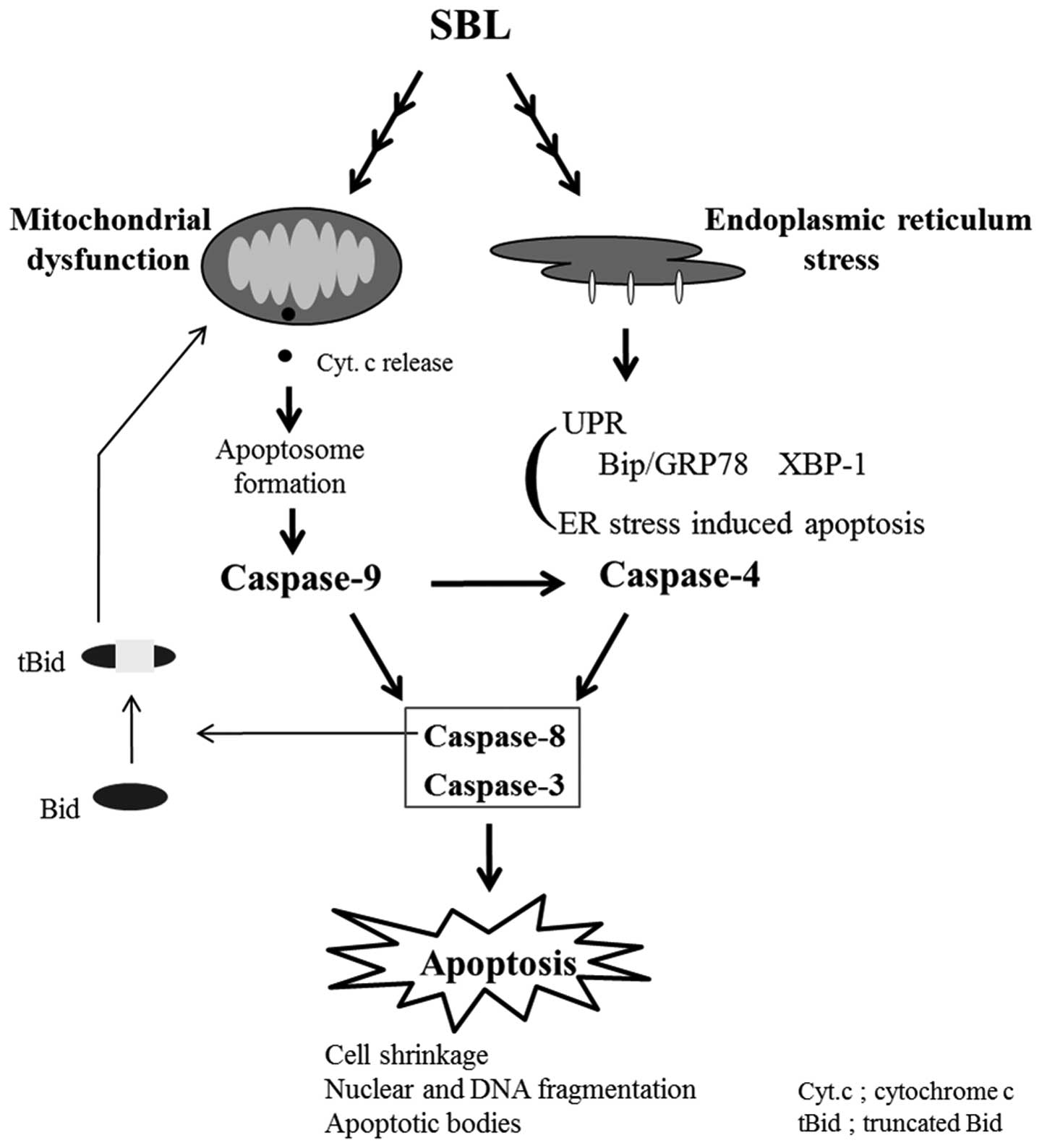
stress induced by SBL. We hypothesized here that mitochondria
perturbation and ER stress may occur independently in SBL-treated
cells and the activation of caspase-9 is partially involved in
activation of caspase-4 (Fig. 7).
To confirm the relationship between activation of caspase-4
attributed to ER stress and mitochondrial perturbation, we assessed
if the reduction of MMP induced by SBL is affected by z-LEVD-fmk
(Fig. 6B), and found that
mitochondrial perturbation is neither affected by z-LEVD-fmk nor
z-LEHD-fmk. These results support our hypothesis that SBL causes
mitochondrial perturbation and ER stress, independently.
Furthermore, when we compared the effects of SBL and TG, the cells
in sub G1 population were observed in SBL-treated cells more than
in TG-treated cells, but ER stress represented by expression of
Bip/GRP78 and active form of XBP-1 was observed more rigidly in
TG-treated cells (Fig. 1 and
2). These results suggested that
SBL causes apoptosis not only by ER stress but also by
mitochondrial pathway and mitochondrial pathway may be intensely
involved in apoptosis induced by SBL.
We analyzed the signaling mechanism of apoptosis
induced by SBL, focusing on induction of ER stress and activation
of caspases, and we concluded that SBL can cause multiple apoptotic
pathways independently. We have recently reported that SBL
activates p38 and JNK MAPKs (32).
It has been reported that MAPKs and other molecules such as
bcl2-family proteins may participate in ER stress (43,44).
The precise antitumor mechanism of SBL and clarification of the
relationship between the effects of SBL and the above molecules
will advance SBL as a potential candidate for development as an
effective anticancer drug.
Acknowledgements
This study was supported in part by
Grant-in-Aid of the ‘Academic Frontier’ Project for Private
Universities from the Ministry of Education, Culture, Sports,
Science and Technology of Japan.
References
|
1.
|
Muller M, Strand S, Hug H, Heinemann EM,
Walczak H, Hofmann WJ, Stremmel W, et al: Drug-induced apoptosis in
hepatoma cells is mediated by the CD95 (APO-1/Fas) receptor/ligand
system and involves activation of wild-type p53. J Clin Invest.
99:403–413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Fulda S, Sieverts H, Friesen C, Herr I and
Debatin KM: The CD95 (APO-1/Fas) system mediates drug-induced
apoptosis in neuroblastoma cells. Cancer Res. 57:3823–3829.
1997.PubMed/NCBI
|
|
3.
|
Kerr JF, Wyllie AH and Currie AR:
Apoptosis: a basic biological phenomenon with wide-ranging
implications in tissue kinetics. Br J Cancer. 26:239–257. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Arends MJ and Wyllie AH: Apoptosis:
mechanisms and roles in pathology. Int Rev Exp Pathol. 32:223–254.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Ashkenazi A and Dixit VM: Apoptosis
control by death and decoy receptors. Curr Opin Cell Biol.
11:255–260. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Ashkenazi A and Dixit VM: Death receptors:
signaling and modulation. Science. 281:1305–1308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Zou H, Henzel WJ, Liu X, Lutschg A and
Wang X: Apaf-1, a human protein homologous to C. elegans CED-4,
participates in cytochrome c-dependent activation of caspase-3.
Cell. 90:405–413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Green DR: Apoptotic pathways: the roads to
ruin. Cell. 94:695–698. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Schroder M and Kaufman RJ: ER stress and
the unfolded protein response. Mutat Res. 569:29–63. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Martinez IM and Chrispeels MJ: Genomic
analysis of the unfolded protein response in Arabidopsis
shows its connection to important cellular processes. Plant Cell.
15:561–576. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Pakula TM, Laxell M, Huuskonen A, Uusitalo
J, Saloheimo M and Penttila M: The effects of drugs inhibiting
protein secretion in the filamentous fungus Trichoderma
reesei. Evidence for down-regulation of genes that encode
secreted proteins in the stressed cells. J Biol Chem.
278:45011–45020. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Harding HP, Zhang Y and Ron D: Protein
translation and folding are coupled by an
endoplasmic-reticulum-resident kinase. Nature. 397:271–274. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Kadowaki H, Nishitoh H and Ichijo H:
Survival and apoptosis signals in ER stress: the role of protein
kinases. J Chem Neuroanat. 28:93–100. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Csordas G, Thomas AP and Hajnoczky G:
Quasi-synaptic calcium signal transmission between endoplasmic
reticulum and mitochondria. EMBO J. 18:96–108. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Imai Y, Soda M, Inoue H, Hattori N, Mizuno
Y and Takahashi R: An unfolded putative transmembrane polypeptide,
which can lead to endoplasmic reticulum stress, is a substrate of
Parkin. Cell. 105:891–902. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Araki E, Oyadomari S and Mori M:
Endoplasmic reticulum stress and diabetes mellitus. Intern Med.
42:7–14. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Nawrocki ST, Carew JS, Dunner K Jr, Boise
LH, Chiao PJ, Huang P, Abbruzzese JL, et al: Bortezomib inhibits
PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis
via ER stress in human pancreatic cancer cells. Cancer Res.
65:11510–11519. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Gills JJ, Lopiccolo J, Tsurutani J,
Shoemaker RH, Best CJ, Abu-Asab MS, Borojerdi J, et al: Nelfinavir,
a lead HIV protease inhibitor, is a broad-spectrum, anticancer
agent that induces endoplasmic reticulum stress, autophagy, and
apoptosis in vitro and in vivo. Clin Cancer Res. 13:5183–5194.
2007. View Article : Google Scholar
|
|
21.
|
Gallerne C, Prola A and Lemaire C: Hsp90
inhibition by PU-H71 induces apoptosis through endoplasmic
reticulum stress and mitochondrial pathway in cancer cells and
overcomes the resistance conferred by Bcl-2. Biochim Biophys Acta.
1833:1356–1366. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Kawauchi H, Sakakibara F and Watanabe K:
Agglutinins of frog eggs: a new class of proteins causing
preferential agglutination of tumor cells. Experientia. 31:364–365.
1975. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Sakakibara F, Kawauchi H, Takayanagi G and
Ise H: Egg lectin of Rana japonica and its receptor
glycoprotein of Ehrlich tumor cells. Cancer Res. 39:1347–1352.
1979.
|
|
24.
|
Nitta K, Takayanagi G, Kawauchi H and
Hakomori S: Isolation and characterization of Rana
catesbeiana lectin and demonstration of the lectin-binding
glycoprotein of rodent and human tumor cell membranes. Cancer Res.
47:4877–4883. 1987.PubMed/NCBI
|
|
25.
|
Titani K, Takio K, Kuwada M, Nitta K,
Sakakibara F, Kawauchi H, Takayanagi G, et al: Amino acid sequence
of sialic acid binding lectin from frog (Rana catesbeiana)
eggs. Biochemistry. 26:2189–2194. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Kamiya Y, Oyama F, Oyama R, Sakakibara F,
Nitta K, Kawauchi H, Takayanagi Y, et al: Amino acid sequence of a
lectin from Japanese frog (Rana japonica) eggs. J Biochem.
108:139–143. 1990.PubMed/NCBI
|
|
27.
|
Nitta K, Oyama F, Oyama R, Sekiguchi K,
Kawauchi H, Takayanagi Y, Hakomori S, et al: Ribonuclease activity
of sialic acid-binding lectin from Rana catesbeiana eggs.
Glycobiology. 3:37–45. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Okabe Y, Katayama N, Iwama M, Watanabe H,
Ohgi K, Irie M, Nitta K, et al: Comparative base specificity,
stability, and lectin activity of two lectins from eggs of Rana
catesbeiana and R. japonica and liver ribonuclease from
R catesbeiana. J Biochem. 109:786–790. 1991.PubMed/NCBI
|
|
29.
|
Nitta K, Ozaki K, Ishikawa M, Furusawa S,
Hosono M, Kawauchi H, Sasaki K, et al: Inhibition of cell
proliferation by Rana catesbeiana and Rana japonica
lectins belonging to the ribonuclease superfamily. Cancer Res.
54:920–927. 1994.PubMed/NCBI
|
|
30.
|
Nitta K, Ozaki K, Tsukamoto Y, Furusawa S,
Ohkubo Y, Takimoto H, Murata R, et al: Characterization of a
Rana catesbeiana lectin-resistant mutant of leukemia P388
cells. Cancer Res. 54:928–934. 1994.
|
|
31.
|
Nitta K, Ozaki K, Tsukamoto Y, Hosono M,
Ogawakonno Y, Kawauchi H, Takayanagi Y, et al: Catalytic lectin
(leczyme) from bullfrog (Rana catesbeiana) eggs. Int J
Oncol. 9:19–23. 1996.PubMed/NCBI
|
|
32.
|
Tatsuta T, Hosono M, Sugawara S, Kariya Y,
Ogawa Y, Hakomori S and Nitta K: Sialic acid-binding lectin
(leczyme) induces caspase-dependent apoptosis mediated
mitochondrial pertubation in Jurkat cells. Int J Oncol.
43:1402–1412. 2013.
|
|
33.
|
Nakamura M, Gotoh T, Okuno Y, Tatetsu H,
Sonoki T, Uneda S, Mori M, et al: Activation of the endoplasmic
reticulum stress pathway is associated with survival of myeloma
cells. Leuk Lymphoma. 47:531–539. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Travers KJ, Patil CK, Wodicka L, Lockhart
DJ, Weissman JS and Walter P: Functional and genomic analyses
reveal an essential coordination between the unfolded protein
response and ER-associated degradation. Cell. 101:249–258. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Shen X, Ellis RE, Sakaki K and Kaufman RJ:
Genetic interactions due to constitutive and inducible gene
regulation mediated by the unfolded protein response in C
elegans. PLoS Genet. 1:e372005. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Yoshida H, Matsui T, Hosokawa N, Kaufman
RJ, Nagata K and Mori K: A time-dependent phase shift in the
mammalian unfolded protein response. Dev Cell. 4:265–271. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Harding HP, Zhang Y, Bertolotti A, Zeng H
and Ron D: Perk is essential for translational regulation and cell
survival during the unfolded protein response. Mol Cell. 5:897–904.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Scheuner D, Song B, McEwen E, Liu C,
Laybutt R, Gillespie P, Saunders T, et al: Translational control is
required for the unfolded protein response and in vivo glucose
homeostasis. Mol Cell. 7:1165–1176. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Ma Y, Brewer JW, Diehl JA and Hendershot
LM: Two distinct stress signaling pathways converge upon the CHOP
promoter during the mammalian unfolded protein response. J Mol
Biol. 318:1351–1365. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Hitomi J, Katayama T, Eguchi Y, Kudo T,
Taniguchi M, Koyama Y, Manabe T, et al: Involvement of caspase-4 in
endoplasmic reticulum stress-induced apoptosis and Abeta-induced
cell death. J Cell Biol. 165:347–356. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Rudy A, Lopez-Anton N, Dirsch VM and
Vollmar AM: The cephalostatin way of apoptosis. J Nat Prod.
71:482–486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Zong WX, Li C, Hatzivassiliou G, Lindsten
T, Yu QC, Yuan J and Thompson CB: Bax and Bak can localize to the
endoplasmic reticulum to initiate apoptosis. J Cell Biol.
162:59–69. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Hung JH, Su IJ, Lei HY, Wang HC, Lin WC,
Chang WT, Huang W, et al: Endoplasmic reticulum stress stimulates
the expression of cyclooxygenase-2 through activation of NF-kappaB
and pp38 mitogen-activated protein kinase. J Biol Chem.
279:46384–46392. 2004. View Article : Google Scholar : PubMed/NCBI
|