Introduction
Colorectal cancer (CRC) represents one of the major
causes of cancer-associated deaths in North America and worldwide
(1). Since the entire intestinal
epithelium is in a constant state of renewal (2), such turnover involves a countless
number of cell divisions, resulting in a non-negligible risk of
genetic alterations. This rapid rate of intestinal epithelial
renewal as well as exposure to toxic substances, notably through
food ingestion, may explain the high prevalence of CRC. CRC is a
heterogeneous disease displaying distinct molecular signatures and
distinct pathological features. There are at least three major
molecular pathways to CRC including the predominant chromosomal
instability (CIN) pathway, the CpG island methylator phenotype
(CIMP) pathway which is the other major pathway to sporadic CRC and
includes sporadic microsatellite instability (MSI) high cancers and
finally the pure MSI pathway resulting from germline mutation in a
DNA mismatch repair (MMR) gene (1,3,4).
One of the most important cellular barriers to
cancer development is the retinoblastoma tumor suppressor (pRB)
pathway, which is inactivated in a wide range of human tumors and
controls cell cycle progression via repression of the E2F/DP
transcription factor family. Indeed, when hypophosphorylated, Rb
proteins bind to E2F/DP dimers, preventing transactivation of their
target genes (5,6). The E2F/DP transcription factor family
currently numbers 8 members (E2F1-8). DNA micro-array analysis
reveals unique sets of target promoters among E2F family members
suggesting that each protein may have a unique role in the cell
cycle (7). Among E2F
transcriptional targets are cyclins, cdks, checkpoints regulators,
DNA repair and replication proteins.
We previously reported that E2F4 silencing in normal
human intestinal epithelial cells markedly reduced the expression
of many cell cycle regulatory genes including thymidine kinase,
c-myc, cdc6 and cyclin A1, slowing their proliferation
rate (8). Moreover, double
staining experiments in vivo and in vitro revealed
that intestinal crypt epithelial cells which expressed high levels
of nuclear E2F4, were positive for Ki67 (9) and cyclin A1 (8). We have also provided evidence that
E2F4 expression is important for growth of colon cancer cell lines.
Indeed, reduction of E2F4 protein expression significantly slowed
the proliferation rate and soft-agar growth of colon cancer cell
lines.
Interestingly, in contrast to other E2F encoding
genes, the E2F-4 gene exhibits frequent tumor-specific
mutations at a coding region of trinucleotide microsatellite (CAG)n
in a subset of human sporadic CRC with high-frequency MSI (MSI-H)
(10–13). Importantly, this trinucleotide
repeat (13 consecutive serines) is localized in the transactivation
domain (14). The two most
frequent mutations observed are the deletion or the addition of one
trinucleotide CAG in the E2F4 serine stretch
[E2F4(Ser)12 and E2F4(Ser)14] (10,12).
However, the functional impact of these molecular alterations on
E2F4 expression, activity and function in colorectal cancer cells
remains to be demonstrated.
Materials and methods
Materials and antibodies
Antibodies against E2F4 (C-20), p130 (C-20), DP-2
(C-20), and HA-probe (F-7) were all purchased from Santa Cruz
Biotechnology Inc. (Santa Cruz, CA, USA). β-actin monoclonal
antibody (clone C-4) was obtained from Millipore (Billerica, MA,
USA). Cycloheximide was purchased from Calbiochem (San Diego, CA,
USA). All other materials were from Sigma-Aldrich (Oakville, ON,
Canada) unless stated otherwise.
E2F4, DP2 and p130 expression
vectors
The expression vectors (pCDNAneo3) encoding for E2F4
and p130 were obtained from Dr C. Sardet (14) (Institut de Génétique Moléculaire,
Montpellier, France). The full length E2F4 cDNA was subcloned into
a pLVX-Tight-Puro (Clontech, CA, USA) expression vector. PCR was
performed to insert the HA-tag using oligonucleotides containing
the HA-tag and a KOZAK sequence: 5′-A GAC TAG GAT CC C ACC ATG TAT
GAT GTT CCT GAT TAT GCT AGC CTC CCG GCG GAG GCC GGG CCA CAG GCG
CCG-3′ and 5′-TCT GTA CTC GAG TCA GAG GTT GAG AAC AGG CAC ATC AAA
GAG GTC-3′. PCR products were next digested and ligated in
BamHI/EcoRI-digested pLVX-Tight-Puro vector. E2F4
mutants (Ser)12 and (Ser)14 were obtained by
site-directed mutagenesis using the following primers,
respectively: 5′-CTG GAC AGC AGC AGC AGC AGC AGC AGC AGC AGC AGC
AGC AGC AAC AGT AAC-3′ and 5′-GTT ACT GTT GCT GCT GCT GCT GCT GCT
GCT GCT GCT GCT GCT GCT GTC CAG-3′; 5′-CTG GAC AGC AGC AGC AGC AGC
AGC AGC AGC AGC AGC AGC AGC AGC AGC AAC AGT AAC-3′ and 5′-GTT ACT
GTT GCT GCT GCT GCT GCT GCT GCT GCT GCT GCT GCT GCT GCT GCT GTC
CAG-3′. pBABE-Puro HA-wtE2F4, HA-E2F4(Ser)12 and
HA-E2F4(Ser)14 vectors were obtained by subcloning from
pLVX-Tight-Puro vectors (BamHI/EcoRI). The pCMVHA-DP2
expression vector was obtained from Dr J.A. Lees (15) (Department of Biology, Massachusetts
Institute of Technology, Cambridge, MA, USA).
Cell culture
Human embryonic kidney 293 cells and all colorectal
cancer cell lines were obtained from ATCC (Manassas, VA, USA). 293T
cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM;
Invitrogen, Burlington, ON, Canada) containing 10% FBS supplemented
with 2 mM glutamine, 10 mM HEPES, 0.5 IU/ml penicillin and 50
μg/ ml streptomycin (all obtained from Wisent, St-Bruno, QC,
Canada). The human colorectal cancer cells were cultured in the
following media: HT-29 and HCT116 in McCoy’s 5A medium; DLD-1 and
COLO205 in RPMI-1640 medium; LoVo in F-12K medium and Caco-2/15 in
DMEM medium. All of the above media were supplemented with 10% FBS,
2 mM glutamine, 10 mM HEPES, 0.5 IU/ml penicillin and 50
μg/ml streptomycin. Non-immortalized human intestinal
epithelial cells (HIEC) were isolated by Perreault and Beaulieu
(16) and cultured in Opti-MEM
(Invitrogen) medium supplemented with 2 mM glutamine (Invitrogen),
5% fetal bovine serum (FBS), 10 mM HEPES, 0.5 IU/ml penicillin, 50
μg/ml streptomycin and 0.2 IU/ml insulin (Connaught Novo
Laboratories, Willowdale, Canada). HIEC, originally generated from
normal human fetal small intestine at mid-gestation, express
typical features of the lower adult crypt region and are unable to
differentiate (16).
Retroviral infections
E2F4 retroviruses were produced by co-transfecting
pBabe-puro HA-wtE2F4, (Ser)12 or (Ser)14
constructs with the pVPack vector system (Agilent Technologies,
Mississauga, ON, Canada) in 293T cells using Lipofectamine 2000
(Invitrogen) according to the manufacturer’s recommendations. Viral
supernatants were harvested 48 h following transfection, filtered
through a 0.45-μm filter and cells were infected following
the instructions of Agilent Technologies.
Protein extraction and
immunoblotting
Cells were washed twice with ice-cold PBS, then
lysed in Triton lysis buffer [1% Triton X-100, 50 mM Tris-HCl pH
7.5, 100 mM NaCl, 5 mM EDTA, 5% glycerol, 40 mM β-glycerophosphate,
50 mM NaF, 200 μM orthovanadate, 5% pepstatin, 5% aprotinin,
5% leupeptin and 5% phenylmethylsulfonyl fluoride (PMSF)] for 30
min under light agitation. Lysates were then cleared by
centrifugation (15,000 g, 10 min) and 4X Laemmli buffer (2.3% SDS,
10% glycerol, 0.005% bromophenol blue and 5% β-mercaptoethanol) was
added to supernatants for gel analysis. Whole cell extracts were
separated on 10% SDS-PAGE gels and then electro-transferred onto
polyvinylidene fluoride (PVDF) membranes (Perkin-Elmer, Montréal,
Québec, Canada). Membranes were blocked for 1 h at 20°C using 0.05%
Tween/PBS containing 5% non-fat dry milk then incubated overnight
in primary antibodies diluted in blocking solution. Membranes were
next incubated with horseradish peroxidase-conjugated goat
anti-mouse or anti-rabbit IgG (GE Healthcare, Baie d’Urfé, Canada)
in blocking solution for 1 h. The blots were visualized using
homemade ECL (Tris-HCl 100 mM pH 8.5, 1.25 mM luminol, 225
μM coumaric acid and 2.9 mM H2O2).
Protein concentrations were measured using the BCA procedure
(Thermo Scientific, Waltham, MA, USA) as described by the
manufacturer, with bovine serum albumin (BSA) as standard.
Transfection, stability, luciferase and
coimmunoprecipitation assays
293T cells were co-transfected with pLVX-Tight-Puro
empty vector, HA-wtE2F4, HA-E2F4(Ser)12 or
HA-E2F4(Ser)14 and pLVX Tet-Off Advanced using
Lipofectamine 2000 (Invitrogen) according to the manufacturer’s
protocol. Cells were cultured in absence of tetracycline to allow
E2F4 expression. For stability assays, cells were transfected for
24 h then treated with cycloheximide for the indicated times. For
luciferase assays, 293T cells were also transfected with
pE2F-TA-Luc luciferase reporter vector (Clontech), pRL-SV40 Renilla
luciferase reporter (Promega, Madison, WI, USA) and pCMV-HA-DP2
vector. Forty-eight hours following transfection, luciferase
activity was measured using the Dual-Luciferase® Assay
Reporter System (Promega) according to the manufacturer’s protocol.
Relative E2F4 luciferase activity was normalized using the Renilla
reporter to account for transfection efficiency. For
co-immunoprecipitation assays, cells were transfected for 48 h and
lysed with Triton lysis buffer. Cleared lysates were incubated with
anti-E2F4 antibody (3 h, 4°C), after which protein A Sepharose
CL-4B beads (GE Healthcare) were added for an additional hour.
Immunocomplexes were washed three times with Triton X-100 lysis
buffer then eluted with Laemmli buffer and loaded on SDS-PAGE gels.
Extensive protocols of 293T cells transfection and luciferase
assays are available upon request.
Characterization of the number of CAG
repeats in the E2F4 microsatellite using capillary
electrophoresis
Genomic DNAs (gDNAs) were isolated from the human
colorectal cancer cells Caco-2/15, COLO205, HT-29, LoVo, DLD-1,
HCT116 et SW48 as well as from normal HIEC using the Spin Doctor
Solution Set (Gerard Biotech, Oxford, OH, USA) according to the
manufacturer’s protocol. The E2F4 microsatellite was amplified by
PCR from obtained gDNAs and PCR products were analyzed by capillary
electrophoresis by the Laboratoire de Génomique Fonctionnelle de
l’Université de Sherbrooke in order to determine the number of CAG
repeats in the E2F4 microsatellite in each of the intestinal cell
lines using control plasmid DNA with 12, 13 or 14 CAG repeats
[pLVX-Tight-Puro E2F4 (Ser)12, wtE2F4 and
E2F4(Ser)14, respectively]. The Agilent 2100 Bioanalyzer
(Agilent Technologies) and the LabChip® 90 (Caliper,
Hopkinton, MA, USA) were used according to the manufacturer’s
recommendations.
RNA extraction and RT-PCR analysis
Total RNA was isolated with the RNeasy mini kit
(Qiagen, Mississauga, ON, Canada). RT-PCR was performed using avian
myeloblastosis virus reverse transcriptase (Roche Diagnostics) and
conventional PCR analysis was conducted using Taq DNA polymerase
according to the manufacturer’s instructions (Qiagen). Real-time
PCR analyses were performed with a LightCycler apparatus (Roche
Diagnostics). Experiments were executed and analyzed with the
LightCycler software 4.0 according to the manufacturer’s
recommendations. Synthesis of double-stranded DNA during PCR cycles
was monitored with SYBR Green I (QuantiTect SYBR Green PCR kit;
Qiagen). All samples were processed in triplicate. E2F4 expression
was quantified relative to β2-microglobulin expression. A standard
calibration curve was prepared for each gene by using serial
dilutions of the calibrator sample, and crossing point values were
plotted vs. the log of the relative concentration of each dilution.
This standard curve was used to correct for differences in PCR
efficiencies. Oligonucleotide primers used for DNA amplification
were synthesized by Integrated DNA Technologies (San Diego, CA,
USA). Primer sequences are available upon request.
Soft agar assays
RPMI medium (Wisent, QC, Canada) was complemented
with 20% FBS, 4 mM glutamine, 20 mM HEPES, 1 IU/ml penicillin and
100 μg/ml streptomycin. This pre-warmed medium was mixed 1:1
(v/v) with autoclaved 1.4% agarose type VII maintained at ∼42°C and
6-well dishes were pre-coated with 1.5 ml/well. A total of 30,000
cells/well were added to the RPMI/agarose mixture. Plates were
allowed to solidify then placed at 37°C under 5% CO2.
Fresh RPMI supplemented with 10% FBS was added on the surface of
the agarose every 2–3 days. After 2–3 weeks, colonies were stained
by adding 1 ml of PBS containing 0.5 mg/ml MTT to each well and
incubated 2 h at 37°C and 5% CO2. Images were acquired
using an AlphaImager camera (Alpha Innotech Corp.). Colonies were
counted and their size assessed using ImageJ software.
Data presentation
Assays were performed in triplicate. Typical western
blots shown are representative of three independent experiments.
Densitometric analyses were performed using ImageJ software.
Results
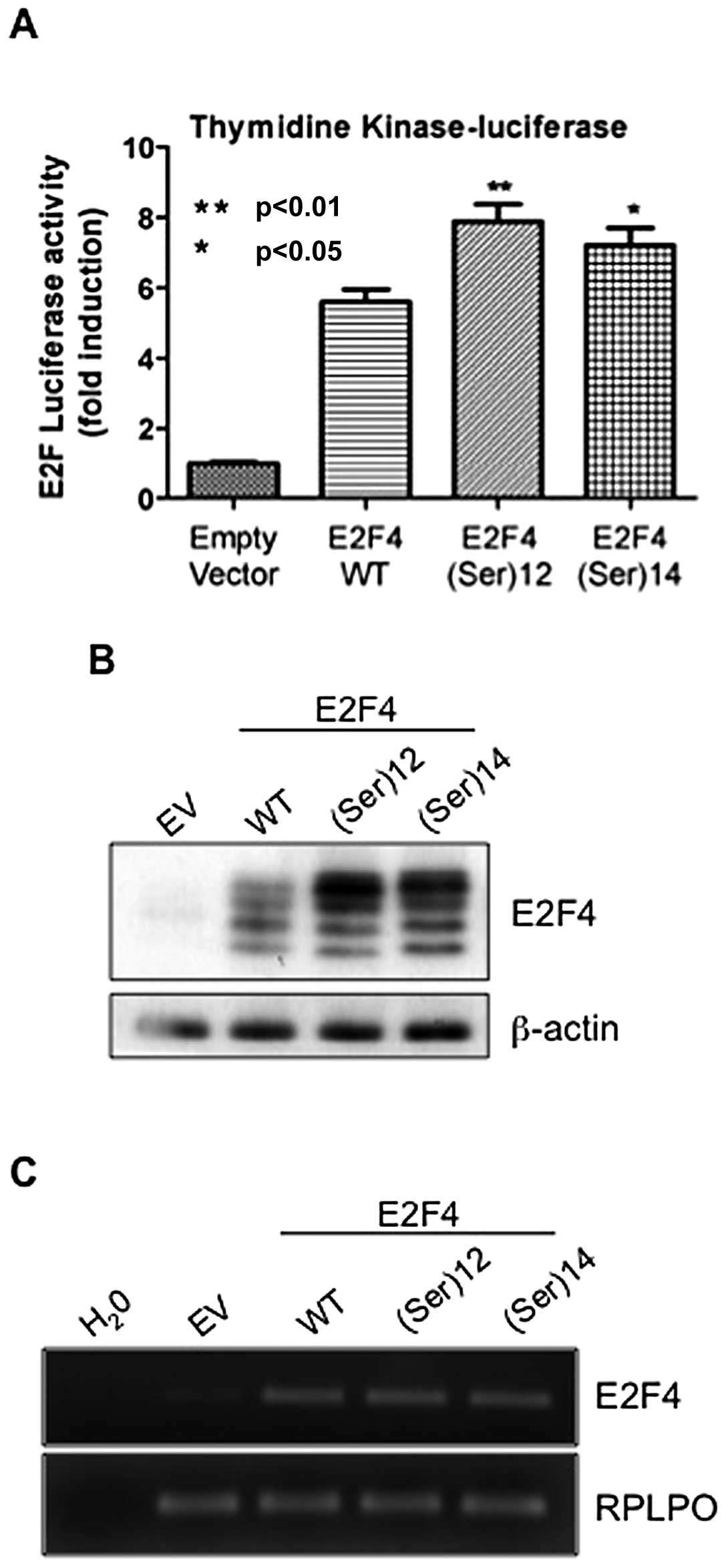
Addition or withdrawal of a single serine
residue in the E2F4 microsatellite increases E2F4 transcriptional
activity and protein expression
To determine the impact of serine stretch-associated
mutations on E2F4 function, both mutant forms of E2F4 reported to
be frequently observed in CRC were generated, namely
E2F4(Ser)12 and E2F4(Ser)14. These mutants as
well as the wild-type form of E2F4 were then cotransfected along
with thymidine kinase luciferase reporter gene. Previous
studies have reported that the gene encoding for this protein
contains E2F4-responsive elements in their promoters (17–19)
and our previous studies have shown that E2F4 silencing in HIEC
markedly reduced thymidine kinase mRNA levels (8). As shown in Fig. 1A, both E2F4(Ser)12 and
E2F4(Ser)14 mutants exhibited significant enhanced
transcriptional activities relative to thymidine kinase gene
compared with wtE2F4. The increased transcriptional activity of
mutants versus wild-type E2F4 prompted us to verify their mRNA and
protein expression by quantitative PCR and western blot analyses
respectively. As shown in Fig. 1B,
protein levels of E2F4(Ser)12 and E2F4(Ser)14
mutants were consistently higher than levels of wild-type E2F4. Of
note however, these increased protein expression levels of mutants
could not be attributed to an increase in mRNA levels which were
similar to those of wild-type E2F4 (Fig. 1C).
Colorectal cancer-associated mutations
increase E2F4 protein stability
Since increased protein but not mRNA levels were
observed for E2F4(Ser)12 and E2F4(Ser)14
mutants, this suggests that colorectal cancer-associated mutations
may affect protein stability of E2F4. Therefore, 293T cells were
transiently transfected with either wild-type E2F4,
E2F4(Ser)12 mutant or E2F4(Ser)14 mutant and
then treated with cycloheximide to inhibit protein synthesis.
Thereafter, cells were lyzed at different time intervals in order
to analyze protein expression levels of E2F4 forms. As shown in
Fig. 2A (lanes 2 versus 7 and 12),
expression of E2F4(Ser)12 and E2F4(Ser)14
mutants was higher than wild-type E2F4 expression in untreated
cells (time 0). Following cycloheximide treatment however,
E2F4(Ser)12 and E2F4(Ser)14 protein levels
decreased much more slowly than that of wild-type E2F4 (Fig. 2A and B, for densitometric
analysis). Specifically, 3 h after cycloheximide addition,
expression of E2F4 protein was drastically decreased while
expression of E2F4(Ser)12 and E2F4(Ser)14
mutants remained at control (time 0) levels. Accordingly, the
half-life of wild-type E2F4 protein was ∼5 h whereas
E2F4(Ser)12 and E2F4(Ser)14 half-lives were
>12 h.
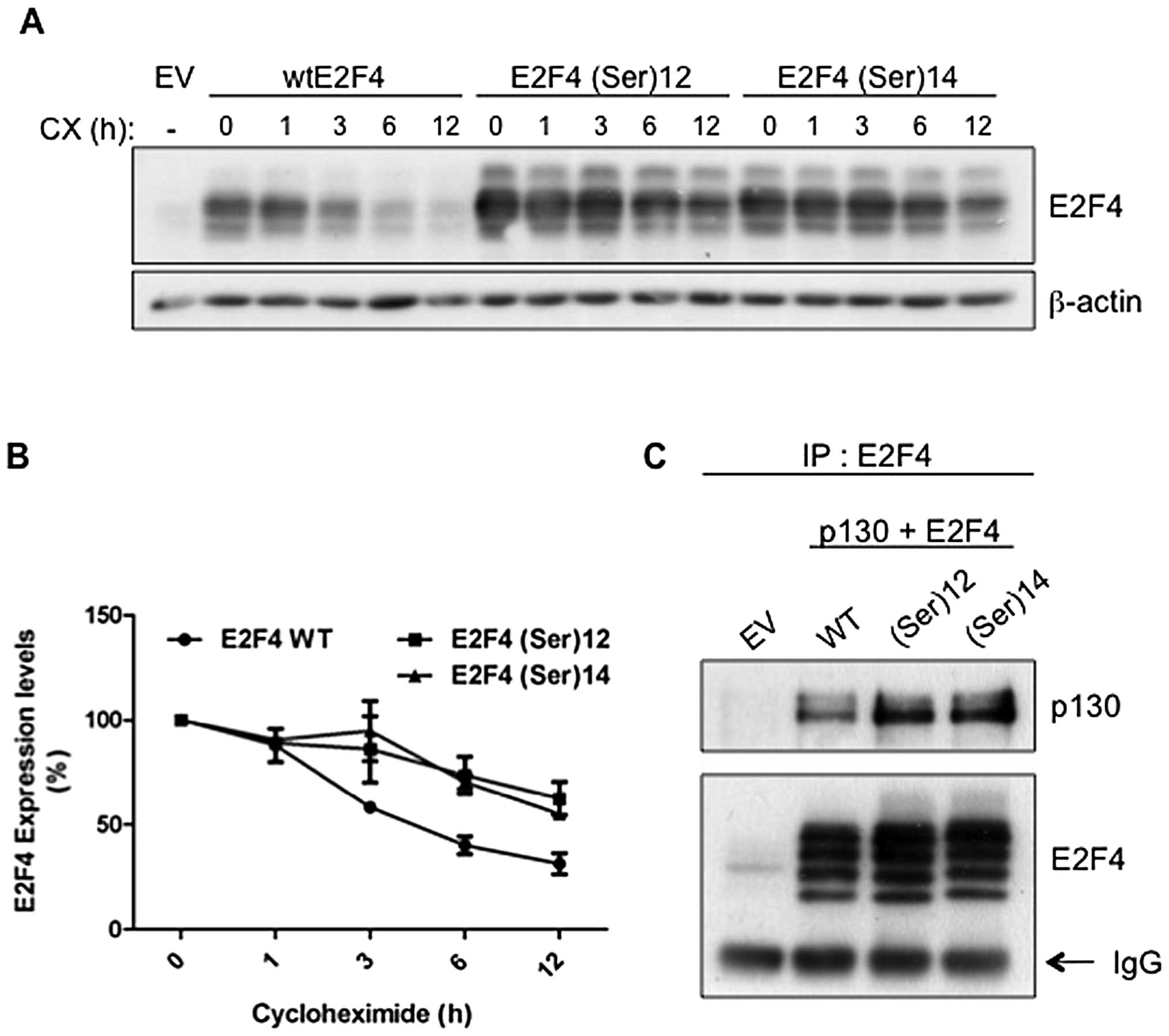 | Figure 2.Colorectal cancer-associated
mutations increase E2F4 protein stability. (A) Twenty-four hours
following transfection, cells were treated with cycloheximide (10
μg/ml) and harvested after 0, 1, 3, 6 or 12 h. All cells
were processed and lysed at the same time. E2F4 and β-actin
expression was analyzed by western blot analysis. (B) Densitometric
analysis of data shown in (C) is represented. E2F4 expression at 0
h of cycloheximide was set at 100%. Relative E2F4 expression levels
were calculated using β-actin as reference. (C) 293T cells were
transfected with either pLVX-Tight-Puro empty vector, HA-wtE2F4,
HA-E2F4(Ser)12 or HA-E2F4(Ser)14 as well as
pLVX-Tet-Off Advanced and pCDNA1-p130 in absence of tetracycline.
Forty-eight hours following transfection, cells were lysed and E2F4
was immunoprecipitated from cleared lysates. E2F4 immunocomplexes
were separated by SDS-PAGE gels and western blot analysis was
performed using specific antibodies against E2F4 and p130. EV,
empty vector; WT, wild-type; IP, immunoprecipitation. |
Furthermore, association of E2F transcription
factors with a pocket protein has been reported to protect E2F
factors against degradation (20–22).
We therefore analyzed whether colorectal cancer-associated
mutations modulate E2F4 interaction with p130/RBL2, the main pocket
protein partner for E2F4 in various cell types including intestinal
epithelial cells (9). Indeed,
association of E2F4 with p130 was found to promote E2F4 stability
(20). Thus, cells were
co-transfected with p130 along with wild-type E2F4,
E2F4(Ser)12 or E2F4(Ser)14 after which E2F4
was immunoprecipitated and the amounts of associated p130 analyzed
by western blotting. As shown in Fig.
2C, neither E2F4(Ser)12 nor E2F4(Ser)14
were found to have an altered capacity to associate with p130 in
comparison to wild-type E2F4 (when normalized to immunoprecipitated
E2F4 levels). These results suggest that the colorectal
cancer-associated E2F4 mutants are more stable than wild-type E2F4,
independently of their capacity to interact with p130.
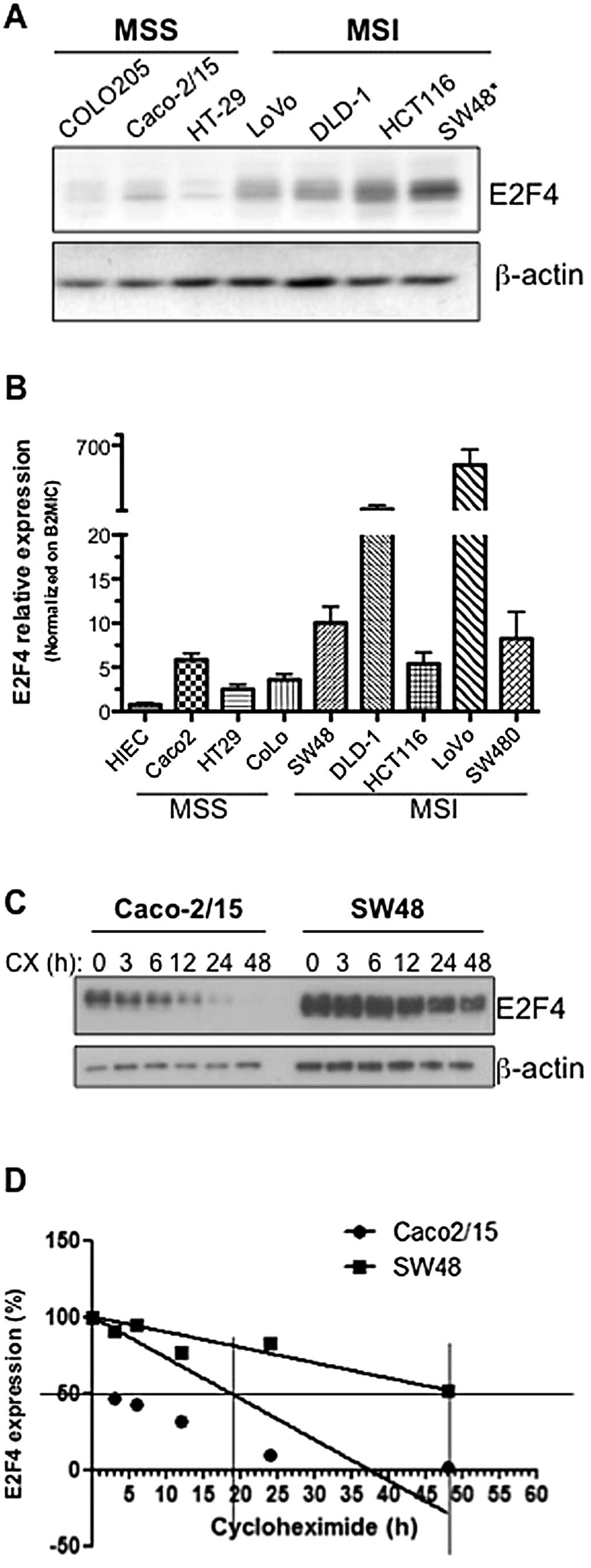
Colorectal cancer cells with MSI exhibit
enhanced E2F4 expression
The CAG triplet repeat in the coding region of the
E2F-4 gene has been reported to be mutated in colorectal
cancers exhibiting a microsatellite instability (MSI) phenotype. We
therefore analyzed E2F4 gene and protein expression patterns in a
panel of colorectal cancer cell lines which are microsatellite
unstable (MSI) (HCT116, SW48, DLD-1, LoVo) and microsatellite
stable (MSS) (COLO205, Caco-2/15 and HT-29) (23,24).
As shown in Fig. 3A and B, E2F4
mRNA and protein levels were observed to be globally enhanced in
colorectal cancer cell lines exhibiting microsatellite
instability.
Given this increased E2F4 expression in MSI
colorectal cancer cells, the number of CAG repeats in the E2F4
microsatellite was determined in each cell line by capillary
electrophoresis as described in Materials and methods. Analysis of
the 13-serine residue microsatellite of E2F4 revealed that only the
SW48 cell line expressed mutated E2F4 with a deletion of one CAG
repeat causing a deletion of one serine residue. Interestingly,
amongst all colorectal cancer cell lines analyzed, SW48 exhibited
the highest expression of E2F4 protein (Fig. 3A). In order to associate this
increased protein expression with increased E2F4 stability,
endogenous E2F4 half-life was analyzed in SW48 cells comparatively
to Caco-2/15, a MSS cell line in which E2F4 is wild-type. SW48 and
Caco-2/15 cells were treated with cycloheximide to inhibit protein
synthesis for various time intervals. As shown in Fig. 3C and D, following cycloheximide
treatment, E2F4 protein levels decreased more rapidly in Caco-2/15
compared to SW48 cells. Specifically, E2F4 half-life in Caco-2/15
cells was established at 19 h whereas E2F4 half-life in SW48 cells
was ∼48 h. These results confirm that the deletion of one serine
residue in the serine stretch increases E2F4 stability resulting in
increased protein levels.
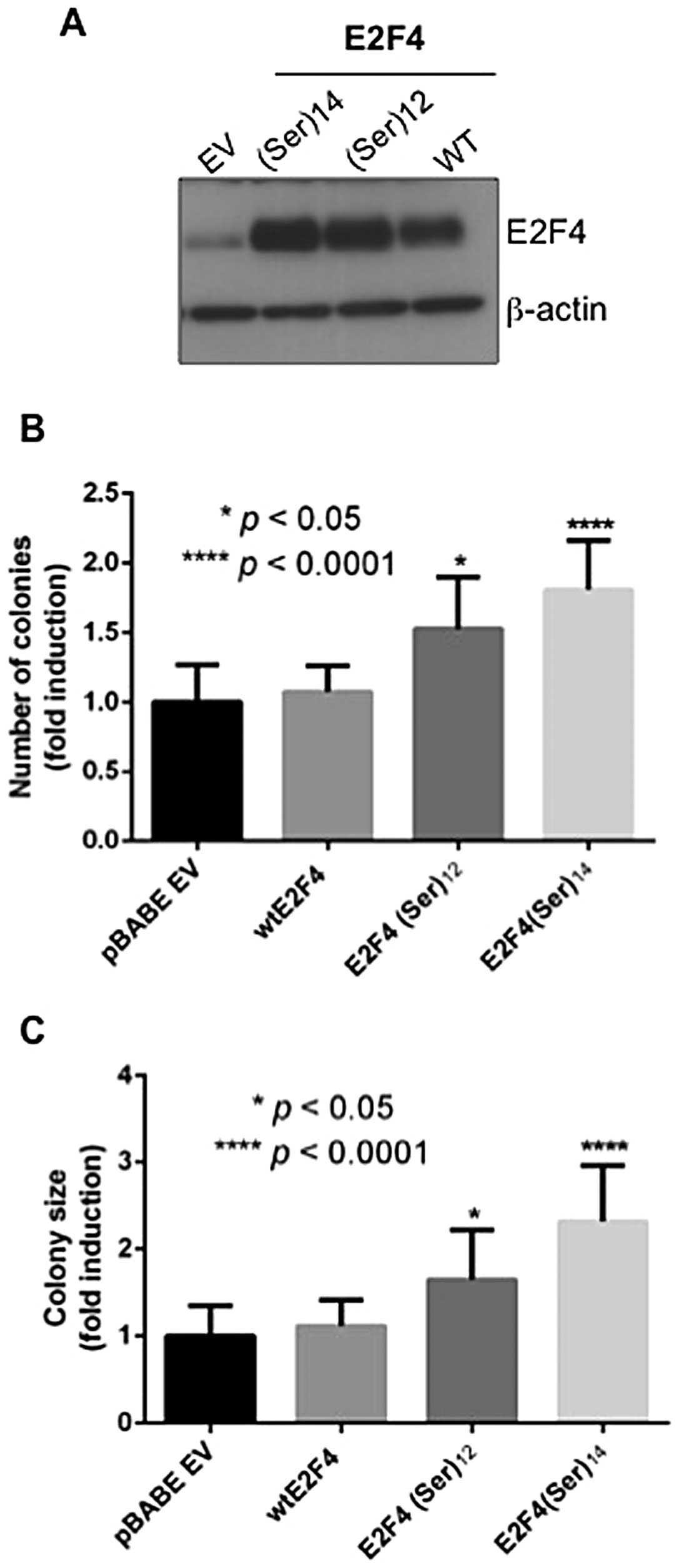
E2F4(Ser)12 or
E2F4(Ser)14 expression enhances the capacity of
colorectal cancer cells to grow in soft agar
Our previous report indicated that E2F4
transcription factor expression is required for anchorage-dependent
and anchorage-independent growth of colorectal cancer cells
(8). Because anchorage-independent
growth potential may better correlate with tumorigenic growth in
vivo, we determined whether E2F4 mutations in the serine
stretch was correlated with stimulation of tumor cell growth in
soft agar. The DLD-1 cell line was chosen because this line
exhibits microsatellite instability and expresses moderate levels
of endogenous wild-type E2F4. As shown in Fig. 4, expression of
E2F4(Ser)12 or E2F4(Ser)14 mutants (Fig. 4A) increased the capacity of DLD-1
cells to form colonies in soft agar (Fig. 4B). Moreover, colonies formed
following the expression of E2F4 mutants were significantly larger
than colonies formed following the expression of wild-type E2F4
(Fig. 4C). Of note, similar
results were obtained with the MSS cell line Caco-2/15 (data not
shown).
Discussion
Microsatellite instability is one of the molecular
pathways leading to colorectal cancer progression (4). Several genes containing
microsatellite and encoding regulators of cell proliferation are
affected by MSI in colorectal cancer (e.g. TGF-β type II
receptor, TCF-4, activin receptor-2, insulin-like growth
factor-2) (4). Among the E2F
transcription factor family, E2F4 is the only member that has a 13
consecutive serine residue microsatellite in its transactivation
domain (14). A number of studies
report that this specific E2F4 microsatellite is frequently mutated
in colorectal cancers bearing microsatellite instability and that
the most common mutations observed are the addition or withdrawal
of a single serine residue (10–13).
These mutants show elevated transactivation of an E2F consensus
promoter sequence and promote proliferation of murine fibroblasts
(25). However, the mechanism and
functional impact of this increased transactivation remains to be
understood in the context of intestinal epithelial cells.
Herein, our results further confirm that the
addition or deletion of a single serine residue in the E2F4
microsatellite increases the transactivation potential of E2F4 on a
target gene that is important for cell proliferation, namely
thymidine kinase. This increased transcriptional activity was also
associated with increased expression of E2F4 protein. Indeed, both
E2F4 mutants exhibited increased stability. This is consistent with
previous results showing that deletion of the carboxy-terminal 112
amino acids of E2F4 led to a dramatic increase in E2F-4 half-life.
Taken together, these results suggest that the E2F4 C-terminus
(which includes its transactivation domain, the pocket protein
interaction domain and the 13 serine stretch) brings about
instability (20). Of note,
several studies have described the importance of pocket protein
interaction for E2F transcription factor stability (20–22).
However, our co-immunoprecipitation studies did not demonstrate any
altered association of E2F4 mutants with p130, the main pocket
protein partner of E2F4 in intestinal epithelial cells. Association
with DP-2 was also analyzed given that DP-2 is known to induce E2F4
nuclear localization and could hence protect E2F4 from cytoplasmic
degradation (26). However, DP-2
association with E2F4(Ser)12 or E2F4(Ser)14
mutants was not affected (data not shown). Interestingly, the E2F4
serine stretch comprises phosphorylation sites for GSK3β, a kinase
known to target many cell cycle regulatory proteins to the
proteasome for degradation (27–29).
Preliminary data obtained in our laboratory suggest that GSK3β can
interact with and phosphorylate E2F4, at least in vitro
(data not shown). Whether endogenous E2F4 is indeed a potent
substrate for GSK3β in cells remains to be established. Further
studies will be needed to firmly elucidate the molecular mechanisms
underlying the increased stability of E2F4 mutants.
In the present study, we showed that both protein
and mRNA expression levels of the E2F4 transcription factor were
higher in CRC cell lines bearing microsatellite instability
compared to both microsatellite-stable cell lines and normal
intestinal epithelial cells. Of note, however, mRNA levels did not
exactly follow protein levels, suggesting the implication of
mechanisms other than transcription in the regulation of E2F4
protein levels in CRC cells. Endogenous sequence analysis of E2F4
in several CRC cell lines further revealed that only SW48 expressed
a mutated E2F4 with 12 serines in its microsatellite. Stability
analysis also confirmed that E2F4 was more stable in SW48 than in
Caco-2/15 cells reinforcing the notion that E2F4 mutations in the
microsatellite sequence increase E2F4 stability. Of particular
interest, the increased expression of E2F4 observed in SW48 cells
could contribute to their higher capacity to form tumors in nude
mice compared to Caco-2/15 cells (30). Furthermore, these findings are in
agreement with our results demonstrating that the expression of
E2F4(Ser)12 or E2F4(Ser)14 mutants in DLD-1
cells significantly increased their capacity to grow in soft
agar.
E2F transcription factors have long been known to
promote transformation featuring anchorage-independent survival and
growth, as well as loss of contact inhibition (31,32).
Accordingly, we previously reported the requirement of E2F4 for
anchorage-independent growth of CRC cells (8). In addition to these observations,
increased nuclear expression of E2F4 in breast cancer has been
associated with poor prognosis with larger tumors, recurrent
disease, distant metastasis and poorer outcome (33). Although such investigation remains
to be performed for colorectal cancer, our results indicate that
mutations of E2F4 promote the capacity of cancer cells to grow
without anchorage, thereby contributing to tumor progression and
metastasis formation (34).
We and others have previously shown that E2F4
protein levels are increased in human colorectal tumors. Since
micro-satellite instability is observed in ∼15% of colorectal
cancers, other mechanisms in addition to mutations should account
for the enhanced expression of E2F4. Nevertheless, our data
demonstrate that, when mutated in its serine stretch, E2F4 protein
is more stable, more readily expressed and more transcriptionally
active. As a result, colorectal cancer-associated E2F4 mutations
may confer increased tumorigenic potential for tumors with
microsatellite instability, thus providing further insight into the
functional impact of these mutations in CRC.
Acknowledgements
We thank Pierre Pothier for the
critical reading of the manuscript. We also thank Anne Vézina for
technical assistance. We gratefully acknowledge the work of Mathieu
Durand, research assistant at the Laboratoire de Génomique
Fonctionnelle de l’Université de Sherbrooke (LGFUS) for the
determination of E2F4 microsatellite mutations in colorectal cancer
cell lines. This study was supported by a grant from the Cancer
Research Society Inc. and from the Canadian Institutes of Health
Research MT-14405 (to N.R.). Marie-Christine Paquin is a recipient
of a NSERC fellowship. Nathalie Rivard is a recipient of a Canadian
Research Chair in Colorectal Cancer and Inflammatory Cell Signaling
and a member of the FRSQ-funded ‘Centre de Recherche Clinique
Étienne LeBel’.
References
|
1.
|
Yang VW, Lewis J, Wang TC and Rustgi AK:
Colon cancer: an update and future directions. Gastroenterology.
138:2027–2028. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
van der Flier LG and Clevers H: Stem
cells, self-renewal, and differentiation in the intestinal
epithelium. Annu Rev Physiol. 71:241–260. 2009.
|
|
3.
|
Worthley DL and Leggett BA: Colorectal
cancer: molecular features and clinical opportunities. Clin Biochem
Rev. 31:31–38. 2010.PubMed/NCBI
|
|
4.
|
Boland CR and Goel A: Microsatellite
instability in colorectal cancer. Gastroenterology.
138:2073–2087.e3. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Tsantoulis PK and Gorgoulis VG:
Involvement of E2F transcription factor family in cancer. Eur J
Cancer. 41:2403–2414. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Chen HZ, Tsai SY and Leone G: Emerging
roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev
Cancer. 9:785–797. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Gaubatz S, Lindeman GJ, Ishida S, Jakoi L,
Nevins JR, Livingston DM and Rempel RE: E2F4 and E2F5 play an
essential role in pocket protein-mediated G1 control. Mol Cell.
6:729–735. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Garneau H, Paquin MC, Carrier JC and
Rivard N: E2F4 expression is required for cell cycle progression of
normal intestinal crypt cells and colorectal cancer cells. J Cell
Physiol. 221:350–358. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Deschenes C, Alvarez L, Lizotte ME, Vezina
A and Rivard N: The nucleocytoplasmic shuttling of E2F4 is involved
in the regulation of human intestinal epithelial cell proliferation
and differentiation. J Cell Physiol. 199:262–273. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Yoshitaka T, Matsubara N, Ikeda M, Tanino
M, Hanafusa H, Tanaka N and Shimizu K: Mutations of E2F-4
trinucleotide repeats in colorectal cancer with microsatellite
instability. Biochem Biophys Res Commun. 227:553–557. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Souza RF, Yin J, Smolinski KN, et al:
Frequent mutation of the E2F-4 cell cycle gene in primary human
gastrointestinal tumors. Cancer Res. 57:2350–2353. 1997.PubMed/NCBI
|
|
12.
|
Ikeda M, Orimo H, Moriyama H, et al: Close
correlation between mutations of E2F4 and hMSH3 genes in colorectal
cancers with microsatellite instability. Cancer Res. 58:594–598.
1998.PubMed/NCBI
|
|
13.
|
Moriyama H, Sasamoto H, Kambara T, et al:
E2F-4 mutation in hereditary non-polyposis colorectal cancer. J Exp
Clin Cancer Res. 21:185–189. 2002.PubMed/NCBI
|
|
14.
|
Sardet C, Vidal M, Cobrinik D, Geng Y,
Onufryk C, Chen A and Weinberg RA: E2F-4 and E2F-5, two members of
the E2F family, are expressed in the early phases of the cell
cycle. Proc Natl Acad Sci USA. 92:2403–2407. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Wu CL, Zukerberg LR, Ngwu C, Harlow E and
Lees JA: In vivo association of E2F and DP family proteins. Mol
Cell Biol. 15:2536–2546. 1995.PubMed/NCBI
|
|
16.
|
Perreault N and Beaulieu JF: Use of the
dissociating enzyme thermolysin to generate viable human normal
intestinal epithelial cell cultures. Exp Cell Res. 224:354–364.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Dou QP, Zhao S, Levin AH, Wang J, Helin K
and Pardee AB: G1/S-regulated E2F-containing protein complexes bind
to the mouse thymidine kinase gene promoter. J Biol Chem.
269:1306–1313. 1994.PubMed/NCBI
|
|
18.
|
Jansen-Durr P, Meichle A, Steiner P, et
al: Differential modulation of cyclin gene expression by MYC. Proc
Natl Acad Sci USA. 90:3685–3689. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Oswald F, Lovec H, Moroy T and Lipp M:
E2F-dependent regulation of human MYC: Trans-activation by cyclins
D1 and A overrides tumour suppressor protein functions. Oncogene.
9:2029–2036. 1994.PubMed/NCBI
|
|
20.
|
Hateboer G, Kerkhoven RM, Shvarts A,
Bernards R and Beijersbergen RL: Degradation of E2F by the
ubiquitin-protea-some pathway: Regulation by retinoblastoma family
proteins and adenovirus transforming proteins. Genes Dev.
10:2960–2970. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Hofmann F, Martelli F, Livingston DM and
Wang Z: The retinoblastoma gene product protects E2F-1 from
degradation by the ubiquitin-proteasome pathway. Genes Dev.
10:2949–2959. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Martelli F and Livingston DM: Regulation
of endogenous E2F1 stability by the retinoblastoma family proteins.
Proc Natl Acad Sci USA. 96:2858–2863. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Heinen CD, Richardson D, White R and
Groden J: Microsatellite instability in colorectal adenocarcinoma
cell lines that have full-length adenomatous polyposis coli
protein. Cancer Res. 55:4797–4799. 1995.PubMed/NCBI
|
|
24.
|
Boyer JC, Umar A, Risinger JI, et al:
Microsatellite instability, mismatch repair deficiency, and genetic
defects in human cancer cell lines. Cancer Res. 55:6063–6070.
1995.PubMed/NCBI
|
|
25.
|
Takashima H, Matsumoto Y, Matsubara N, et
al: Effect of naturally occurring E2F-4 alterations on
transcriptional activation and proliferation in transfected cells.
Lab Invest. 81:1565–1573. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Martelli F, Hamilton T, Silver DP, et al:
p19ARF targets certain E2F species for degradation. Proc Natl Acad
Sci USA. 98:4455–4460. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Vlach J, Hennecke S and Amati B:
Phosphorylation-dependent degradation of the cyclin-dependent
kinase inhibitor p27. EMBO J. 16:5334–5344. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Diehl JA, Cheng M, Roussel MF and Sherr
CJ: Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis
and subcellular localization. Genes Dev. 12:3499–3511. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Vandel L and Kouzarides T: Residues
phosphorylated by TFIIH are required for E2F-1 degradation during
S-phase. EMBO J. 18:4280–4291. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Dunn EF, Iida M, Myers RA, et al:
Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab.
Oncogene. 30:561–574. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Chen C and Wells AD: Comparative analysis
of E2F family member oncogenic activity. PLoS One. 2:e9122007.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Xu G, Livingston DM and Krek W: Multiple
members of the E2F transcription factor family are the products of
oncogenes. Proc Natl Acad Sci USA. 92:1357–1361. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Rakha EA, Pinder SE, Paish EC, Robertson
JF and Ellis IO: Expression of E2F-4 in invasive breast carcinomas
is associated with poor prognosis. J Pathol. 203:754–761. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Mori S, Chang JT, Andrechek ER, et al:
Anchorage-independent cell growth signature identifies tumors with
metastatic potential. Oncogene. 28:2796–2805. 2009. View Article : Google Scholar : PubMed/NCBI
|