Introduction
Breast cancer, the most common cancer in women all
over the world (1), is generally
managed with success thank to available treatments (2). Regrettably, resistance to
conventional therapy and increasing in metastatic and aggressive
disease are emerging problems (3,4). A
particular aggressive subtype of breast cancer is the
triple-negative breast cancer (TNBC) that is defined by the absence
of ER, PR and Her2/neu (5,6). TNBC generally occurs more frequently
in younger women, frequently presents early metastatic spread and
has poor overall prognosis (7,8).
In breast cancer, as well as in other tumour types,
epigenetic modifications are considered a crucial mechanism
involved in cancer growth, dedifferentiation and aggressiveness;
indeed, epigenetic deregulation can alter a number of molecular
pathways involved in the control of cell function. Epigenetic
drugs, able to restore normal epigenome in cancer cells, are under
extensive pharmacological research (9). In breast cancer, histone acetylation
state is considered of great importance. Histone acetylation and
deacetylation, controlled by the enzymes histone acetyltransferases
(HATs) and histone deacetylases (HDACs), affect chromatin
conformation and, therefore, gene transcription, DNA repair and
replication, and cell cycle checkpoints (10). Altered expression of HDACs has been
reported in breast cancer by several authors (11,12).
Therefore, HDAC inhibitors (DCI) are considered valuable
therapeutic tools and have been under extensive evaluation. DCI
vorinostat (13), for example, in
association with paclitaxel and bevacizumab induced a partial or
complete response in >50% of patients with metastatic breast
cancer. The anti-proliferative and re-differentiating effect of
several DCI was reported in vitro by a number of
laboratories (14–17). In recent years, the pan-deacetylase
inhibitor panobinostat (LBH589) has been taken into consideration.
Our laboratory demonstrated for the first time that LBH589 in
nanomolar concentration is a potent antiproliferative agent in
ER-positive and -negative breast cancer cells, and that its
anticancer activity is sustained by H4 histone acetylation
(18).
It is so well known that ERα is the hallmark of
breast cancer estrogen sensitivity and good response to tamoxifen
therapy (19) and that it can be
lost during disease progression, giving rise to hormone-independent
and aggressive phenotype (20).
Epigenetic deregulation has also been considered one of the
possible causes of ERα loss in breast cancer (e.g., histone tail
deacetylation and methylation or methylation of the CpG islands in
the ER promoter) (21). Hence, DCI
were exhaustively studied also for their ability to restore ERα and
its pathway in ER-negative breast cancer cells. VPA, for example,
was demonstrated by us (16) and
by other authors (14) to restore
estrogen and, thus, antiestrogen sensitivity in MDA-MB-231 cells
considered a good model of TNBC; LBH589 was shown either to induce
ERα in MDA-MB-231 cells (22), or
not to have any detectable effect (23,24).
Estradiol sensitivity is also strictly linked to
aggressiveness of breast cancer cells. Aggressive breast tumours
are characterized by E-cadherin downregulation that switches on the
phenomenon called epithelial-to-mesenchimal transition (EMT), the
corner stone of tumour spreading and metastasis (25). E-cadherin downregulation is mainly
due to epigenetic alterations of CDH1 gene promoter
(26,27) or by epigenetic controlled
overexpression of several E-cadherin transcriptional repressors,
such as Snail/SNAI1, Slug/SNAI2, SIP1/ZEB2 or Twist (28–32).
In breast cancer, estradiol (E2)/ERα and
MTA3/Snail/E-cadherin signalling pathways are intimately linked
(33). Estradiol-activated ERα
induces MTA3, a member of the histone deacetylase Mi-2/NuRD
macro-complex, that down-regulates Snail, upregulating E-cadherin
(34). The absence of estrogen
receptor or MTA3 leads to aberrant expression of the
transcriptional repressor Snail and consequent inhibition of
E-cadherin (30).
E-cadherin and its regulators are considered
attractive therapeutic targets, in order to inactivate cell
invasion and metastasis.
The aim of the present study was to evaluate the
effect of the pan-deacetylase inhibitor panobinostat (LBH589) on
expression and function of ERα and its cognate proteins PR
(progesterone receptor) and FoxA1 (forkhead box A1), and of
E-cadherin and its repressors Snail and Slug, in TNBC MDA-MB-231
cells.
Materials and methods
Cell lines and reagents
Triple-negative breast cancer cell line MDA-MB-231
and estrogen receptor-positive MCF-7 cells were purchased from
ECACC (Salisbury, UK), which certifies the origin and identity of
the cells. Moreover, none of the cell lines are included in the
database of cross-contaminated or misidentified cell lines
(http://www.hpacultures.org.uk/services/celllineidentityverification/misidentifiedcelllines.jsp).
Cells were routinely maintained at 37°C, in 5% CO2 and
95% humidity, in RPMI-1640 (Sigma, St. Louis, MO, USA) with 100
IU/ml penicillin and 100 μg/ml streptomycin added,
supplemented with 10% heat-inactivated FCS (Euroclone, Wetherby,
UK). LBH589 was provided by Novartis Pharma AG (Basel,
Switzerland), prepared as a 5 mM stock solution in DMSO and stored
at −20°C.
Gene expression: evaluation with
real-time PCR
Cells (1×106) were seeded in
75-cm2 flasks and treated with LBH589 (5–50 nM). Total
RNA was extracted using TRIzol reagent (Invitrogen Ltd., Paisley,
UK), as previously described. DNase I was added to remove remaining
genomic DNA. Total RNA (1 μg) was reverse-transcribed with
iScript cDNA Synthesis kit (Bio-Rad Laboratories, Inc.), following
the manufacturer’s protocol.
Primers (Table I)
were designed using Beacon Designer 5.0 software according to
parameters outlined in the Bio-Rad iCycler manual. Specificity of
primers was confirmed by BLAST analysis. Real-time PCR was
performed using a Bio-Rad iQ iCycler Detection system (Bio-Rad
Laboratories, Inc.) with SYBR green fluorophore. Reactions were
performed in a total volume of 25 μl, including 12.5
μl IQ SYBR Green Supermix (Bio-Rad Laboratories, Inc.), 1
μl of each primer at 10 μM concentration, and 5
μl of the previously reverse-transcribed cDNA template.
Protocol for primer set was optimized using seven serial 5X
dilutions of template cDNA obtained from cells in basal
conditions.
 | Table I.Primers for real-time PCR. |
Table I.
Primers for real-time PCR.
| CDH1 | Sense: |
5′-TTGAAAGAGAAACAGGATGGCTG-3′ |
| Antisense: |
5′-TCATTCTGATCGGTTACCGTGAT-3′ |
| ERα | Sense: | 5′-TGT GTC CAG CCA
CCA ACC AG-3′ |
| Antisense: | 5′-TTC AAC ATT CTC
CCT CCT CTT CGG-3′ |
| PR | Sense: | 5′-GCA TCA GGC TGT
CAT TAT GGT GTC-3′ |
| Antisense: | 5′-CAT AAG TAG TTG
TGC TGC CCT TCC-3′ |
| FoxA1 | Sense: | 5′-GGG TGG CTCCAG
GAT GTT AGG-3′ |
| Antisense: | 5′-GGG TCA TGT TGC
CGC TCG TAG-3′ |
| MTA3 | Sense: |
5′-GTCGGAGATTATGTCTACTTTGAG-3′ |
| Antisense: |
5′-CAGTCTTGTTGAGTTCTTCTATCC-3′ |
| SNAIL | Sense: |
5′-CCTGCGTCTGCGGAACCTG-3′ |
| Antisense: |
5′-GGAGCGGTCAGCGAAGGC-3′ |
| SLUG | Sense: |
5′-AAACTACAGCGAACTGGACACAC-3′ |
| Antisense: |
5′-GTGGTATGACAGGCATGGAGTAAC-3′ |
| β-ACT | Sense: | 5′-GCG AGA AGA TGA
CCC AGA TC-3′ |
| Antisense: | 5′-GGA TAG CAC AGC
CTG GAT AG-3′ |
|
β2-microglobulin | Sense: | 5′-AGA TGA GTA TGC
CTG CCG TGT G-3′ |
| Antisense: | 5′-TCA ACC CTC CAT
GAT GCT GCT TAC-3′ |
| L13A | Sense: | 5′-GCA AGC GGA TGA
ACA CCA ACC-3′ |
| Antisense: | 5′-TTG AGG GCA GCA
GGA ACC AC-3′ |
The protocol used is as follows: denaturation (95°C
for 5 min), amplification repeated 40 times (95°C for 15 sec, 60°C
for 1 min). A melting curve analysis was performed following every
run to ensure a single amplified product for every reaction. All
reactions were carried out at least three times for each sample.
Every gene expression level was normalized on the expression level
of three house-keeping genes (β-actin, L13A,
β-2-microglobulin).
Immunoblotting
Sub-confluent breast cancer cells were treated with
LBH589 (25 nM) for 24 h and then they were harvested and lysed in
the presence of lysis buffer (0.5% Triton X-100, 2.5 mM EGTA, 5 mM
MgCl2, 50 mM NaH2PO4) for 2 min on
ice. Soluble cytosolic fraction was recovered and stored at −80°C.
The insoluble membrane fraction was dissolved in SDS sample buffer
(TrizmaBase 0.2 M, glycerol 50%, SDS 10%), recovered and stored at
−80°C.
SDS-PAGE was performed on gels, loading 30 μg
protein/well. Separated proteins were electro-transferred onto PVDF
membrane and probed with anti-E-cadherin antibody (1:1,000
dilution, Sigma). PVDF membrane was then stripped and re-probed
with an anti-α-tubulin antibody (clone 6-11B-1, 1:2,000 dilution,
Sigma) to check protein loading. Proteins were detected with Pierce
Super Signal chemiluminescent substrate following the
manufacturer’s instructions. Bands were photographed and analyzed
using Kodak 1D Image analysis software.
Immunofluorescence microscopy
Cells (4×103) were seeded in 96-well
plates and treated with 25 nM LBH589 for 24 h. After treatment,
cells were fixed in PFA 1% and incubated with polyclonal
anti-E-cadherin antibody (1:100 dilution, Sigma) at 4°C overnight.
Then cells were washed with PBS containing 0.5% Triton and 0.05%
NaN3 followed by detection with anti-rabbit
Cy3-conjugated secondary antibody (1:1,000 dilution, GE Healthcare
Europe, GmbH, Milan, Italy) in PBS plus 0.5% Triton and 0.05%
NaN3 for 2 h. Nuclear staining was obtained by treating
cells with Hoechst 33258 (500 ng/ml in DMSO) in PBS. Cells were
washed twice with distilled water and mounted with 50% glycerol-PBS
media.
Scratch wound assay
Cells (2×105) were seeded in 6-well
plates. Cells were treated with 25 nM LBH589 for 24 h and
afterwards cell monolayer was gently wounded by scratching. The
cells were washed twice with cooled PBS and incubated for further
24 h either in the absence or presence of 25 nM LBH589. For each
wound, pictures were taken in the same field and the distance
between the wound edges was analyzed using the ImageJ 1.42
software. For each condition, the percentage of the wound recovery
in respect to the wound area at 0 h, was calculated.
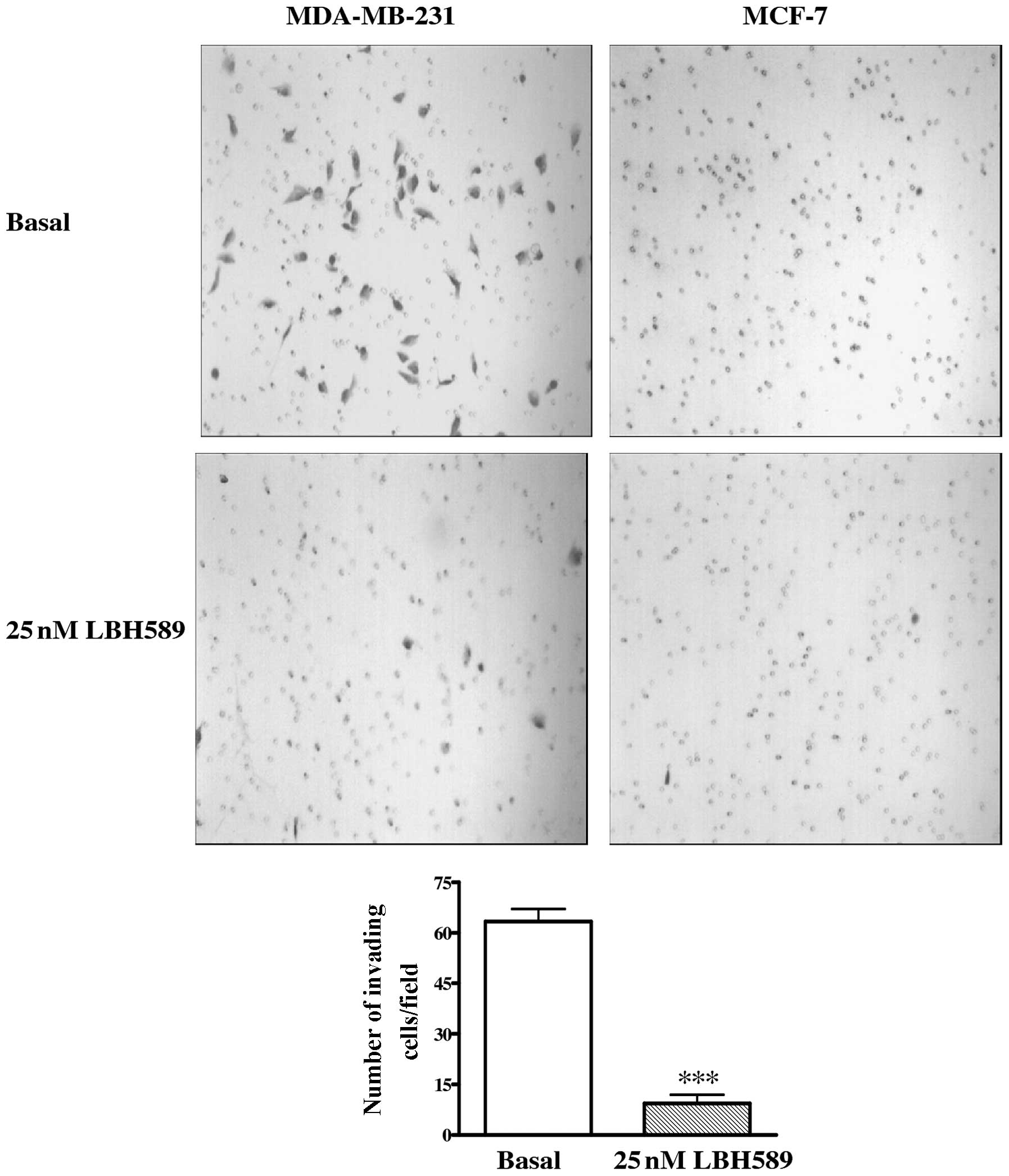
Invasion assay
Cells (5×105) were seeded in
75-cm2 flasks and treated with LBH589 (25 nM). After 24
h, 2×105 cells were seeded in the BD BioCoat™ BD
Matrigel™ invasion chamber (BD Biosciences Discovery Labware, Two
Oak Park, Bedford, MA, USA) and stimulated with 25 nM LBH589 for 24
h. At completion, the lower surfaces of the membrane were fixed
with methanol and stained with crystal violet solution, to point
out cells that migrated across the Matrigel and invaded the
inferior face of the membranes. The number of cells that had
migrated to the basal side of the membrane was quantified by
counting 12 independent fields under the microscope.
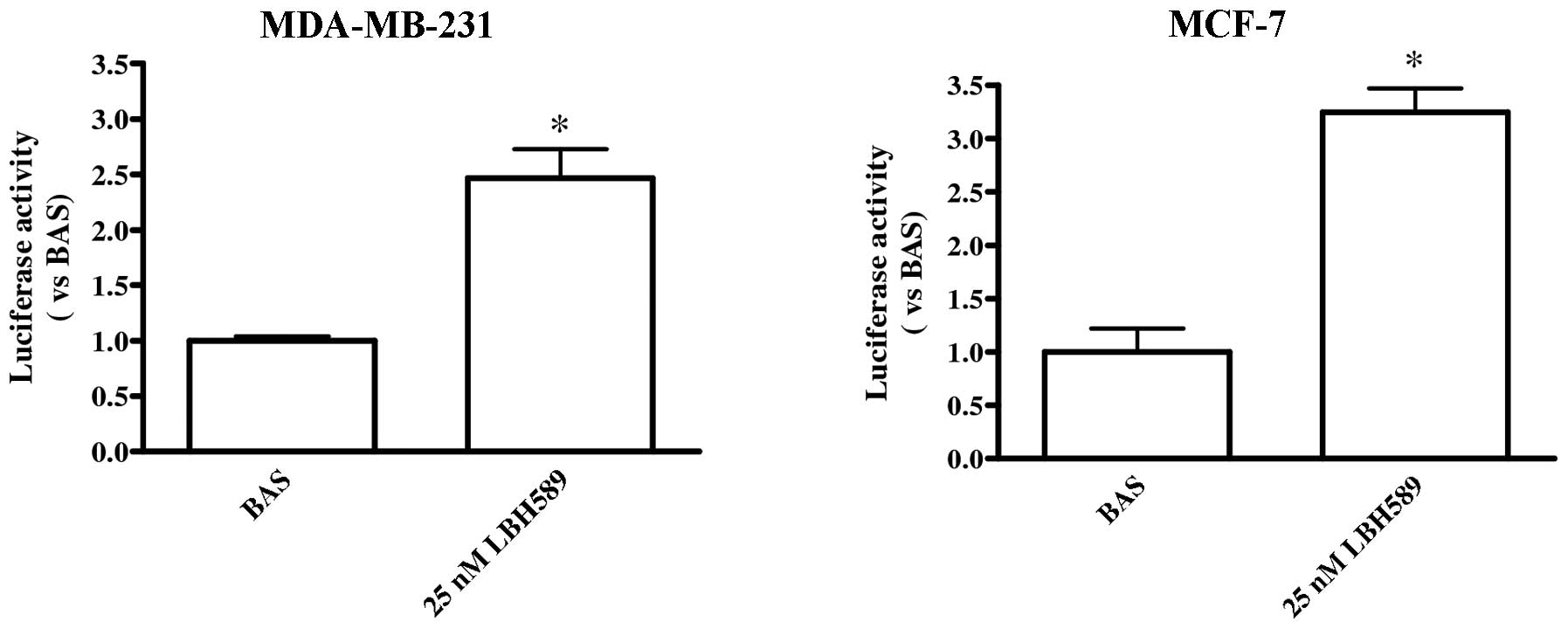
Luciferase assay
Cells (2.5×105) were seeded in 6-well
plates. After 24 h, cells were transfected with 1 μg of
pGL2Basic-EcadK1 plasmid/well using Lipofectin reagent (Invitrogen
Ltd.). The plasmid (Promega Italia, Milan, Italy) contains the
human CDH1 promoter sequences from −108 to +125 linked to
the luciferase gene as reporter (17). After transient transfection, cells
were treated with 25 nM LBH589 for 24 h. Luciferase activity was
assayed with the Luciferase assay system (Promega Corp., Madison,
WI, USA).
Statistical analysis
Data are expressed throughout as the means ± SD,
calculated from at least three different experiments. Comparison
between groups was performed with analysis of variance (one-way
ANOVA) and the threshold of significance was calculated with the
Bonferroni test. Statistical significance was set at P<0.05.
Results
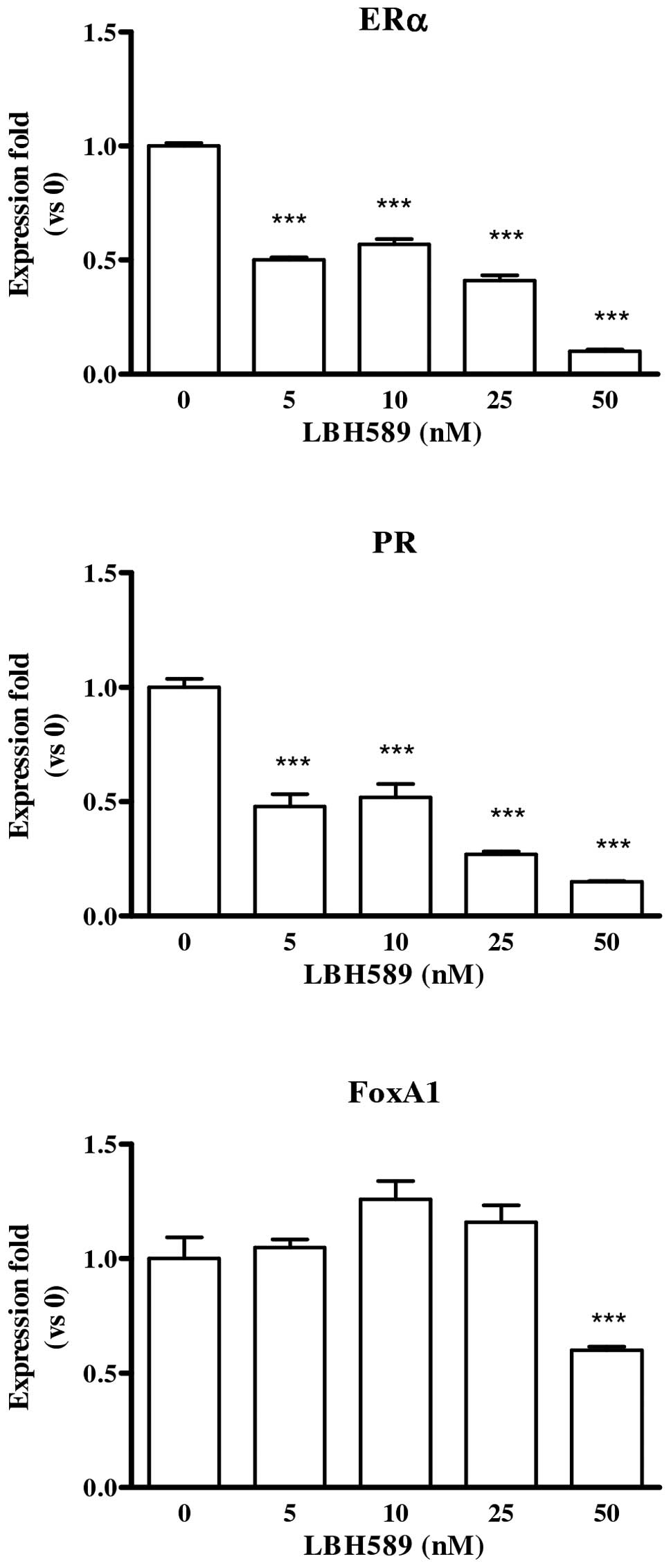
LBH589 effect on ERα, PR and FoxA1
First of all, the effect of LBH589 on the expression
of ERα and cognate PR (progesterone receptor) and FoxA1 (forkhead
box A1) was studied. In estrogen-sensitive MCF-7 cells, LBH589
reduced the level of expression of all the three genes (Fig. 1), while MDA-MB-231 cells did not
express any of the genes under study, neither in basal condition
nor after LBH589 treatment.
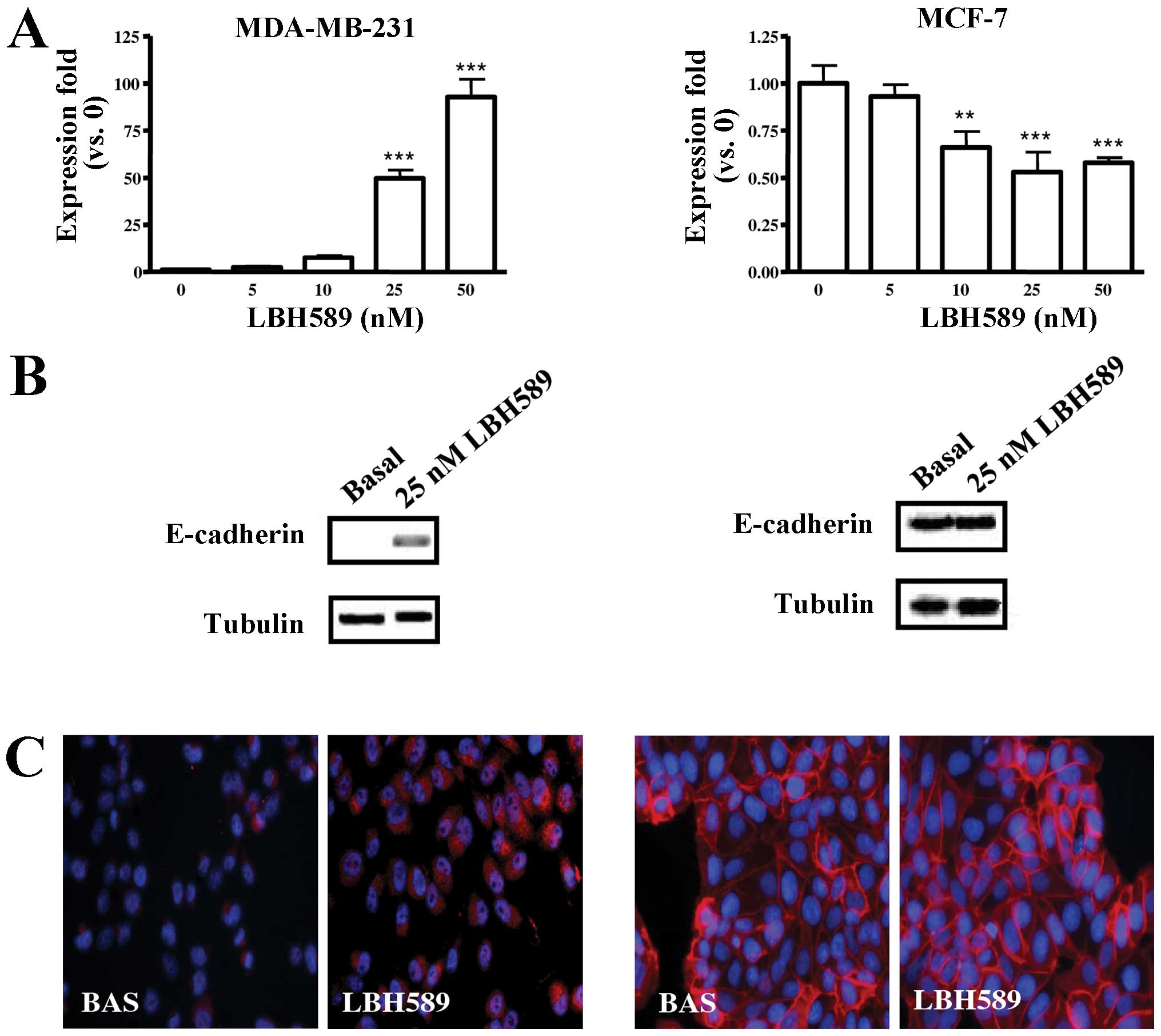
LBH589 effect on E-cadherin
expression
E-cadherin was not expressed in untreated MDA-MB-231
cells; LBH589 treatment increased E-cadherin gene expression, its
effect being evident at doses ≥25 nM; a slight reduction of gene
expression in E-cadherin positive MCF-7 cells was also observed
(Fig. 2A). Western blotting for
E-cadherin, reported in Fig. 2B,
confirmed that increased gene expression resulted in the appearance
of the protein in MDA-MB-231 cells; on the other hand, the
reduction of E-cadherin gene expression observed in MCF-7 cells was
not followed by a significant reduction of the protein level.
LBH589 treatment determined also a correct expression of E-cadherin
on the MDA-MB-231 surface (Fig.
2C).
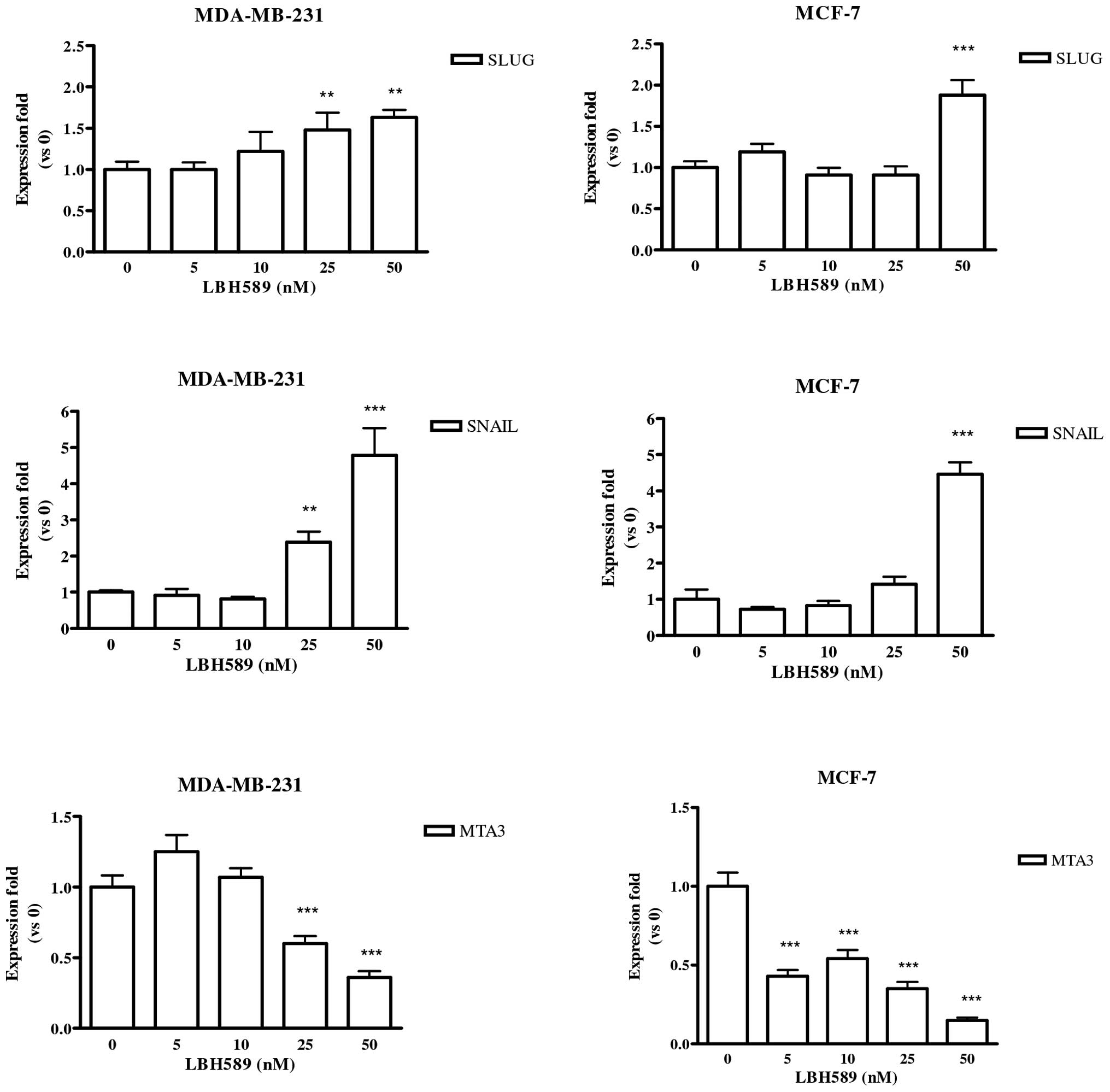
LBH589 effect on Slug, Snail and
MTA3
Both E-cadherin repressors, Slug and Snail, were
evaluated. As reported in Fig. 3,
the expression of Slug and Snail was significantly enhanced by
LBH589 treatment in both cell lines. Lastly, the Mi-2/NuRD
macro-complex member MTA3 was studied. We observed that LBH589
significantly reduced the level of expression of MTA3 in both cell
lines (Fig. 3, last row).
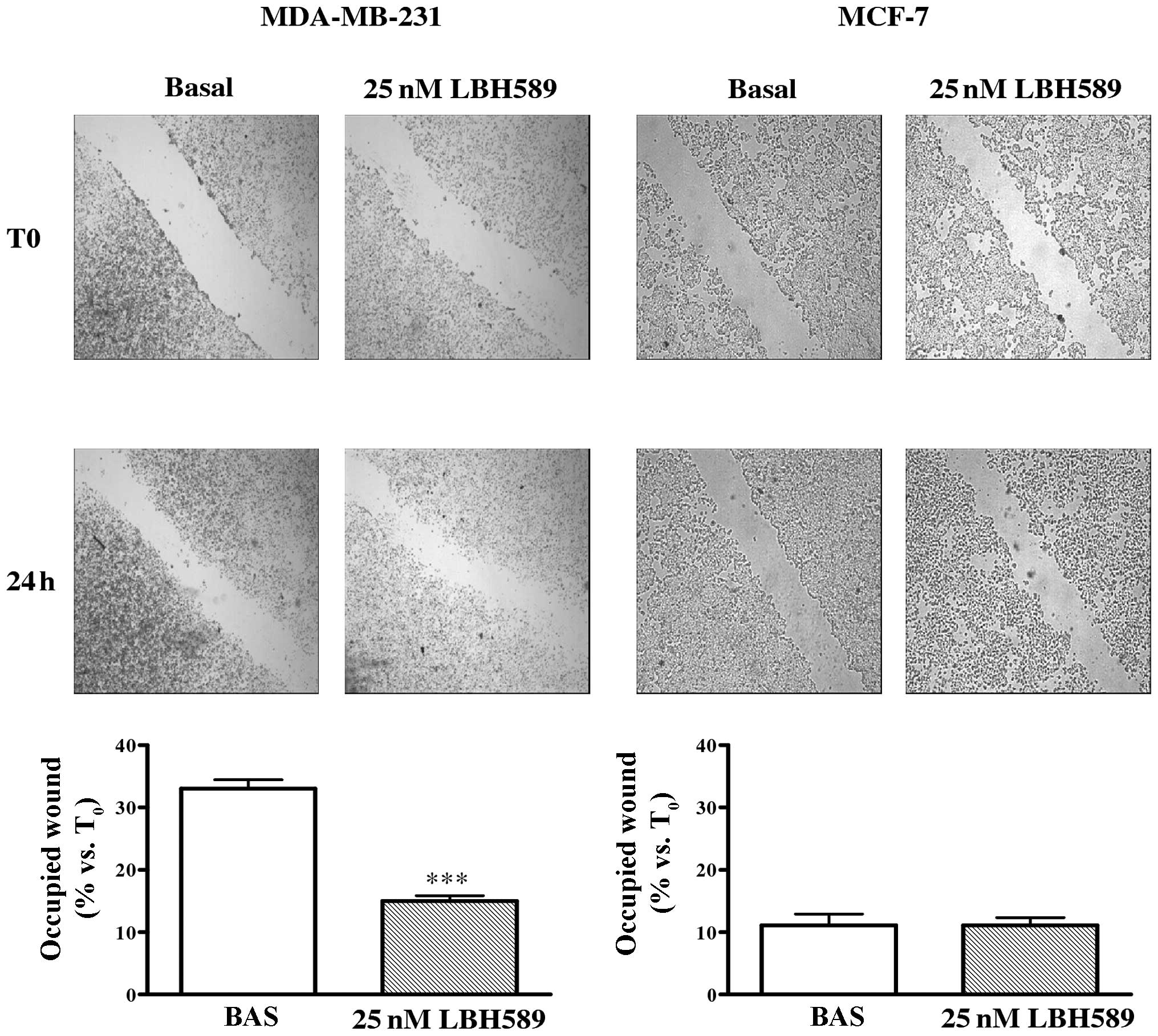
LBH589 effect on cell migration and
invasion
To determine whether LBH589 had a role in MDA-MB-231
cell migration, a scratch wound assay was carried out. As shown in
Fig. 4, the migration of treated
cells was significantly reduced with respect to untreated controls.
After 24 h, cells treated with 25 nM LBH589 occupied only 15% of
the wound compared to 33% occupation of untreated cells
(P<0.001). Moreover, the role of LBH589 on the invasive
potential of MDA-MB-231 cells was assayed through the use of a
transwell invasion chamber (Fig.
5). We observed that invasion of treated cells was markedly
reduced compared with untreated cells after 24 h (P<0.001). As
reported also in Figs. 4 and
5, migration and invasion of MCF-7
cells were not affected by LBH589 treatment.
LBH589 effect on CDH1 promoter
To evaluate whether LBH589 directly acts on
CDH1 promoter inducing transcription, a luciferase assay
using a construct containing CDH1 promoter was performed. We
observed a significant increase in CDH1 promoter transcription
level homogeneous with the increase of E-cadherin we reported above
in MDA-MB-231 cells; the direct effect of LBH589 on CDH1
promoter was observed even in MCF-7 cells (Fig. 6).
Discussion
The pan-deacetylase inhibitor LBH589 is a powerful
anti-tumour agent even at low nanomolar concentrations in a number
of hematologic and solid tumours. It was, in addition, reported to
induce re-expression of previously silenced genes modulating thus
the behaviour of cancer cells (35–37).
The present report follows our previous observation
in breast cancer cells where we observed a potent cytotoxic
activity of LBH589 in both estrogen-sensitive and -insensitive
cells (16). Our present data
demonstrate that LBH589 induces E-cadherin gene expression in
aggressive TNBC cells. E-cadherin gene expression resulted,
furthermore, in the correct appearance of the cognate protein on
MDA-MB-231 surface and, the re-expression of E-cadherin caused a
clear modification of MDA-MB-231 cell behaviour. In fact, after
LBH589 treatment, cell migration and invasive potential were
greatly reduced.
Our previous observation on the anti-proliferative
and pro-apoptotic effects of LBH589 in ER-negative breast cancer
cells has been recently confirmed by other authors (38). The same authors reported that the
most induced gene by LBH589 treatment in MDA-MB-231 cells is the
CDH1 gene that codes for E-cadherin, in total agreement with
our observation on the increase of the CDH1 gene promoter
activity we report in the present study and consequent E-cadherin
induction. Tate et al also noted an increase in the
CDH1 gene expression on the periphery of the primary tumour
from MDA-MB-231 xenografts, and hypothesized that the induction of
CDH1 expression by LHB589 at the invasive edge may be
indicative of decreased metastatic potential. Actually, our
observation in vitro of reduced migration and invasion of
MDA-MB-231 cells treated with LBH589 substantiates their
hypothesis, confirming that E-cadherin induction by LBH589 has a
functional meaning and could be exploited for therapeutic
application.
It is also interesting to note that in ER-positive
MCF-7 cells that express high level of E-cadherin, LBH589 is a
clear anti-proliferative and proapoptotic agent even at low
nanomolar doses (16), but does
not modify the E-cadherin expression and cell migration/invasion
leaving unaffected the low metastatic potential of these cells.
As reported, in breast cancer MTA3/Snail/E-cadherin
and estradiol (E2)/ERα signalling are closely linked (34). The appearance of E-cadherin in
MDA-MB-231 cells treated with LBH589 could be due to the
reactivation of ERα pathway in TNBC cells since other DCI were
reported to act similarly in several studies. For example, DCI in
association with demethylating agents like 5-aza-2′-deoxycytidine
were shown to reactivate ERα expression in ER-negative breast
cancer cells (22,39,40);
the DCI valproic acid was demonstrated to enhance the efficacy of
the anti-estrogen tamoxifen through increasing ER-mediated
transcription (16) and also to
induce ERα, PR, pS2 and FoxA1, giving to MDA-MB-231 cells an
estrogen-sensitive ‘phenotype’ and restoring their sensitivity to
anti-estrogen therapy (18).
Controversial results on LBH589 effect on ERα
expression in MDA-MB-231 cells, either receptor induction (23) or no effect at all (24), have been reported.
The present data confirm that LBH589 does not induce
ERα expression and ER-pathway restoration in MDA-MB-231 cells.
Moreover, LBH589 reduced MTA3 levels, and significantly increased
both Snail and Slug, whose levels are inversely related to
E-cadherin expression, either in ER-negative MDA-MB-231 or in
ER-positive MCF-7 cells. Furthermore, in MCF-7 cells LBH589
significantly decreased the expression level of ERα, PR and FoxA1,
as previously reported (24). The
effects of LBH589 on ER pathway are not consistent with those
observed on E-cadherin gene and protein expression in both cell
lines. It is thus conceivable that LBH589 can control E-cadherin
independently of estradiol and this is not affected by the level of
expression of Snail, in agreement with recent observations in
ovarian carcinoma (41). Snail
requires histone deacetylase activity to repress E-cadherin
promoter and it has already been demonstrated that treatment with
trichostatin A is sufficient to block this effect (42). Our data suggest that LBH589 in
MDA-MB-231 breast cancer acts directly on the CDH1 promoter
and does not need to modify E-cadherin transcriptional repressors
to induce E-cadherin expression and to modify the cell aggressive
attitude. In MCF-7 cells, even though LBH589 smoothens down the ER
pathway, reducing MTA3 and increasing Snail and Slug, its direct
action on CDH1 promoter is also present, the final effect on
E-cadherin protein expression is irrelevant, so that migration and
invasion of MCF-7 cells are not affected.
In conclusion, LBH589 is able to induce E-cadherin
in highly aggressive TNBC cells reducing their migration and
invasion, by-passing E-cadherin transcriptional repressors such as
Snail and Slug and without any detectable effect on ERα expression
and pathway. This compound can, therefore, be proposed for
treatment of aggressive breast cancer, refractory to hormonal
therapy exploiting its antiproliferative and anti-invasive
properties. At least eight different trials on advanced breast
cancer treatment with LBH589 both in monotherapy and in association
with trastuzumab, capecitabine, lapatinib, or paclitaxel,
respectively, are now listed on the site www.clinicaltrials.gov. We are looking forward to
their conclusions and strongly hope LBH589 will be available soon
for advanced breast cancer treatment.
Acknowledgements
We thank Novartis Pharma AG, Basel,
Switzerland for providing us with LBH589; and Daniela Taverna and
Francesca Orso, Dipartimento di Biotecnologie Molecolari e Scienze
della Salute, Università di Torino and M.B.C., Torino, Italy, for
helping us with luciferase experiments.
References
|
1.
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|
|
2.
|
Berry DA, Cronin KA, Plevritis SK, Fryback
DG, Clarke L, Zelen M, Mandelblatt JS, Yakovlev AY, Habbema JD and
Feuer EJ; Cancer Intervention and Surveillance Modeling Network
(CISNET) Collaborators: Effect of screening and adjuvant therapy on
mortality from breast cancer. N Engl J Med. 353:1784–1792. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Cardoso F, Bedard PL, Winer EP, Pagani O,
Senkus-Konefka E, Fallowfield LJ, Kyriakides S, Costa A, Cufer T
and Albain KS: ESO-MBC Task Force, International guidelines for
management of metastatic breast cancer: combination vs. sequential
single-agent chemotherapy. J Natl Cancer Inst. 101:1174–1181. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Gonzalez-Angulo AM, Morales-Vasquez F and
Hortobagyi GN: Overview of resistance to systemic therapy in
patients with breast cancer. Adv Exp Med Biol. 608:1–22. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Dent R, Trudeau M, Pritchard KI, Hanna WM,
Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P and Narod SA:
Triple-negative breast cancer: clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Schneider BP, Winer EP, Foulkes WD, Garber
J, Perou CM, Richardson A, Sledge GW and Carey LA: Triple-negative
breast cancer: risk factors to potential targets. Clin Cancer Res.
14:8010–8018. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Anders CK and Carey LA: Biology,
metastatic patterns, and treatment of patients with triple-negative
breast cancer. Clin Cancer Res. 9:S73–S81. 2009.PubMed/NCBI
|
|
8.
|
Elias AD: Triple-negative breast cancer: a
short review. Am J Clin Oncol. 33:637–645. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Rodríguez-Paredes M and Manel Esteller M:
Cancer epigenetic reaches mainstream oncology. Nat Med. 17:330–339.
2011.PubMed/NCBI
|
|
10.
|
Sawan C, Vaissière T, Murr R and Herceg Z:
Epigenetic drivers and genetic passengers on the road to cancer.
Mutat Res. 642:1–13. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Ellis L, Atadja PW and Johnstone RW:
Epigenetics in cancer: targeting chromatin modifications. Mol
Cancer Ther. 8:1409–1420. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Wong ST: Emerging treatment combinations:
integrating therapy into clinical practice. Am J Health Syst Pharm.
66:S9–S14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Göttlicher M, Minucci S, Zhu P, Krämer OH,
Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG
and Heinzel T: Valproic acid defines a novel class of HDAC
inhibitors inducing differentiation of transformed cells. EMBO J.
20:6969–6978. 2001.
|
|
14.
|
Hodges-Gallagher L, Valentine CD, Bader SE
and Kushner PJ: Inhibition of histone deacetylase enhances the
anti-proliferative action of antiestrogens on breast cancer cells
and blocks tamoxifen-induced proliferation of uterine cells. Breast
Cancer Res Treat. 105:297–309. 2007. View Article : Google Scholar
|
|
15.
|
Fortunati N, Bertino S, Costantino L,
Bosco O, Vercellinatto I, Catalano MG and Boccuzzi G: Valproic acid
is a selective antiproliferative agent in estrogen-sensitive breast
cancer cells. Cancer Lett. 259:156–164. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Fortunati N, Catalano MG, Marano F, Mugoni
V, Pugliese M, Bosco O, Mainini F and Boccuzzi G: The pan-DAC
inhibitor LBH589 is a multi-functional agent in breast cancer
cells: cytotoxic drug and inducer of sodium-iodide symporter (NIS).
Breast Cancer Res Treat. 124:667–675. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Hajra KM, Ji X and Fearon ER: Extinction
of E-cadherin expression in breast cancer via a dominant repression
pathway acting on proximal promoter elements. Oncogene.
18:7274–7279. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Fortunati N, Bertino S, Costantino L, De
Bortoli M, Compagnone A, Bandino A, Catalano MG and Boccuzzi G:
Valproic acid restores ER alpha and antiestrogen sensitivity to ER
alpha-negative breast cancer cells. Mol Cell Endocrinol. 314:17–22.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Katzenellenbogen BS and Frasor J:
Therapeutic targeting in the estrogen receptor hormonal pathway.
Semin Oncol. 31:28–38. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Ring A and Dowsett M: Mechanisms of
tamoxifen resistance. Endocr Relat Cancer. 11:643–658. 2004.
View Article : Google Scholar
|
|
21.
|
Jovanovic J, Rønneberg JA, Tost J and
Kristensen V: The epigenetics of breast cancer. Mol Oncol.
4:242–254. 2010. View Article : Google Scholar
|
|
22.
|
Zhou Q, Atadja P and Davidson NE: Histone
deacetylase inhibitor LBH589 reactivates silenced estrogen receptor
alpha (ER) gene expression without loss of DNA hypermethylation.
Cancer Biol Ther. 6:64–69. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Fiskus W, Ren Y, Mohapatra A, Bali P,
Mandawat A, Rao R, Herger B, Yang Y, Atadja P, Wu J and Bhalla K:
Hydroxamic acid analogue histone deacetylase inhibitors attenuate
estrogen receptor-alpha levels and transcriptional activity: a
result of hyperacetylation and inhibition of chaperone function of
heat shock protein 90. Clin Cancer Res. 13:4882–4890. 2007.
View Article : Google Scholar
|
|
24.
|
Hayashi A, Horiuchi A, Kikuchi N, Hayashi
T, Fuseya C, Suzuki A, Konishi I and Shiozawa T: Type-specific
roles of histone deacetylase (HDAC) overexpression in ovarian
carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates
cell migration with downregulation of E-cadherin. Int J Cancer.
127:1332–1346. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelialmesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Lombaerts M, van Wezel T, Philippo K,
Dierssen JWF, Zimmerman RME, Oosting J, van Eijk R, Eilers PH, van
De Water B, Cornelisse CJ and Cleton-Jansen AM: E-cadherin
transcriptional downregulation by promoter methylation but not
mutation is related to epithelial-to-mesenchymal transition in
breast cancer cell lines. Br J Cancer. 94:661–671. 2006.PubMed/NCBI
|
|
27.
|
Nam JS, Ino Y, Kanai Y, Sakamoto M and
Hirohashi S: 5-Aza-2′-deoxycytidine restores the E-cadherin system
in E-cadherin-silenced cancer cells and reduces cancer metastasis.
Clin Exp Metastasis. 21:49–56. 2004.
|
|
28.
|
Blanco MJ, Moreno-Bueno G, Sarrio D,
Locascio A, Cano A, Palacios J and Nieto MA: Correlation of Snail
expression with histological grade and lymph node status in breast
carcinomas. Oncogene. 21:3241–3246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Cheng CW, Wu PE, Yu JC, Huang CS, Yue CT,
Wu CW and Shen CY: Mechanisms of inactivation of E-cadherin in
breast carcinoma: modification of the two-hit hypothesis of tumor
suppressor gene. Oncogene. 20:3814–3823. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Elloul S, Elstrand MB, Nesland JM, Trope
CG, Kvalheim G, Goldberg I, Reich R and Davidson B: Snail, Slug,
and Smad-interacting protein 1 as novel parameters of disease
aggressiveness in metastatic ovarian and breast carcinoma. Cancer.
103:1631–1643. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Rosivatz E, Becker I, Specht K, Fricke E,
Luber B, Busch R, Hofler H and Becker KF: Differential expression
of the epithelialmesenchymal transition regulators Snail, SIP1, and
Twist in gastric cancer. Am J Pathol. 161:1881–1891. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Yang J, Mani SA, Donaher JL, Ramaswamy S,
Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and
Weinberg RA: Twist, a master regulator of morphogenesis, plays an
essential role in tumor metastasis. Cell. 117:927–939. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Fearon ER: Connecting estrogen receptor
function, transcriptional repression, and E-cadherin expression in
breast cancer. Cancer Cell. 3:307–310. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Fujita N, Jaye DL, Kajita M, Geigerman C,
Moreno CS and Wade PA: MTA3, a Mi-2/NuRD complex subunit, regulates
an invasive growth pathway in breast cancer. Cell. 113:207–219.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Catalano MG, Pugliese M, Gargantini E,
Grange C, Bussolati B, Asioli S, Bosco O, Poli R, Compagnone A,
Bandino A, Mainini F, Fortunati N and Boccuzzi G: Cytotoxic
activity of the histone deacetylase inhibitor Panobinostat (LBH589)
in anaplastic thyroid cancer in vitro and in vivo. Int J Cancer.
130:694–704. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Maiso P, Carvajal-Vergara X, Ocio EM,
López-Pérez R, Mateo G, Gutiérrez N, Atadja P, Pandiella A and San
Miguel JF: The histone deacetylase inhibitor LBH589 is a potent
antimyeloma agent that overcomes drug resistance. Cancer Res.
66:5781–5789. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Shao W, Growney JD, Feng Y, O’Connor G, Pu
M, Zhu W, Yao YM, Kwon P, Fawell S and Atadja P: Activity of
deacetylase inhibitor Panobinostat (LBH589) in cutaneous T-cell
lymphoma models: defining molecular mechanisms of resistance. Int J
Cancer. 127:2199–2208. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Tate CR, Rhodes LV, Segar HC, Driver JL,
Pounder FN, Burow ME and Collins-Burow BM: Targeting
triple-negative breast cancer cells with the histone deacetylase
inhibitor panobinostat. Breast Cancer Res. 14:R792012. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Keen JC, Yan L, Mack KM, Pettit C, Smith
D, Sharma D and Davidson NE: A novel histone deacetylase inhibitor,
scriptaid, enhances expression of functional estrogen receptor
alpha (ER) in ER negative human breast cancer cells in combination
with 5-aza 2′-deoxycytidine. Breast Cancer Res Treat. 81:177–186.
2003.PubMed/NCBI
|
|
40.
|
Sharma D, Saxena NK, Davidson NE and
Vertino PM: Restoration of tamoxifen sensitivity in estrogen
receptor-negative breast cancer cells: tamoxifen-bound reactivated
ER recruits distinctive corepressor complexes. Cancer Res.
66:6370–6378. 2006. View Article : Google Scholar
|
|
41.
|
Peinado H, Ballestar E, Esteller M and
Cano A: Snail mediates E-cadherin repression by the recruitment of
the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell
Biol. 24:306–319. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Kim IA, No M, Lee JM, Shin JH, Oh JS, Choi
EJ, Kim IH, Atadja P and Bernhard EJ: Epigenetic modulation of
radiation response in human cancer cells with activated EGFR or
HER-2 signaling: potential role of histone deacetylase 6. Radiother
Oncol. 92:125–132. 2009. View Article : Google Scholar : PubMed/NCBI
|