Introduction
The tumor suppressor protein p53 is a transcription
factor that orchestrates anti-carcinogenesis programs such as cell
cycle arrest, apoptosis, DNA repair and senescence in response to
genotoxic and non-genotoxic cellular injuries (1,2).
Although various transcriptional target genes of p53 contribute to
the suppression of tumor development, they are not always
compatible in terms of cell survival. For example, several p53
target genes that are involved in cell cycle arrest and DNA repair
such as p21WAF1 and 14-3-3σ, an inhibitor of G1-S and G2-M
transition, respectively, and Ku70, a non-homologous end joining
repair gene, inhibit DNA damage-induced p53-dependent apoptosis
(3–5). In addition, transcriptional target
genes of p53 that protect various cells from apoptosis have been
identified. In γ-irradiated hematopoietic progenitors, p53 induces
SLUG, which functions to repress PUMA, thereby inhibiting
p53-induced apoptosis (6). PIDD,
which is induced by p53 upon double strand DNA breaks, can activate
NF-κB, thus contributing to the inhibition of apoptosis and cancer
cell survival (7). p53 also
activates cell survival signaling such as the Ras-Raf-MEK1/2-ERK1/2
pathway and PI3K/AKT via the transcriptional induction of HB-EGF
and DDR1, the blocking of which results in the augmentation of
genotoxic stress-induced apoptosis (8,9).
These survival target genes of p53 are simultaneously induced with
the induction of apoptotic target genes of p53, resulting in
fine-tuning or constituting the negative feedback loop of
p53-induced apoptosis. Therefore, it is possible that p53 may
induce cell survival signaling as well as apoptosis. Nutlin-3, a
cis-imidazoline analog, was initially developed as an antagonist of
MDM2, an E3 ubiquitin ligase of proteins of the p53 family, that
ubiquitinates and directs them to proteasomal degradation (10).
Nutlin-3 interferes with the binding between MDM2
and p53 by preoccupying the p53 binding pocket of MDM2. In
vitro and in vivo treatments of nutlin-3 induce p53
functions, such as cell cycle arrest and apoptosis (11,12).
Nutlin-3 does not induce damage to genomic DNA to activate the
p53-dependent pathway so that the adverse effect on non-transformed
cells and the risk of tumor development secondary to nutlin-3
treatment would be expected to be much less than conventional
chemotherapeutic agents which induce DNA damage (13). However, it has been reported that
nutlin-3 predominantly induces cell cycle arrest in cancer cells
particularly derived from solid tumors rather than apoptosis
(14). Nutlin-3 induces prominent
p21WAF1 expression by upregulating hnRNP K, a coactivator of the
transcriptional activity of p53, and downregulating HIPK2, an
activator of p53-induced apoptosis, that phosphorylates Ser 46 on
p53 (15,16). In addition to these genetic
interactions, it is likely that nutlin-3 activates other cell
survival pathways as well as apoptosis and such cell survival
pathways may inhibit the nutlin-3-induced apoptotic signaling
pathway. During our studies directed at cell survival pathways that
are modulated by nutlin-3, we found that, along with inducing
apoptosis, nutlin-3 activates ERK1/2 (17). It should be noted that
nutlin-3-induced ERK1/2 activation was independent of the
transcriptional activity of p53. Instead, nutlin-3 induces the
mitochondrial translocation of p53 and subsequently ROS
accumulation, which activates the MEK1/2-ERK1/2 pathway to confer
survival characteristics to cancer cells.
Heme oxygenase-1 (HO-1, EC1:14.99.3) is a microsomal
enzyme that catalyzes the degradation of heme derived from
heme-containing proteins (18).
The degradation of heme results in the production of biliverdin,
ferrous iron and carbon monoxide (CO). Because these metabolites
have anti-inflammatory and anti-apoptotic effects, it is possible
that HO-1 has the potential for anti-inflammatory activity, which
could protect cells against cellular insults, including oxidative
stress. Chronic inflammation and oxidative stress are frequently
associated with the initiation and progression of cancer and it can
therefore be assumed that HO-1 would be elevated in cancer tissues
and if so, HO-1 could endow cancer cells with growth-enhancing
characteristics by inhibiting apoptosis (19). The expression of HO-1 was reported
to be higher in various cancer tissues than in normal tissues
(20). Moreover, the knockdown of
HO-1 results in cancer cells being more susceptible to anticancer
drug treatment (21,22). The expression of HO-1 is usually
regulated at the transcriptional level, which occurs via a
three-layered pathway i.e. challenging of the stimuli-activation of
MAPK-activation of transcription factors. The transcription of HO-1
can be induced by a wide range of stimuli including oxidative and
pro-inflammatory stress, various chemicals and a change in
extracellular pH (18,23). These stimuli activate one or more
of the MAPKs such p38 MAPK, JNK, and ERK1/2 and PI3K/AKT, which, in
turn, direct various transcription factors to bind to their cognate
sites in the promoter of HO-1. Among the transcription factors,
Nrf2, AP-1, NF-κB and HSF are the most responsible for activating
HO-1 transcription (24).
Interestingly, a recent report showed that p53 induces the
expression of HO-1 and thus protects cells against oxidative
stress-induced apoptosis, suggesting that p53 itself contributes to
the survival of cancer cells, but not cell death (25).
This report prompted us to speculate that nutlin-3
may also stimulate the expression of HO-1, and the possibility
exists that this induction could inhibit the nutlin-3-induced
apoptosis, resulting in predominant cell cycle arrest. To address
this hypothesis, we examined the induction of HO-1 in
nutlin-3-treated cells and analyzed the underlying mechanism of
HO-1 responsible for induction by nutlin-3.
Materials and methods
Cell culture
Human osteosarcoma U2OS cells and human colon cancer
RKO cells were purchased from the American Type Culture Collection
(Manassas, VA, USA) and were maintained in DMEM supplemented with
10% fetal bovine serum (HyClone, Logan, UT, USA), 100 U/ml
penicillin/streptomycin (HyClone) and glutamate (Invitrogen,
Calsbad, CA, USA) at 37°C under 5% CO2. Cells were
sub-cultured or refreshed with media every 3 days.
Chemicals
Nutlin-3 was purchased from Selleck Chemicals
(Houston, TX, USA). Pifithrin (PFT)-α, PFT-μ and TEMPO were
obtained from Sigma-Aldrich (St. Louis, MO, USA). Inhibitors of
MAPK including SB203580, SP600125 and U0126 were obtained from
TOCRIS Bioscience (Bristol, UK) or AdooQ Bioscience (Irvine, CA,
USA). Other chemicals were obtained from Sigma-Aldrich, unless
otherwise specified.
Immunoblot analysis
For immunoblot analysis, U2OS cells were treated as
described in the figure legends and were lysed with RIPA buffer.
Following a protein assay, equal amounts of proteins of each sample
were separated by SDS-PAGE and transferred to nitrocellulose
membranes, which were submerged in TBS-T (Tris-buffered saline with
0.025% Tween-20) containing 5% skim-milk for 30 min. The NC
membranes were then processed sequentially for incubation with
primary antibodies against proteins of interest, washing with
TBS-T, incubation with secondary antibodies, and washing with
TBS-T. Finally, the protein that reacted with the primary antibody
of interest was visualized by an enhanced chemiluminescence
detection method (ECL, GE Healthcare, Buckinghamshire, UK).
Real-time quantitative
reverse-transcription PCR (QRT-PCR)
Transcripts of HO-1 and p21WAF1 were measured by
QRT-PCR using GAPDH as the reference gene following a previous
report (17). Briefly, first
strand cDNA synthesis from total RNA and subsequent QRT-PCR were
performed using a PrimeScript™ RT reagent Kit (Takara Bio Inc.,
Shiga, Japan) and SYBR Premix Ex Taq (KAPA), respectively. All
reactions were performed in triplicate in an ABI 7300 Real-Time PCR
System (Applied Biosystems, Carlsbad, CA, USA). Relative changes of
transcripts level were calculated by the ΔΔCt method (26).
Luciferase reporter assay
A human HO-1 promoter (hHO-1) cloned into a basic
pGL3 plasmid was obtained from Professor J. Alam at the Ochsner
Medical Center, New Orleans, LA, USA. Cells were transfected with
0.3 μg of hHO-1 promoter-luciferase and 0.03 μg of
Renilla luciferase (Promega, Madison, WI, USA) using FuGENE
HD (Roche Applied Science, Indianapolis, IN, USA) for 24 h, and
cells were treated with nutlin-3. At the indicated times after the
treatments, the activities of firefly and Renilla luciferase
were determined using the Dual Luciferase kit (Promega) and the
data are expressed as relative luciferase activity (RLA) of three
independent experiments performed in triplicate. Renilla
luciferase activity was for the normalization of transfection
efficiency.
Transfection of siRNA
Small interfering RNA against p53 was purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA), and siRNA against
HO-1 and scrambled siRNA were from Sigma-Aldrich. They were
dissolved in RNase-free H2O and diluted with siRNA
diluent. SiRNAs were transfected into cells using Lipofectamine
RNAiMAX™ (Invitrogen) following the manufacturer’s instruction.
Measurement of ROS
Intracellular ROS levels were measured using
H2DCF-DA dye (Invitrogen). After cells were incubated in
the presence of 20 μM H2DCF-DA, the intensity of
the fluorescence in cells was observed by means of a fluorescence
inverted microscope (Olympus IX71, Tokyo, Japan) and was quantified
by flow cytometry.
Assessment of cell death
To measure the translocation of phosphatidylserine
in the cytoplasmic membranes, treated cells were stained with
propidium iodide (PI) and FITC-labeled Annexin V using ApoScan Kit
(BioBud, Gyunggido, Korea), followed by flow cytometry analysis as
described previously (27).
Emissions of Annexin V-FITC and PI were measured in the FL1 and FL3
channels with emission filters of 488 and 635 nm, respectively.
Results
Nutlin-3 induces the expression of HO-1
at the transcriptional level
It was recently reported that p53 directly activates
the transcription of HO-1 in response to treatment with
H2O2 (25).
We therefore attempted to observe whether the activation of p53 by
nutlin-3, an antagonist of MDM2, stimulates the expression of HO-1.
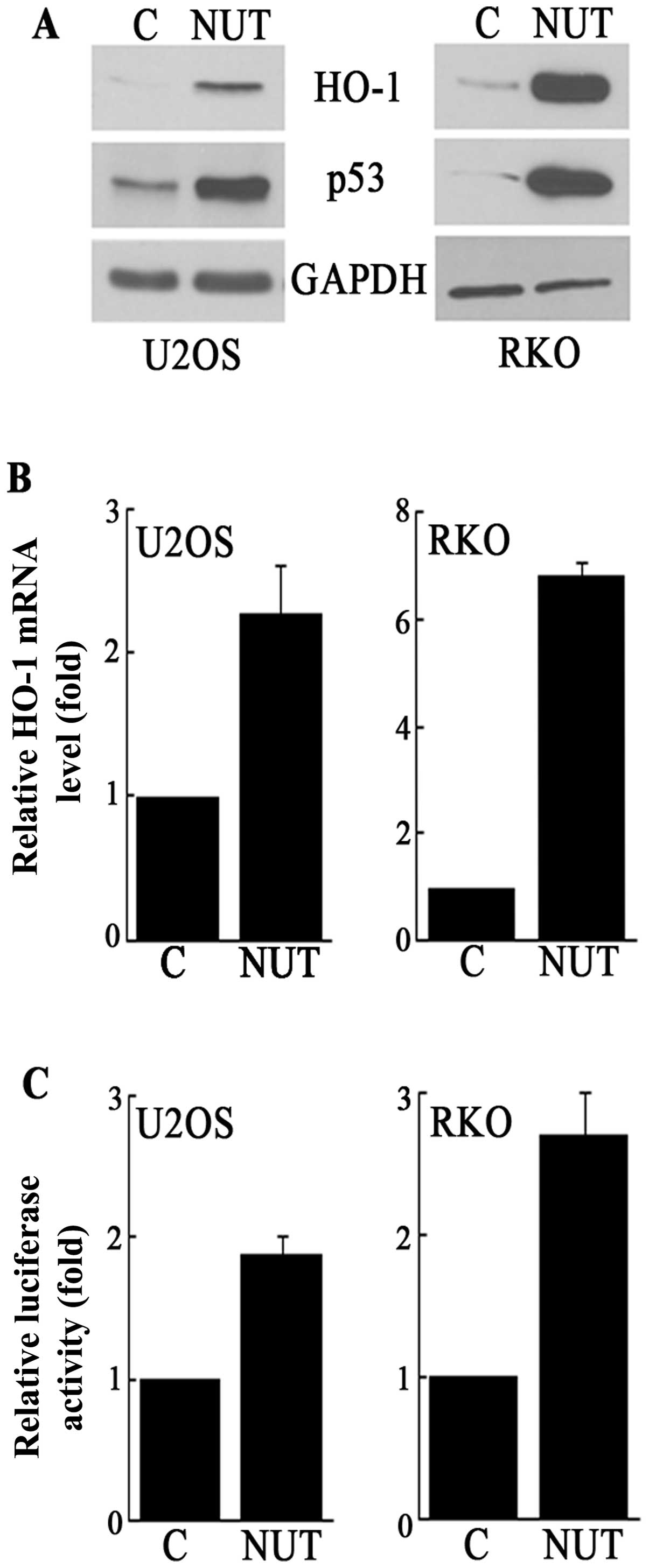
As shown in Fig. 1A, the nutlin-3
treatment resulted in increased levels of the HO-1 protein as well
as the p53 protein in both U2OS (human osteosarcoma) and RKO (human
colon cancer) cells. This increase in HO-1 protein levels was
accompanied by an increase in HO-1 mRNA and HO-1 promoter activity
(Fig. 1B and C). Therefore, these
data demonstrate that nutlin-3 induces HO-1 expression at the level
of transcription.
Nutlin-3-induced HO-1 is dependent on p53
but not the transcriptional activity of p53
Next, since nutlin-3 is an antagonist of MDM-2, a
ubiquitin ligase of proteins of the p53 family, and in fact,
nutlin-3 was also reported to enhance the function of p73 (28), it became necessary to determine the
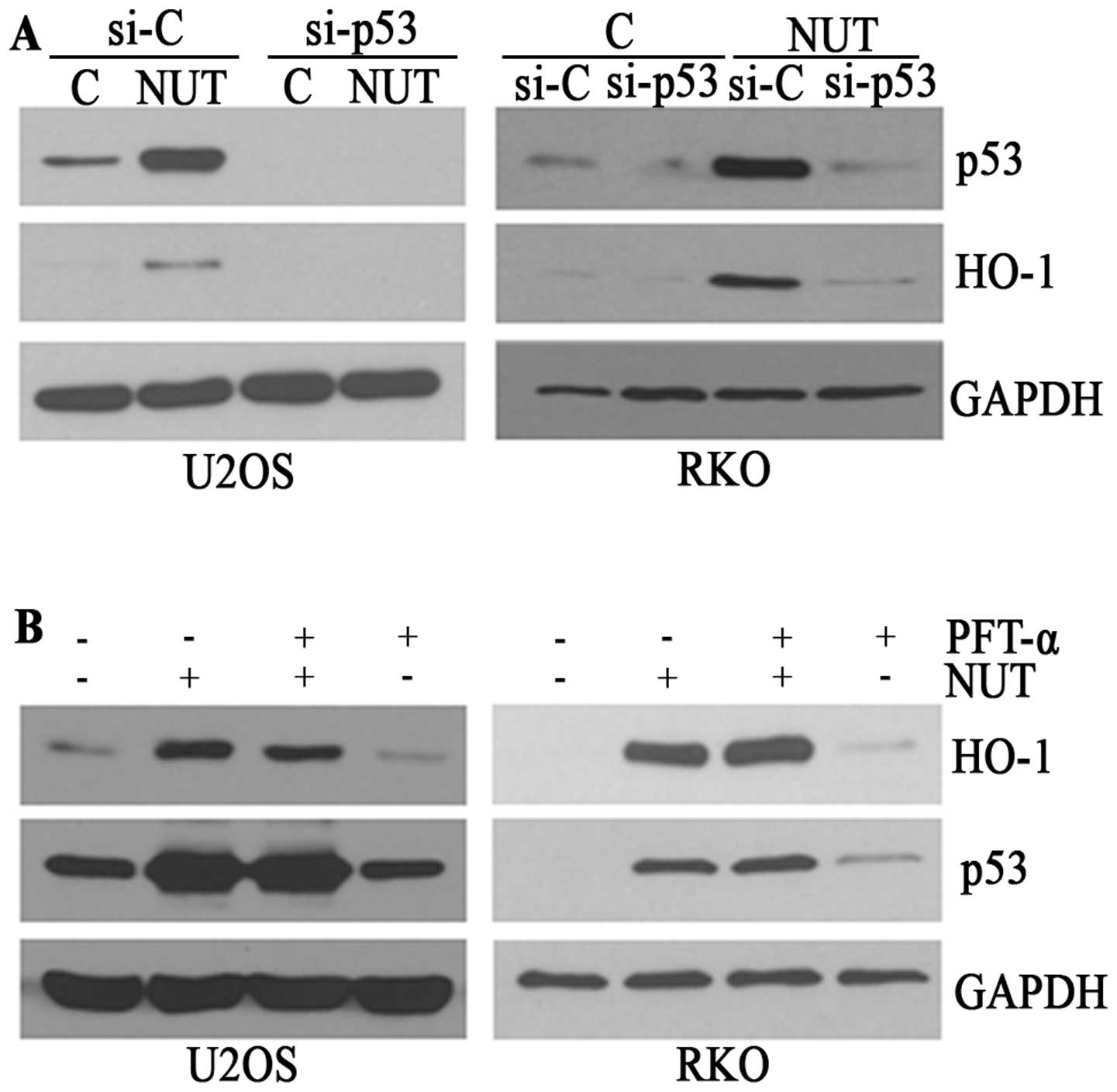
role of p53 in this HO-1 induction. In experiments using siRNA
against p53, nutlin-3 failed to induce HO-1 expression in
p53-knocked down cells (Fig. 2A)
as well as SAOS cells in which p53 was mutated (data not shown),
indicating that p53 is indispensible for this nutlin-3-induced HO-1
expression. However, PFT-α, an inhibitor of the transcriptional
activity of p53 did not interfere with the increase in HO-1 levels
in nutlin-3-treated cells (Fig.
2B). Collectively, these data suggest that the nutlin-3-induced
HO-1 expression is dependent on functions other than the
transcriptional activity of the p53 protein and that HO-1 may not
be a direct transcriptional target of p53 in nutlin-3-treated
cells.
Nutlin-3-induced HO-1 is dependent on JNK
and P38 MAPK activation
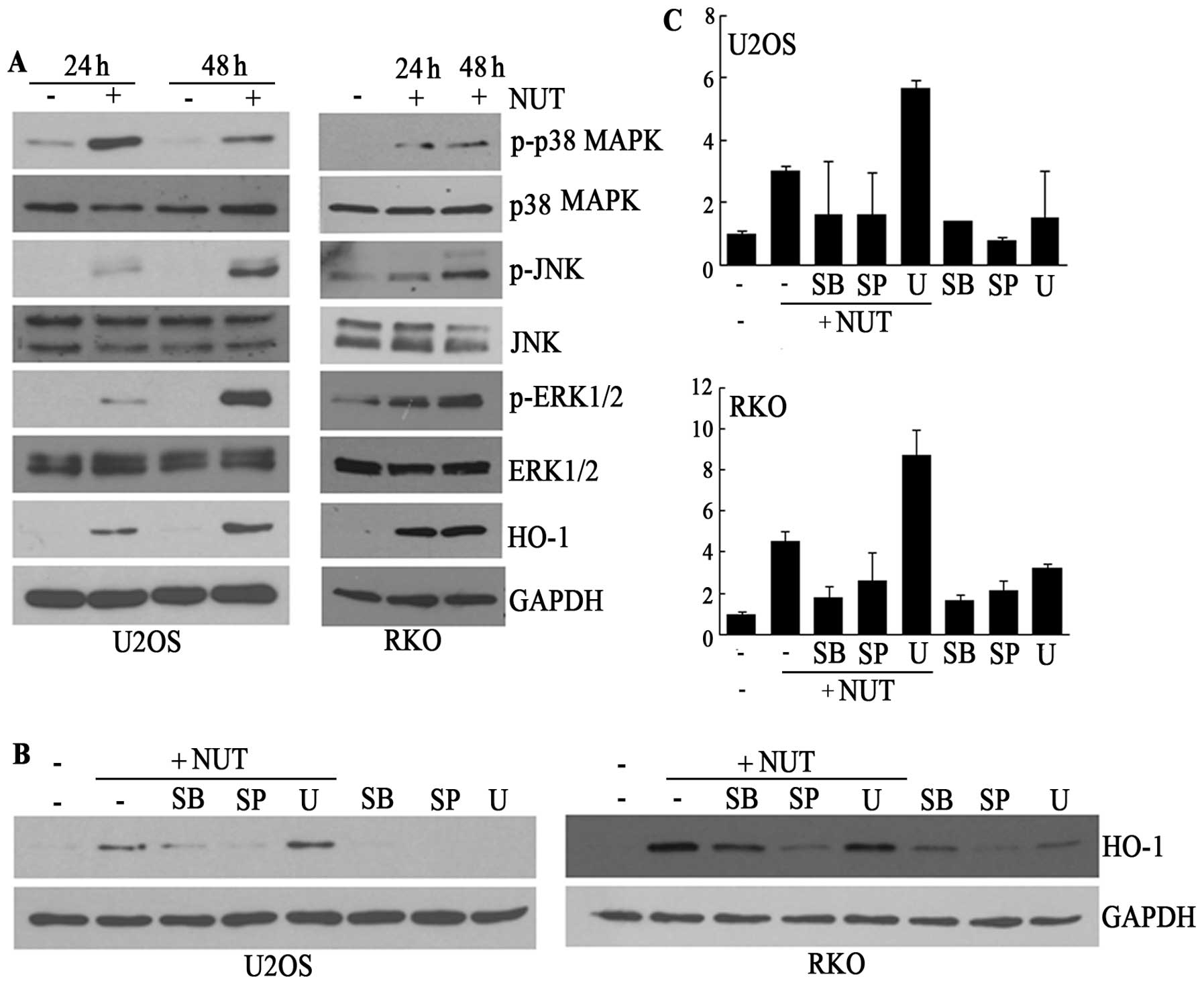
Since MAPK has been shown to be a critical mediator
of HO-1 induction in many models and moreover, nutlin-3 can
activate ERK1/2, independent of the transcriptional activity of p53
(17), we speculated that
nutlin-3-activated MAPKs, including ERK1/2, may mediate the
induction of HO-1 transcription. Based on this speculation, we
analyzed the activation of MAPK by nutlin-3. As shown in Fig. 3A, nutlin-3 clearly induced the
phosphorylation of p38 MAPK, JNK, and ERK1/2 accompanied by the
induction of HO-1 and p53. The phosphorylation of MAPK, except for
p38 MAPK, was dependent on incubation time up to 48 h, which was
also the case for HO-1 and p53, implying that JNK and ERK1/2 might
be mediators of HO-1 transcription. However, contrary to this
expectation, the inhibition of JNK and p38 MAPK, but not ERK1/2, by
chemical inhibitors prior to the nutlin-3 treatment suppressed the
elevation of both HO-1 protein and mRNA (Fig. 3B and C). These findings, therefore,
suggest that the p53 protein levels that were elevated as the
result of the nutlin-3 treatment induced the transcription of HO-1
via the activation of JNK and p38 MAPK.
Nutlin-3-induced HO-1 and JNK activation
but not p38 MAPK activation is dependent on ROS generation
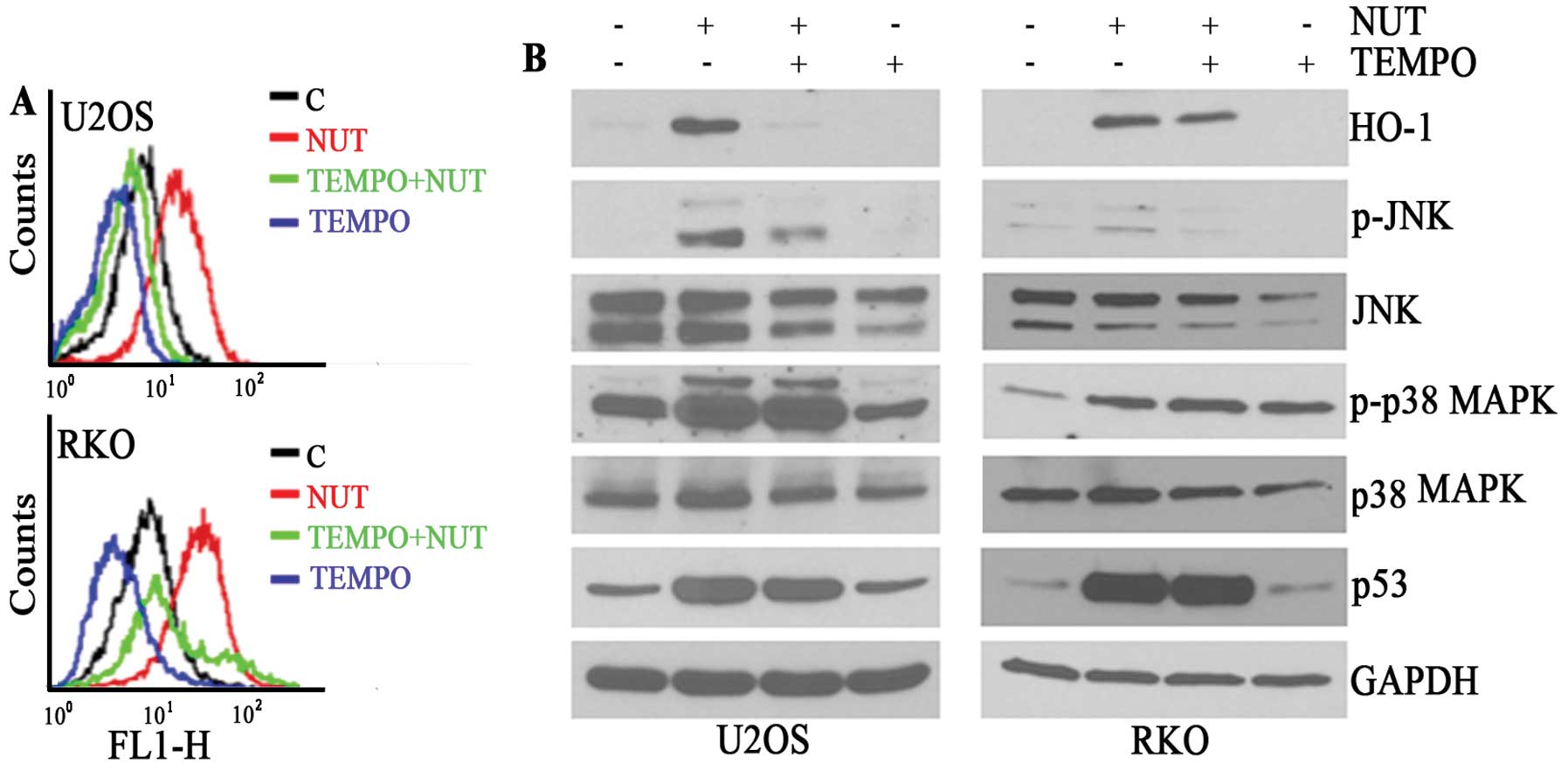
In a previous report, we showed that
nutlin-3-induced ERK1/2 activation was due to ROS, which prompted
us to analyze the effect of ROS on HO-1 induction. As reported
previously, nutlin-3 was found to induce the accumulation of ROS in
both U2OS and RKO cells (Fig. 4A).
In addition, TEMPO, a ROS scavenger, suppressed the induction of
HO-1 protein expression as well as ROS accumulation (Fig. 4B). Under these conditions, the
phosphorylation of JNK but not p38 MAPK was inhibited (Fig. 4B). These findings suggest that the
nutlin-3-upregulated p53 may induce ROS generation, which, in turn,
would activate JNK, which mediates the transcriptional induction of
HO-1, and that the activation of p38 MAPK occur via a different
mechanism than that for JNK activation, irrespective of whether ROS
is present or not.
Nutlin-3-induced HO-1 is dependent on
mitochondrial translocation of p53
The above data showing that the nutlin-3-induced
formation of HO-1 is dependent on p53-induced ROS generation
regardless of the transcriptional activity of p53 led us to examine
the generation of ROS by mitochondrial p53. We and others recently
reported that p53 moves to mitochondria, where it stimulates the
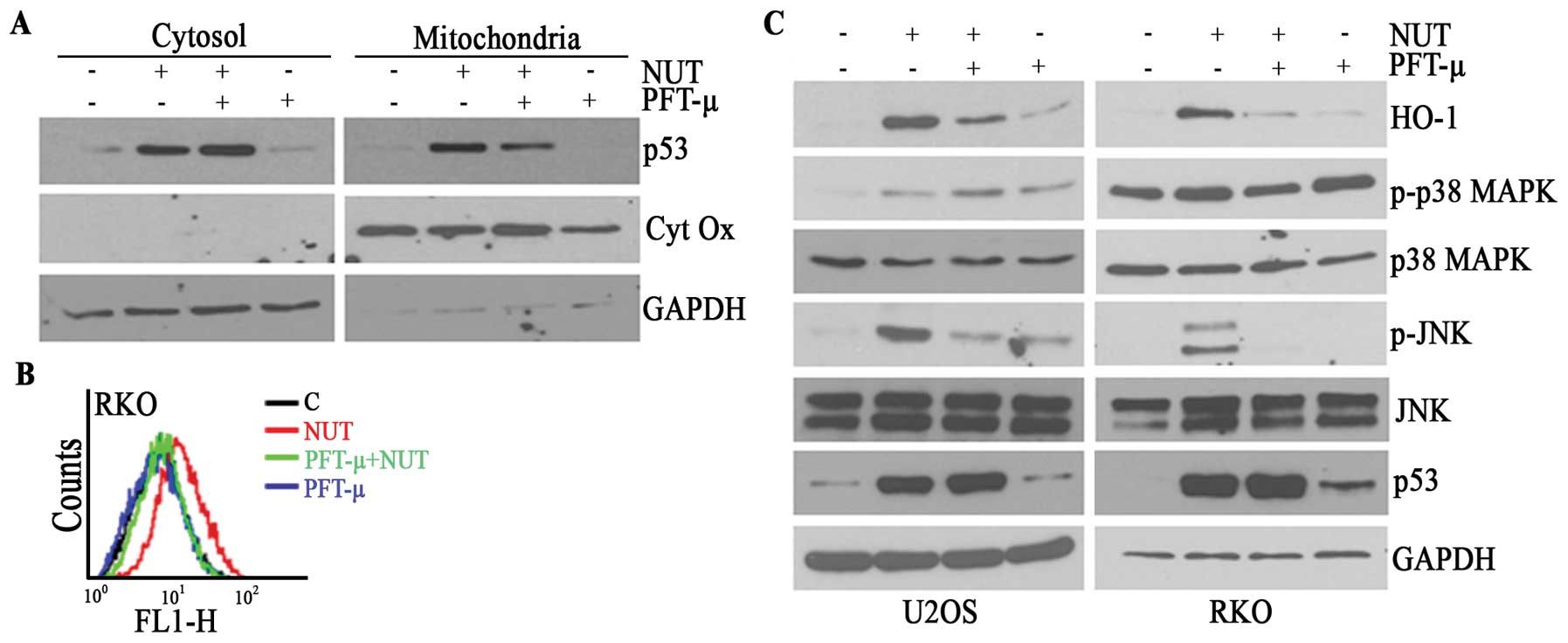
generation of ROS. Also in this model, nutlin-3 induced the
mitochondrial translocation of p53, which was prevented by PFT-μ
pretreatment in U2OS (data not shown) and RKO (Fig. 5A) cells. PFT-μ also prevented the
accumulation of ROS in these cells (Fig. 5B), implying that the mitochondrial
translocation of p53 plays a pivotal role in ROS generation.
Consistent with the suppressive effect of TEMPO on the
phosphorylation of JNK and the resulting HO-1 expression, PFT-μ
reduced the nutlin-3-induced phosphorylation of JNK as well as the
level of HO-1 expression (Fig.
5C). These findings suggest that both nutlin-3-induced HO-1
expression and the phosphorylation of JNK can be attributed to the
ROS generated subsequent to the mitochondrial translocation of
p53.
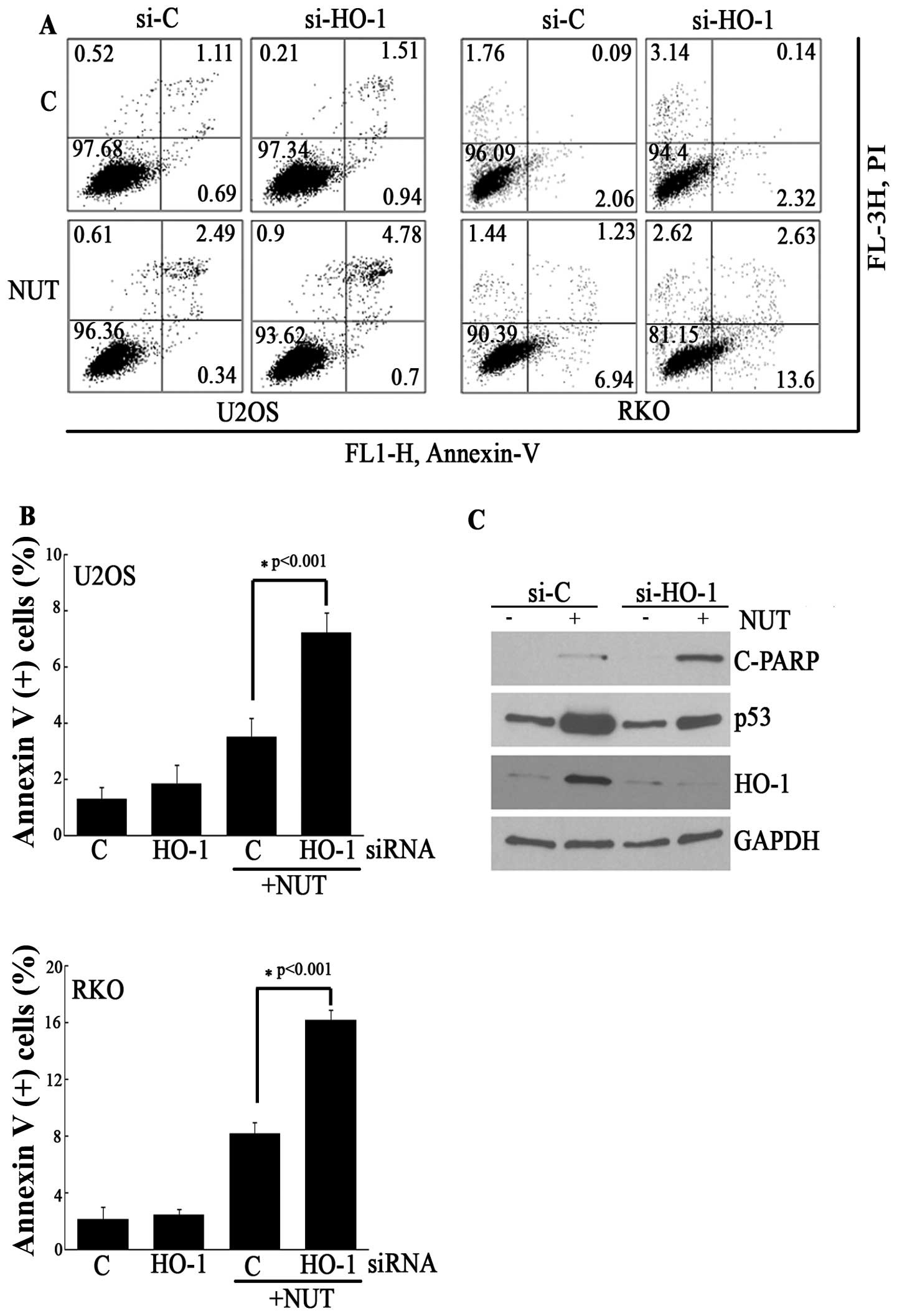
The effect of HO-1 on the
nutlin-3-induced apoptosis
Because HO-1 is an anti-apoptotic protein, we
examined the effect of HO-1 induction on apoptosis in this model.
As expected, the knockdown of HO-1 using siRNA against HO-1
significantly increased Annexin V-positive cells in
nutlin-3-treated U2OS and RKO cells (Fig. 6A and B). Furthermore, HO-1
siRNA-transfected U2OS cells increased the nutlin-3-induced
cleavage of poly (ADP-ribose) polymerase-1 (PARP) to a greater
extent than control siRNA-transfected U2OS cells (Fig. 6C). Based on these findings, it can
be suggested that HO-1 induced by nutlin-3 plays a role in
protecting cancer cells from p53-induced apoptosis.
Discussion
In addition to its cell death-inducing activity, p53
has the potential to increase cell survival as well. The cell
survival effect of p53 is mediated by target genes of p53 such as
EGFR ligands (HB-EGF), and anti-apoptotic transcription factors
(SLUG), thus being dependent on its transcriptional activity
(6–9). Recently, HO-1, an anti-apoptotic gene
was added to the target gene list of p53 (25). Based on this report, it would be
expected that nutlin-3 could induce the expression of HO-1 in a
transcription-dependent manner of p53. As expected, nutlin-3
induced the expression of HO-1 at the transcriptional level in
cancer cells such as U2OS and RKO cells. However, the
transcriptional activity of p53 was not involved and instead, the
activities of JNK and p38 MAPK played critical roles in
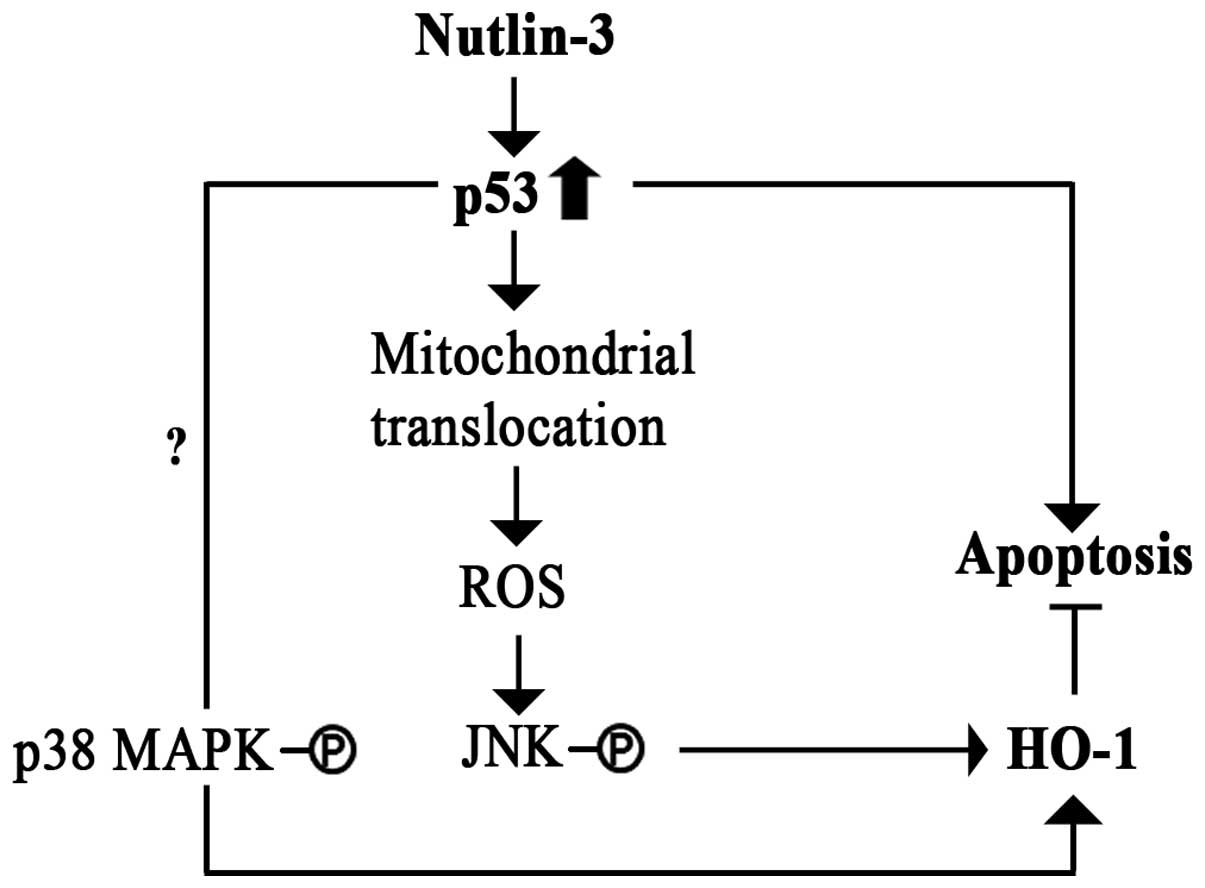
nutlin-3-induced HO-1 expression. As summarized in Fig. 7, the results reported herein
demonstrate that the nutlin-3 treatment induced both p53 protein
levels and the mitochondrial translocation of p53 in cancer cells.
Mitochondrial p53 induces the generation of ROS, which, in turn,
activates JNK, which mediates HO-1 transcription. In the meanwhile,
nutlin-3-induced p38 MAPK activation was not prevented by the
presence of either TEMPO, a ROS scavenger, or PFT-μ, a blocker of
the translocation of p53 to mitochondria, suggesting that ROS and
mitochondrial p53 is not involved in p38 MAPK activation and that
this process is controlled by an alternative mechanism (Figs. 4 and 5). However, since the respective
treatment of TEMPO or the PFT-μ stimulated activation of p38 MAPK,
the involvement of ROS and mitochondrial p53 in p38 MAPK activation
cannot be confirmed, yet.
Mitochondrial p53 has been reported to induce ROS
generation by virtue of its interaction with MnSOD (SOD2), thereby
inhibiting its activity (29). In
this model, however, whereas nutlin-3-induced HO-1 expression was
augmented by MnSOD siRNA, an MnSOD mimetic such as Mn(III)TMPyP
[Mn(III)tetrakis(1-methyl-4-pyridyl) porphyrin pentachloride,
C44H36MnN8•5HCl] failed to prevent
the induction of HO-1 (data not shown). It can therefore be
suggested that the ROS-scavenging effect of MnSOD modulates the
nutlin-3-induced HO-1 but MnSOD itself may not be a critical
regulator of nutlin-3-induced HO-1 expression in these cells. In
support of this, no evidence was found for an interaction between
p53 and MnSOD at the endogenous level (data not shown). The
potentiating effect of MnSOD siRNA on HO-1 induction can be
considered to constitute additional evidence for the contribution
of ROS to the induction of HO-1 expression.
It has been reported that mitochondrial p53 induces
apoptosis in many models, and that this process occurs via a
transcription-independent apoptotic mechanism (30,31).
P53 interacts with the anti-apoptotic proteins, BCL-2 and BCL-xL in
the mitochondria, relieving their inhibitory effects on the
apoptotic protein BAK (30).
Mitochondrial p53 can directly interact with BAK (32). Mitochondrial p53 stimulates BAK
oligomerization via these pathways, leading to mitochondrial
outermembrane permeabilization, a rate-limiting step in intrinsic
apoptosis. The interaction of p53 with Mn-SOD in mitochondria also
activates apoptosis by initiating a ROS-dependent pathway (29). It was recently reported that p53,
after being translocated to mitochondria as the result of oxidative
stress, interacts with cyclophilin D and thus induces mitochondrial
permeability transition, resulting in the necrosis of neuronal
cells (33). Taken together,
mitochondrial p53 can be solely regarded as an inducer of cell
death such as apoptosis and necrosis. To be consistent with the
effects of the genetic expression of p53 and DNA damage-induced
p53, the non-genotoxic activation of p53 by nutlin-3 was also
reported to induce apoptosis by the mitochondrial translocation of
p53 in cancer cells including leukemia and lymphoma cells (34). However, p53, when translocated to
mitochondria in response to γ-irradiation, was not found to induce
apoptosis in various cancer cells (35), and moreover, as reported in our
previous study, the nutlin-3-induced mitochondrial trans location
of p53 induced a cell survival pathway consisting of ROS and ERK1/2
(17). Therefore, it appears that
mitochondrial p53 has the ability to induce different pathways such
as apoptosis, necrosis and cell survival, depending on the type of
toxic stress and cellular context. These different effects may be
due to the interaction of different proteins with p53 in
mitochondria. In this model, although we were not able to identify
the protein that binds to mitochondrial p53 and is responsible for
ROS accumulation, MnSOD, BCL-xL and cyclophilin D were not detected
in the protein complexes that were immunoprecipitated with p53 in
mitochondria (data not shown), implying the presence of
unidentified proteins being involved in the accumulation of ROS to
induce MAPK activation.
The mitochondrial translocation of p53 appears to
occur by nutlin-3 treatment in all cancer cells tested in our
experiments including leukemia, colon cancer and glioma cells.
However, ROS generation was observed in subsets of these cells such
as U2OS, RKO, A172 and U87 cells but not in leukemic cells and
HCT116 colon cancer cells (data not shown), suggesting that various
proteins that interact with p53 responsible for ROS generation are
expressed by different cell types. Although ROS generation by
nutlin-3 differs with according to cell types, the location of
mitochondrial p53 could be a contributing factor. For example,
BCL-xL and cyclophilin D reside in the outer mitochondrial
membranes and the mitochondrial matrix, respectively, implying the
precise location of mitochondrial p53 could explain the dependency
of the different effects of p53 according to cell type, and the
mechanism underlying the different location of p53 could help to
dissect the biological functions of mitochondrial p53.
Nutlin-3 is known to predominantly induce cell cycle
arrest in some solid cancer cell lines including U2OS and RKO
cells, as shown in this study (14). Although the induction of cell cycle
arrest can inhibit the growth of cancer cells, it has the potential
to suppress apoptosis initiated by chemotherapeutic agents and thus
to confer cancer cells with resistance to chemotherapeutic agents
(36). This preference of nutlin-3
for growth arrest has been explained by hnRNPK expression and HIPK2
activation (15,16). In our previous report, we proposed
a mitochondrial p53-ROS-MEK1/2-ERK1/2 activation pathway as being
responsible for the inhibition of nutlin-3-induced apoptosis
(17). In addition to this
pathway, we proposed an alternate pathway involving mitochondrial
p53-ROS-JNK-HO-1 expression, which would inhibit the
nutlin-3-induced apoptosis found in this study. These two pathways
may constitute a negative feedback loop for nutlin-3-induced
apoptosis, implying modulators of these two pathways may be
therapeutic targets capable of enhancing the anticancer effect of
nutlin-3. It can also be speculated that ROS generation during
nutlin-3 treatment may be a critical mechanism for inducing the
cell survival pathway through diverse mechanisms that counteract
nutlin-3-induced apoptosis. Therefore, the mechanism responsible
for ROS generation by mitochondrial p53 needs to be clarified to
increase the apoptosis-inducing activity of nutlin-3 and thus for
the use of nutlin-3 in future anticancer treatments.
Abbreviations:
|
DDR1
|
discoidin domain receptor 1;
|
|
H2DCF-DA
|
2′,7′-dichlorodihydrofluorescein
diacetate;
|
|
ERK
|
extracellular signal-regulated
kinases;
|
|
HB-EGF
|
heparin-binding epidermal growth
factor-like growth factor;
|
|
HO-1
|
heme oxygenase-1;
|
|
JNK
|
c-jun N-terminal kinase;
|
|
MAPK
|
mitogen-activated protein kinase;
|
|
MDM2
|
murine double minute 2;
|
|
MnSOD
|
manganese superoxide dismutase;
|
|
PFT
|
pifithrin;
|
|
ROS
|
reactive oxygen species;
|
|
TEMPO
|
2,2,6,6-tetramethyl-1-piperidinyloxy
|
Acknowledgements
This research was supported by a grant
(2012R1A5A2047939) and the Basic Science Research Program
(2010-0025420) through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and
Technology.
References
|
1.
|
Menendez D, Inga A and Resnick MA: The
expanding universe of p53 targets. Nat Rev Cancer. 9:724–737. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Riley T, Sontag E, Chen P and Levine A:
Transcriptional control of human p53-regulated genes. Nat Rev Mol
Cell Biol. 9:402–412. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Gartel AL and Tyner AL: The role of the
cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer
Ther. 1:639–649. 2002.PubMed/NCBI
|
|
4.
|
Chan TA, Hermeking H, Lengauer C, Kinzler
KW and Vogelstein B: 14-3-3s is required to prevent mitotic
catastrophe after DNA damage. Nature. 401:616–620. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Cohen HY, Lavu S, Bitterman KJ, et al:
Acetylation of the C terminus of Ku70 by CBP and PCAF controls
Bax-mediated apoptosis. Mol Cell. 13:627–638. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Wu WS, Heinrichs S, Xu D, et al: Slug
antagonizes p53-mediated apoptosis of hematopoietic progenitors by
repressing puma. Cell. 123:641–653. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Janssens S, Tinel A, Lippens S and Tschopp
J: PIDD mediates NF-kappaB activation in response to DNA damage.
Cell. 123:1079–1092. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Fang L, Li G, Liu G, Lee SW and Aaronson
SA: p53 induction of heparin-binding EGF-like growth factor
counteracts p53 growth suppression through activation of MAPK and
PI3K/Akt signaling cascades. EMBO J. 20:1931–1939. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Ongusaha PP, Kim JI, Fang L, et al: p53
induction and activation of DDR1 kinase counteract p53-mediated
apoptosis and influence p53 regulation through a positive feedback
loop. EMBO J. 22:1289–1301. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Wade M, Li YC and Wahl GM: MDM2, MDMX and
p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 13:83–96.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Vassilev LT, Vu BT, Graves B, et al: In
vivo activation of the p53 pathway by small-molecule antagonists of
MDM2. Science. 303:844–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kojima K, Konopleva M, McQueen T, O’Brien
S, Plunkett W and Andreeff M: Mdm2 inhibitor Nutlin-3a induces
p53-mediated apoptosis by transcription-dependent and
transcription-independent mechanisms and may overcome Atm-mediated
resistance to fludarabine in chronic lymphocytic leukemia. Blood.
108:993–1000. 2006. View Article : Google Scholar
|
|
13.
|
Vassilev LT: MDM2 inhibitors for cancer
therapy. Trends Mol Med. 13:23–31. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Tovar C, Rosinski J, Filipovic Z, et al:
Small-molecule MDM2 antagonists reveal aberrant p53 signaling in
cancer: implications for therapy. Proc Natl Acad Sci USA.
103:1888–1893. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Enge M, Bao W, Hedstrom E, Jackson SP,
Moumen A and Selivanova G: MDM2-dependent downregulation of p21 and
hnRNP K provides a switch between apoptosis and growth arrest
induced by pharmacologically activated p53. Cancer Cell.
15:171–183. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Rinaldo C, Prodosmo A, Siepi F, et al:
HIPK2 regulation by MDM2 determines tumor cell response to the
p53-reactivating drugs nutlin-3 and RITA. Cancer Res. 69:6241–6248.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Lee SY, Shin SJ and Kim HS: ERK1/2
activation mediated by the nutlin3-induced mitochondrial
translocation of p53. Int J Oncol. 42:1027–1035. 2013.PubMed/NCBI
|
|
18.
|
Ryter SW, Alam J and Choi AM: Heme
oxygenase-1/carbon monoxide: from basic science to therapeutic
applications. Physiol Rev. 86:583–650. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Yang JD, Nakamura I and Roberts LR: The
tumor microenvironment in hepatocellular carcinoma: current status
and therapeutic targets. Semin Cancer Biol. 21:35–43. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Was H, Dulak J and Jozkowicz A: Heme
oxygenase-1 in tumor biology and therapy. Curr Drug Targets.
11:1551–1570. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Berberat PO, Dambrauskas Z, Gulbinas A, et
al: Inhibition of heme oxygenase-1 increases responsiveness of
pancreatic cancer cells to anticancer treatment. Clin Cancer Res.
11:3790–3798. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Rushworth SA and MacEwan DJ: HO-1
underlies resistance of AML cells to TNF-induced apoptosis. Blood.
111:3793–3801. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Guan J, Wu X, Arons E and Christou H: The
p38 mitogen-activated protein kinase pathway is involved in the
regulation of heme oxygenase-1 by acidic extracellular pH in aortic
smooth muscle cells. J Cell Biochem. 105:1298–1306. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Alam J and Cook JL: How many transcription
factors does it take to turn on the heme oxygenase-1 gene? Am J
Respir Cell Mol Biol. 36:166–174. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Nam SY and Sabapathy K: p53 promotes
cellular survival in a context-dependent manner by directly
inducing the expression of haeme-oxygenase-1. Oncogene.
30:4476–4486. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Jang JY, Kim MK, Jeon YK, Joung YK, Park
KD and Kim CW: Adenovirus adenine nucleotide translocator-2 shRNA
effectively induces apoptosis and enhances chemosensitivity by the
down-regulation of ABCG2 in breast cancer stem-like cells. Exp Mol
Med. 44:251–259. 2012. View Article : Google Scholar
|
|
28.
|
Lau LM, Nugent JK, Zhao X and Irwin MS:
HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73
function. Oncogene. 27:997–1003. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Zhao Y, Chaiswing L, Velez JM, et al: p53
translocation to mitochondria precedes its nuclear translocation
and targets mitochondrial oxidative defense protein-manganese
superoxide dismutase. Cancer Res. 65:3745–3750. 2005. View Article : Google Scholar
|
|
30.
|
Mihara M, Erster S, Zaika A, et al: p53
has a direct apoptogenic role at the mitochondria. Mol Cell.
11:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Palacios G, Crawford HC, Vaseva A and Moll
UM: Mitochondrially targeted wild-type p53 induces apoptosis in a
solid human tumor xenograft model. Cell Cycle. 7:2584–2590. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Leu JI, Dumont P, Hafey M, Murphy ME and
George DL: Mitochondrial p53 activates Bak and causes disruption of
a Bak-Mcl1 complex. Nat Cell Biol. 6:443–450. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Vaseva AV, Marchenko ND, Ji K, Tsirka SE,
Holzmann S and Moll UM: p53 opens the mitochondrial permeability
transition pore to trigger necrosis. Cell. 149:1536–1548. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Vaseva AV, Marchenko ND and Moll UM: The
transcription-independent mitochondrial p53 program is a major
contributor to nutlin-induced apoptosis in tumor cells. Cell Cycle.
8:1711–1719. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Essmann F, Pohlmann S, Gillissen B, Daniel
PT, Schulze-Osthoff K and Janicke RU: Irradiation-induced
trans-location of p53 to mitochondria in the absence of apoptosis.
J Biol Chem. 280:37169–37177. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Moreno CS, Matyunina L, Dickerson EB, et
al: Evidence that p53-mediated cell-cycle-arrest inhibits
chemotherapeutic treatment of ovarian carcinomas. PLoS One.
2:e4412007. View Article : Google Scholar : PubMed/NCBI
|