Introduction
Pancreatic cancer is one of the deadliest human
cancer types with an estimated 45,220 new cases and 38,460 deaths
in 2013 in US (1). Although
pancreatic cancer accounts for ∼7% of all cancer deaths and ranks
4th as a cause of cancer death, the incidence and mortality rates
increased for the overall US during 2000–2009 (1). In addition, the five-year survival
rate is estimated as <5–6% (2).
Around 90% of all pancreatic cancers are adenocarcinomas that
originate in the epithelial cells of the pancreatic duct (3,4).
Since the early stage pancreatic cancer usually has no detectable
symptoms, only ∼15 to 20% of pancreatic cancer cases are diagnosed
early enough to be eligible for surgery that provides the only
chance of cure for pancreatic cancer patients (1). Gemcitabine, the recommended
first-line chemotherapeutics, can be given alone or in combination
with other drugs (1,5–7);
however chemotherapy of pancreatic cancers is limited by innate or
acquired resistance (8).
The nuclear factor erythroid 2-related factor 2
(NRF2) is a master regulator of genes involved in oxidative stress
response, detoxification and drug resistance (9–12).
As a member of basic region leucine zipper transcription factors,
NRF2 binds to a DNA sequence called antioxidant response element
(ARE) and induces transcription of target genes (9–12).
Under normal reducing conditions, NRF2 is repressed by Kelch-like
erythroid cell-derived protein with CNC homology-associated protein
1 (KEAP1)-dependent ubiquitination-proteasomal degradation
(9–12). In addition, NRF2 is also
downregulated by CR6-interacting factor 1 (CRIF1) under both
reducing and oxidative stress conditions (13). While NRF2 decreases tumor
susceptibility in most carcinogenesis models, constitutive
activation of NRF2 may enhance tumor cell proliferation and/or
protection against chemotherapy (10–12,14).
In fact, NRF2 is upregulated in many types of tumors through
somatic mutations that disrupt NRF2-KEAP1 regulation (10–12,15).
More recently, it has been reported that several oncogenes,
including K-Ras, B-Raf and Myc, increased the transcription of Nrf2
gene in primary murine cells to activate antioxidant and
detoxification programs preferable for oncogenesis (16). Genetic targeting of
K-RasG12D-driven Nrf2 impaired in vivo
tumorigenesis (16). Silencing
NRF2 by RNA interference also inhibited tumor growth and increased
efficacy of chemotherapy (17) or
EGF-driven proliferation (18) in
non-small cell lung cancer models and reduced the proliferation and
drug-resistance in human lung cancer cells (19) or human pancreatic cancer cells
(20,21). Taken together, NRF2 pathway is a
plausible therapeutic target for cancer therapy.
In this study, we identified PIK-75 as an agent to
down-regulate NRF2 protein level and demonstrated its application
in combination with gemcitabine to further reduce in vivo
tumor growth of human pancreatic cancer.
Materials and methods
Cell culture and reagents
MIA PaCa-2 cells were purchased from American Type
Culture Collection (Manassas, VA, USA) and AsPC-1 cells were
obtained from Tissue Culture Shared Resource of Georgetown
University Medical School. MIA PaCa-2 cells were maintained in
Dulbecco’s modified Eagle’s medium (DMEM) containing 10%
heat-inactivated fetal bovine serum (HI-FBS; HyClone, Logan, UT,
USA), 2.5% horse serum (HS) and 100 U/ml penicillin/streptomycin.
AsPC-1 cells were cultured in RPMI-1640 media supplemented with 20%
HI-FBS, 100 U/ml penicillin/streptomycin and 1 mM sodium pyruvate.
Cell culture reagents were purchased from BioWhittaker
(Walkersville, MD, USA), Lonza (Basel, Switzerland), Invitrogen
(Carlsbad, CA, USA) or Cellgro (Manassas, VA, USA). Viable cells
were monitored by the Luna Automated Cell Counter (Logos
Biosystems, Gyunggi-do, Korea). Small molecule compounds were
purchased from the following sources: PIK-75, PI-103, brivanib,
TAE-684, XL-880, enzastaurin, GDC-0879, deforolimus and TGX221 from
Selleck Chemicals (Houston, TX, USA); BMS-754807 from MedKoo
(Chapel Hill, NC, USA); dasatinib, everolimus and ZSTK474 from LC
Labs (Woburn, MA, USA); and tertbutylhydroquinone (tBHQ) and MG132
from Sigma (St. Louis, MO, USA). Compounds were dissolved in
dimethyl sulfoxide (DMSO) and stored at −20°C in small aliquots.
Gemcitabine was obtained from Sigma and dissolved in
phosphate-buffered saline (PBS).
Cell proliferation assay
Cells in 6-well plates were transfected with 100 nM
of either control- or NRF2-siRNA (20) by Lipofectamine 2000 reagent
(Invitrogen). Four hours after transfection, equal volume of fresh
media were added to each well. The cells were trypsinized and the
number of viable cells was counted by trypan blue dye exclusion
assay every day. After counting, the cell lysates from harvested
cells were subjected to western blot analysis.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
A total of 2,000 human pancreatic cancer cells (MIA
PaCa-2 or AsPC-1) per well were plated in 96-well flat-bottom
plates and then treated with either gemcitabine, PIK-75 alone or in
combination of both drugs with indicated concentrations. At the
indicated times, 20 μl of 1 mg/ml MTT (Sigma) in PBS was
added to each well and further incubated for ∼4 h. After
centrifugation and removal of the medium, 150 μl of DMSO
(Sigma) was added to each well to dissolve the formazan crystals.
The absorbance was measured at 562 nm using an ELx808 absorbance
microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
Absorbance of untreated cells was designated as 100%, and the
relative viable cells were expressed as a percentage of this value.
The drug interaction was evaluated by using the combination index
(CI) according to the method of Chou and Talalay (22) using CompuSyn software (ComboSyn,
Inc., Paramus, NJ, USA).
Transfection and reporter gene assay
Cell culture, seeding and DNA plasmid transfection
were performed as previously reported (23,24).
The luciferase activity was measured according to manufacturer’s
instruction (Promega, Madison, WI, USA) using Victor2 plate reader
(Perkin-Elmer, Waltham, MA, USA) at the Genomics and Epigenomics
Shared Resource of Georgetown University Medical Center and
normalized to β-galactosidase activities.
Clonogenic assay
MIA PaCa-2 or AsPC-1 cells (2×105 cells)
were seeded in 60-mm dishes. Twenty-four hours after plating,
various concentrations of PIK-75 were added to each dish. After
treatment for 24 h, cells were trypsinized and re-seeded in 60-mm
dishes at a density of 500 cells per dish in triplicate. The cells
were further incubated for 14 days and stained with 0.5% crystal
violet in PBS containing 25% methanol. Colonies were examined under
a light microscope and counted after capturing images by scanner.
Relative colony numbers were calculated as a percentage of the
untreated cells (25).
Western blot analysis
MIA PaCa-2 or AsPC-1 cells were grown to ∼70%
confluency and treated with drugs as indicated. Cells were lysed by
lysis buffer containing 20 mM Tris-HCl, 0.5 M NaCl, 0.25% Triton
X-100, 1 mM EDTA, 1 mM EGTA, 10 mM β-glycerophosphate, 10 mM NaF,
300 μM Na3VO4, 1 mM benzamidine, 2
μM PMSF and 1 mM DTT. The protein concentration was
determined by the BCA protein assay (Thermo Scientific, Rockford,
IL, USA). Proteins were separated on SDS-PAGE, transferred on to
PVDF membrane, blocked in 1X blocking buffer (Sigma) and probed
with the following antibodies: phospho-AKT (S473), phospho-GSK3β
(S9), AKT, GSK3β, X-linked Inhibitor of Apoptosis Protein (XIAP),
and Poly-ADP-Ribose-Polymerase (PARP) from Cell Signaling
Technology (Boston, MA, USA); NRF2, multi-drug resistance
associated protein 5 (MRP5), and survivin from Santa Cruz
Biotechnology (Santa Cruz, CA, USA); Glutamate-Cysteine Ligase
Catalytic subunit (GCLC) from Novus Biologicals (Littleton, CO,
USA); Heme Oxygenase-1 (HO-1) from Stressgen Biotechnologies
(Victoria, BC, Canada); NAD(P)H:quinone oxidoreductase 1 (NQO1) and
GFP from Abcam (Cambridge, MA, USA); and FLAG (M2) and β-actin from
Sigma. Then, the membranes were incubated with horseradish
peroxidase (HRP)-conjugated secondary antibodies (Sigma), incubated
with a chemiluminescence reagent (Santa Cruz Biotechnology)
according to the manufacturer’s recommendation, and exposed to
X-ray film (American X-ray and Medical Supply, Jackson, CA,
USA).
DNA binding assay
AsPC-1 cells treated with various concentrations of
PIK-75 for 24 h were used to prepare nuclear extracts. The DNA
binding activity was determined by TransAM NRF2 assay kit (Active
Motif, Carlsbad, CA, USA) according to the manufacturer’s
protocol.
Caspase-3/7 activity assay
MIA PaCa-2 or AsPC-1 cells were treated as indicated
and caspase-3/7 activity was measured from cell lysates with
Caspase-3/7 Glo Assay kit (Promega) according to the manufacturer’s
protocol. Luminescence was measured using Victor X or
Victor2 multilable plate readers (Perkin-Elmer Life
Sciences, Boston, MA, USA). Relative luminescence units were
determined by calculating luminescence values from samples as a
percentage of values from vehicle-treated control samples. The
experiments were performed in triplicate and repeated on two
separately initiated cultures.
Reverse transcriptase (RT)-PCR and
quantitative real-time PCR (qRT-PCR)
RT-PCR and qRT-PCR were performed as described
previously (23) with an Applied
Biosystems-Prism Sequence Detector System 7900HT at the Genomics
and Epigenomics Shared Resource of Georgetown University Medical
Center. The following primer were used: NRF2 forward, 5′-aaa cca
ccc tga aac gac ag-3′ and reverse, 5′-agc ggc ttg aat gtt tgt c-3′;
GCLC forward, 5′-ctg ggg agt gat ttc tgc at-3′ and reverse, 5′-agg
agg ggg ctt aaa tct ca-3′; HO-1 forward, 5′-agg tca tcc cct aca cac
ca-3′ and reverse, 5′- tgt tgg gga agg tga aga ag-3′; MRP5 forward,
5′-acc cgt tgt tgc cat ctt ag-3′ and reverse, 5′-tct gtc aac agc
cac tga gg-3′; β-actin forward, 5′-gct atc cct gta cgc ctc tg-3′
and reverse, 5′-ata tct gct gga agg tgg ac-3′; GAPDH forward,
5′-gta tga caa cga att tgg cta cag -3′ and reverse, 5′-agc aca ggg
tac ttt att gat ggt-3′.
Tumor xenograft study
Animal use procedures were approved by the
Institutional Animal Care and Use Committee of Georgetown
University Medical Center. MIA PaCa-2 cells (∼1.7×106
cells/mouse) mixed with Matrigel (BD Biosciences, San Jose, CA,
USA) were injected subcutaneously into the flank of male athymic
nude (Foxn1nu) mice aged 6-weeks (Harlan Laboratories, Frederick,
MD, USA). Gemcitabine (50 mg/ml) was dissolved in PBS and PIK-75
(20 mg/ml) was dissolved in DMSO. Injection solution was made as
10% of Cremophor® EL (Sigma) and 3% of poly(ethylene
glycol) 400 (Sigma) in sterile water. Before administration of
compounds, gemcitabine was further diluted in PBS and DMSO or
PIK-75 was further diluted in the injection solution and sterilized
by 0.2 μm filter unit. These diluents were mixed with 1:1
ratio and administered into peritoneal cavity of the mouse.
Gemcitabine (20 mg/kg) or gemcitabine (20 mg/kg)/PIK-75 (2 mg/kg)
combination was administered twice per week and vehicle control and
PIK-75 (2 mg/kg) were administered 5 times per week. The body
weights and tumor sizes were measured 3 times per week. Tumor
volumes were calculated as width (mm) × length (mm) × height
(mm)/2.
Statistical analysis
For multiple comparisons, analysis of variance
(ANOVA) using Tukey’s multiple comparison adjustments and
subsequent two-sample t-tests were conducted for comparison. All
statistical tests were two-tailed and employed at a significance
level of 5% to determine whether a significant difference exists in
the assigned experiments. Data were analyzed using SAS version 9.3.
*P≤0.05; **P≤0.01; and
***P≤0.001.
Results
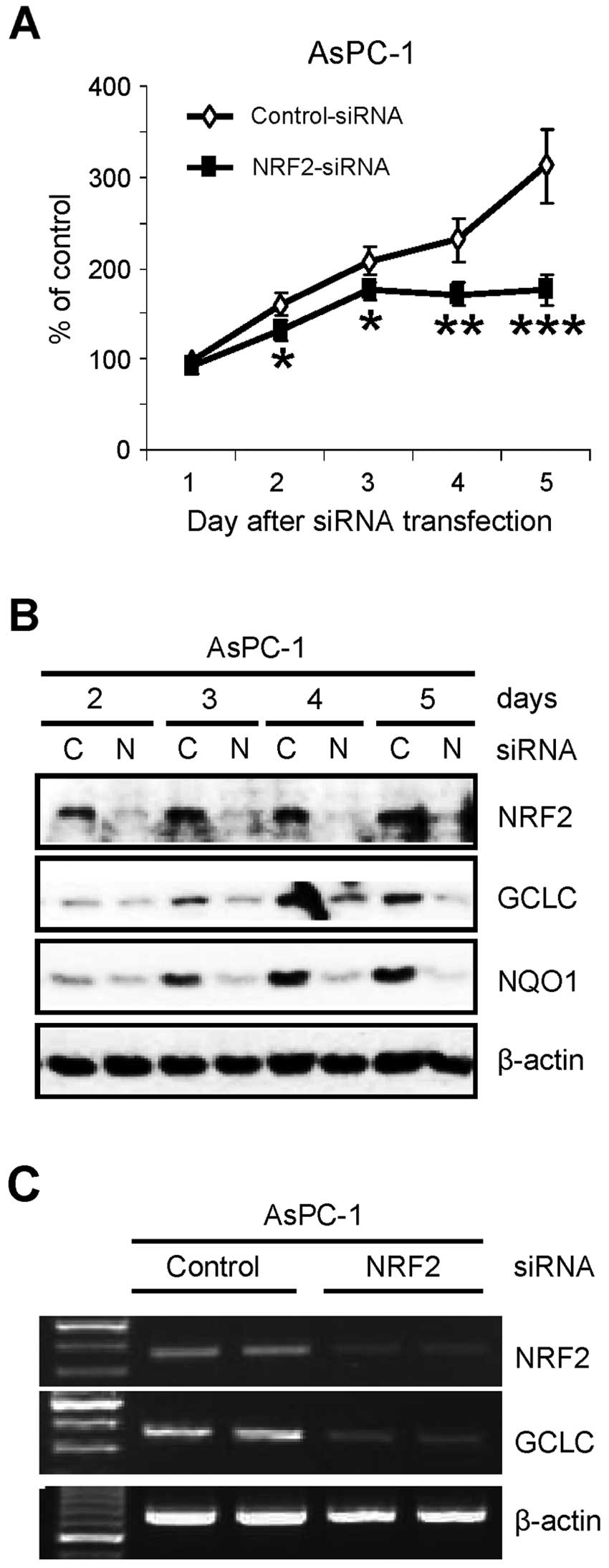
NRF2 is essential for the proliferation
of pancreatic cancer AsPC-1 cells
Previously, we found that NRF2 protein is abnormally
elevated in pancreatic cancer tissues and cell lines (20). To further investigate the role of
NRF2 in pancreatic cancer cells, we determined the proliferation of
pancreatic cancer cells after knockdown of NRF2. After transfection
of siRNA, the number of viable cells was determined by trypan blue
dye exclusion assay. Similar to recent reports in lung cancer cells
(17–19), knockdown of NRF2 (NRF2-KD) by siRNA
reduced the proliferation of AsPC-1 pancreatic cancer cells over
the times tested (Fig. 1A). As
confirmation, the cell lysates from the same experiment were
analyzed by western blot analysis. NRF2-KD was evident on the day
after siRNA transfection and maintained up to 5 days after
transfection (Fig. 1B). As
expected, NRF2-KD reduced the protein level of its downstream genes
such as GCLC and NQO1 (Fig. 1B).
Suppression of NRF2-dependent transcription in NRF2-KD cells was
further analyzed by RT-PCR analysis. The reduction of GCLC protein
(Fig. 1B) was well correlated with
the reduction of its mRNA in NRF2-KD cells (Fig. 1C).
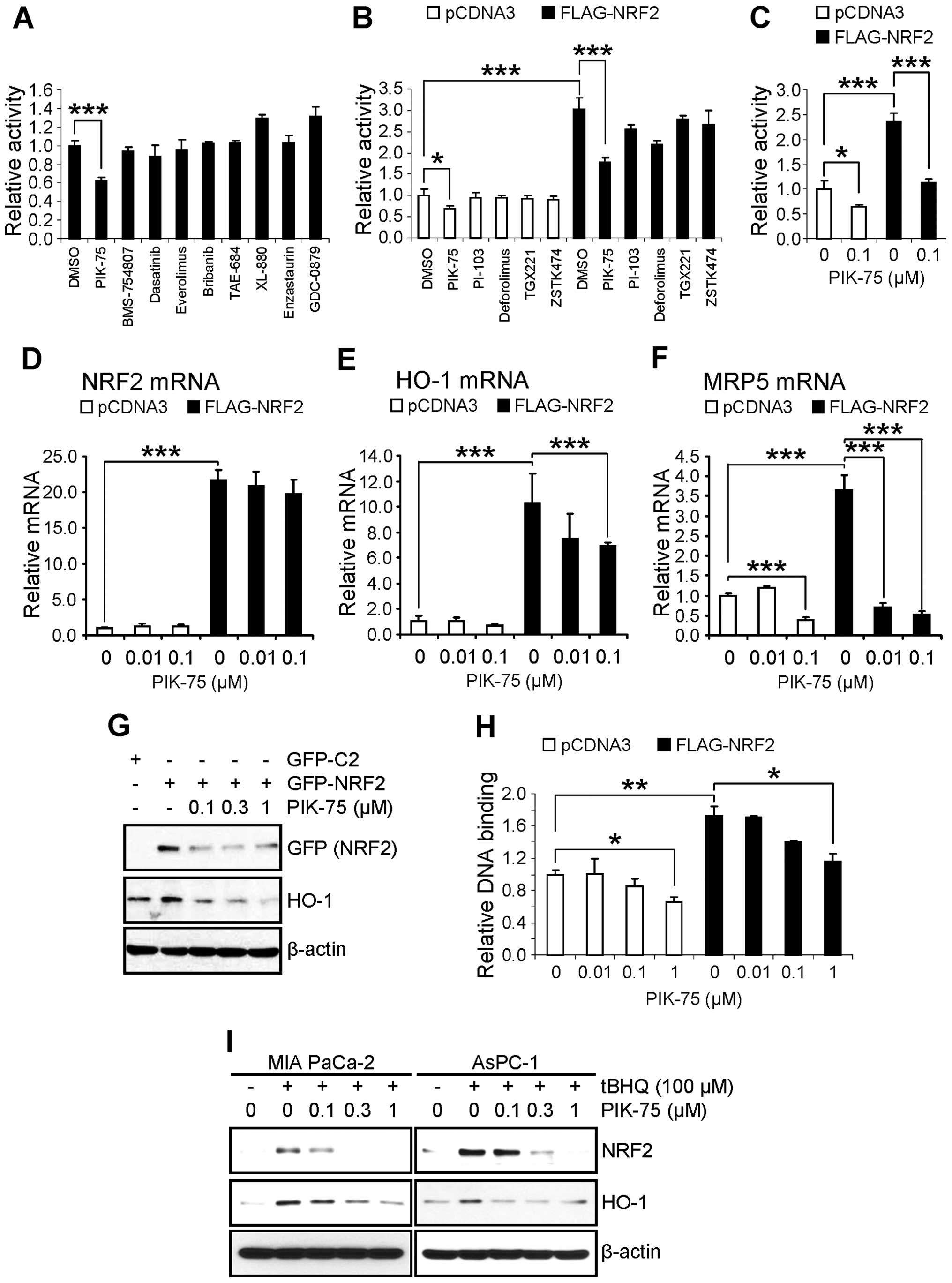
Identification of PIK-75 as an inhibitor
of NRF2-dependent transcription
NRF2 is tightly regulated by proteasomal degradation
that mediated by KEAP1 (9–12) or CRIF1 (13). Since NRF2 can be activated by
post-translational modification such as phosphorylation by various
protein kinases (11), it is
plausible that activation of NRF2 is mediated by oncogenic
activation of upstream signaling pathway in cancer cells. We
postulated that NRF2 could be downregulated by small molecule
kinase inhibitors targeting the proper signaling pathway. To
identify the NRF2 downregulating inhibitor, AsPC-1 cells were
transfected with a luciferase reporter gene construct containing
the ARE from NQO1 gene (ARE-Luc) and treated with various kinase
inhibitors (0.1 μM) for 24 h. Interestingly, PIK-75, known
as a PI3K/DNA-PK inhibitor (26),
significantly reduced the ARE-Luc activity in AsPC-1 cells
(Fig. 2A). We further compared a
series of inhibitors (0.1 μM) targeting PI3K/AKT/mTOR
pathway in the AsPC-1 cells transfected with FLAG-NRF2 and ARE-Luc.
Kinase inhibitors targeting this pathway reduced the ARE-Luc
reporter activity in the presence of FLAG-NRF2 to certain degree,
but PIK-75 was the most potent inhibitor that reduced the ARE-Luc
reporter expression upto ∼50% either in the absence or presence of
exogenous FLAG-NRF2 (Fig. 2B). The
PIK-75-mediated suppression of NRF2 transactivation was further
confirmed in another pancreatic cancer cell, MIA PaCa-2. The
suppression of NRF2-mediated ARE-Luc activation by PIK-75 was as
rapid as 8 h post-treatment in MIA PaCa-2 cells (Fig. 2C).
PIK-75 inhibits NRF2-target gene
expression by reducing NRF2 protein level
The effect of PIK-75 on the mRNA expression of NRF2
and its target gene was assessed by qRT-PCR in the cDNAs from
AsPC-1 cells transfected with FLAG-NRF2 and treated with PIK-75. As
expected, overexpression of FLAG-NRF2 (Fig. 2D) markedly increased the mRNA
expression of NRF2 target gene HO-1 (Fig. 2E) and MRP5 (Fig. 2F). Under these conditions, PIK-75
reduced the NRF2-mediated expression of HO-1 and MRP5 mRNA
(Fig. 2E and F). On the contrary,
the effect of PIK-75 on the NRF2 mRNA was limited (Fig. 2D) and the NRF2 mRNA was slightly
reduced at 1 μM concentration of PIK-75 (data not
shown).
The effect of PIK-75 on the level of NRF2 protein
was also determined by western blot analysis. AsPC-1 cells were
transfected with GFP-NRF2 and then treated with increasing amount
of PIK-75 for 8 h. As results, the level of overexpressed GFP-NRF2
was decreased by PIK-75 in a dose-dependent manner (Fig. 2G). Consistent with mRNA expression,
the level of HO-1 protein was also decreased by PIK-75 treatment
under these conditions (Fig.
2G).
The effect of PIK-75 on the NRF2 activation was
confirmed by NRF2-DNA binding activity. Nuclear extracts from
AsPC-1 cells, transfected with FLAG-NRF2 and treated with PIK-75,
were subject to the ELISA-based DNA binding assay. As shown in
Fig. 2H, PIK-75 reduced the
DNA-binding activity of the overexpressed FLAG-NRF2 as well as
endogenous NRF2.
The effect of PIK-75 on the endogenous NRF2 was
further determined by tBHQ activation model. Two pancreatic cancer
cells, MIA PaCa-2 and AsPC-1 were activated by tBHQ for 1 h
followed by PIK-75 treatment for 8 h. Again, PIK-75 reduced the
levels of tBHQ-activated NRF2 and its downstream targets, HO-1 as
early as 8 h post-treatment in both cells (Fig. 2I). Taken together, PIK-75 represses
NRF2-target gene expression through downregulation of the NRF2
protein.
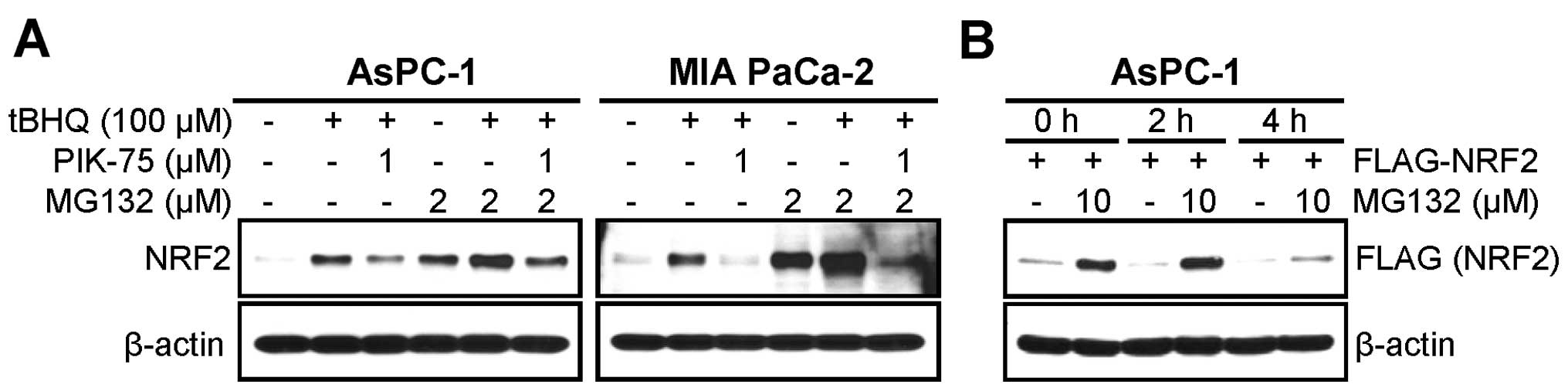
PIK-75 induces proteasomal degradation of
NRF2
NRF2 is actively regulated by proteasomal
degradation. Since PIK-75 reduced both endogenous and exogenous
NRF2 protein, we further tested the PIK-75-mediated NRF2
downregulation in the presence of proteasome inhibitor. AsPC-1
cells were activated by tBHQ for 1 h followed by treatment of
PIK-75 for 4 h in the absence or presence of the proteasome
inhibitor MG132. Western blot analysis showed that treatment of
MG132 alone induced the level of NRF2 protein similarly to that by
tBHQ treatment (Fig. 3A left
panel, lanes 2 vs. 4). This indicates that NRF2 is actively
degraded by proteasome in this cell line. Indeed, co-treatment of
tBHQ and MG132 further increased the NRF protein. Under this
condition MG132 treatment was repressed the PIK-75-mediated
reduction of NRF2 (Fig. 3A left
panel, lanes 3 vs. 6). Inhibition of proteasome by MG132 also
recovered by the PIK-75-mediated reduction of NRF2 in MIA PaCa-2
cells (Fig. 3A right panel).
AsPC-1 cells transfected with FLAG-NRF2 were briefly
treated with PIK-75 in the absence or presence of MG132.
Interestingly, it was evident that overexpressed FLAG-NRF2 was also
actively regulated by proteasome (Fig.
3B, lanes 1 vs. 2). Under this experimental setting, the
PIK-75-mediated downregulation was very rapid, at 2 h
post-treatment. Within this time period, treatment of MG132 almost
completely blocked the NRF2 degradation by PIK-75 (Fig. 3B, lanes 3 vs. 4). Prolonged (4 h)
PIK-75 treatment reduced the effect of MG132 (Fig. 3B, lanes 5 vs. 6). All these results
suggest that PIK-75 rapidly induces NRF2 degradation by
proteasome.
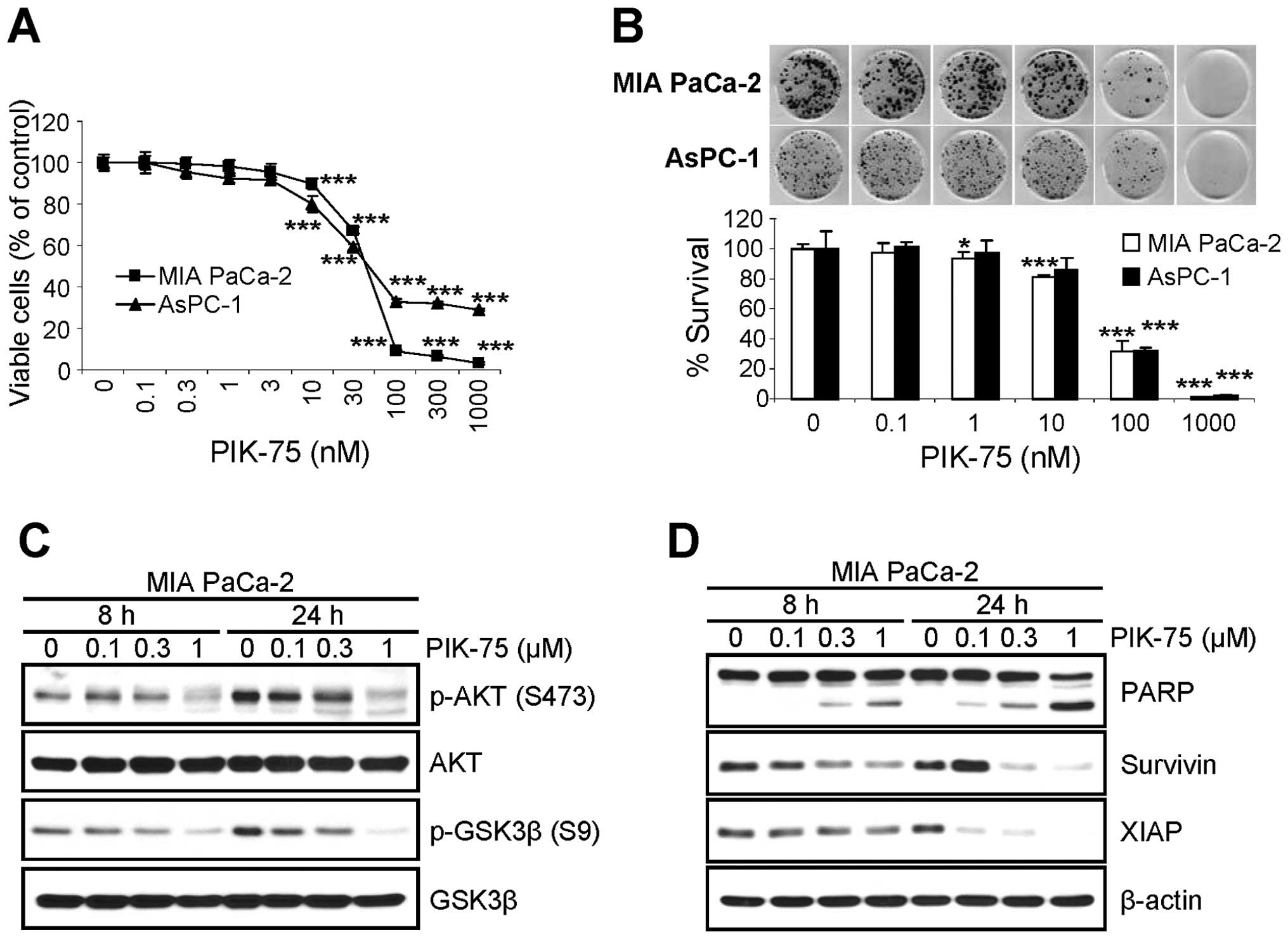
PIK-75 inhibits the proliferation of
pancreatic cancer cells via apoptotic cell death
Since the depletion of NRF2 reduced the
proliferation of pancreatic cancer cells (Fig. 1) and PIK-75 reduced the level of
NRF2 protein in pancreatic cancer cells (Fig. 2), we tested the effect of PIK-75 on
the proliferation of pancreatic cancer cells. Submicromolar
concentration of PIK-75 was sufficient to inhibit the proliferation
of pancreatic cancer, MIA PaCa-2 and AsPC-1 cells after 48-h
treatment (Fig. 4A). The effect of
PIK-75 on the survival of these pancreatic cancer cells were
further evaluated by clonogenic assay. Consistently, PIK-75 also
reduced the colony formation of pancreatic cancer MIA PaCa-2 and
AsPC-1 cells (Fig. 4B).
The effect of PIK-75 on the PI3K/AKT signal
transduction was determined in MIA PaCa-2 cells. The cells were
treated with different concentrations of PIK-75 and western blot
analysis was performed. As expected, PIK-75 reduced the levels of
phospho-AKT (S473) and its substrate phospho-GSK3β (S9) in a
dose-dependent manner within 8 h post-treatment (Fig. 4C).
To determine the markers for apoptotic cell death,
MIA PaCa-2 cells were treated with increasing amount of PIK-75 and
the cell lysates were subjected to western blot analysis. The PARP
cleavage was evident as early as 8 h post-treatment in a
dose-dependent manner (Fig. 4D).
The levels of anti-apoptotic proteins including survivin and XIAP
were also reduced by PIK-75 in a dose- and time-dependent manner
(Fig. 4D).
PIK-75 enhances the cytotoxicity of
gemcitabine through downregulation of MRP5
Since we found that NRF2 confers resistance of
pancreatic cancer cells to various chemotherapeutic agents
(20), we assessed the effect of
NRF2-KD on the cytotoxicity of gemcitabine in pancreatic cancer
cells. AsPC-1 cells were transfected with siRNA (either control or
NRF2), then treated with gemcitabine for 48 h and viable cells were
determined by MTT assay. Similar to other chemotherapeutic agents
(20), the cytotoxic effect of
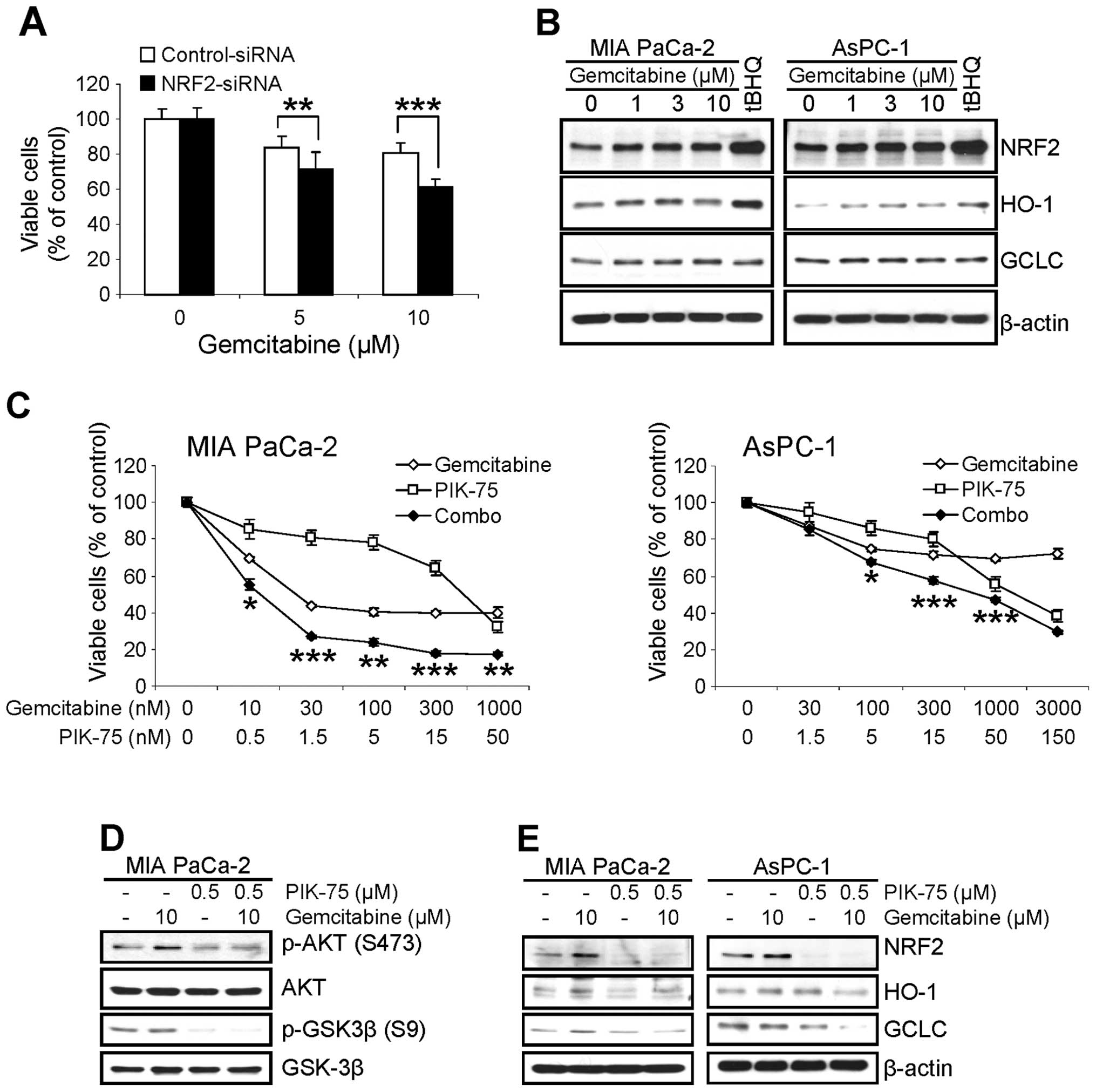
gemcitabine was profound in NRF2-KD cells (Fig. 5A).
Next, we determined the effect of gemcitabine on the
level of NRF2. MIA PaCa-2 and AsPC-1 cells were treated with
increasing amount of gemcitabine for 8 h and the level of NRF2 and
its downstream targets were determined by western blot analysis. As
a control for NRF2 induction, cells were treated with 100 μM
of tBHQ for 8 h. Interestingly treatment of gemcitabine slightly
increased the level of NRF2 in both cell lines within 8 h (Fig. 5B). The level of HO-1 protein was
also slightly increased in both cells. On the contrary, slight
increase of GCLC protein was only observed in MIA PaCa-2 cells
(Fig. 5B).
To determine any beneficial effect by
PIK-75-mediated downregulation of NRF2 in pancreatic cancer cells
to cytotoxicity of gemcitabine, the effect of PIK-75/gemcitabine
combination on the proliferation of pancreatic cancer cells was
performed by MTT cell viability assay. The cells were treated with
either gemcitabine or PIK-75 alone, or in combination of both drugs
for 48 h and the viable cells were determined. As shown in Fig. 5C, co-treatment of PIK-75 profoundly
enhanced the cytotoxic effect of gemcitabine in both cells with
CI50 values of 0.1 for MIA PaCa-2 and 0.41 for AsPC-1,
respectively.
The effect of PIK-75/gemcitabine combination was
further analyzed by western blot analysis. Consistent with previous
report (25), gemcitabine induced
the phospho-AKT (S473) in MIA PaCa-2 cells after 24 h treatment
(Fig. 5D). Under these conditions,
PIK-75 reduced the gemcitabine-induced phospho-AKT (S473) in MIA
PaCa-2 cells (Fig. 5D).
To determine the effect of PIK-75/gemcitabine
combination on the NRF2 pathway, pancreatic cancer cells were
treated with either drug as single agents or combination of both
drugs for 8 h and western blot analysis was performed. Treatment of
PIK-75 reduced the level of NRF2 and its downstream targets, HO-1
and GCLC even in the presence of gemcitabine in both cell types
(Fig. 5E).
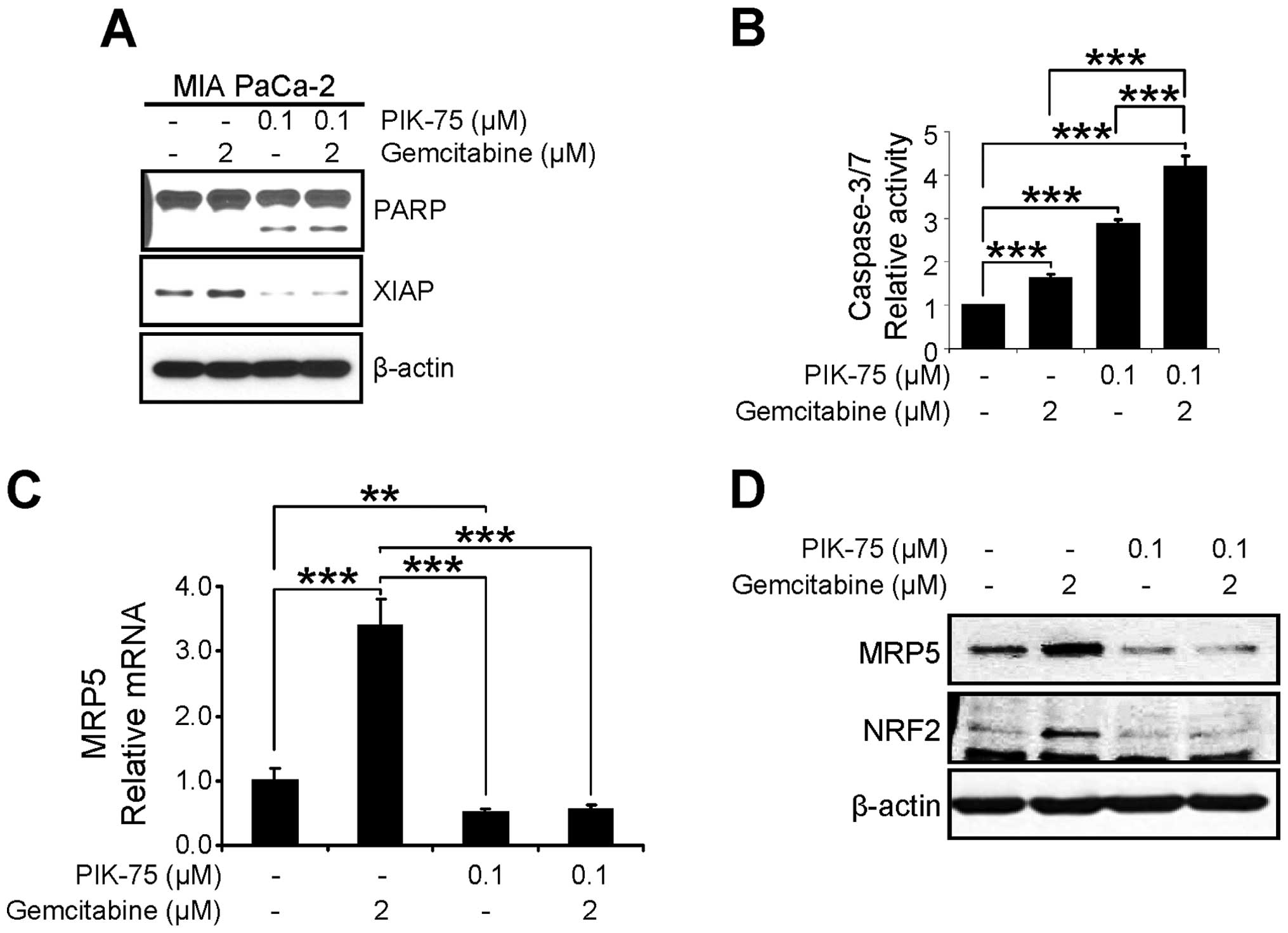
The anti-proliferative effect of PIK-75/gemcitabine
combination was further assessed by western blot analysis of
apoptotic markers. MIA PaCa-2 cells were treated with either drug
or combination of both drugs for 24 h and western blot analysis was
performed. As shown in Fig. 6A,
PIK-75 alone induced the PARP cleavage and combination of both
drugs further induced PARP cleavage. In addition, the level of the
anti-apoptotic protein XIAP, was reduced by PIK-75 treatment. We
further demonstrated the apoptotic cell death by measuring
caspase-3/7 activity. MIA PaCa-2 cells were treated with minimal
amount of gemcitabine (2 μM), PIK-75 (0.1 μM), or
combination of both for 12 h and the caspase-3/7 activity was
determined. Either gemcitabine, or PIK-75 alone induced significant
activity of caspase-3/7 within 12 h (Fig. 6B). Again, PIK-75/gemcitabine
combination further enhanced caspase-3/7 activity in MIA PaCa-2
cells.
The effect of PIK-75/gemcitabine on the expression
of MRP5 was determined by qRT-PCR and western blot analysis. MIA
PaCa-2 cells were treated with either drug as single agents or
combination for 24 h. Under these conditions, gemcitabine markedly
induced both mRNA and protein levels of MRP5 and co-treatment of
PIK-75 reduced the gemcitabine-induced MRP5 expression (Fig. 6C and D).
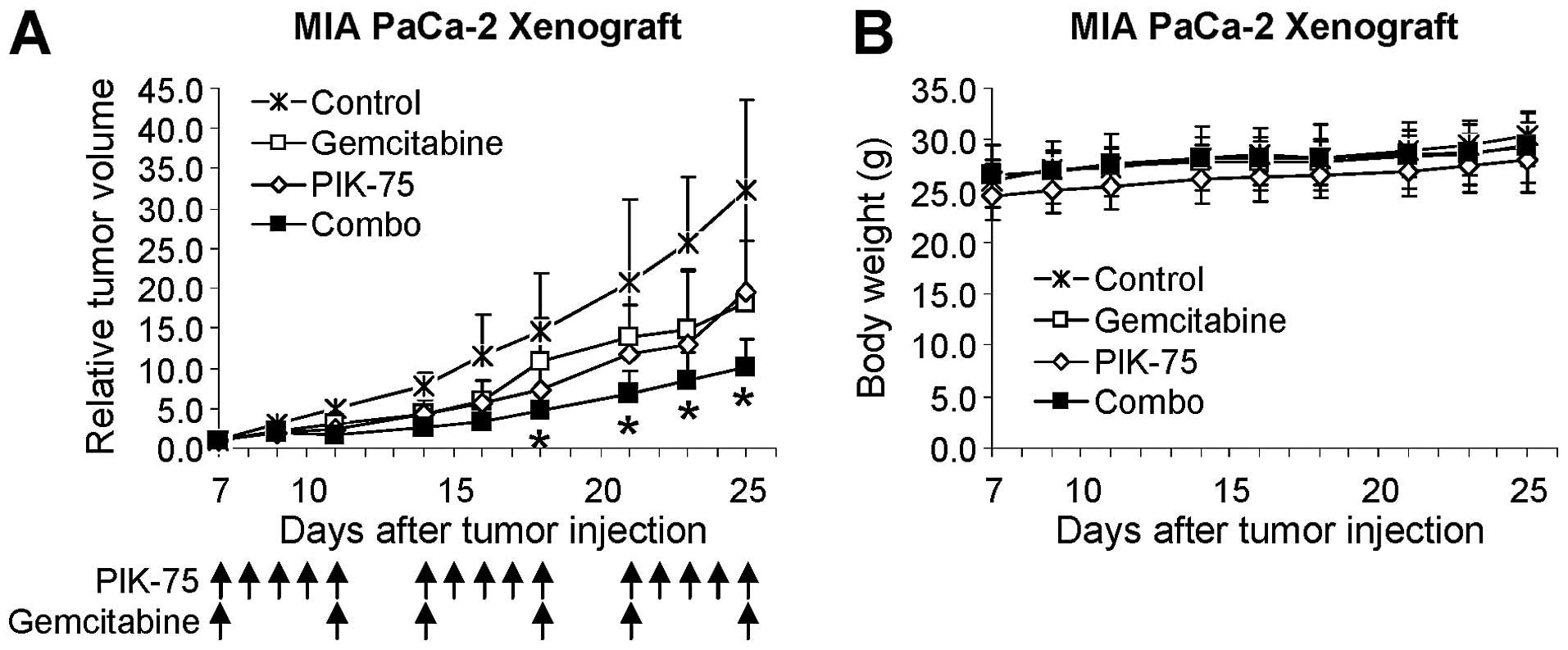
PIK-75 potentiates anticancer activity of
gemcitabine in vivo
The effect of PIK-75/gemcitabine combination was
further demonstrated by in vivo mouse xenograft model. Mice
bearing tumors of MIA PaCa-2 were administered with gemcitabine (20
mg/kg), PIK-75 (2 mg/kg), or combination of both drugs. Since
PIK-75 is a reversible inhibitor, PIK-75 was administered 5 times
per week to ensure maintaining sufficient inhibitory effects.
Gemcitabine was administered twice per week. As shown in Fig. 7A, gemcitabine or PIK-75 reduced the
tumor growth to similar degree. Beneficial effect of
PIK-75/gemcitabine was evident as this combination markedly reduced
the tumor growth in vivo without affecting the body weights
of mice (Fig. 7B).
Discussion
In the present study, we demonstrated that PIK-75 is
a potent inhibitor of NRF2 by inducing its proteasomal degradation
in human pancreatic cancer cells. In addition, PIK-75 potentiated
gemcitabine-induced antitumor effect through downregulation of MRP5
in vitro.
NRF2, by inducing expression of multiple genes that
have roles in oxidative stress, detoxification, drug resistance and
cell survival, functions either as a tumor protector or oncogene
(27–30). In cancer cells, NRF2 induces
resistance of cancer cells to chemotherapeutic drugs by
upregulating transcription of various drug resistant genes such as
anti-apoptotic proteins and drug transporters (12,31,32).
While various activators of the NRF2 pathway have been reported to
increase the level of NRF2 (33),
small molecules that inhibit NRF2 activity are less defined. Since
proteins that negatively regulate the NRF2 level also regulate
various signaling pathways (32),
it is possible that small molecule kinase inhibitors affect the
level of NRF2. As an example, the MEK inhibitor AZD6244 decreased
K-RasG12D-induced expression of Nrf2 gene and its target
genes in primary murine cells (16). As described in the present study,
our initial attempt identified PIK-75 as an inhibitor of NRF2 from
a small set of kinase inhibitors. Since the stability of NRF2 is
regulated by PI3K/AKT/GSK3β pathway, reducing NRF2 by a PI3K/AKT
pathway inhibitor can be predictable as an educated guess, in some
respect. The distinct potency of PIK-75 in downregulation of NRF2,
however, was unpredictable as compared to other PI3K/AKT pathway
inhibitors such as PI-103. Both PIK-75 and PI-103 was reported to
commonly inhibit PI3K p110α, p110δ and DNA-PK with similar degree
(26). Downregulation of NRF2 by 1
μM of PIK-75 was rapid at 8 h post-treatment, whereas other
inhibitors required prolonged incubation time (data not shown).
Further investigation will be needed to address the pathway and
target responsible to this unique feature of PIK-75.
Our present study raises additional questions as to
how gemcitabine induces NRF2 level in pancreatic cancer cells.
Gemcitabine is known to activate various protein kinases such as
ERK (34), AKT (25,34),
EGFR and HER3 (34) in pancreatic
cancer cells, and PKC (35) in
ovarian cancer cells. Interestingly, all these kinases have
implications in the regulation of NRF2 stability and/or induction:
i) the MEK inhibitor AZD6244, that inhibits MEK-ERK pathway
(36), reduced the
K-RasG12D-mediated Nrf2 induction in primary murine
cells (16); ii) AKT
phosphorylates GSK3β (S9) (37,38)
to inhibit its kinase activity that phosphorylates NRF2 and induces
its degradation by β-TrCP-dependent ubiquitination (39). GSK3β is also known to be activated
by phosphorylation of Y216 by unknown upstream tyrosine kinase.
Active GSK3β (Y216) phosphorylates and activates SRC family kinases
that induce nuclear exclusion and ubiquitin-mediated degradation of
NRF2 through its phosphorylation of Y568 (40,41);
iii) activation of EGFR by EGF lead to induction of NRF2 in
non-small cell lung cancer cells (18); and iv) phosphorylation of NRF2
serine 40 by PKCδ is required for stabilization and nuclear
localization of NRF2 (42).
Alternatively, gemcitabine induces reactive oxygen species (ROS) in
pancreatic cancer cells (43,44).
Increased ROS may induce stabilization/activation of NRF2 (9–12).
In the present study, induction of NRF2 by
gemcitabine enhanced the expression of MRP5. It has been reported
that MRP5 is a target gene of NRF2 in mouse liver by micro-array
analysis (31) and knockdown of
NRF2 reduced the MRP5 mRNA in pancreatic cancer cells (20,21).
MRP5 is a member of MRP-related ABCC family (45). MRP5 contains two membrane-spanning
domains and is known to confer resistance to cyclic nucleotides,
acyclic nucleoside phosphates and monophosphorylated nucleoside
analogs (46). Notably, high dose
(20 μM) of gemcitabine has been reported to induce
expression of MRP5 mRNA and MRP5-overexpression contributed to
gemcitabine resistance in HEK293 and PANC-1 cells (47). In addition, silencing MRP5 by shRNA
potentiated the cytotoxicity of gemcitabine in PANC-1 pancreatic
cancer cells (47). MRP5 was also
reported to confer gemcitabine resistance in non-small cell lung
cancer cells (48). Consistently,
we found that inhibiting NRF2 by PIK-75 resulted in the reduction
of MRP5 expression and potentiation of gemcitabine toxicity in
pancreatic cancer cells. Importantly, this synergism showed marked
reduction of in vivo tumor growth in a mouse xenograft
model.
In conclusion, our data suggest that blocking the
NRF2 pathway by small molecule inhibitors is a promising
therapeutic approach to treat pancreatic cancers. While several
studies suggest the potential benefit of genetic silencing of NRF2
by RNA interference to reduce proliferation and/or resistance of
cancer cells to chemotherapeutics, its immediate application is
hampered by inefficient delivery of nucleic acids into cells. In
this aspect, small molecules are preferable for clinical
applications. Notably a recent study on urethane-induced lung
carcinogenesis in Nrf2−/− mouse model has also suggested NRF2
inhibitors as rational tools to prevent malignant progression of
lung cancer (49). In addition,
recently it has been reported that the natural compound
trigonelline inhibiting NRF2 activity with unknown mechanism,
enhanced antitumor effect of etoposide in mouse xenograft models of
pancreatic cancers (50). Further
investigations addressing more detailed mechanisms of PIK-75 in
NRF2 downregulation could increase the specificity and avoid the
potential side-effects of NRF2-targeting drugs.
Acknowledgements
This study was supported by National
Institutes of Health (1R03CA152530), by the Lombardi Comprehensive
Cancer Center, Georgetown University (P30-CA051008) and by the
National Research Foundation of Korea (R31-10069 WCU program).
References
|
1.
|
American Cancer Society: Cancer Facts and
Figures 2013. American Cancer Society; Atlanta, GA: 2013
|
|
2.
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar
|
|
3.
|
Cowgill SM and Muscarella P: The genetics
of pancreatic cancer. Am J Surg. 186:279–286. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Li D and Jiao L: Molecular epidemiology of
pancreatic cancer. Int J Gastrointest Cancer. 33:3–14. 2003.
View Article : Google Scholar
|
|
5.
|
Burris HA III, Moore MJ, Andersen J, et
al: Improvements in survival and clinical benefic with gemcitabine
as first-line therapy for patients with advanced pancreas cancer: a
randomized trial. J Clin Oncol. 15:2403–2413. 1997.PubMed/NCBI
|
|
6.
|
Cunningham D, Chau I, Stocken DD, et al:
Phase III randomized comparison of gemcitabine versus gemcitabine
plus capecitabine in patients with advanced pancreatic cancer. J
Clin Oncol. 27:5513–5518. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Conroy T, Desseigne F, Ychou M, et al:
FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N
Engl J Med. 364:1817–1825. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Matthaios D, Zarogoulidis P, Balgouranidou
I, Chatzaki E and Kakolyris S: Molecular pathogenesis of pancreatic
cancer and clinical perspective. Oncology. 81:259–272. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Bryan HK, Olayanju A, Goldring CE and Park
BK: The Nrf2 cell defence pathway: Keap1-dependent and -independent
mechanisms of regulation. Biochem Pharmacol. 85:705–717. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Mitsuishi Y, Motohashi H and Yamamoto M:
The Keap1-Nrf2 system in cancers: stress response and anabolic
metabolism. Front Oncol. 2:2002012. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Ma Q: Role of Nrf2 in oxidative stress and
toxicity. Annu Rev Pharmacol Toxicol. 53:401–426. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Niture SK, Khatri R and Jaiswal AK:
Regulation of NRF2-an update. Free Radic Biol Med. View Article : Google Scholar : 2013.
|
|
13.
|
Kang HJ, Hong YB, Kim HJ and Bae I:
CR6-interacting factor 1 (CRIF1) regulates NF-E2-related factor 2
(NRF2) protein stability by proteasome-mediated degradation. J Biol
Chem. 285:21258–21268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Martín-Montalvo A, Villalba JM, Navas P
and de Cabo R: NRF2, cancer and calorie restriction. Oncogene.
30:505–520. 2011.PubMed/NCBI
|
|
15.
|
Hayes JD and McMahon M: NRF2 and KEAP1
mutations: permanent activation of an adaptive response in cancer.
Trends Biochem Sci. 34:176–188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
DeNicola GM, Karreth FA, Humpton TJ, et
al: Oncogene-induced Nrf2 transcription promotes ROS detoxification
and tumorigenesis. Nature. 475:106–109. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Singh A, Boldin-Adamsky S, Thimmulappa RK,
et al: RNAi-mediated silencing of nuclear factor erythroid-2
related factor 2 gene expression in non-small cell lung cancer
inhibits tumor growth and increases efficacy of chemotherapy.
Cancer Res. 68:7975–7984. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Yamadori T, Ishii Y, Homma S, et al:
Molecular mechanisms for the regulation of Nrf2-mediated cell
proliferation in non-small-cell lung cancers. Oncogene.
31:4568–4577. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Homma S, Ishii Y, Morishima Y, et al: Nrf2
enhances cell proliferation and resistance to anticancer drugs in
human lung cancer. Clin Cancer Res. 15:3423–3432. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Hong YB, Kang HJ, Kwon SY, et al: Nuclear
factor (erythroid-derived 2)-like 2 regulates drug resistance in
pancreatic cancer cells. Pancreas. 39:463–472. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Lister A, Nedjadi T, Kitteringham NR, et
al: Nrf2 is overexpressed in pancreatic cancer: implications for
cell proliferation and therapy. Mol Cancer. 10:372011. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: the combined effects of
multiple drugs and enzyme inhibitors. Adv Enz Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Kang HJ, Kim HJ, Kim SK, et al: BRCA1
modulates xenobiotic stress-inducible gene expression by
interacting with ARNT in human breast cancer cells. J Biol Chem.
281:14654–14662. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Kang HJ, Kim HJ, Rih JK, et al: BRCA1
plays a role in the hypoxic response by regulating HIF-1alpha
stability and by modulating vascular endothelial growth factor
expression. J Biol Chem. 281:13047–13056. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Duong HQ, Kim HJ, Kang HJ, Seong YS and
Bae I: ZSTK474, a PI3K inhibitor, suppresses proliferation and
sensitizes human pancreatic adenocarcinoma cells to gemcitabine.
Oncol Rep. 27:182–188. 2012.PubMed/NCBI
|
|
26.
|
Knight ZA, Gonzalez B, Feldman ME, et al:
A pharmacological map of the PI3-K family defines a role for p110α
in insulin signaling. Cell. 125:733–747. 2006.PubMed/NCBI
|
|
27.
|
Loboda A, Was H, Jozkowicz A and Dulak J:
Janus face of Nrf2-HO-1 axis in cancer - friend in chemoprevention,
foe in anticancer therapy. Lung Cancer. 60:1–3. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Kensler TW and Wakabayashi N: Nrf2: friend
or foe for chemoprevention? Carcinogenesis. 31:90–99. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Müller T and Hengstermann A: Nrf2: friend
and foe in preventing cigarette smoking-dependent lung disease.
Chem Res Toxicol. 25:1805–1824. 2012.PubMed/NCBI
|
|
30.
|
Sporn MB and Liby KT: NRF2 and cancer: the
good, the bad and the importance of context. Nat Rev Cancer.
12:564–571. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Hayes JD, McMahon M, Chowdhry S and
Dinkova-Kostova AT: Cancer chemoprevention mechanisms mediated
through the Keap1-Nrf2 pathway. Antioxid Redox Signal.
13:1713–1748. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Shelton P and Jaiswal AK: The
transcription factor NF-E2-related factor 2 (Nrf2): a
protooncogene? FASEB J. 27:414–423. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Hur W and Gray NS: Small molecule
modulators of antioxidant response pathway. Curr Opin Chem Biol.
15:162–173. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Yotsumoto F, Fukami T, Yagi H, et al:
Amphiregulin regulates the activation of ERK and Akt through
epidermal growth factor receptor and HER3 signals involved in the
progression of pancreatic cancer. Cancer Sci. 101:2351–2360. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Cartee L and Kucera GL: Gemcitabine
induces programmed cell death and activates protein kinase C in
BG-1 human ovarian cancer cells. Cancer Chemother Pharmacol.
41:403–412. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Huynh H, Soo KC, Chow PK and Tran E:
Targeted inhibition of the extracellular signal-regulated kinase
kinase pathway with AZD6244 (ARRY-142886) in the treatment of
hepatocellular carcinoma. Mol Cancer Ther. 6:138–146. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Cross DA, Alessi DR, Cohen P, Andjelkovich
M and Hemmings BA: Inhibition of glycogen synthase kinase-3 by
insulin mediated by protein kinase B. Nature. 378:785–789. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Srivastava AK and Pandey SK: Potential
mechanism(s) involved in the regulation of glycogen synthesis by
insulin. Mol Cell Biochem. 182:135–141. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Chowdhry S, Zhang Y, McMahon M, Sutherland
C, Cuadrado A and Hayes JD: Nrf2 is controlled by two distinct
β-TrCP recognition motifs in its Neh6 domain, one of which can be
modulated by GSK-3 activity. Oncogene. 32:3765–3781. 2013.
|
|
40.
|
Jain AK and Jaiswal AK: GSK-3β acts
upstream of Fyn kinase in regulation of nuclear export and
degradation of NF-E2 related factor 2. J Biol Chem.
282:16502–16510. 2007.
|
|
41.
|
Niture SK, Jain AK, Shelton PM and Jaiswal
AK: Src subfamily kinases regulate nuclear export and degradation
of transcription factor Nrf2 to switch off Nrf2-mediated
antioxidant activation of cytoprotective gene expression. J Biol
Chem. 286:28821–28832. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Niture SK, Jain AK and Jaiswal AK:
Antioxidant-induced modification of INrf2 cysteine 151 and
PKC-δ-mediated phosphorylation of Nrf2 serine 40 are both required
for stabilization and nuclear translocation of Nrf2 and increased
drug resistance. J Cell Sci. 122:4452–4464. 2009.PubMed/NCBI
|
|
43.
|
Donadelli M, Costanzo C, Beghelli S, et
al: Synergistic inhibition of pancreatic adenocarcinoma cell growth
by trichostatin A and gemcitabine. Biochim Biophys Acta.
1773:1095–1106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Duong HQ, Hong YB, Kim JS, et al:
Inhibition of checkpoint kinase 2 (CHK2) enhances sensitivity of
pancreatic adenocarcinoma cells to gemcitabine. J Cell Mol Med.
17:1261–1270. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Haimeur A, Conseil G, Deeley RG and Cole
SP: The MRP-related and BCRP/ABCG2 multidrug resistance proteins:
biology, substrate specificity and regulation. Curr Drug Metab.
5:21–53. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Dallas S, Schlichter L and Bendayan R:
Multidrug resistance protein (MRP) 4- and MRP 5-mediated efflus of
9-(2-phosphonylmethoxyethyl)adenine by microglia. J Pharmacol Exp
Ther. 309:1221–1229. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Hagmann W, Jesnowski R and Löhr JM:
Interdependence of gemcitabine treatment, transporter expression,
and resistance in human pancreatic carcinoma cells. Neoplasia.
12:740–747. 2010.PubMed/NCBI
|
|
48.
|
Oguri T, Achiwa H, Sato S, et al: The
determinants of sensitivity and acquired resistance to gemcitabine
differ in non-small cell lung cancer: a role of ABCC5 in
gemcitabine sensitivity. Mol Cancer Ther. 5:1800–1806. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Satoh H, Moriguchi T, Takai J, Ebina M and
Yamamoto M: Nrf2 prevents initiation but accelerates progression
through the Kras signaling pathway during lung carcinogenesis.
Cancer Res. 73:4158–4168. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Arlt A, Sebens S, Krebs S, et al:
Inhibition of the Nrf2 transcription factor by the alkaloid
trigonelline renders pancreatic cancer cells more susceptible to
apoptosis through decreased proteasomal gene expression and
proteasome activity. Oncogene. 32:4825–4835. 2013. View Article : Google Scholar
|