Introduction
Epstein-Barr virus (EBV), also called human
herpesvirus 4, is widespread in >95% of the adult population and
persists asymptomatically in all infected individuals for their
entire lives (1). Acute EBV
infection is best known as the cause of infectious mononucleosis
and its latent state is involved in several malignant disorders,
including Burkitt’s lymphoma, Hodgkin’s lymphoma, non-Hodgkin’s
lymphoma, immunoblastic B lymphoma associated with HIV, T cell
lymphoma, gastric carcinoma, nasopharyngeal carcinoma, rheumatoid
arthritis, Sjogren’s syndrome and multiple sclerosis (2)
Sorafenib (SRF; BAY43-9006, Nexavar) is a small
molecule multi-kinase inhibitor that targets Raf kinases as well as
diverse receptor tyrosine kinases including VEGFR1, VEGFR2, PDGFR,
FLT-3 and c-Kit (3,4). SRF was recently approved by the FDA
for the treatment of hepatocellular carcinoma (5) and renal cell carcinoma (6) and is now undergoing phase II/III
clinical trials (7,8). SRF-based combination therapy has also
been examined in several cancer clinical trials and studies
(9,10). Previous studies demonstrated that
SRF has anti-proliferative, anti-angiogenic, and antitumor activity
against various types of cancer and human xenograft models such as
multiple myeloma, chronic myelogenous leukemia, chronic lymphocytic
leukemia, acute myeloid leukemia, breast, renal, ovarian, colon,
pancreatic, melanoma and non-small cell lung cancers (11). The apoptotic effects and molecular
signaling mechanisms of SRF differed among these cancer cell lines.
Generally, SRF inhibited the MEK/ERK pathway and decreased levels
of the anti-apoptotic protein Mcl-1 (12), which is involved in resistance to
anticancer drugs and is over-expressed in diverse leukemia and
lymphoma. Furthermore, SRF-induced inhibition of the ERK pathway
contributed to a decrease in Bcl-XL expression (13). SRF inhibited Hif-1α/VEGF and
downregulated the phosphorylation of mTOR/ERK in hepatocellular
carcinoma (14).
Mitogen-activated protein kinases (MAPKs) comprise
ERK, JNK, and p38-MAPK, and mediate various signaling transduction
pathways (15). The JNK/p38-MAPK
pathway plays a central role in apoptosis, especially oxidative
stress-induced apoptosis, whereas ERK is involved in cell
proliferation, cell migration, cell differentiation and cell
survival. These pathways play crucial roles in chemical-induced
apoptosis. For instance, acanthoic acid leads to apoptosis of
leukemia HL-60 cells by activating p38-MAPK without activating ERK
or JNK (16). Curcumin provokes
tumor cell death via activation of MAPKs (17). Berberine elicits apoptosis of HepG2
cells through p38-MAPK activation (18). However, it is unclear whether
JNK/p38-MAPK signaling in involved in SRF-induced apoptosis.
Although SRF is an inhibitor of Raf/MEK/ERK, very
little is known about whether or not SRF can induce apoptosis by
activating alternative kinase pathways. It has been reported that
SRF was able to elicit apoptotic cell death of human leukemia cells
via a mechanism involving ER stress (19), but detailed molecular mechanisms
were not elucidated. Therefore, we set out to investigate whether
SRF can induce apoptosis through MEK/ERK-independent pathways in
EBV-transformed B cells. We report the novel finding that SRF can
induce apoptosis of EBV-transformed B cells by reactive oxygen
species (ROS) generation, JNK/p38-MAPK activation, and Bax
translocation.
Materials and methods
Preparation of EBV infectious culture
supernatant and generation of EBV-transformed B cells
Cell-free EBV virions were prepared from culture
supernatant of the B95-8 marmoset cell line. To establish EBV
infection of B cells from normal peripheral blood mononuclear cells
(PBMCs), PBMCs were isolated from peripheral blood of a healthy
donor by Ficollpaque (Amersham Life Science, Buckinghamshire, UK)
gradient centrifugation. PBMCs were added to EBV virion stock in a
culture flask, and after a 2-h incubation at 37°C, RPMI-1640
culture medium (Hyclone) and 1 mg/ml of cyclosporine A
(Sigma-Aldrich, St. Louis, MO, USA) were added to cells
(1×106 cells/ml). Cultures were incubated for 2–4 weeks.
This study was approved by the Institutional Bioethics Review Board
of the Medical College of Inje University and all donors provided
informed consent.
Proliferation measurement by
AlamarBlue
Cells (5×104 cells/well) were cultured in
medium containing 10% FBS in 96-well plates. After 24 h, cell
proliferation was measured by AlamarBlue (Serotec Ltd., Kidlington,
Oxford, UK) assay. AlamarBlue was added (10% by volume) to each
well and relative fluorescence was determined 9 h later using a
SpectraMax M2e Multi-Detection Microplate reader (Molecular
Devices, Sunnyvale, CA, USA; excitation, 530 nm; emission, 590 nm).
Relative fluorescence unit (RFU) values are expressed as means ±
SEMs of three determinations.
Quantification of apoptotic cells by flow
cytometry
The level of SRF (BAY43-9006, Nexavar; LC
Laboratories, Woburn, MA, USA)-induced apoptosis in human
EBV-transformed B cells (4 weeks, 5×105 cells/ml) and
normal PBMCs was measured by flow cytometry using FITC-labeled
Annexin-V (BD Biosciences, San Diego, CA, USA) and 7-AAD (BD
Biosciences). In brief, to determine optimal conditions,
experiments were performed using various concentrations of SRF (0,
1, 2.5, 5 and 10 μM) and various incubation periods (2, 4,
8, 16 and 24 h). To inhibit generation of ROS, cells were
pretreated with NAC (N-acetyl-1-cysteine, 10 mM; Sigma-Aldrich) for
1 h. To block activation of JNK or p38-MAPK, cells were pretreated
with SP600125 (25 μM; Calbiochem, San Diego, CA, USA) or
SB203580 (10 μM; Calbiochem) for 1 h. Cells were then
harvested, washed in PBS, and incubated with Annexin-V and 7-AAD in
Annexin-V binding buffer at room temperature for 15 min in the
dark. Stained cells were analyzed using a FACSCalibur flow
cytometer (BD Biosciences) running CellQuestpro software (BD
Biosciences).
Detection of mitochondria membrane
potential (ΔΨm) and intracellular reactive oxygen
species (ROS) generation
We detected changes in mitochondrial membrane
potential (ΔΨm) using DiOC6
(3,3′-dihexyloxacarboxyanine iodide; Molecular Probes, Eugene, OR,
USA). Cells were treated with SRF or DMSO for 24 h, harvested,
washed twice in PBS, re-suspended in PBS supplemented with
DiOC6 (20 nM), incubated at 37°C for 15 min in the dark,
and then analyzed immediately by flow cytometry. Intracellular
accumulation of ROS was examined by flow cytometry after staining
with the fluorescent probe, DCFH-DA
(2′,7′-dichlorodihydro-fluorescein diacetate, 10 μM;
Molecular Probes). DCFH-DA is deacetylated in cells by esterase to
a non-fluorescent compound, DCFH, which remains trapped within the
cell and is cleaved and oxidized by ROS in the presence of
endogenous peroxidases to a highly fluorescent compound, DCF
(2′,7′-dichlorofluorescein). Briefly, EBV-transformed B cells were
seeded in 6-well plates (5×105 cells/ml) and pretreated
with 10 μM DCFH-DA for 30 min at 37°C. Cells were then
washed, re-suspended in RPMI-1640 media, and incubated with SRF or
DMSO.
Western blot analysis
After treatment, cells were harvested and lysed in
NP-40 buffer (Elpis Biotech, Daejeon, Korea) containing a protease
inhibitor cocktail (AEBSF, aprotinin, bestatin hydrochloride, E-64,
EDTA and leupeptin hemisulfate salt; Sigma-Aldrich). To address
phosphorylation events, an additional set of phosphatase inhibitors
(Cocktail II, sodium orthovanadate, sodium molybdate, sodium
tartrate, and imidazole; Sigma-Aldrich) was added to NP-40 buffer
(Elpis Biotech, Daejeon, Korea). Protein concentration was
determined using a BCA assay kit (Pierce, Rockford, IL, USA).
Proteins (10 μg/sample) were then heated for 5 min at 100°C.
Total cell lysates (5×106 cells/sample) were subjected
to SDS-PAGE on 15% (w/v) acrylamide gels under reducing conditions.
Separated proteins were transferred to nitrocellulose membranes
(Millipore Corp., Billerica, MA, USA), and membranes were blocked
with 5% skim milk followed by commercial western blot analysis.
Chemiluminescence was detected using an ECL kit (Advansta Corp.,
Menlo Park, CA, USA) and the multiple Gel DOC system (Fujifilm).
Primary antibodies against the following proteins were used:
caspase-8, caspase-3, caspase-9, PARP, β-actin, Bcl-2, Bax,
phospho-JNK (Thr183/Tyr185), JNK,
phospho-p38-MAPK (Thr180/Tyr182), p38-MAPK,
phospho-ERK1/2 (Thr202/Tyr204), ERK1/2,
phospho-PI3K p85 (Tyr458), PI3K p85, phospho-Akt
(Ser473), Akt (Cell Signaling Technology, Beverly, MA,
USA), COX-IV (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and
β-tubulin (BD Biosciences). Data were analyzed using ImageJ 1.38
software.
Measurement of Bax translocation
Following treatment, mitochondrial and cytosol
cellular fractions were prepared using a Cytosol/Mitochondria
Fractionation kit (Calbiochem). Approximately 1×107
treated or untreated cells were harvested by centrifugation at 600
× g for 5 min at 4°C and washed twice with cold PBS. Afterward,
cells were re-suspended in 250 μl cytosol extraction buffer
containing protease inhibitor cocktail and 1 mM dithiothreitol
(DTT). After incubation on ice for 10 min, cells were homogenized
on ice using a Dounce tissue homogenizer. Homogenized cells were
centrifuged at 700 × g for 10 min at 4°C and supernatants were
collected. Supernatants were then centrifuged again at 10,000 × g
for 30 min at 4°C. The resulting supernatants were harvested and
designated cytosolic fractions while pellets were re-suspended in
50 μl mitochondria extraction buffer containing a protease
inhibitor cocktail and 1 mM DTT and designated mitochondrial
fractions. All fractions were stored at −80°C until use.
Results
SRF selectively induces apoptosis in
EBV-transformed B cells but not in normal PBMCs
To investigate the effects of SRF on the
proliferation of EBV-transformed B cells, cells were treated with
various doses of SRF (1, 2.5, 5, 10, 20 or 50 μM) for 24 h
and then subjected to AlamarBlue assay. In the presence of SRF,
EBV-transformed B cell proliferation decreased in a dose-dependent
manner, suggesting that SRF has potential anticancer activity
(Fig. 1A). SRF suppressed
proliferation by ∼50% at a dose of 10 μM. We performed
experiments to check whether this inhibitory effect of SRF on cell
growth resulted from apoptotic cell death. As shown in Fig. 1B (upper panel), it is clear that
treatment of cells with SRF (0, 1, 2.5, 5, 10, 20 or 50 μM)
for 24 h increased the percentage of cell undergoing apoptosis
(Annexin-V+/7-AAD+; 9.21, 13.95, 32.82,
43.04, and 65.76% respectively) compared with DMSO-treated cells
(9.80%), whereas up to 10 μM SRF had no cytotoxic effects on
normal human PBMCs (Fig. 1D).
Fig. 1C (upper panel) shows cells
that were treated with SRF for various time intervals; the
percentages of Annexin-V and 7-AAD positive cells after incubation
times of 2, 4, 8, 16 or 24 h were 6.75, 6.84, 6.07, 21.44 and
39.90%, respectively. Moreover, SRF disrupted ΔΨm
significantly (Fig. 1B, lower
panel), especially between 16 and 24 h (Fig. 1C, lower panel; from 46.93 to
64.18%). Because the optimal dose and time of ΔΨm
treatment were 10 μM and 24 h, we chose these conditions to
examine protein alterations in SRF-induced apoptosis. Together,
these results indicate that SRF preferentially targets cancerous
EBV-transformed B cells.
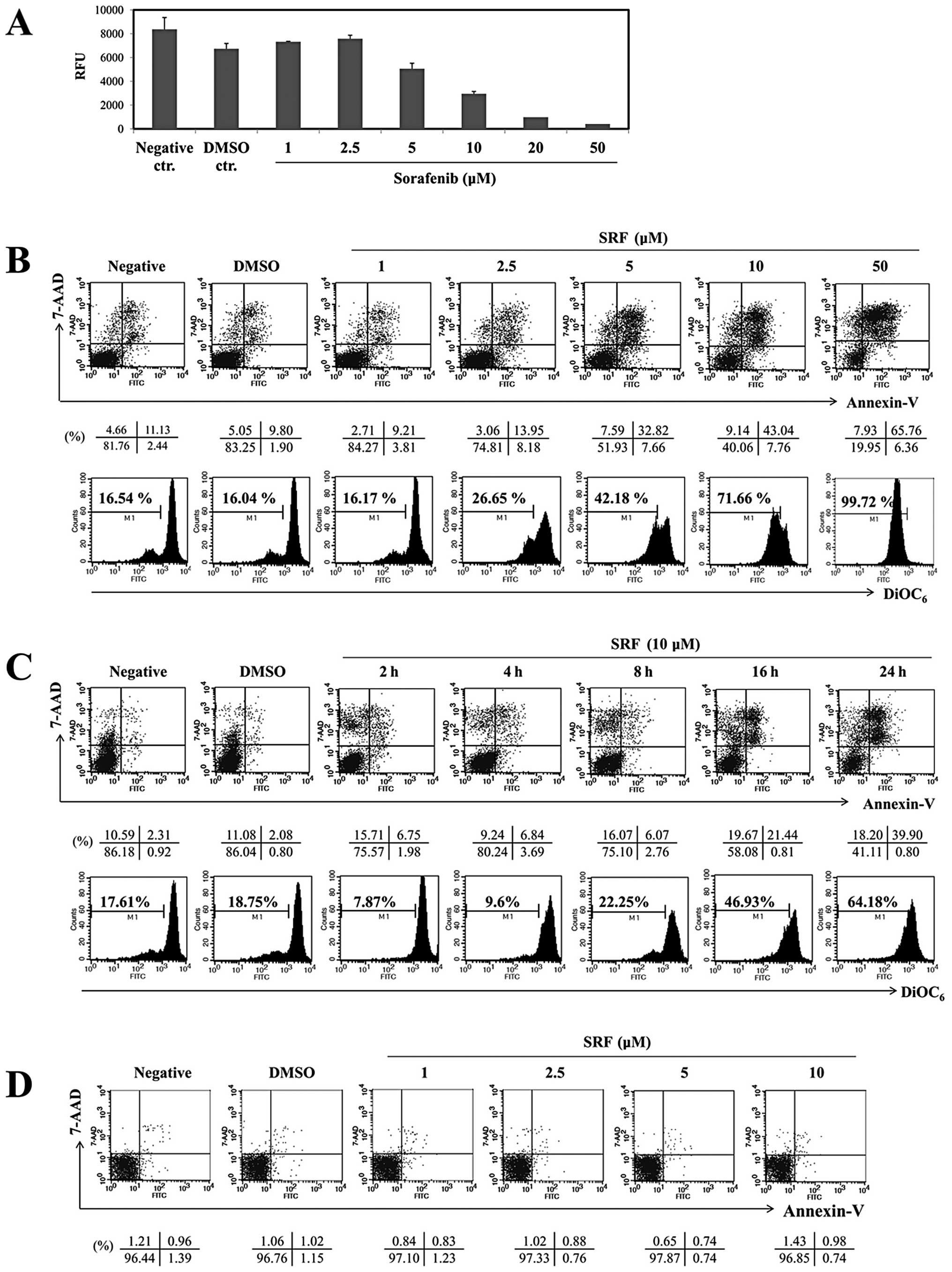 | Figure 1.SRF induces apoptosis in a dose- and
time-dependent manner in EBV-transformed B cells. (A)
EBV-transformed B cells (5×104 cells/well) were cultured
in 96-well plates and exposed to SRF (1, 2.5, 5, 10, 20 and 50
μM) or DMSO. After 24 h, cell proliferation was measured by
AlamarBlue assay. RFU, relative fluorescence units. EBV-transformed
B cells (B and C) and PBMCs (D) were treated with 1, 2.5, 5, 10 and
50 μM SRF for 2, 4, 8, 16 and 24 h. The percentage of
apoptotic cells was estimated by Annexin-V/7-AAD staining. Dot plot
graphs showing the percentage of viable cells
(Annexin-V−/7-AAD−), early-phase apoptotic
cells (Annexin-V+/7-AAD−), late-phase
apoptotic cells (Annexin-V+/7-AAD+), and
necrotic cells (Annexin-V−/7-AAD+). To
measure disruption of ΔΨm, cells were stained
DiOC6. Diminished DiOC6 fluorescence
indicates ΔΨm disruption. Percentages indicate the cell
proportion in each bar. Results are representative of three
independent experiments. |
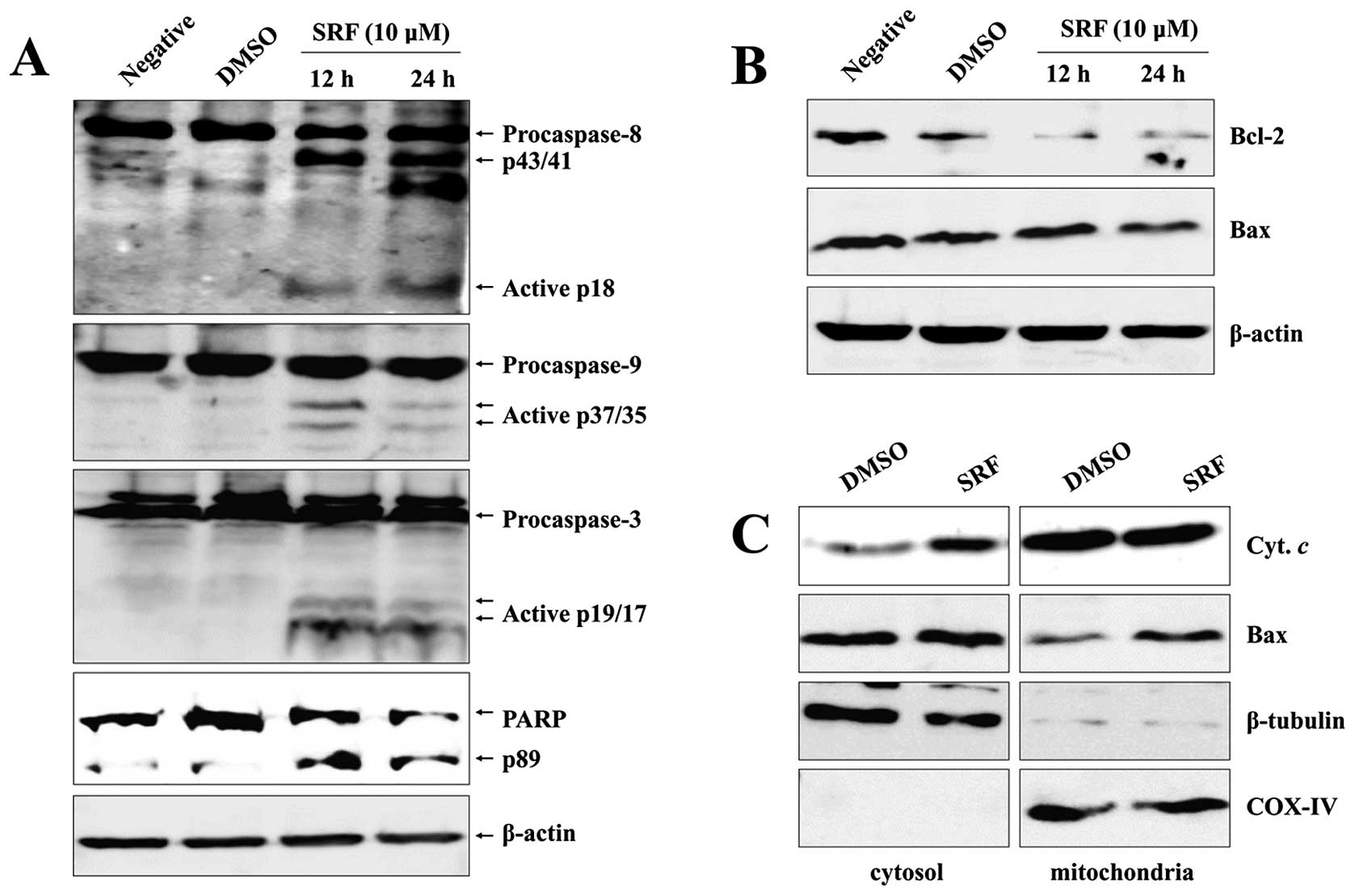
SRF leads to caspase-dependent apoptosis
in EBV-transformed B cells
To examine whether the strong apoptotic effect of
SRF involved caspase activation, we analyzed cleavage of caspases
and PARP by western blot analyses. SRF induced dose-dependent
cleavage of caspase-8, -9, and -3, followed by executioner capase-3
activation and caspase-mediated cleavage of PARP, indicating that
multiple caspases were activated in SRF-treated EBV-transformed B
cells (Fig. 2A).
SRF-induced apoptosis involves
alterations in the intracellular distribution of Bcl-2 and Bax
translocation
Several genes have been reported to play an
important role in modulating apoptosis. Abnormal expression of
anti- and pro-apoptotic molecules after stimulation is one of the
main mechanisms by which cell fate is determined in an apoptotic
system. Accumulating evidence suggests that Bcl-2 family members
play key roles in controlling apoptosis by acting as enhancers
(e.g., Bax) or inhibitors (e.g., Bcl-2) of cell death (20–22).
Accordingly, we monitored the expression of these molecules
following SRF treatment using immunoblot analysis. We observed that
SRF significantly downregulated protein levels of Bcl-2, whereas it
had no effect on Bax expression (Fig.
2B). We separated mitochondrial and cytosolic fractions
following SRF treatment to assess Bax translocation by western blot
analysis. As depicted in Fig. 2C,
there was a significant enhancement in translocation of Bax from
the cytosol to mitochondria after SRF treatment compared with the
control. Western blot analysis revealed that SRF caused an increase
in the translocation of Bax to the mitochondria and an increase in
the release of cytochrome c to the cytoplasm, thus
confirming the disintegration of ΔΨm after SRF exposure
(Fig. 2C).
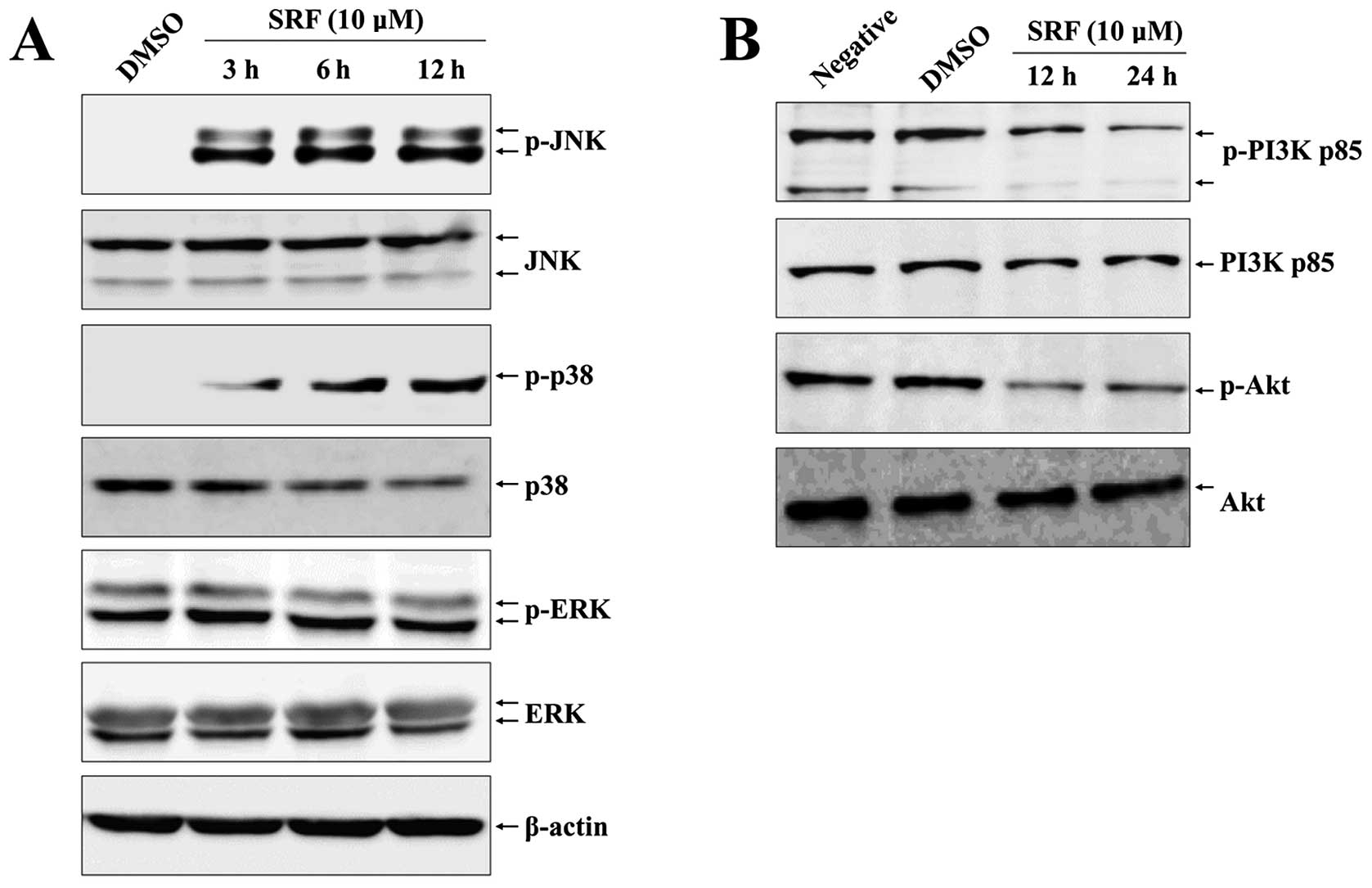
SRF induces sustained JNK and p38-MAPK
activation and inhibits the PI3K/Akt survival pathway in
EBV-transformed B cells
MAPK signaling is associated with various cellular
stresses and stimuli and has been shown to contribute to induction
of apoptosis (23), whereas the
PI3K/Akt pathway plays a critical role in the survival of various
cancer cells, including EBV-transformed B cells (24). We thus examined the effects of SRF
on MAPK and PI3K/Akt signaling and the role of these pathways in
SRF-induced apoptosis of EBV-transformed B cells. Cells were
exposed to SRF and the activity of ERK1/2, p38-MAPK, and JNK was
assessed. Fig. 3A shows that SRF
clearly induced activation of JNK and p38-MAPK after 3 h and
phosphorylation was sustained for up to 12 h in a time-dependent
manner, whereas the level of ERK1/2 phosphorylation did not change
after SRF exposure. These results suggest that JNK and p38-MAPK are
mediators of SRF-induced apoptosis. Moreover, we detected
constitutive activation of the PI3K/Akt pathway in the control
group, but SRF treatment decreased the phosphorylation of PI3K and
Akt (Fig. 3B). To confirm the role
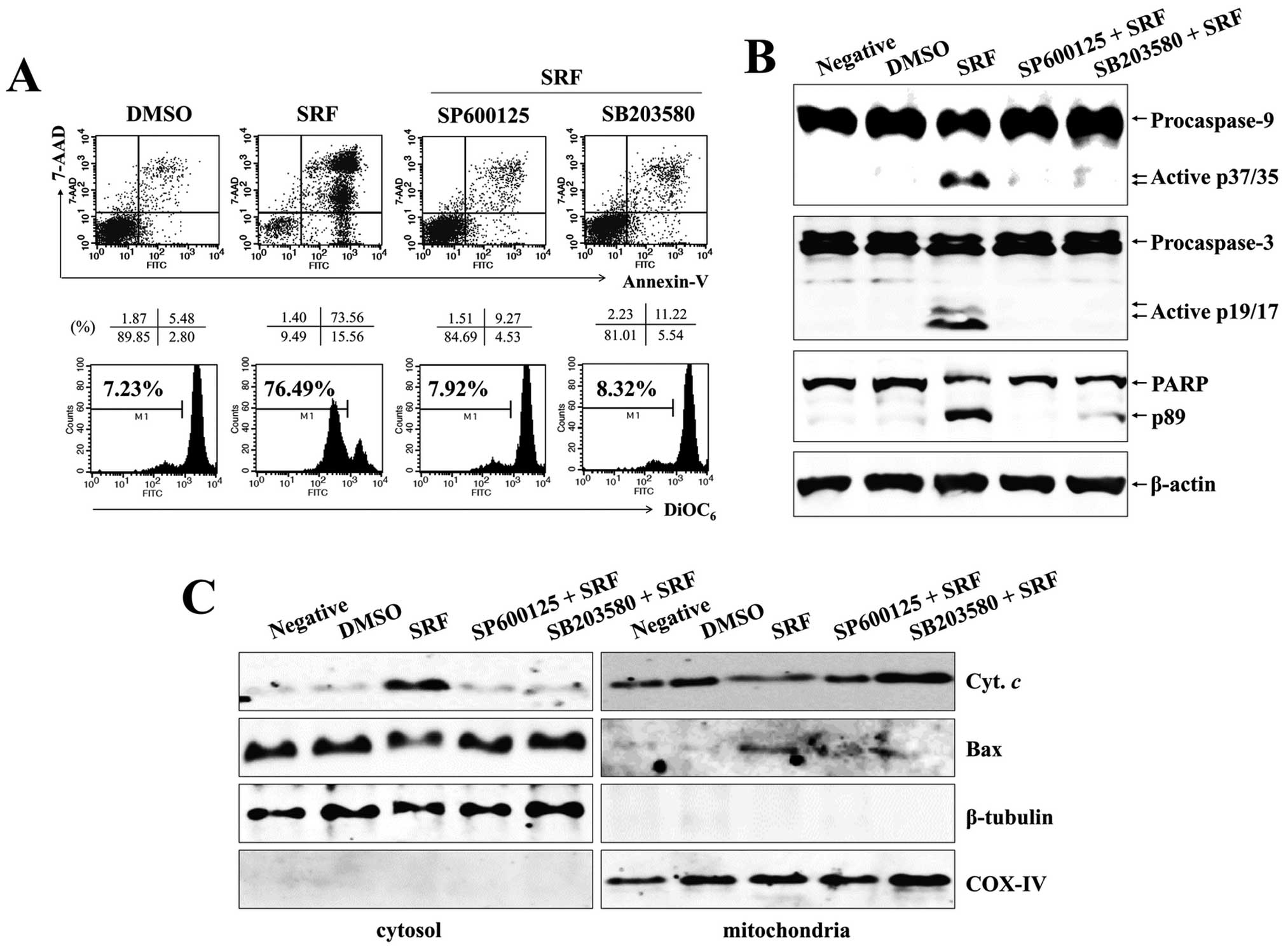
of JNK and p38-MAPK in SRF-induced apoptosis, cells were exposed to
SRF either alone or in combination with a specific JNK inhibitor
(SP600125) and p38-MAPK inhibitor (SB203580) for 24 h. As
illustrated in Fig. 4A, both
SP600125 and SB203580 pretreatment suppressed SRF-induced apoptosis
(SRF, 73.56%; with SP600125, 9.27%; with SB203580, 11.22%) and
ΔΨm disruption (SRF, 76.49%; with SP600125, 7.92%; with
SB203580, 8.32%) effectively. These inhibitors completely abolished
the activation of caspase-8, -9 and -3, as well as degradation of
PARP after SRF treatment (Fig. 4B)
and blocked the translocation of Bax to mitochondria and the
release of cytochrome c to the cytosol (Fig. 4C). These data indicate that
apoptosis caused by SRF treatment is dependent on the JNK/p38-MAPK
pathway.
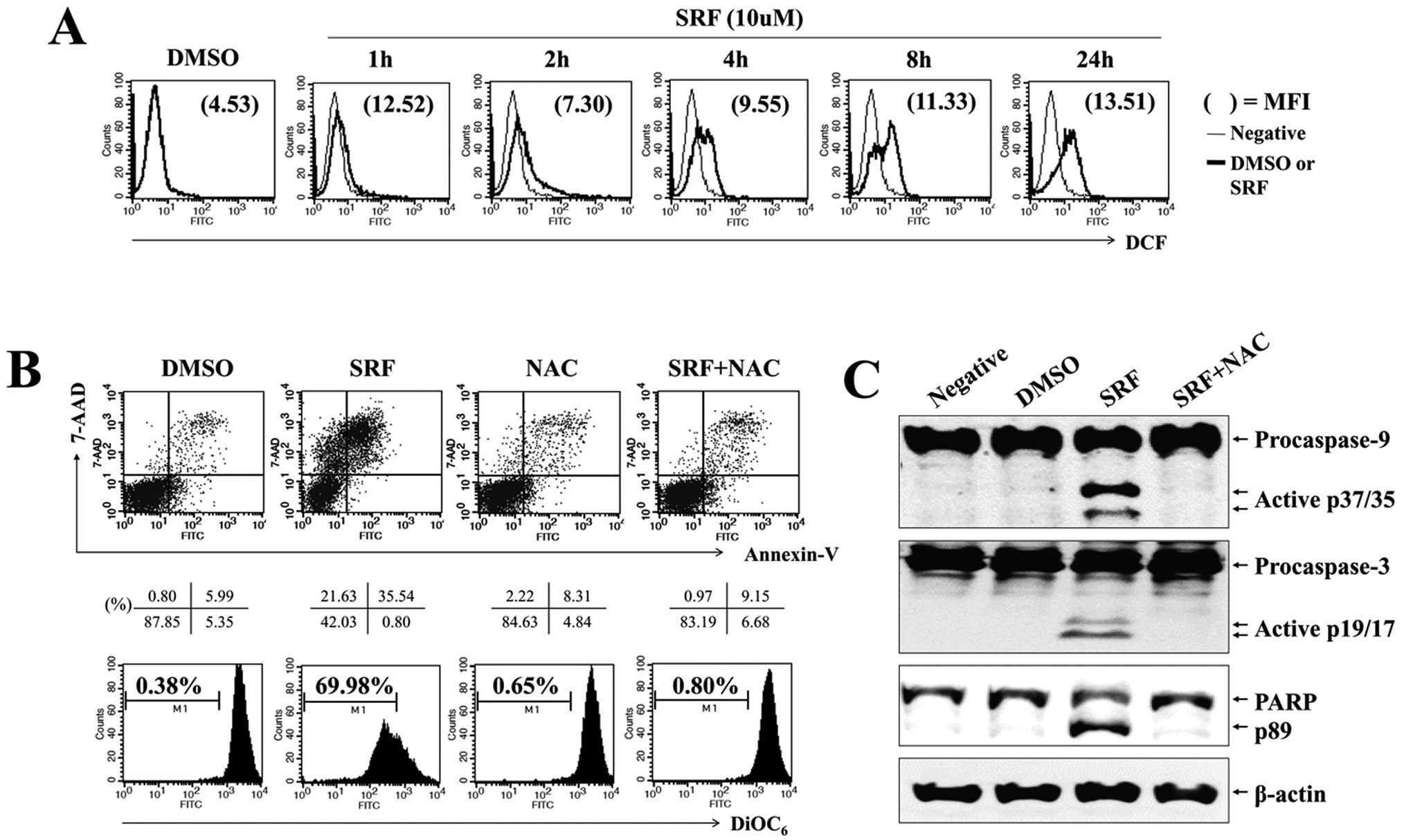
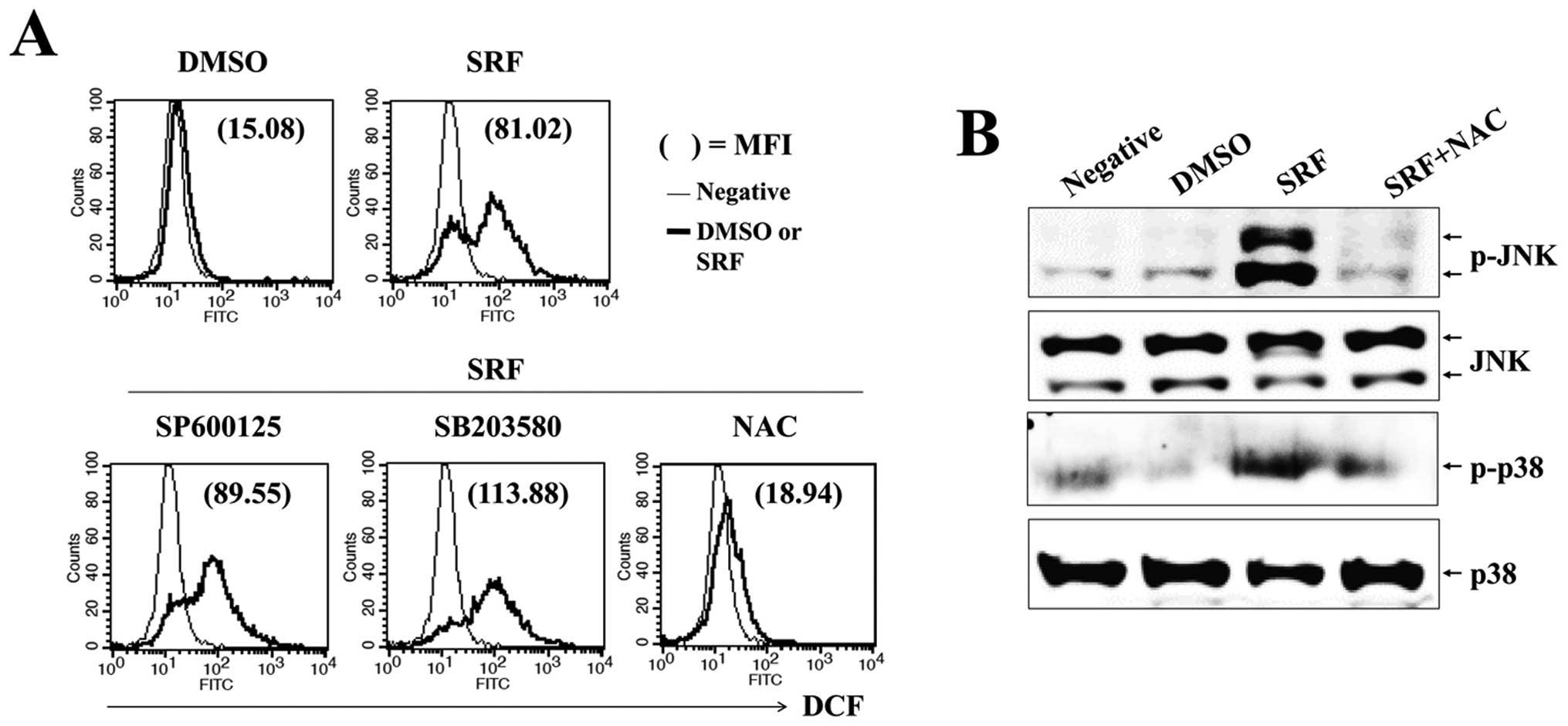
ROS is responsible for sustained
activation of JNK and p38-MAPK and mitochondrial disruption by
SRF
Reactive oxygen species, an early signal of
apoptosis (25), are directly
involved in the activation of caspases and MAPKs (26) and are responsible for the antitumor
effects of several antitumor drugs. We evaluated whether SRF
triggered intracellular ROS production and examined if ROS mediated
JNK/p38-MAPK and PI3K/Akt signaling. Cells were exposed to 10
μM SRF for the indicated time periods, followed by loading
with DCFH-DA to measure intracellular ROS levels. We found that SRF
elicited a significant increment in DCF fluorescence within 1–2 h,
and SRF-induced ROS levels were maintained for up to 24 h (Fig. 5A). To verify the effect of
SRF-induced ROS on the sustained phosphorylation of JNK/p38-MAPK
and caspases during apoptosis, we pretreated cells with NAC, a
scavenger of ROS, prior to SRF exposure. As shown in Fig. 5B, NAC significantly abrogated
SRF-induced apoptosis (SRF, 35.54%; with NAC, 9.15%) and
ΔΨm disruption (SRF, 69.98%; with NAC, 0.80%). NAC also
attenuated the activation of caspase-9 and -3, and the cleavage of
PARP (Fig. 5C). SRF-induced ROS
generation was blocked by NAC pretreatment, but not SP600125 or
SB203580 treatment (Fig. 6A). NAC
suppressed the SRF-induced phosphorylation of JNK/p38-MAPK
(Fig. 6B). These findings indicate
that ROS plays an important role in SRF-induced activation of
JNK/p38-MAPK as well as apoptosis.
Discussion
It is well-established that extrinsic (death
receptor) pathways and/or intrinsic (mitochondria) pathways
contribute to chemotherapeutic drug-induced apoptosis in many
cancer cells (27). One of the key
events in the induction of mitochondrial pathway-mediated apoptosis
is the disintegration of ΔΨm, which induces the release
of cytochrome c and elicits caspase-9 activation. This event
is regulated by Bcl-2 family proteins (28). In particular, Bax translocation to
mitochondria can change ΔΨm. Bax plays an important role
in inducing apoptosis in response to several stimuli (29). Bax is mostly cytosolic, but
relocates to the mitochondria in response to stimuli (30). After translocation to the
mitochondria, Bax, together with other pro-apoptotic Bcl-2 family
members, including truncated Bid, Bad, and Bak, induces the release
of cytochrome c, AIF, endonuclease G, and Smac/DIABLO either
by making pore channels by oligomerization in the outer
mitochondrial membrane or by opening other channels (28,31).
Bax may be activated by phosphorylation of JNK and/or p38-MAPK or
by modifications in intracellular pH. Due to its many tyrosine
kinase targets, SRF has been reported to inhibit growth and induce
apoptosis in preclinical models of human cancer (3). Here we found that SRF treatment
resulted in the release of cytochrome c from the
mitochondria and induction of caspase-9 and -3 activation in
EBV-transformed B cells. SRF decreased Bcl-2 expression and
promoted Bax translocation to mitochondria (Fig. 2). These findings suggest that SRF
treatment results in the generation of mitochondrial injury and
caspase-dependent apoptosis in EBV-transformed B cells.
Although originally identified as an inhibitor of
the Raf/MEK/Erk pathway (4), SRF
is now known to function through diverse mechanisms in various
tumor systems. We set out to scrutinize the mechanisms by which SRF
induces apoptosis of EBV-transformed B cells. We demonstrated, for
the first time, that SRF treatment induced apoptosis through
activation of p38-MAPK in an ERK1/2-independent manner. Our results
show that SRF treatment caused persistent activation of JNK and
p38-MAPK rather than ERK1/2 in EBV-transformed B cells (Fig. 3A). Use of the JNK inhibitor
SP600125 and p38-MAPK inhibitor SB203580 potently attenuated Bax
translocation and afforded significant protection against
SRF-induced apoptosis (Fig. 4).
Proteins in the MAPK family contribute to various cellular
responses. In particular, p38-MAPK and JNK play a pivotal role in
the transmission of apoptotic signals (32,33).
Because the release of cytochrome c from injured
mitochondria represents a critical step in caspase activation, our
finding that JNK and p38-MAPK activity was necessary for caspase
activation implies that JNK and p38-MAPK may control some other
mitochondrial-associated factor (e.g., Bax). In EBV-transformed B
cells, SRF treatment caused downregulation of PI3K/Akt
phosphorylation (Fig. 3B), similar
to results obtained in human neuroblastoma cells and prostate
cancer cells (34,35). However, contrary to our
expectations, there was no reduction in the phosphorylation level
of ERK1/2. These results are consistent with those of recent
studies that reported that SRF could induce apoptosis in melanoma
and hepatocellular carcinoma cells through a MEK/ERK-independent
mechanism (36,37). Thus, it is clear that SRF has
antitumor effects irrespective of its ability to inhibit
p-ERK1/2.
Recent studies suggested that ROS may play a key
role in apoptosis induction (25,26).
Oxidative stress can be elicited by sustained or aberrant ROS
production and is associated with several biological events
including apoptosis (27,28). ROS is an important modulator of
cellular signaling related to proliferation, apoptosis, and
senescence (26). Several
chemotherapeutic drugs exert their cytotoxic effects through the
generation of ROS as a key mediator. Recent studies have reported
that SRF might also be associated with ROS production (29,30).
Ample evidence indicates that chemically-mediated ROS production
results in alteration of cellular functions and eventual apoptosis
(26). Dysfunction of mitochondria
induced by excessive ROS generation leads to dissipation of
ΔΨm and apoptosis (26). ROS are also known to activate MAPKs
(25). Our data indicated that
sustained phosphorylation of JNK and p38-MAPK was caused by ROS
generation after SRF treatment (Fig.
6). It is noteworthy that inhibition of ROS by NAC pretreatment
ameliorated the effect of SRF on JNK and p38-MAPK phosphorylation,
suggesting that SRF stimulates the production of ROS, which
subsequently activate JNK and p38-MAPK, resulting in the
translocation of Bax to mitochondria.
Taken together, we found that SRF inhibited cell
growth and induced apoptosis in EBV-transformed B cells.
SRF-induced apoptosis involved a reduction in Bcl-2 expression and
induction of Bax translocation to mitochondria, resulting in
disruption of ΔΨm in EBV-transformed B cells. Our
results also indicated that SRF elicited the activation of
caspase-9 as the initiator caspase, followed by activation of
caspase-3 and -8. More importantly, ROS, JNK, p38-MAPK, and Bax
participated in SRF-induced apoptosis. Consistent with this
finding, ROS induced by SRF may act as upstream mediators of JNK
and p38-MAPK signaling in EBV-transformed B cells treated with SRF
(Fig. 5A). Furthermore, there is
increasing evidence within the literature that ROS contribute to
apoptosis caused by diverse stimuli. In conclusion, we demonstrated
that SRF induces apoptosis of EBV-transformed B cells through
ROS-dependent JNK/p38-MAPK signaling in an ERK-independent
manner.
Abbreviations:
|
EBV
|
Epstein-Barr virus;
|
|
SRF
|
sorafenib;
|
|
ROS
|
reactive oxygen species;
|
|
NAC
|
N-acetyl-l-cysteine
|
Acknowledgements
This study was supported by the 2014
Inje University Research Grant.
References
|
1.
|
Young LS and Rickinson AB: Epstein-Barr
virus: 40 years on. Nat Rev Cancer. 4:757–768. 2004.PubMed/NCBI
|
|
2.
|
Kuppers R: B cells under influence:
transformation of B cells by Epstein-Barr virus. Nat Rev Immunol.
3:801–812. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Wilhelm S, Carter C, Lynch M, et al:
Discovery and development of sorafenib: a multikinase inhibitor for
treating cancer. Nat Rev Drug Discov. 5:835–844. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Wilhelm SM, Carter C, Tang L, et al: BAY
43-9006 exhibits broad spectrum oral antitumor activity and targets
the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in
tumor progression and angiogenesis. Cancer Res. 64:7099–7109. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Abou-Alfa GK, Schwartz L, Ricci S, et al:
Phase II study of sorafenib in patients with advanced
hepatocellular carcinoma. J Clin Oncol. 24:4293–4300. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kane RC, Farrell AT, Saber H, et al:
Sorafenib for the treatment of advanced renal cell carcinoma. Clin
Cancer Res. 12:7271–7278. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Llovet JM and Bruix J: Molecular targeted
therapies in hepatocellular carcinoma. Hepatology. 48:1312–1327.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Yang SY, Cui CL, Chi ZH, et al: Phase II
clinical trial of sorafenib plus local chemotherapy in the
treatment of metastatic renal cell carcinoma with pleural effusion.
Zhonghua Yi Xue Za Zhi. 92:2998–3000. 2012.(In Chinese).
|
|
9.
|
Galanis E, Anderson SK, Lafky JM, et al:
Phase II study of bevacizumab in combination with sorafenib in
recurrent glioblastoma (N0776): a north central cancer treatment
group trial. Clin Cancer Res. 19:4816–4823. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Cervello M, Bachvarov D, Lampiasi N, et
al: Novel combination of sorafenib and celecoxib provides
synergistic anti-proliferative and pro-apoptotic effects in human
liver cancer cells. PLoS One. 8:e655692013. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Wilhelm SM, Adnane L, Newell P, et al:
Preclinical overview of sorafenib, a multikinase inhibitor that
targets both Raf and VEGF and PDGF receptor tyrosine kinase
signaling. Mol Cancer Ther. 7:3129–3140. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Huber S, Oelsner M, Decker T, et al:
Sorafenib induces cell death in chronic lymphocytic leukemia by
translational downregulation of Mcl-1. Leukemia. 25:838–847. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Lu X, Tang X, Guo W, et al: Sorafenib
induces growth inhibition and apoptosis of human chondrosarcoma
cells by blocking the RAF/ERK/MEK pathway. J Surg Oncol.
102:821–826. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Liu LP, Ho RL, Chen GG and Lai PB:
Sorafenib inhibits hypoxia-inducible factor-1α synthesis:
implications for antiangiogenic activity in hepatocellular
carcinoma. Clin Cancer Res. 18:5662–5671. 2012.
|
|
15.
|
Reddy KB, Nabha SM and Atanaskova N: Role
of MAP kinase in tumor progression and invasion. Cancer Metastasis
Rev. 22:395–403. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Kim KN, Ham YM, Moon JY, et al: Acanthoic
acid induces cell apoptosis through activation of the p38 MAPK
pathway in HL-60 human promyelocytic leukaemia. Food Chem.
135:2112–2117. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Han X, Xu B, Beevers CS, et al: Curcumin
inhibits protein phosphatases 2A and 5, leading to activation of
mitogen-activated protein kinases and death in tumor cells.
Carcinogenesis. 33:868–875. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Hyun MS, Hur JM, Mun YJ, et al: BBR
induces apoptosis in HepG2 cell through an
Akt-ASK1-ROS-p38MAPKs-linked cascade. J Cell Biochem. 109:329–338.
2010.PubMed/NCBI
|
|
19.
|
Rahmani M, Davis EM, Crabtree TR, et al:
The kinase inhibitor sorafenib induces cell death through a process
involving induction of endoplasmic reticulum stress. Mol Cell Biol.
27:5499–5513. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Reed JC: Bcl-2 family proteins: regulators
of apoptosis and chemoresistance in hematologic malignancies. Semin
Hematol. 34:9–19. 1997.PubMed/NCBI
|
|
21.
|
Adams JM and Cory S: The Bcl-2 protein
family: arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Chao DT and Korsmeyer SJ: BCL-2 family:
regulators of cell death. Annu Rev Immunol. 16:395–419. 1998.
View Article : Google Scholar
|
|
23.
|
Wada T and Penninger JM: Mitogen-activated
protein kinases in apoptosis regulation. Oncogene. 23:2838–2849.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Fresno Vara JA, Casado E, de Castro J, et
al: PI3K/Akt signalling pathway and cancer. Cancer Treat Rev.
30:193–204. 2004.PubMed/NCBI
|
|
25.
|
Chiu WH, Luo SJ, Chen CL, et al: Vinca
alkaloids cause aberrant ROS-mediated JNK activation, Mcl-1
downregulation, DNA damage, mitochondrial dysfunction, and
apoptosis in lung adenocarcinoma cells. Biochem Pharmacol.
83:1159–1171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Matsuzawa A and Ichijo H:
Stress-responsive protein kinases in redox-regulated apoptosis
signaling. Antioxid Redox Signal. 7:472–481. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Budihardjo I, Oliver H, Lutter M, et al:
Biochemical pathways of caspase activation during apoptosis. Annu
Rev Cell Dev Biol. 15:269–290. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Green DR: At the gates of death. Cancer
Cell. 9:328–330. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Wei MC, Zong WX, Cheng EH, et al:
Proapoptotic BAX and BAK: a requisite gateway to mitochondrial
dysfunction and death. Science. 292:727–730. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Wolter KG, Hsu YT, Smith CL, et al:
Movement of Bax from the cytosol to mitochondria during apoptosis.
J Cell Biol. 139:1281–1292. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Kuwana T, Mackey MR, Perkins G, et al:
Bid, Bax, and lipids cooperate to form supramolecular openings in
the outer mitochondrial membrane. Cell. 111:331–342. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Ghatan S, Larner S, Kinoshita Y, et al:
P38 MAP kinase mediates Bax translocation in nitric oxide-induced
apoptosis in neurons. J Cell Biol. 150:335–347. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Tsuruta F, Sunayama J, Mori Y, et al: JNK
promotes Bax translocation to mitochondria through phosphorylation
of 14-3-3 proteins. EMBO J. 23:1889–1899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Chai H, Luo AZ, Weerasinghe P, et al:
Sorafenib downregulates ERK/Akt and STAT3 survival pathways and
induces apoptosis in a human neuroblastoma cell line. Int J Clin
Exp Pathol. 3:408–415. 2010.PubMed/NCBI
|
|
35.
|
Oh SJ, Erb HH, Hobisch A, et al: Sorafenib
decreases proliferation and induces apoptosis of prostate cancer
cells by inhibition of the androgen receptor and Akt signaling
pathways. Endocr Relat Cancer. 19:305–319. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Sánchez-Hernández I, Baquero P, Calleros
L, et al: Dual inhibition of (V600E)BRAF and the PI3K/AKT/mTOR
pathway cooperates to induce apoptosis in melanoma cells through a
MEK-independent mechanism. Cancer Lett. 314:244–255.
2012.PubMed/NCBI
|
|
37.
|
Ou DL, Shen YC, Yu SL, et al: Induction of
DNA damage-inducible gene GADD45beta contributes to
sorafenib-induced apoptosis in hepatocellular carcinoma cells.
Cancer Res. 70:9309–9318. 2010. View Article : Google Scholar : PubMed/NCBI
|