Introduction
Glioblastoma multiforme (GBM) remains one of the
most common and biologically aggressive tumors in adults. Despite
standard therapeutic protocols, which include maximal surgical
resection followed by regional fractionated radiation and
chemotherapy with temozolomide, the prognosis of patients with GBM
remains dismal, with median survival of less than 15 months
(1). As they are invariably fatal
due, in part, to resistance to postsurgical therapy, identification
of genetic alterations which can predict efficient management and
areas of targets for therapy are continuously being sought
(2,3).
A broad range of human malignancies is associated
with non-random 1p36 deletions, suggesting the existence of tumor
suppressors encoded in this region (4). Allelic loss involving 1p36 is a
common chromosomal aberration observed in oligodendroglioma and has
been associated with radiation and chemosensitivity, as well as
enhanced tumor infiltration (5).
We recently discovered the frequent loss of expression of adherens
junction-associated protein 1 (AJAP1) in GBM via multiple
genome-wide studies. Located on 1p36, AJAP-1 may have tumor
suppressor-like qualities and may play an important role in
regulation of cell migration and adhesion (6). However, the role of AJAP1 in cell
cycle arrest or apoptosis and resistance to chemotherapy remain
largely unexplored.
Temozolomide (TMZ) is the first-line
chemotherapeutic drug agent for GBM with excellent CNS
bioavailability (7,8). It is an alkylating cytostatic drug,
which causes DNA damage and induces cell death. However, clinical
response toward TMZ is generally short-lived, possibly due to
multiple factors such as tumor cell heterogeneity, resistant glioma
stem cells, rapid transformation, genetic instability and selective
pressures (9,10). The available data suggest that
neither MGMT protein expression levels nor MGMT promoter
methylation status is a sufficiently accurate predictor of the
sensitivity of the glioma cells to TMZ (11). More recently, further studies
indicated that additional pathways, such as activation of DNA
damage checkpoint responses and the changes in mitochondria DNA and
electron transport chain may contribute to TMZ resistance (12,13).
Therefore, the intrinsic and acquired resistance towards TMZ makes
it crucial to find new therapeutic strategies aimed at improving
the prognosis of patients suffering from GBM. In the present study
the concomitant treatment of GBM cells with AJAP1-restoration and
TMZ in overcoming TMZ resistance was investigated.
MAGEA2 protein induces a novel p53 inhibitory loop
involving recruitment of histone deacetylase 3 (HDAC3) to a
MAGEA2-p53 complex (14). This
complex formation strongly downregulates the p53 transactivation
function in melanoma without changing p53 expression levels
(15). Other data suggest
potential involvement of MAGEA family proteins in modulating cell
survival (16). The correlation
between MAGEA2 expression and resistance to apoptosis has been
validated in various kinds of tumors partly through its interaction
with the p53 pathway, where high MAGEA2 levels reestablish p53
response and increase chemoresistance (17–19).
In this study, we investigated AJAP1 function in the induction of
cell apoptosis and potentiation of TMZ-induced apoptosis in glioma
cells through the transcriptional downregulation of MAGEA2.
Materials and methods
Cell culture and drug treatments
HEK293, U87MG, U251MG, D373MG and D409MG cells were
maintained in DMEM - High Glucose medium (Gibco, Carlsbad, CA, USA)
supplemented with 10% FBS (Gibco) and incubated in a humidified
incubator at 37°C with 5% CO2. U87MG, U251MG, D373MG and
D409MG cells were previously stably transfected with the GFP-AJAP1
plasmid (6). Similar cells were
also transfected with GFP-N1 vector using identical transfection
techniques to be used as a control. Clones were maintained in 100
μg/ml geneticin. For temozolomide (TMZ) studies, glioma
cells were seeded in 10 cm round Petri dishes, 24- or 96-well
plates, allowed to attach overnight, then treated with 100
μM temozolomide (TMZ, Sigma-Aldrich Co., St. Louis, MO, USA)
for 48 h. In control groups, the same concentration of dimethyl
sulfoxide (DMSO) was used to treat cells. Fresh drug and medium
were changed every 24 h.
RNA preparation and GeneChip array
assay
GBM cell lines (D373) stably expressing GFP-N1
(vector control) or GFP-AJAP1 were assessed for their gene
expression profile using a high-density oligonucleotide array
(GeneChip Human Genome U133 Plus 2.0 Array; Affymetrix, Santa
Clara, CA, USA). Total RNA was extracted from freshly prepared
confluent cells. cDNA was generated from the total RNA using
Qiagen’s RNeasy system (Quiagen, Valencia, CA, USA), then
fragmented and hybridized to a DNA oligonucleotide expression array
(Affymetrix Human Genome U133A 2.0 Array). The hybridized probe
array was washed and stained following the manufacturer’s
instructions. The probe array was then scanned with a confocal
laser scanner (GeneChip Scanner 3000; Affymetrix). The expression
array analysis for each cell line was run in triplicate. The
expression level of each gene (22,277 probe sets for approximately
14,500 human genes) was calculated to identify discriminative genes
by Partek (St. Louis, MO, USA).
Real-time quantitative RT-PCR
Quantification of ten candidate genes was performed
to confirm Affymetrix data by the real-time quantitative reverse
transcription-PCR method. Total RNA was extracted from GBM cell
lines as described previously. The real-time PCR reaction mixture
was prepared using a TaqMan Universal Master Mix (Applied
Biosystems, Foster City, CA, USA), consisting of 120 nM of primer,
200 nM probes, and 2.5 μl of cDNA sample. PCR conditions
were as follows: 95°C for 10 min; 45 cycles at 95°C for 30 sec,
62°C for 30 sec, and 72°C for 30 sec, then 72°C for 1 min. Each
reaction was run in triplicate, and we generated a standard curve
for each probe using serial dilutions from reference cDNA to
quantify relative gene expression. The glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) gene was used as an endogenous reference, and
each sample was normalized to its GAPDH content.
Isolation of the proximal promoter of the
human MAGEA2 gene and plasmid construction
The region of the human MAGEA2 gene 986 bp upstream
(−896 to +90), containing the complete proximal promoter regulatory
elements, was amplified by PCR from genomic DNA. The primer pairs
were MAGEA2 5′-GGG GTA CC accaaaggtttgccaagtgtag-3′ with a
KpnI site at the 5′-end and MAGEA2 5′-CCC AAG CTT
ctgccctggtcacacaaag-3′ with a HindIII site at the 5′-end.
The PCR was conducted at 95°C for 1 min followed by 30 cycles at
95°C for 30 sec, 58°C for 1.5 min, 72°C for 1 min. The amplified
fragment was isolated and purified following agarose
electrophoresis using a Gel Extract Kit (Thermo Scientific,
Rockford, IL, USA), digested with KpnI and HindIII
(New England Biolabs, Ipswich, MA, USA), and ligated into the
equivalent sites of the pGL3-enhancer vector (Promega, Madison, WI,
USA) to generate the pGL3-MAGEA2 construct. The resulting construct
was confirmed by restriction enzyme analysis and DNA sequence
analysis.
Transient transfection
For the promoter activity assay, HEK293, U87 and
U251 cells were transfected with lipofectamine 2000 (Invitrogen,
Carlsbad, CA, USA) in 24-well plates. Each well included
0.5×105 cells, 1.0 μg pGL3-MAGEA2, 1 ng internal
control vector pRL-CMV (Promega), 2 μl lipofectamine 2000
and 500 μl Opti-MEM I reduced serum media (Gibco). For
co-transfection experiments of the GFP-AJAP1 with pGL3-MAGEA2
plasmid, HEK293 cells were transfected with lipofectamine 2000 in
24-well plates, and each well included 0.5×105 cells,
0.5 μg pGL3-MAGEA2, 0.5 μg GFP-AJAP1, 1 ng pRL-CMV, 2
μl lipofectamine 2000 and 500 μl Opti-MEM I reduced
serum media. Empty pGL3-enhancer vector and GFP-N1 vector were used
as control.
Dual-luciferase reporter assays
Twenty-four hours after transfection, the activities
of firefly luciferase in pGL3-constructs and Renilla
luciferase in pRL-CMV were determined by the dual-luciferase
reporter assays following manufacturer’s protocol (Promega). The
cells were rinsed with PBS, and lysed with 1X passive lysis buffer.
A total of 20 μl of cell lysate was transferred into the
luminometer plates containing 100 μl luciferase assay
reagent II. Firefly luciferase activity was measured first, and
then Renilla luciferase activity was measured after the
addition of 100 μl Stop & Glo Reagent. Results were
expressed as a ratio of firefly luciferase activity to
Renilla luciferase activity.
Western blot analysis
Cultured cells were harvested by scraping in
ice-cold PBS and spun down at 2,500 rpm in a 4°C centrifuge for 5
min. Cell pellets were then lysed in lysis buffer (25 mM Tris pH
7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol) containing 1%
of protease inhibitor cocktail (Thermo Scientific). Protein
concentration was determined by standard BCA assay (Pierce, Thermo
Scientific), and total cell lysates were separated by
electrophoresis 10% precast polyacrylamide gel (Bio-Rad, Hercules,
CA, USA). Gel loading was normalized for equal GAPDH (25 μg
protein per lane). Proteins were then transferred onto Hybond-P
polyvinylidene difluoride membranes (Amersham Biosciences,
Piscataway, NJ, USA), and the membranes were probed with each
antibody. Rabbit polyclonal anti-MAGEA2 (1:100), rabbit monoclonal
anti-Bcl2 (1:2000) and rabbit polyclonal anti-Bax (1:1,000) were
obtained from Abcam Biotechnology. Rabbit polyclonal anti-AJAP1
(1:2000) was obtained from Sigma-Aldrich Co. Rabbit monoclonal
anti-GAPDH (1:1,000) and chicken anti-rabbit IgG-HRP (1:2,000) were
purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Immunoblots were detected using a Supersignal West Femto substrate
kit (Thermo Scientific) and visualized by autoradiography.
Caspase 3/7 activity assay
A total of 104 U87 MG cells and U251 MG
cells overexpressing AJAP1 or control (GFP-N1) were seeded into
96-well plates overnight. Room temperature Caspase-Glo 3/7 Reagent
(100 μl) was added to each well of a white-walled 96-well
plate containing 100 μl of blank, DMSO (negative control)
cells or TMZ treated cells in culture medium. Wells were gently
mixed on a plate shaker at 500 rpm for 30 sec, incubated at room
temperature for 1 h, then luminescence measured in a plate-reading
luminometer.
Confocal TUNEL assay
A total of 105 cells were seeded onto
12-mm poly-L-lysine coated coverslips placed in the bottom of
24-well plates. After treatment, cells were fixed with 4%
paraformaldehyde for 1 h at room temperature. After incubating in
0.1% Triton X-100 for 2 min on ice, a terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labelling (TUNEL) reaction
solution containing TMR red-labeled nucleotides (Roche, Mannheim,
Germany) was added. Transferase was excluded in identical mixtures
as negative control. Slides were placed in a humidified atmosphere
overnight at 4°C in the dark, rinsed with PBS three times, then
cover slips were mounted in mounting medium with counterstaining
cell nuclei using 4’,6-diamidino-2-phenylindole (DAPI) (Vector
Laboratories, Burlingame, CA, USA) overnight. Using an excitation
wavelength in 540 nm and detection in 620 nm, confocal images were
taken with a microscope (Zeiss 780 upright confocal; Carl Zeiss,
Göttingen, Germany). Images were collected using the Laser-Sharp
2000 software.
Statistical analysis
All experiments were done in triplicate. Statistical
significance was set at P<0.05. Analyses were done with
Microsoft Excel and SAS E-guide statistical packages (SAS Inc.,
Cary, NC, USA).
Results
Identification of AJAP1-associated gene
changes that may augment TMZ-induced cytotoxicity
To investigate the possible transcriptional
regulatory role of AJAP1, microarray-based gene expression analysis
of GBM cell lines (D373) stably expressing EGFP-N1 (vector control)
or EGFP-AJAP1 identified a set of novel candidate genes being
upregulated or downregulated following AJAP1 overexpression
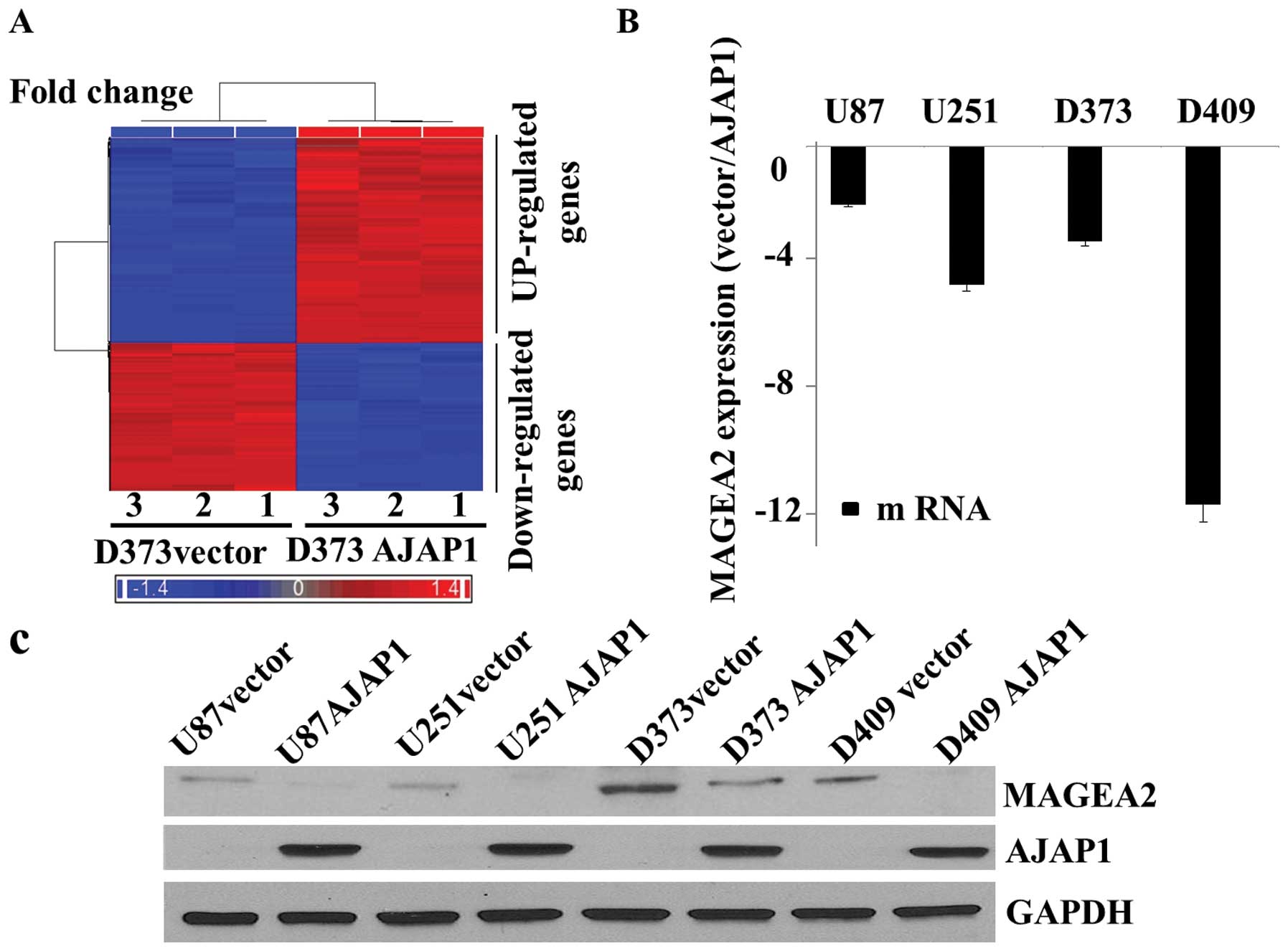
(Fig. 1A). MAGEA2 was identified
as a likely candidate that may alter cytotoxicity responses. The
fold change in MAGEA2 (116.931-fold decrease) was one of the most
significant in the genome after AJAP1 overexpression (Table I). Quantitative real-time PCR
analyzed MAGEA2 in U87, U251, D373MG and D409MG cells stably
transfected with EGFP-AJAP1 or EGFP-N1 plasmid to validate the
microarray findings. MAGEA2 expression was decreased in all AJAP1
positive glioma cell lines tested in triplicate (Fig. 1B), as demonstrated by western blot
analysis (Fig. 1C).
 | Table I.Summary of fold change of gene
expression level between AJAP1 cells (DS) and vector cells (DV) by
microarray screening. |
Table I.
Summary of fold change of gene
expression level between AJAP1 cells (DS) and vector cells (DV) by
microarray screening.
| Gene title | Gene symbol | Fold change (DS vs.
DV) |
|---|
| Melanoma antigen
family A, 2///2B | MAGEA2///2B | −116.93 |
| Melanoma antigen
family A, 3 | MAGEA3 | −111.46 |
| Proteolipid protein
1 | PLP1 | −37.40 |
| Attractin-like
1 | ATRNL1 | −27.24 |
|
Butyrylcholinesterase | BCHE | −25.53 |
| MACRO domain
containing 2 | MACROD2 | −19.80 |
| Hypothetical
protein LOC169834 | LOC169834 | −17.40 |
| KIAA1239 | KIAA1239 | −15.41 |
| ADAM
metallopeptidase with thrombospondin type 1 mmotif, 3 |
ADAMTS3(3)(7)(10) | −12.95 |
| Tetraspanin 8 | TSPAN8 | −10.73 |
| Protein kinase,
AMP-activated, alpha 2 catalytic subunit | PRKAA2(6) | 12.30 |
| MARCKS-like 1 | MARCKSL1 | 12.89 |
| Secreted
frizzled-related protein 1 | SFRP1 | 14.10 |
| Fibulin 1 | FBLN1(3) | 14.27 |
| Secreted
frizzled-related protein 1 | SFRP1 | 17.63 |
| Paraneoplastic
antigen MA2 | PNMA2 | 19.78 |
| Neurofascin homolog
(chicken) | NFASC(2) | 21.93 |
| Protein kinase,
AMP-activated, alpha 2 catalytic subunit | PRKAA2 | 22.64 |
| Kynureninase
(L-kynurenine hydrolase) | KYNU | 24.08 |
| Docking protein
5 | DOK5 | 24.52 |
| Peptidase inhibitor
15 | PI15 | 29.53 |
| Adherens junctions
associated protein 1 | AJAP1 | 40.79 |
| Transmembrane
protein 47 | TMEM47 | 106.87 |
Construction of the luciferase reporter
plasmid with the MAGEA2 gene promoter
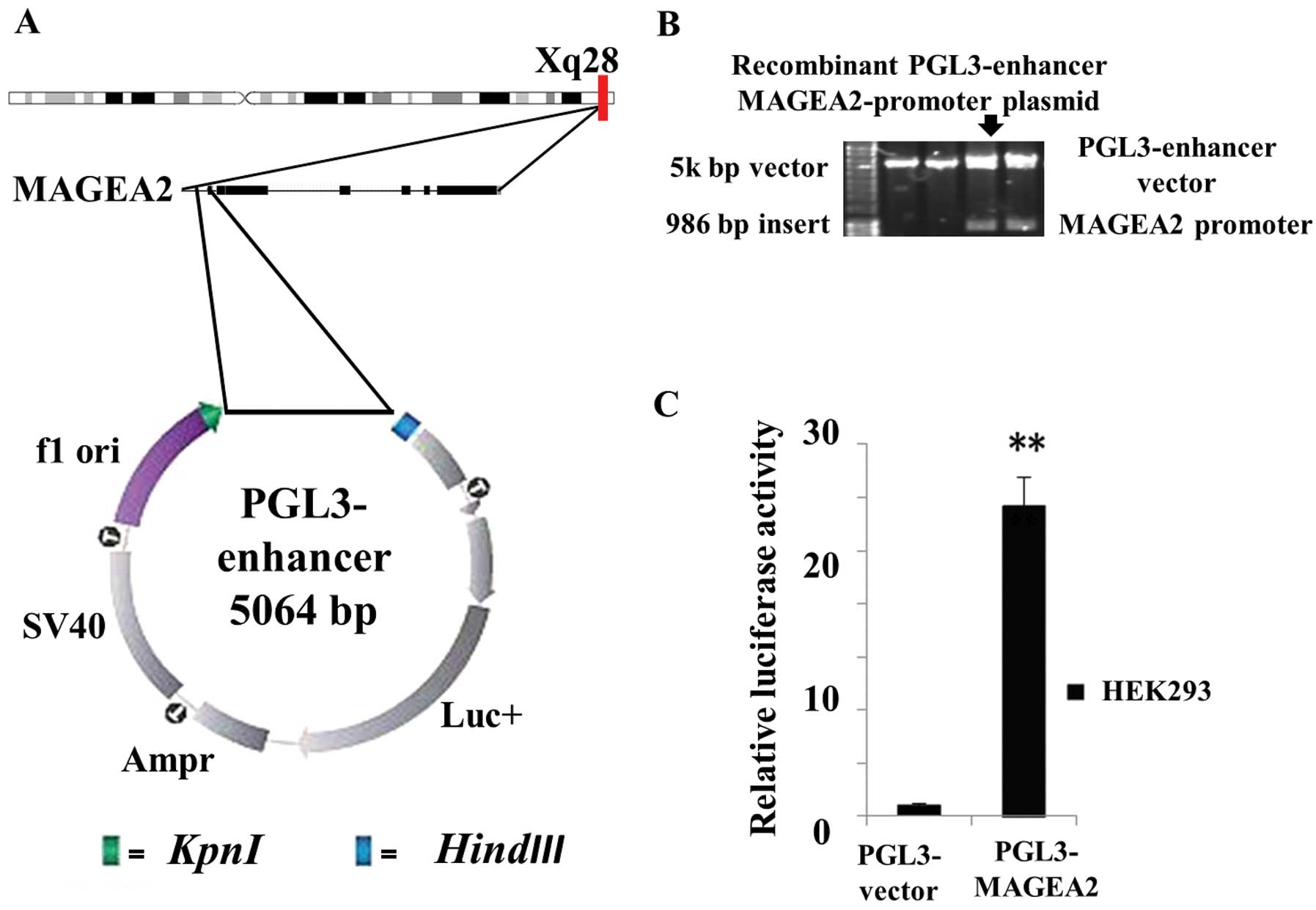
Promoter fragment (1 kb) of the MAGEA2 gene was
amplified by PCR and inserted into a pGL3-enhancer vector to form
the MAGEA2 promoter-luciferase reporter plasmid designated as
pGL3-MAGEA2 (Fig. 2A). The
recombinant plasmid was confirmed by double restriction enzyme
digestion (Fig. 2B) and DNA
sequence analysis. A significant increase (over 24-fold greater,
P<0.01, Fig. 2C) in relatively
luciferase activity was observed in HEK293 cells co-transfected
PGL3-MAGEA2 compared with vector control. Thus, the
recombinant PGL3-MAGEA2 luciferase reporter plasmid was
determined to be able to drive the luciferase expression.
AJAP1 transcriptional downregulated the
expression of MAGEA2
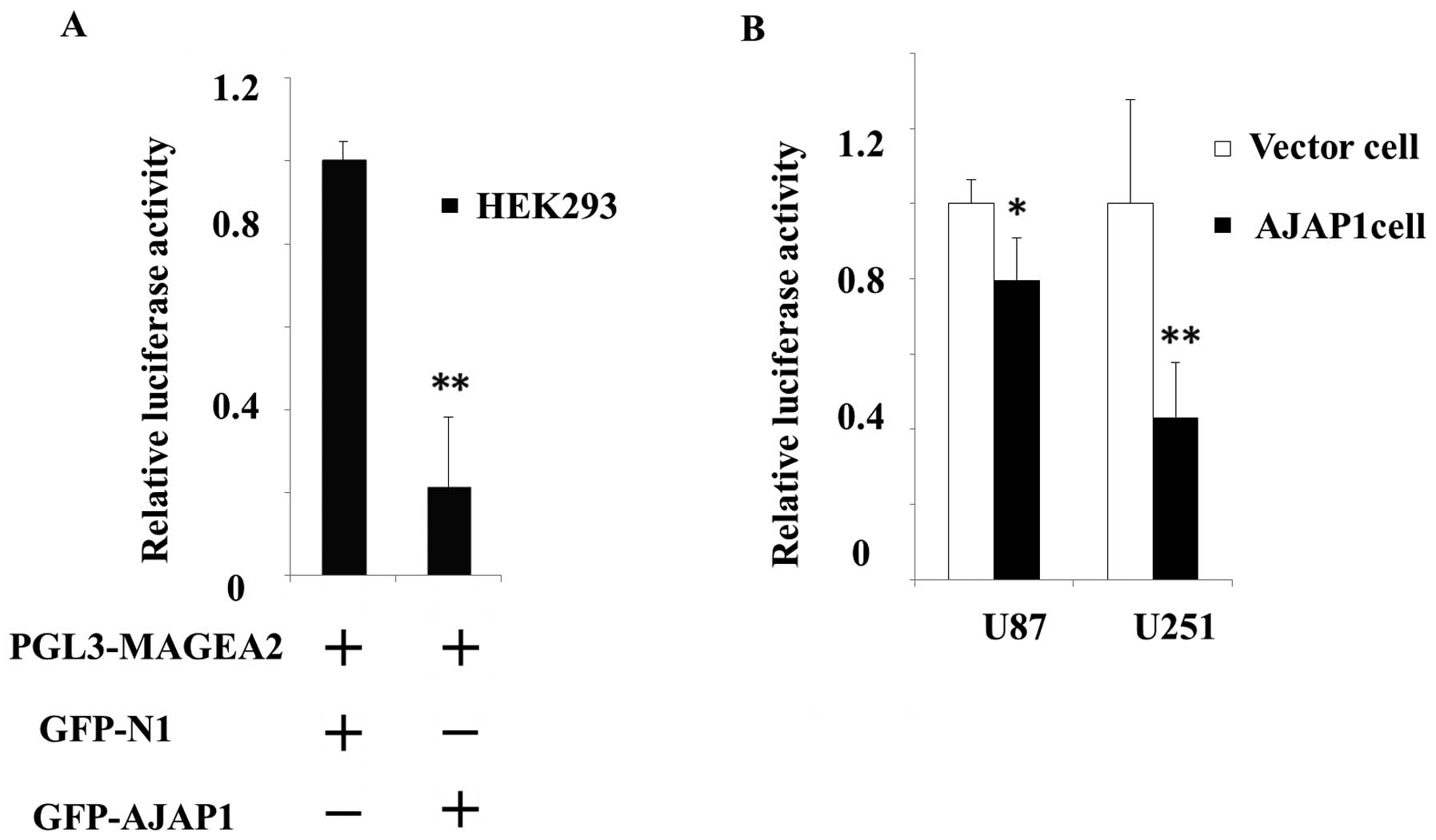
To explore the effects of AJAP1 on MAGEA2 gene
expression, HEK293 cells were harvested 24 h after co-transfection
with pGL3-MAGEA2 and EGFP-AJAP1. The control cells were
co-transfected with pGL3-MAGEA2 and GFP-N1 plasmid. The
MAGEA2 promoter activity was considerably blocked (78.7%,
P<0.01) in HEK293 cells with GFP-AJAP1 co-transfection, compared
with control cells (Fig. 3A). To
further confirm the transcriptional relationship between AJAP1 and
MAGEA2, glioma cell lines stable expressing AJAP1 gene or empty
vector were transfected with pGL3-MAGEA2. We observed a
significant decrease (20.5% in U87 AJAP1 cells, P<0.05; 56.8% in
U251 AJAP1 cells, P<0.05) in relative luciferase activity,
compared with respective vector cells (Fig. 3B). These data support that
expression of AJAP1 repressed MAGEA2 expression by physical
interaction with the MAGEA2 gene promoter.
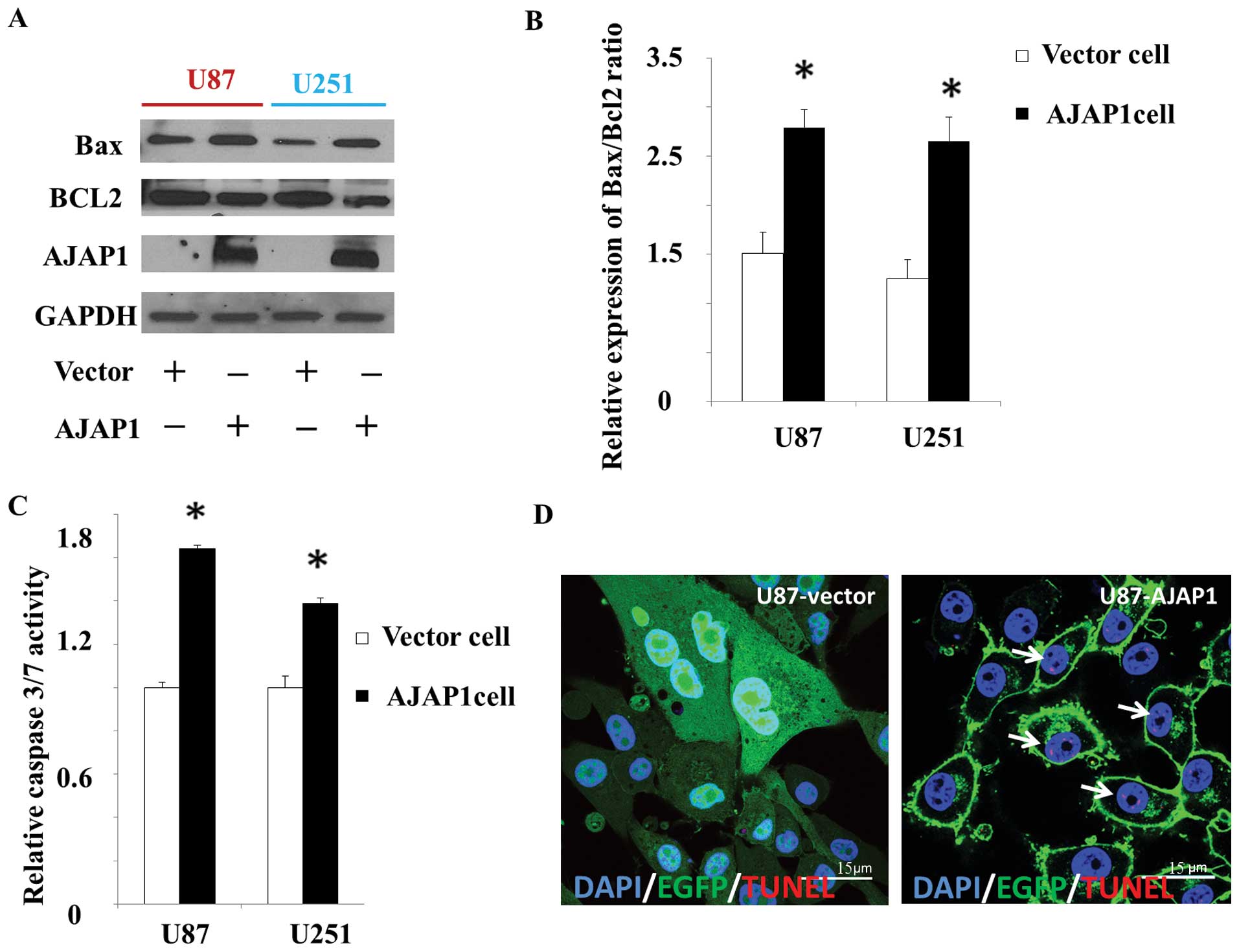
AJAP1 moderately induces cell apoptosis
in GBM cells
The decrease in Bax/Bcl-2 ratio is thought to
contribute to the resistance of glioma cells to therapy by
modulating the apoptotic cascade (20). In this study, we measured the
levels of Bax and Bcl-2 by western blot analysis to detect the
putative pro-apoptosis function of AJAP1. We observed 39.6 and
42.2% increase in Bax/Bcl-2 ratio in cells overexpressing AJAP1,
compared to the control cells (P<0.05) (Fig. 4A and B). Caspase proteins are
cysteine proteases that act downstream of the Bcl-2 family by
initiating cellular breakdown during apoptosis (21). Among the effector caspases,
caspase-3/7 is most frequently involved in tumor cellular apoptosis
(22). To determine whether
caspase-3/7 is activated after AJAP1 involvement, caspase-3/7
activity was measured by the Caspase-Glo 3/7 functional assay. In
this study, results demonstrated a 34.1% increase (P<0.05) in
U87 AJAP1 cells and 39.0% increase (P<0.05) in U251 AJAP1 cells,
compared with vector cells (Fig.
4C). Labeling of degraded DNA by TUNEL suggests apoptotic
mechanisms. Distribution of apoptotic nuclei in U87 cell lines
showed a tendency towards an accumulation of apoptotic cells in the
AJAP1 overexpressed group compared with vector control (Fig. 4D).
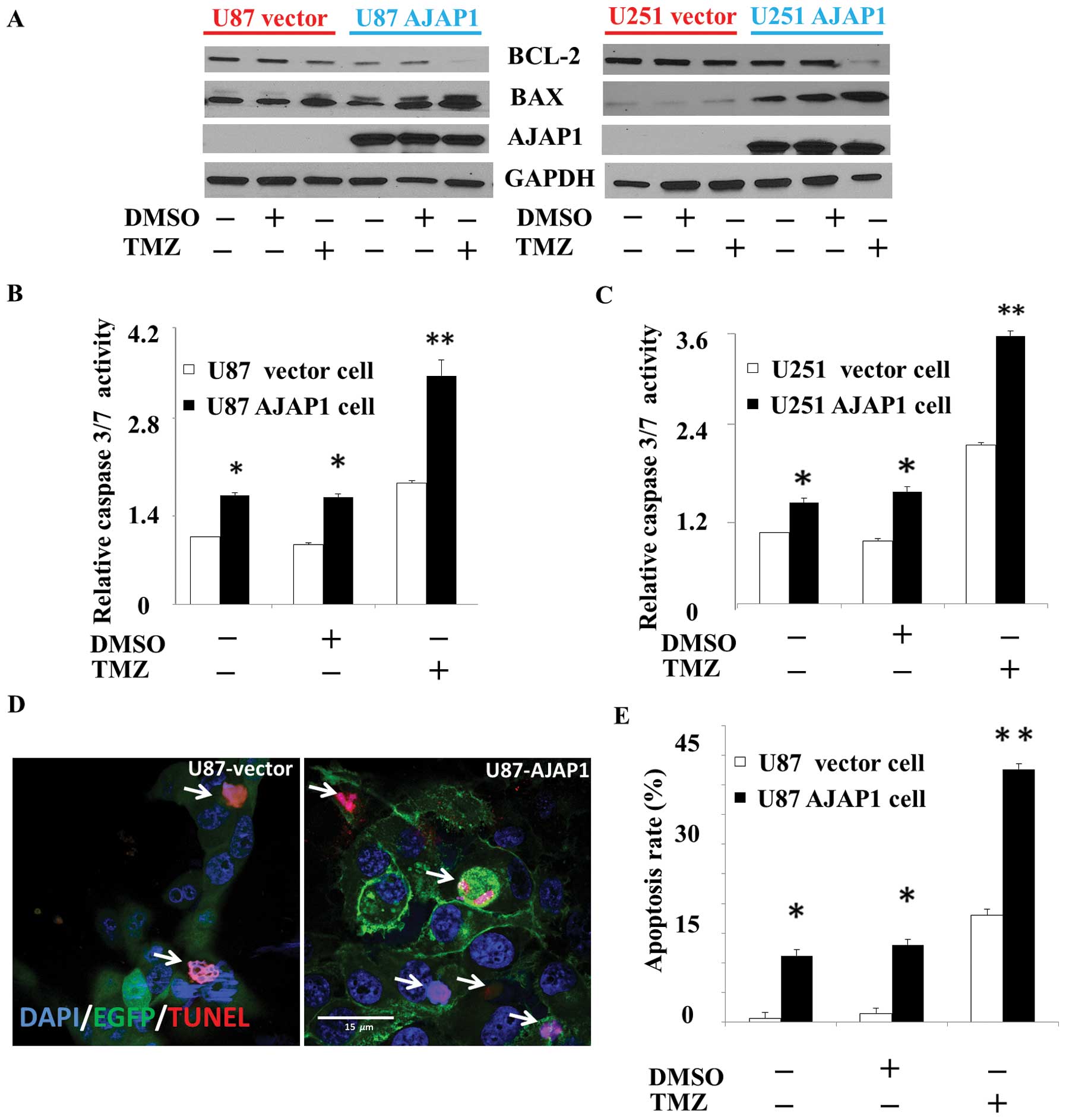
AJAP1 potentiates TMZ-induced
cytotoxicity in GBM cells
To investigate the consequence of AJAP1 expression
on the sensitivity to TMZ-induced cytotoxicity, the levels of Bax
and Bcl-2 were measured by western blot analysis in all treatment
groups. In this study, a dramatic increase in Bax/Bcl-2 ratio was
observed in U87 and U251 cells exposed to AJAP1 overexpressed plus
TMZ, compared to control cells not exposed to AJAP1 overexpression
(P<0.01) (Fig. 5A). In AJAP1
overexpressed cells treated with TMZ, the caspase-3/7 activity was
found dramatically increased compared with control group in U87
cells (68.5%, P<0.01) and U251 cells (88.9%, P<0.01)
(Fig. 5B and C). TUNEL labeling
showed that the U87 AJAP1 cells contain significantly more
apoptotic nuclei 48 h after TMZ treatment compared with control
group (36.7%, P<0.01) (Fig. 5D and
E). Moreover, U87 AJAP1 cells with TMZ treatment presented more
typical morphological apoptotic changes, such as nuclear
condensation, nuclear fragmentation and shrinkage of the cells
(Fig. 5D). Taken together, these
results showed that AJAP1 sensitized U87 and U251 cells to TMZ, as
demonstrated by cellular Bax/Bcl-2 ratio, caspase-3/7 activity and
TUNEL labeling.
Discussion
The results summarized here strongly suggest that
AJAP1 is a negative transcriptional regulator of MAGEA2, by
possibly interacting with the promoter of MAGEA2 in GBM cells,
resulting in downregulation of MAGEA2 expression by mRNA and
protein analyses. AJAP1 was initially described as a novel
component of adherens junctions in polarized epithelial cells,
which co-localized and co-immunoprecipitated with endogenous
E-cadherin (23). Direct
interaction between β-catenin and AJAP1 in an in vitro
pull-down assay suggested that AJAP1 might be linked to the
E-cadherin–mediated junctional complexes via β-catenin (24). β-catenin contains armadillo repeats
and is able to bind to other proteins such as transcription factors
(25). The ability of β-catenin to
bind to other proteins is regulated by tyrosine kinases (26) and serine kinases such as GSK-3
(27). Various signals such as the
Wnt signaling pathway can inhibit GSK-3-mediated phosphorylation of
β-catenin, (28) allowing
β-catenin to translocate to the cell nucleus, interact with
transcription factors and regulate gene transcription. We
hypothesize that AJAP1 may be translocated to the nucleus, possibly
via its interaction with β-catenin complexes, where it then can
bind and inhibit MAGEA2 transcription. The in-depth mechanism of
how AJAP1 regulates MAGEA2 needs further investigation.
We previously demonstrated the frequent loss of
AJAP1 in glioblastoma cell lines and primary tumors with
high-resolution genome-wide mapping (29). AJAP1 is a putative tumor
suppressor-like gene that maps to the 1p36 region, where
alterations are commonly associated with cancer (4) and has been implicated in cancer cell
invasion, adhesion and migration (6,30–32).
This study explores its potential role in gene regulation and
cytotoxicity. Using extensive microarray analyses, we identified
genetic alterations that occur in the context of glioma cell AJAP1
expression. We found that MAGEA2 expression is significantly
decreased when AJAP1 is expressed. MAGEA2 provides a potent
anti-apoptotic function to cancer cells partly through
downregulation of the p53 pathway (14). Using in vitro approaches, we
showed that AJAP1 expression produced proapoptotic results in
glioma cells. Apoptosis is regulated by several protein families,
including the upstream Bcl-2 family (i.e. the anti-apoptotic Bcl-2
and pro-apoptotic Bax) and the downstream caspase family (i.e.
caspase-3/7) (33). Thus, we
investigated the levels of expression of Bax and Bcl-2 proteins in
U87 and U251 GBM cells following restoration of AJAP1. Glioma cells
with restoration of AJAP1 increase the expression of Bax and
decrease the expression of Bcl-2, which suggests a regulatory
effect of AJAP1 in Bax/Bcl-2 ratios. Bcl2 is known to inhibit
cytochrome c release from mitochondria, thus blocking
caspase activation, while Bax has the opposite function, which in
turn promotes the release of cytochrome c into the cytosol
from mitochondria to activate caspase-3/7 (34). In our study, caspase-3/7 activity
increased in the presence of AJAP1, compared with the vector
control. These results indicated that AJAP1 could, at least in
part, induce the mitochondria-related apoptosis pathways.
The resistance of GBM to standard of care
chemotherapy (TMZ) may be more complex than simple dependence on
MGMT levels (35,36). It is likely a multifactorial
phenomenon involving several major mechanisms, such as increased
repair of DNA damage, reduced apoptosis, ectopic microRNA
expression and increased energy-dependent efflux of
chemotherapeutic drugs (37–39).
Here, we show for the first time the enhancement of TMZ-induced
apoptosis by AJAP1 in human glioma cell lines through MAGEA2
transcriptional regulation. Several studies have established that
MAGE-A proteins confer resistance to chemotherapeutic drugs that
act via p53-mediated apoptosis (15,40,41).
But the complete role of p53 in TMZ resistance is incompletely
known. A trend toward increased TMZ sensitivity in patients with
p53 mutations has also been suggested (39). We hypothesize that overexpression
of AJAP1 in GBM may represent an alternative mechanism, aside from
p53, to activate a tumor suppressive-like function of the p53
pathway.
In summary, AJAP1 appears to play a role in inducing
apoptosis by directly downregulating MAGEA2 in GBM. In addition,
AJAP1 can potentiate the sensitivity of TMZ in GBM cells.
Therefore, AJAP1 may serve as a candidate biomarker for
chemotherapy in GBM, but also a putative therapy opportunity for
GBM.
Acknowledgements
We thank Dr Jing Mi (Department of
Surgery, Duke University Medical Center) and Dr Hailan Piao
(Department of Pathology, Duke University Medical Center) for
expert technological assistance. This project was supported by
Award no. I01BX007080 from the Biomedical Laboratory Research and
Development Service of the VA Office of Research and
Development.
References
|
1.
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, et al: Effects of radiotherapy with concomitant
and adjuvant temozolomide versus radiotherapy alone on survival in
glioblastoma in a randomised phase III study: 5-year analysis of
the EORTC-NCIC trial. Lancet Oncol. 10:459–466. 2009.
|
|
2.
|
Gravendeel LA, Kouwenhoven MC, Gevaert O,
de Rooi JJ, Stubbs AP, et al: Intrinsic gene expression profiles of
gliomas are a better predictor of survival than histology. Cancer
Res. 69:9065–9072. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Verhaak RG, Hoadley KA, Purdom E, Wang V,
Qi Y, et al: Integrated genomic analysis identifies clinically
relevant subtypes of glioblastoma characterized by abnormalities in
PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 17:98–110. 2010.
View Article : Google Scholar
|
|
4.
|
Henrich KO, Schwab M and Westermann F:
1p36 tumor suppression - a matter of dosage? Cancer Res.
72:6079–6088. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Zlatescu MC, TehraniYazdi A, Sasaki H,
Megyesi JF, Betensky RA, Louis DN, et al: Tumor location and growth
pattern correlate with genetic signature in oligodendroglial
neoplasms. Cancer Res. 61:6713–6715. 2001.
|
|
6.
|
Lin N, Di C, Bortoff K, Fu J, Truszkowski
P, Killela P, Duncan C, McLendon R, Bigner D, Gregory S and Adamson
DC: Deletion or epigenetic silencing of AJAP1 on 1p36 in
glioblastoma. Mol Cancer Res. 10:208–217. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Aoki T, Hashimoto N and Matsutani M:
Management of glioblastoma. Expert Opin Pharmacother. 8:3133–3146.
2007. View Article : Google Scholar
|
|
8.
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, et al: Mirimanoff Radiotherapy plus concomitant and adjuvant
temozolomide for glioblastoma. N Engl J Med. 352:987–996. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Salvati M, D’Elia A, Formichella AI and
Frati A: Insights into pharmacotherapy of malignant glioma in
adults. Expert Opin Pharmacother. 10:2279–2290. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Giese A, Bjerkvig R, Berens ME and
Westphal M: Cost of migration: invasion of malignant gliomas and
implications for treatment. J Clin Oncol. 21:1624–1636. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Kitange GJ, Carlson BL, Schroeder MA,
Grogan PT, Lamont JD, Decker PA, Wu W, James CD and Sarkaria JN:
Induction of MGMT expression is associated with temozolomide
resistance in glioblastoma xenografts. Neuro Oncol. 11:281–289.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Frosina G: DNA repair and resistance of
gliomas to chemotherapy and radiotherapy. Mol Cancer Res.
7:989–999. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Oliva CR, Nozell SE, Diers A, McClugage
SG, et al: Acquisition of temozolomide chemoresistance in gliomas
leads to remodeling of mitochondrial electron transport chain. J
Biol Chem. 285:39759–39767. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Marcar L, Maclaine NJ, Hupp TR and Meek
DW: Mage-A cancer/testis antigens inhibit p53 function by blocking
its interaction with chromatin. Cancer Res. 70:10362–10370. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Monte M, Simonatto M, Peche LY, Bublik DR,
Gobessi S, Pierotti MA, Rodolfo M and Schneider C: MAGE-A tumor
antigens target p53 transactivation function through histone
deacetylase recruitment and confer resistance to chemotherapeutic
agents. Proc Natl Acad Sci. 103:11160–11165. 2006. View Article : Google Scholar
|
|
16.
|
Yang B, O’Herrin SM, Wu J, Reagan-Shaw S,
Ma Y, Bhat KM, Gravekamp C, Setaluri V, Peters N, Hoffmann FM, Peng
H, Ivanov AV, Simpson AJ and Longley BJ: MAGE-A, MAGE-B, and MAGE-C
proteins form complexes with KAP1 and suppress p53-dependent
apoptosis in MAGE-positive cell lines. Cancer Res. 67:9954–9962.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Bhan S, Negi SS, Shao C, Glazer CA, Chuang
A, Gaykalova DA, Sun W, Sidransky D, Ha PK and Califano JA: BORIS
binding to the promoters of cancer testis antigens, MAGEA2, MAGEA3,
and MAGEA4, is associated with their transcriptional activation in
lung cancer. Clin Cancer Res. 17:4267–4276. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Glazer CA, Smith IM, Bhan S, Sun W, Chang
SS, Pattani KM, Westra W, Khan Z and Califano JA: The role of
MAGEA2 in head and neck cancer. Arch Otolaryngol Head Neck Surg.
137:286–293. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Nardiello T, Jungbluth AA, Mei A,
Diliberto M, Huang X, Dabrowski A, Andrade VC, Wasserstrum R, Ely
S, Niesvizky R, Pearse R, Coleman M, Jayabalan DS, Bhardwaj N, Old
LJ, Chen-Kiang S and Cho HJ: MAGE-A inhibits apoptosis in
proliferating myeloma cells through repression of Bax and
maintenance of survivin. Clin Cancer Res. 17:4309–4319. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Sawada M, Nakashima S, Banno Y, Yamakawa
H, Hayashi K, Takenaka K, Nishimura Y, Sakai N and Nozawa Y:
Ordering of ceramide formation, caspase activation, and Bax/Bcl-2
expression during etoposide-induced apoptosis in C6 glioma cells.
Cell Death Differ. 7:761–772. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Tanaka K, Asanuma M and Ogawa N: Molecular
basis of anti-apoptotic effect of immunophilin ligands on hydrogen
peroxide-induced apoptosis in human glioma cells. Neurochem Res.
29:1529–1536. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Stegh AH, Chin L, Louis DN and DePinho RA:
What drives intense apoptosis resistance and propensity for
necrosis in glioblastoma? A role for Bcl2L12 as a multifunctional
cell death regulator. Cell Cycle. 7:2833–2839. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Shore EM and Nelson WJ: Biosynthesis of
the cell adhesion molecule uvomorulin (E-cadherin) in Madin-Darby
canine kidney epithelial cells. J Biol Chem. 266:19672–19680.
1991.PubMed/NCBI
|
|
24.
|
Bharti S, Handrow-Metzmacher H,
Zickenheiner S, Zeitvogel A, Baumann R and Starzinski-Powitz A:
Novel membrane protein shrew-1 targets to cadherin-mediated
junctions in polarized epithelial cells. Mol Biol Cell. 15:397–406.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Gottardi CJ and Peifer M: Terminal regions
of beta-catenin come into view. Structure. 16:336–338. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Lilien J and Balsamo J: The regulation of
cadherin-mediated adhesion by tyrosine
phosphorylation/dephosphorylation of β-catenin. Curr Opin Cell
Biol. 17:459–465. 2005.PubMed/NCBI
|
|
27.
|
Castellone MD, Teramoto H, Williams BO,
Druey KM and Gutkind JS: Prostaglandin E2 promotes colon cancer
cell growth through a Gs-axin-beta-catenin signaling axis. Science.
310:1504–1510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Liu X, Rubin JS and Kimmel AR: Rapid
Wnt-induced changes in GSK3beta associations that regulate
beta-catenin stabilization are mediated by Galpha proteins. Curr
Biol. 15:1989–1997. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Hino S, Tanji C, Nakayama KI and Kikuchi
A: Phosphorylation of β-catenin by cyclic AMP-dependent protein
kinase stabilizes β-catenin through inhibition of its
ubiquitination. Mol Cell. 25:9063–9072. 2005.
|
|
30.
|
McDonald JM, Dunlap S, Cogdell D, et al:
The SHREW1 gene, frequently deleted in oligodendrogliomas,
functions to inhibit cell adhesion and migration. Cancer Biol Ther.
5:300–304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Schreiner A, Ruonala M, Jakob V, et al:
Junction protein shrew-1 influences cell invasion and interacts
with invasion promoting protein CD147. Mol Biol Cell. 18:1272–1281.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Cogdell D, Chung W, Liu Y, McDonald JM,
Aldape K, Issa JP, Fuller GN and Zhang W: Tumor-associated
methylation of the putative tumor suppressor AJAP1 gene and
association between decreased AJAP1 expression and shorter survival
in patients with glioma. Chin J Cancer. 30:247–253. 2011.
View Article : Google Scholar
|
|
33.
|
Jarskog LF, Selinger ES, Lieberman JA and
Gilmore JH: Apoptotic proteins in the temporal cortex in
schizophrenia: high bax/bcl-2 ratio without caspase-3 activation.
Am J Psychiatry. 161:109–115. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Ryan KM, Phillips AC and Vousden KH:
Regulation and function of the p53 tumor suppressor protein. Curr
Opin Cell Biol. 13:332–337. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Kaina B and Christmann M: DNA repair in
resistance to alkylating anticancer drugs. Int J Clin Pharmacol
Ther. 40:354–367. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Bredel M, Bredel C, Juric D, et al: Tumor
necrosis factor-alpha induced protein 3 as a putative regulator of
nuclear factorkappaB- mediated resistance to O6-alkylating agents
in human glioblastoma. J Clin Oncol. 24:274–287. 2006. View Article : Google Scholar
|
|
37.
|
Prokopenko O and Mirochnitchenko O:
Ischemia-reperfusion-inducible protein modulates cell sensitivity
to anticancer drugs by regulating activity of efflux transporter.
Am J Physiol Cell Physiol. 296:1086–1097. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Chekhun VF, Lukyanova NY, Kovalchuk, et
al: Epigenetic profiling of multidrug-resistant human MCF-7 breast
adenocarcinoma cells reveals novel hyper- and hypomethylated
targets. Mol Cancer Ther. 6:1089–1098. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Roberti A, Sala DL and Cinti C: Multiple
genetic and epigenetic interacting mechanisms contribute to
clonally selection of drug-resistant tumors: current views and new
therapeutic prospective. J Cell Physiol. 207:571–581. 2006.
View Article : Google Scholar
|
|
40.
|
Duan Z, Duan Y, Lamendola DE, Yusuf RZ, et
al: Overexpression of MAGE/GAGE genes in paclitaxel/doxorubicin
resistant human cancer cell lines. Clin Cancer Res. 9:2778–2785.
2003.PubMed/NCBI
|
|
41.
|
Weeraratne SD, Amani V, Neiss A, et al:
MiR-34a confers chemosensitivity through modulation of MAGE-A and
p53 in medulloblastoma. Neuro Oncol. 13:165–175. 2011. View Article : Google Scholar : PubMed/NCBI
|