Contents
Introduction
Apoptosis
HSF1
Targeting HSP27
Targeting HSP70
Targeting HSP90
Conclusions
Introduction
Stress or heat shock proteins (HSPs) are a family of
highly conserved proteins induced in response to a wide variety of
physiological and environmental insults such as hypoxia, hyperoxia,
exposure to UV light and chemicals, viral agents, surgical stress,
nutritional deficiencies (e.g. glucose deprivation), emotional and
mechanical stress, or other stresses, thus helping maintain
cellular homeostasis under stress or allowing the cell to survive
to lethal conditions (1–4).
Mammalian HSPs have been classified into six
families according to their molecular size: HSP100, HSP90, HSP70,
HSP60, HSP40 and small HSPs (15 to 30 kDa) including HSP27. Family
members of HSPs are expressed either constitutively or regulated
inductively, and are present in different subcellular compartments
(5). High molecular weight HSPs
are ATP-dependent chaperones, whereas small HSPs act in an
ATP-independent fashion (5). As
molecular chaperones, the function of HSPs is to regulate protein
folding, transport, translocation and assembly, especially helping
in the refolding of misfolded proteins or assisting in their
elimination if they become irreversibly damaged after various
stresses or environmental insults.
Cancer cells, with higher metabolic requirements and
more abundant signal transduction pathways than normal cells,
thereby have a higher need of chaperones than non-transformed cells
to maintain cancer cells survival. In addition, by commanding over
the folding and stabilization of relevant oncoproteins, HSPs stand
at the crossroads of multiple important oncogenic pathways.
Inhibition of HSPs hereby offers the unique advantage of depleting
multiple oncoproteins while simultaneously attacking several
pathways necessary for tumor progression (6). The most studied stress-inducible HSPs
are HSP90, HSP70 and HSP27. Indeed, the expression and/or activity
of the three HSPs is abnormally high in cancer cells and further
increased after many different death stimuli (5,7).They
are powerful anti-apoptotic proteins, associating with key
apoptotic factors, and thereby blocking this cell death process at
different levels (8). Preclinical
trials have proved that overexpression of the HSPs increases tumor
growth, metastatic potential, and resistance to chemotherapy in
rodent models. The inhibition of HSP90, HSP70 and/or HSP27 is thus
emerging as a novel strategy for cancer therapy. In this review, we
will present our view on the role of HSP90, HSP70 and HSP27 in
apoptosis (Fig. 1) and the
emerging strategies that are being developed for cancer therapy in
clinic based on the inhibition of these three HSPs.
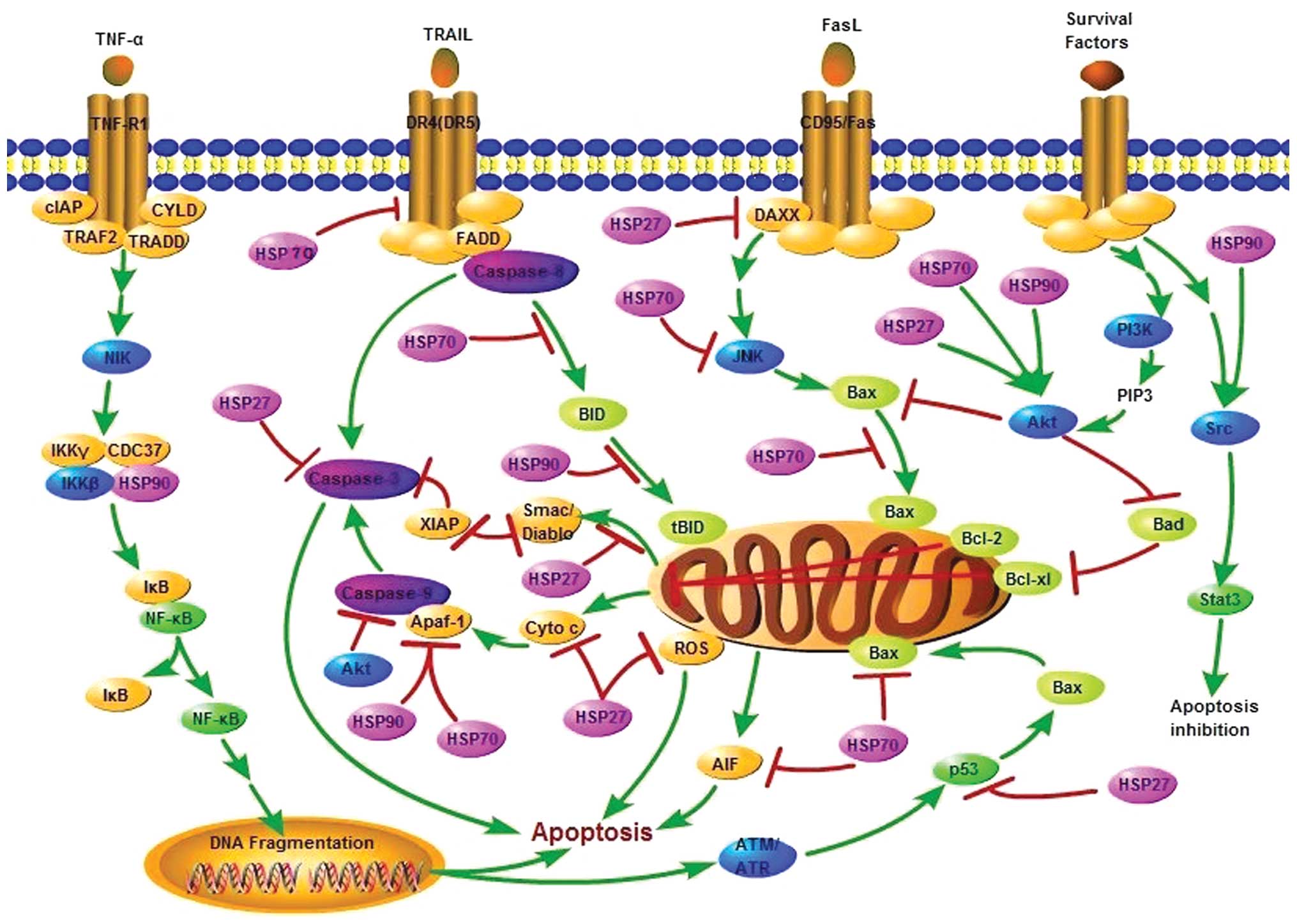
Apoptosis
Mainly, two pathways of apoptosis can be
distinguished, although crosstalk between the two signal
transducing cascades is present: the intrinsic or mitochondrial
pathway and the extrinsic or death receptors pathway. The two
signal-transducing cascades meet at the point of caspase-3, an
effector caspase that leads to the typical morphologic and
biochemical changes of the apoptotic cell.
The intrinsic pathway involves intracellular stress
signals that provoke the permeabilization of the outer
mitochondrial membrane, resulting in the release of proapoptotic
molecules normally confined to the intermembrane space. Outer
mitochondrial membrane permeabilization leads to the release of
caspase activators under control of the Bcl-2 (B-cell
lymphocytic-leukaemia proto-oncogene) family of proteins. Bcl-2
proteins include antiapoptotic members such as Bcl-2 and Bcl-xL,
multi-domain proapoptotic members mainly Bax and Bak (9,10)
and a series of BH3 domain-only proapoptotic proteins, such as Bid,
that function upstream of Bax and Bak (11). One of the released mitochondrial
molecules is cytochrome c, which interacts with cytosolic
apoptosis protease-activating factor-1 (Apaf-1) and procaspase-9 to
form the caspase-3 activation complex, apoptosome (12). Apoptosis inducing factor (AIF) and
the DNase, endonuclease G (EndoG), are other mitochondria
intermembrane proteins released upon an apoptotic stimulus. They
translocate to the nucleus and trigger caspase-independent nuclear
changes (13). Two additional
mitochondrial proteins, Smac/Diablo and Htra2/Omi, activate
apoptosis by neutralizing the inhibitory activity of the IAPs
(inhibitory apoptotic proteins) that associate with and inhibit
some of the activated caspases (14).
The extrinsic pathway is triggered through plasma
membrane proteins of the tumor necrosis factor (TNF) receptor
family known as death receptors, and leads to the direct activation
of caspases, starting with the receptor-proximal caspase-8 or
caspase-10 in the death-inducing signalling complex (DISC).
Caspase-8 either directly activates the downstream cascade of
caspases or cleaves Bid into an active truncated form named tBid
that connects the extrinsic to the intrinsic apoptotic pathways
through mitochondria permeabilization (15).
HSF1
The rapid induction of HSPs in response to multiple
stress is collectively referred to as the heat shock response
(HSR)(16). The HSR is mediated at
the transcriptional level by heat shock transcription factors
(HSFs), the upstream transcriptional regulators of HSPs (17). So far, the vertebrates HSFs that
have been identified include HSF1, 2, 3, 4 and HSFY, all of which
exhibit a similar structure with a highly conserved amino-terminal
helix-turn-helix DNA-binding domain and a carboxy-terminal
transactivation domain (18–20).
Different HSFs are differently regulated and have a different
impact on transcriptional responses, which suggests their
specialized functions in response to distinct stimulation (21,22).
Among them, HSF1 is considered as the master transcription factor
for the HSR (17,23). It not only regulates the expression
of HSPs but also orchestrates the survival of cells in response to
different forms of cellular stress (21,23).
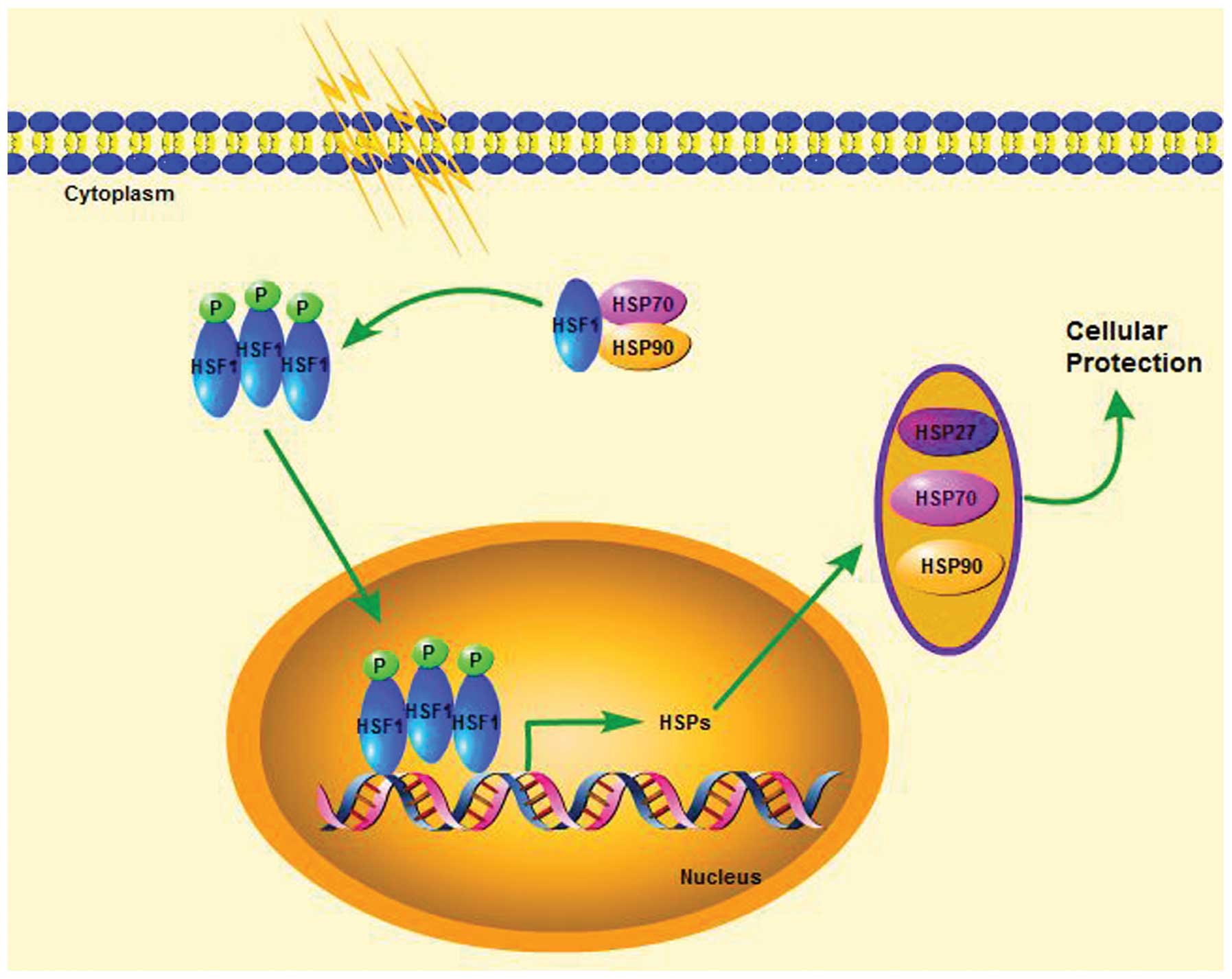
In physiological conditions HSF1 exists as an inactive monomer.
HSP90 and HSP70 can bind to HSF1 in unstressed state to abrogate
the transcription function of HSF1 and dissociate from it under the
exterior cellular stress to activate HSF1 (24). Then the monomeric HSF1 trimerizes,
phosphorylates and translocates to the nucleus. In the nucleus,
HSF1 binds cis-acting DNA elements, termed heat shock
elements (HSEs), which are present in heat shock genes, and
activate transcription of the Hsp genes, e.g. HSP27, HSP70 and
HSP90 (6) (Fig. 2). The inhibition of HSP90
expression therefore should activate HSF1 with the increased
expression of HSP27 and HSP70 (24). Because both HSP27 and HSP70 are
anti-apoptotic proteins with tumorigenic properties, the overall
anticancer effect of the HSP90 inhibitors might be impaired (see
below). Considering this fact, targeting HSF1 should enable the
simultaneous downregulation of several HSPs. Additionally, tumor
cells are more dependent on HSF1 than normal cells for
proliferation and survival, as confirmed by both cell-based and
clinically relevant examples (23). Consequently, the targeting of HSF1
can be considered as a potentially efficient strategy to combat
cancer. However, lacking of specificity for HSF1 inhibitors
simultaneously is its greatest deficiency.
Targeting HSP27
HSP27 structure
HSP27 (HSPB1) belongs to a member of the small heat
shock proteins (sHSP). The primary structure of HSP27 is highly
homologous to other members of the sHSP family, containing the
conserved α-crystallin domain and differing in the C- and
N-terminal regions. HSP27 is expressed in all human tissues,
including astrocytes and primary neuronal cells but mainly in
skeletal, smooth and cardiac muscles (25) and shares with other members of the
small HSP family the capacity to phosphorylated and oligomerize.
Human Hsp27 can be phosphorylated on three serine residues 15, 78
and 82 and on threonine (Thr143) by a large number of kinases
including MAPKAP kinases 2 and 3, p90Rsk, PKC, PKD and PKG
(26). The phosphorylation is a
reversible event that modulates the oligomerization of the protein:
its dephosphorylation favors the formation of large oligomers
(27,28). However, some studies have found
that the ability to oligomerize is reduced in in vitro cell
based assays by phosphorylation, and that in vivo
oligomerization has been tied to cell-cell contact and is
independent of phosphorylation status (26,29).
HSP27 can form oligomers of up to 1,000 kDa. This oligomerization
is a highly dynamic process that seems to play a central role in
regulating the chaperone activity of HSP27, the multimer being the
binding competent state for affinity for client proteins (30). The dimer of HSP27 is the ‘building
block’ for multimeric complexes. Particularly, phosphorylated and
small oligomers of HSP27 are efficient in binding to F-actin and
Daxx (31) and it is the
phosphorylated form of HSP27 that protects from neurotoxicity
(32).
The function of HSP27 in cancer
HSP27 has a strong protective effect on cells. High
levels of HSP27 have been observed in many cancer types, and the
tumorigenic potential of HSP27 has been observed in experimental
models (33). Many clinical trials
have also shown its association with promoting drug resistance,
aggressive cancers, metastasis, and poor patient outcomes (34–37).
The strong protective effect of HSP27 is mainly due to its vital
function at apoptosis regulation. HSP27 is able to block apoptosis
at different stages, because of its interaction with a number of
partners implicated in the apoptotic pathways.
Numerous studies describe that HSP27 inactivates the
caspase cascade through its binding with caspase-3 and cytochrome
c released from mitochondria (38–40).
Knockdown of HSP27 by small interfering RNAs displayed increased
caspase-3 activation, thereby inducing more apoptosis (41). Other data also confirmed that HSP27
prevents apoptosis and induces resistance to chemotherapy through
sequestration of cytochrome c when released from the
mitochondria into the cytosol (38).
High intracellular levels of HSP27 can inhibit
caspase activation by interfering upstream of the mitochondria
(42). This effect seems to have
connection with the ability of HSP27 to stabilize cytoskeletal
elements including actin microfilaments, such as F-actin, to
prevent the cytoskeletal disruption and Bid intracellular
redistribution that precede cytochrome c release, which is
also required for the activation of matrix metalloproteinase 2
(MMP2) (42). In myeloma cell
lines, it has been reported that HSP27 activation blocks release of
Smac (second mitochondria-derivated activator of caspase) from
mitochondria (43). In stressed
cells, HSP27 has also shown its importance in Akt activation,
through binding the protein kinase Akt (7). In renal epithelial cells, HSP27
indirectly inactivates Bax and its translocation to mitochondria.
This is due to an increase of PI3-kinase activity that activates
Akt and promotes interaction between Akt and Bax (38,44).
The phosphorylated form of HSP27 directly interacts with
death-domain-associated protein (Daxx), which connects Fas
signaling to the protein kinase Ask1 that mediates
caspase-independent cell death (7,31).
Large oligomers of HSP27 have also been described to
display anti-oxidant property, which is related to its ability to
maintain glutathione in its reduced (non-oxidized) form to abolish
the production of the potentially lethal burst of intracellular
reactive oxygen species (ROS) that can occur (45,46).
These anti-oxidant properties of HSP27 particularly contribute to
its cytoprotection in neuronal cells.
The function of HSP27 and the role that it plays in
cancer were recently reviewed (38). These numerous reports account for
the role of HSP27 in apoptotic cell death inhibition, which also
emphasizes its properties in cancer therapy (44,47).
The inhibition of HSP27 in cancer
therapy
The strong cyto-protective function of HSP27,
together with the fact that this protein is overexpressed in most
cancers, makes this chaperone an attractive target in cancer
therapy. Depletion of HSP27 in various animal models induces the
regression of tumors (48). Though
starting late, Hsp27 therapies have produced only few advances
after tremendous efforts.
The antisense oligonucleotide OGX-427 is the only
known specific inhibitor of HSP27 that can be safely administered
in patients and is currently in phase II clinical trials
(http://www.oncogenex.ca/). OGX-427 targets the
human hsp27 translation initiation site
(5′-GGGACGCGGCGCTCGGTCAT-3′) and prevents the translation of hsp27
mRNA, thereby decreasing the expression of the protein compared to
untreated cells (49).
Less specific, the chemical molecule RP101 (also
known as bromovinyldeoxyuridine, BVDU, brivudine) was reported to
improve the efficacy of chemotherapy in pancreatic cancer through
its interaction with HSP27 (50).
RP101 is a nucleoside that binds via π-stacking with Phe29 and
Phe33 of Hsp27 thereby inhibiting its function (50). Functioning as a chemosensitizing
agent and preventing the development of resistance, RP101 recently
completed a phase II clinical trial for the treatment of pancreatic
cancer in combination with gemcitabine (Hidalgo M; http://clinicaltrials.gov/ct2/show/NCT00550004?term=NCT00550004andrank=1,
2011). However, overdosing caused an increase of the toxic
side-effects of gemcitabine and thus the combination provided a 25%
increase in survival only for patients that had a body surface area
(BSA) ≥1.85 m2 compared with gemcitabine combined with
placebo (50). There were no
side-effects caused by RP101, and more accurate dosing would likely
improve the survival rates for all patients regardless of size
(50). Development of
second-generation candidates of RP101 are ongoing.
A strategy of peptide aptamers has also been used to
target HSP27. Protein aptamers, small amino acid sequences, are
designed to bind to a specific protein domain, thus inhibiting its
function (51). Gibert et
al have shown that peptide aptamers (PA11 and PA50) that
specifically interact with HSP27 are able to disturb the
dimerization and oligomerization of the chaperone, thereby acting
as negative regulators of HSP27 anti-apoptotic and cytoprotective
properties (52). PA11 prevents
the HSP27 oligomerization, which leads to the inability of HSP27 to
inhibit early stage protein aggregation and induces proteotoxic
stress that ends in cell death. PA50, through a different
mechanism, mainly inhibits HSP27 dimerization, disrupting the
ability of HSP27 to participate in cell-signaling events thereby
interfering with processes essential for cell survival. In
xenograft models these peptide aptamers strongly reduced tumor
cells growth (52). Similar to the
small molecule inhibitors of HSP27, peptide aptamers are not
effective on their own but are used to sensitize cancers to other
therapies. The pre-clinical success of peptide aptamers suggests
this avenue of cancer therapy has potential.
Different kind of inhibitors, which have been
experimentally tested, like the flavonoid quercetin and the
diterpene triepoxide, triptolide (24), act at the level of the HSF1 to
block the transcription of heat shock proteins genes, thus
inhibiting the heat shock response. However, such approaches are
non-specific since through HSF1 inhibition, all the
stress-inducible heat shock proteins can be blocked affecting
important housekeeping functions in normal cells.
Targeting HSP70
Human Hsp70: structure and general
function
HSP70 refers to a family of chaperone proteins that
are 70 kDa. The HSP70 human genome superfamily consists of at least
13 members (53). There are four
major proteins: constitutively expressed HSC70 (HSP73 or HSPA8),
endoplasmic reticulum-localized GRP78/Bip, mitochondrial mtHSP70
and stress-inducible HSP70 (HSP72 or HSPA1) (called here simply
HSP70) (54).
HSC70 is ubiquitously expressed in practically all
organs and tissues. Under normal conditions, it functions as
ATP-dependent molecular chaperone that assists the folding of newly
synthesized polypeptides, the assembly of multi-protein complexes
and the transport of proteins across cellular membranes (55–57).
Its levels are also increased under stress conditions showing the
involvement in stress response (57). On the contrary, the expression of
HSP70 is often not observed under non-stress conditions. Under
stressful conditions, elevated HSP70 levels allow cells to cope
with increased concentrations of unfolded or denatured proteins
(58).
All of the proteins share homology and contain two
distinct functional domains: a C-terminal peptide-binding domain
(PBD) and the N-terminal ATPase domain (ABD), which were connected
through a hydrophobic linker and both domains are important for
substrate binding and stabilization. The PBD, which include a
carboxyl-terminal chaperone EEVD motif, is responsible for
substrate binding and refolding. The ABD, containing the ATPase
pocket and binding J-domain-containing proteins, such as HSP40 that
regulate the HSP70 ATPase activity, in turn, facilitates the
release of the client protein after ATP hydrolysis. A conserved
proline in the ATPase domain is essential to alternate HSP70
conformations in response to ATP binding and hydrolysis (59,60).
HSP70 chaperone activity is regulated by distinct
co-chaperones, e.g. Hip, CHIP or Bag-1. These co-chaperones bind to
HSP70 and modulate its chaperone function by increasing or
decreasing HSP70 affinity for substrates through the stabilization
of the ADP or ATP bound state of HSP70. They can be classified into
three groups. i) The J-domain co-chaperones, like HSP40, are a
relatively large group that binds to the HSP70 ABD and stimulate
the low ATPase activity of this chaperone (1). ii) The nucleotide exchange factor
co-chaperones catalyze the release of ADP which is required for the
completion of the HSP70 ATPase cycle. Members of this group are
Bag-1, HSP110, or HSPBP1. iii) The TPR domain co-chaperones (Hop,
CHIP) bind to the C-terminal EEVD motif presented in both HSP70 and
HSP90. They are essential for combinational assembly of HSP70 and
HSP90 complexes, required for the stabilization of HSP90 client
proteins. CHIP, with ubiquitin ligase activity, has been implicated
in the ubiquitination of at least some HSP client proteins
(5,61).
The function of HSP70 in cancer
Similar to HSP27, HSP70 is also abundantly expressed
in many tumor forms and is accompanied by increased cell
proliferation, metastases and poor response to chemotherapy.
Constitutively high expression of HSP70 enhances the ability of the
cancer cells to survive to a range of lethal conditions. The
cytotoxic effect of HSP70 down-modulation is particularly strong in
transformed cells yet undetectable in normal, non-transformed cell
lines or primary cells (62). This
fact has been interpreted by assuming that tumor cells, as compared
to their normal counterparts, exhibit a constitutively stressed
phenotype with an enhanced dependency on the cytoprotective action
of HSP70. HSP70 exerts the cytoprotective action probably through
its ability to inhibit apoptosis. Gene ablation studies demonstrate
that HSP70 plays an important role in apoptosis. Cells lacking
hsp70.1 and hsp70.3, the two genes that code for inducible HSP70,
are highly sensitive to apoptosis induced by a wide range of lethal
stimuli (62). Ablation of the
testis specific isoform of HSP70 (hsp70.2) results in germ cell
apoptosis (63).
HSP70 can regulate apoptosis at the different levels
from death receptors signaling to executors of cell death program
affecting both upstream and downstream of the death-associated
mitochondrial events.
At the level of death receptors, HSP70 can bind to
the death receptors DR4 and DR5, thereby inhibiting the
TNF-α-related apoptosis-inducing ligand (TRAIL)-induced assembly
and activity of death inducing signaling complex (DISC) (64). HSP70 also appears to affect the
Bid-dependent apoptotic pathway. HSP70 inhibits TNF-α-induced cell
death and this protective effect is lost in Bid homozygous-deleted
MEF cells. HSP70 can block the cleavage of Bid by activated
caspase-8 (65).
At the premitochondrial level, HSP70 inhibits
stress-activated kinases, such as apoptosis signal regulating
kinase 1 (Ask1). In NIH3T3 cells, it was shown that downregulation
of HSP70 facilitates H2O2-induced Ask1
activation and subsequent apoptosis (66). HSP70 also negatively interferes
with MAPK family kinase activity, in particular, the p38 kinase and
the c-Jun N-terminal kinase (JNK) (67). Studies have found that HSP70
inhibits the apoptosis induced by hyperosmolarity modulating JNK
and ERK phosphorylation (68).
HSP70 has been shown to contribute to stabilize the
stress-activated kinases, such as non-phosphorylated protein kinase
C (PKC) and Akt, by means of binding to the non-phosphorylated
kinase via the kinase unphosphorylated carboxyl-terminus, priming
the kinase for rephosphorylation and stabilizing the protein
(69).
HSP70 has also been shown to affect some
transcription factors involved in the expression of the Bcl-2
family. Bcl-2 family of proteins, playing a critical role in the
regulation of apoptosis through controling the release of caspase
activators, are transcriptional targets of the tumor suppressor
protein p53. The transcription of Bcl-2 is repressed by p53,
whereas that of Bax is induced. HSP70 can form stable complexes
with mutated p53, thus inducing apoptosis in response to DNA
damage. HSP70 can also cover the nuclear localization sequence of
p53, thereby preventing its nuclear import (70).
At the mitochondrial level, HSP70 blocks
heat-induced apoptosis by binding to Bax to prevent its
translocation to the mitochondria (71), thus preventing outer mitochondrial
membrane permeabilization and inhibiting the release of
mitochondrial apoptogenic molecules, such as cytochrome c
and AIF (72). This HSP70 function
relies on both its chaperone HSP40, and its ATP hydrolytic
domains.
At the post-mitochondrial level, downstream of the
release of cytochrome c and upstream of the activation of
caspase-3, HSP70 has been demonstrated to directly bind to Apaf-1
to prevent the recruitment of procaspase-9 to the apoptosome, thus
inhibiting apoptosis (73,74). This interaction depends on the
ATPase domain of HSP70 (74).
HSP70 can prevent cell death under caspase
inactivation. That is HSP70 can also prevent caspase-independent
pathways (75,76). Indeed, HSP70 directly binds to AIF
and inhibits AIF nuclear translocation, thereby inhibiting
AIF-induced chromatin condensation (76–78).
The ATPase function of HSP70 was described to be necessary for this
interaction, which also depends on a region between amino acids 150
and 228 of AIF (76). HSP70 can
also indirectly associate with EndoG to prevent DNA fragmentation
through affecting AIF (79).
Moreover, HSP70 can also rescue cells from a later
phase of apoptosis. During the final phases of apoptosis, the main
characteristic is nuclear condensation and fragmentation, and the
chromosomal DNA fragmentation is digested by the DNase CAD (caspase
activated DNase) following activation by caspase-3. It has been
reported that HSP70 has an important influence on the enzymatic
activity and proper folding of CAD, which also depends on its
cochaperones: HSP40 and the inhibitor of CAD(ICAD), suggesting that
HSP70 plays a role in maintaining DNA integrity (80,81).
Some studies also found that HSP70 could protect GATA-1, another
final target of caspase-3, from caspase-3 cleavage (82). However, specific mechanism remains
to be further studied.
HSP70 has been shown to promote cancer cell
viability by safeguarding lysosomal integrity. In cysteine
cathepsin-dependent death, HSP70 acts to inhibit lysosomal membrane
permeabilization, thereby preventing the release of lysosomal
constituents into the cytosol, which contains a group of proteases
that are involved in apoptosis (83,84).
HSP70 is a crucial negative regulator of the
mitochondrial pathway of apoptosis that can block cell death at
several levels from death receptors signaling to executors of cell
death program affecting both upstream and downstream of the
death-associated mitochondrial events.
The inhibitors of HSP70
Despite the critical role of HSP70, as discussed
above, in protein regulation and cancer progression, tremendous
efforts have produced few advances in hsp70 inhibitors.Here, we
explore HSP70 inhibitors though three basic categories: small
molecule inhibitors, protein aptamers, and antibody treatments,
also, the targets of drugs - targeting PBD, targeting ABD and
targeting HSP70 co-chaperones are also discussed.
Small molecule inhibitors: a) Targeting the
peptide binding domain (PBD). A small molecule inhibitor called
2-phenylethynesulfonamide (PES) or pifithrin-μ interacts with the
C-terminal PBD of HSP70, disrupting the association between HSP70
and several of its cofactors such as HSP40 and client proteins,
including pro-apoptotic proteins: APAF-1, p53 and others (85). This disruption leads to the
aggregation of misfolded proteins, and the destabilization of
lysosome membranes, thus inducing cell death (85). PES has been proven as a potent
antitumor agent.
Neutralization of HSP70 functions could be achieved
with peptides that mimic a domain of the AIF which is required for
HSP70 binding. The AIF-derived peptides were designed carrying the
AIF region from amino acid 150 to 228, which was previously defined
as required for HSP70 binding in itsPBD and lack AIF pro-apoptotic
function (77). These peptides
bind HSP70 and block its function (62,76).
Experiments in vitro carried out on different cell lines,
such as leukemia, colon and breast cancer lines, demonstrated that
several of these peptides increase sensitivity to chemotherapy
(62). Experiments in vivo:
in syngeneic rat colon cancer and mouse melanoma models,
demonstrated that AIF-derived decoy for HSP70 (ADD70), an inhibitor
of HSP70, reduced the tumor size and metastatic potential, and led
to a complete and permanent cure after treatment with cisplatin
(86).
b) Targeting the amino-terminal ATPase domain
(ABD). ATP hydrolysis and ADP/ATP exchange play a central role
in HSP70 chaperone activity. Therefore, disruption of HSP70-ATP
interaction could lead to the inability of HSP70 to perform its
functions.
15-Deoxyspergualin (15-DSG), a natural
immunosuppressive agent, disrupting HSP70-ATP interaction through
binding to HSP70 and stimulating its ATPase activity, was the first
compound described by Nadeau et al in 1994 (87). It binds ABD with its main
structure, the dihydropyrimidine group. Screening for inhibitors of
HSP70 ATPase activity and a subset of the National Cancer Institute
drug collection brought about the identification of NSC630668, a
dihydropyrimidine, which also effectively blocked protein
translocation mediated by yeast HSC70 in vitro (88). Noteworthy is the second generation
compound MAL3-101 and its subsequent modifications, which was
described inhibiting HSP70 ATPase activity and blocking
proliferation of SK-BR-3 cancer cells (89). Fortuitously, MAL2-11B, an
intermediate in the synthesis of MAL3-101, was also shown to
interfere with the activity of a viral J-domain of a chaperone-like
protein, T antigen, suggesting that it may be a new class of
polyoma-virus inhibitors (90).
However, the exact action mechanism of these molecules remains
unclear.
VER-155008 is an adenosine-derived compound. It
functions to inhibit the chaperone activity of HSP70 and other
family members by binding the ATPase domain. Although further
studies are necessary to determine its specificity and potency,
some in vitro results are encouraging: inducing
caspase-dependent apoptosis in BT474 breast cancer cells and
non-caspase-dependent cell death in HCT116 colon cancer cells
(91). This product has undergone
pharmacokinetics studies in mice, but efficacy studies have yet to
be reported.
Azure C, methylene blue and myricetin have been
identified as inhibitors of HSP70 through a high-throughput
screening for ATP turnover mediated by human HSP70, but their
specificity for inducible HSP70 family has not yet been analyzed
(92).
MKT-077, a cationic rhodacyanine dye analog, can
also bind to the ABD of Hsp70 (93). Mechanistic studies indicate that
MTK-077 localizes in the mitochondria where it inhibits the
deleterious interaction of mitochondrial HSP70 with p53 by binding
to the mt-HSP70 ABD (94). Studies
on MKT-077 have generated significant excitement about this product
and it has been explored on phase I clinical trial as an antitumor
agent (95). However, MKT-077 was
found to be nephrotoxic in solid tumor-bearing patients due to lack
of binding specificity (95).
Although the product does not specifically bind to HSP70 (i.e., it
also binds to actin), such a drug-like compound deserves further
investigation.
Apoptozole was discovered to induce apoptosis in the
human embryonic carcinoma cell line while looking for small
molecules that induced apoptosis in the imidazole compound library
(96). It was shown to inhibit the
ATPase activity of HSC70, but further information is required to
define the precise molecular mode of action and the selectivity of
this compound (97).
Sphingolipids, another group of HSP70 ABD binding
agents, can bind and specifically inhibit HSP70 ATPase activity
in vitro depending on the rate of ATP hydrolysis (98).
Protein aptamers. Peptide aptamers, targeting
the ATP binding domain of HSP70 to attenuate the HSP70 function,
were recently demonstrated as promising drugs in cancer therapy
(51). The most potent aptamer,
A17, binds to the ABD of HSP70 and disrupts the function of HSP70
in a biochemical assay in vitro (99). A17 increases the sensitivity to
apoptosis induction by anticancer drugs (cisplatin and
5-fluorouracil) and, in vivo, has a strong antitumor effect
(99).
Antibody treatments. The most promising
strategy reported for developing HSP70 inhibitors utilizes the
immune system, and it is the only HSP70-targeted therapy currently
in clinical trials (clinicaltrials.gov). However, they are limited
by the lack of tumor-specific markers (100). A recently developed monoclonal
antibody, cmHsp70.1, successfully recognizes the extracellular
motif, TKDNNLLGRFELSG (TKD) of membrane bound HSP70 (101). Furthermore, tumors express HSP70
in the membrane while normal (non-transformed) cells do not, thus
making the TKD motif an excellent tumor-specific biomarker
(101). CmHsp70.1 has
successfully passed through a safety and efficacy phase I trial and
it is currently in a phase II clinical trial for non-small cell
lung cancer in combination with radio chemotherapy (102).
Targeting HSP90
HSP90 structure
HSP90, a highly abundant chaperone protein expressed
by all eukaryotic cells, belongs to another important class of the
HSP family (103). It is highly
conserved throughout evolution and accounts for 1–2% of total
cellular proteins, increasing upon induction from baseline levels
to 4–6% (104). It is an
ATP-dependent chaperone with various isoforms among which the most
prominent members in humans are HSP90α (inducible form) and HSP90β
(constitutive form) isoforms (now also called HSPC1 and HSPC3,
respectively) which are encoded by separate, but highly conserved
genes, and have different roles (105). The hsp90α was shown to be
constitutively expressed at low level but strongly heat inducible.
In contrast, the hsp90β gene (hsp90αβ1) is expressed constitutively
at a much higher level and is only weakly inducible following a
heat shock (105). HSP90 exists
as a homodimer and contains three major regions (104,106): i) the amino (N)-terminal domain,
with an adenosine triphosphate (ATP)-binding and hydrolyzing
pocket, is responsible for the protein’s ATPase activity, ii) the
charged middle linker region involved in co-chaperones and client
proteins recognition/binding, and iii) the carboxy (C)-terminal
dimerization domain which directs HSP90 dimerization contains the
tetratricopeptide repeat-binding (TRP) motif, EEVD. TPR-containing
co-chaperones such as Hop (HSP organizing protein), bind to this
motif regulating the ATPase function of HSP90 (17). ATP is required for HSP90 activity
and it is possible to determine a potential conformational
equilibrium of HSP90 (108,109). The available structural
information for HSP90s shows that the C-terminal domains are
involved in dimerization and that the dimer formation by these
domains is independent of nucleotide binding or client proteins or
co-chaperone interactions (108).
On the other hand, the dimerization of the N-terminal domains is
dependent on binding. The binding of ATP triggers the dimerization
of the N-terminal domains and enables client protein
binding/loading. HSP90-bound ATP is then hydrolyzed, and the energy
released by ATP hydrolysis enables client protein folding (104). ATP hydrolysis results in a
conformational changing from an elongated orientation in which the
N-terminal domains dimerize to a wide, open V-shaped orientation
releasing the client protein (108). There are a few contacts between
the middle domains, a gap remains between the two middle domains,
although each makes contact with the N-terminal domain of the other
protomer upon dimerization of the N-termini (110).
The function of HSP90 in cancer
As a molecular chaperone, like HSP27 and HSP70,
HSP90 helps nascent proteins adopt their biologically active
conformations, correct the conformation of misfolded proteins, and
helps incorrigibly misfolded proteins to be removed and degraded by
the ubiquitin-proteo-some system (104).
HSP90 functions as part of a multichaperone complex
via association with a variety of co-chaperones and client proteins
that rely on the complex for acquiring active conformation. It
facilitates the maturation, stability, activity and intracellular
sorting of more than 200 client proteins (104,111). These client proteins covering
almost all cellular processes have been identified (for an updated
list, see http://www.picard.ch/downloads/Hsp90facts.pdf). Many
of these client proteins are involved in critical cellular
functions that promote cell growth, proliferation and cell survival
which are also important to maintain the cancer phenotype. HSP90 is
overexpressed in cancer cells and several of its client proteins
are signaling oncoproteins that represent nodal points in multiple
oncogenic signaling pathways, including mutant cKIT, human
epidermal growth factor receptor 2 (HER2)/neu, mutant epidermal
growth factor receptor (EGFR), the BCR-ABL fusion protein and BRAF
(111–113). HSP90 client proteins are also
involved in other hallmark processes of cancer, including induction
of angiogenesis, mediation of apoptosis, and promotion of tissue
invasion and metastasis (114).
For example, HSP90 influences angiogenesis by chaperoning
hypoxia-inducible factor-1α (HIF-1α) and vascular endothelial
growth factor receptor (VEGFR) in addition to governing nitric
oxide synthase upregulation. HSP90 chaperones client proteins that
are apoptotic mediators, including Bcl-2, Apaf-1, the
serinethreonine protein kinase AKT/PKB and surviving (114). Also, HSP90 may promote tissue
invasion and metastasis through MMP-2 activation, digesting
extracellular matrix proteins (114). Other client proteins of HSP90
that play a role in cell signaling processes include FAK (integrin
pathway), IL6R (JAK/STAT3 pathway), IκB kinases (NFκB pathway), CDK
4, 6, 9, hTERT (cell cycle), p53 (tumor suppressor genes), and the
steroid hormone receptors (estrogen receptor and androgen receptor)
(115).
Because these oncogenic proteins substantially rely
on the function of HSP90 for their maturation and/or stabilization,
as well as regulation of their activated states (116), inhibition of HSP90 provides the
unique advantage of causing depletion of multiple oncogenic client
proteins, while simultaneously leading to blockade of many key
cancer causing pathways, and hence leads to potent anticancer
effect.
Over 20 co-chaperones regulate HSP90 activity mainly
through the modulation between the interconvertion of the ATP- and
ADP-bound states. Some of these inhibit HSP90 ATPase activity, thus
to be involved in client loading or the formation of mature HSP90
complexes, such as HSP70/HSP90 organizing protein (HOP), cell
division cycle protein 37 (CDC37) and p23. Whereas, others enhance
it, such as activator of HSP90 ATPase 1 (AHA1) and cyclophilin-40
(Cpr6 and Cpr7), hence leading to their use as activators of the
HSP90 conformational cycle (104,117,118).
Lessons learned in oncology clinical
trials and future directions for oncology drug development of HSP90
inhibitors
Many of HSP90 client proteins hold important
functions in the development and promotion of cancer, as described
above, thus, Hsp90 has a putative role in numerous cancers and
deserves to be an attractive target for therapeutics. Targeting
HSP90 as a therapeutic approach in treating cancer began with
geldanamycin (GM), which exhibits antiproliferative activity by
binding to the ATP-binding site of HSP90 and thereby preventing its
function. However, GM has limited therapeutic potential owing to
its hepatotoxicity. The discovery of GM sparked much interest in
the inhibition of HSP90 as a strategy for the treatment of cancer,
resulting in intense efforts from both industry and academic
research institutes to develop clinically viable HSP90 inhibitors
(119). Although the exact
antitumor action of HSP90 inhibitors remains largely unknown,
substantial number of molecules are currently in preclinical and
clinical evaluations, and some have promising results. These
inhibitors are summarized in Table
I.
 | Table I.HSP inhibitors in clinical
development as mono- or combination-therapy. |
Table I.
HSP inhibitors in clinical
development as mono- or combination-therapy.
| Drug (HSP90
inhibitors) | Disease type | Stage of
development |
|---|
| Geldanamycin
analogues | | |
| Tanespimycin
(17-AAG) | | |
| 17-AAG | Kidney tumors in
Von Hippel-Lindau disease; relapsed or refractory anaplastic large
cell lymphoma, mantle cell lymphoma, or Hodgkin’s lymphoma | II |
| 17-AAG +
trastuzumab | Breast cancer | II |
| 17-AAG +
bortezomib | Multiple
myeloma | II/III |
| 17-AAG +
gemcitabine | Recurrent advanced
ovarian epithelial or peritoneal cavity cancer | II |
| 17-AAG +
bortezomib | Advanced solid
tumors or lymphoma | I |
| Alvespimycin
(17-DMAG) | | |
| 17-DMAG | Melanoma | I |
| Prostate | I |
| 17-DMAG +
trastuzumab | Breast cancer | I |
| 17-DMAG
(KOS-1022) + trastuzumab | Ovarian | I |
| Retaspimycin
hydrochloride (IPI-504) | | |
| IPI-504 | Hormone-resistant
prostate cancer | II |
| Relapsed and
relapsed refractory multiple myeloma | I |
| Relapsed/refractory
stage IIIb, or stage IV NSCLC | I/II |
| IPI-504 +
docetaxol | Advanced solid
tumors | I |
| IPI-504 +
everolimus | KRAS mutant
NSCLC | I/II |
| IPI-504 +
trastuzumab | HER2+
breast cancer (study terminated) | II |
| IPI-493 | Advanced
malignancies; hematologic malignancies(study terminated) | I |
| Resorcinol
derivatives | | |
| Ganetespib
(STA-9090) | | |
| STA-9090 | Patients with
unresectable stage III or stage IV melanoma who received prior
tyrosine kinase inhibitor treatment (has 2 arms-mutant V600E BRAF
arm and a wild-type BRAF arm); metastatic hormone-resistant
prostate cancer previously treated with docetaxel-based
chemotherapy; previously untreated metastatic HER2+ or
triple negative breast cancer; stage IIIB/IV NSCLC; metastatic
ocular melanoma; metastatic or unresectable GIST; refractory
metastatic colorectal cancer | II |
| Advanced
hepatocellular carcinoma; solid tumors, STA-9090 administered
twice-weekly | I |
| AML, ALL and
blast-phase CML | I/II |
| STA-9090 as
second-or third-line therapy | Metastatic
pancreatic cancer | II |
| STA-9090 +
docetaxel | Solid tumors | I |
| STA-9090 +
dutasteride |
Castration-resistant prostate cancer | II |
| STA-9090 +
fulvestrant | HR+,
metastatic breast cancer | II |
| AUY922 | | |
| AUY922 | Advanced solid
malignancies in older patients (≥75 years) | I |
| Lymphoma;
metastatic pancreatic cancer resistant to first line chemotherapy;
NSCLC patients who received 2 previous lines of chemotherapy;
refractory GIST | II |
| HER2+
trastuzumab-resistant breast cancer [imaging component using
89Zr-trastuzumab positron emission tomography (PET) to study the
effect of HSP90 inhibition on HER2 expression] | I/II |
| ER+
hormone therapy refractory breast cancer (to study the effect of
HSP90 inhibition by AUY922 on VEGF using 89Zr-bevacizumab PET) | I/II |
| AUY922 +
capecitabine | Patients with
advanced solid tumors (active not recruiting) | I |
| AUY922 +
cetuximab | KRAS wild-type
metastatic colorectal cancer | I |
| AUY922 +
erlotinib hydrochloride | Stage IIIB/IV
NSCLC | I/II |
| AUY922 +
trastuzumab | Patients with
HER2+ advanced gastric cancer, who had received
trastuzumab plus chemotherapy as first line treatment | II |
| AT-13387 | Refractory solid
tumors | I |
| KW-2478 | | |
| KW-2478 +
bortezomib | Relapsed or
refractory multiple myeloma | I/II |
| Purine
analogues | | |
| BIIB021 (CNF
2024) | | |
| BIIB021 | Advanced solid
tumors; advanced breast cancer (PK/PD study) | I |
| GIST | II |
| BIIB021 +
aromasin | Hormone receptor
positive metastatic breast cancer | II |
| MPC-3100 | Refractory or
relapsed cancer | I |
| Debio 0932
(CUDC-305) | Advanced solid
tumors or lymphoma | I |
| PU-H71 | Refractory solid
tumor and low-grade non-Hodgkin’s lymphoma; advanced malignancies
(metastatic solid tumor, lymphoma) | I |
| Other
compounds | | |
| SNX-5422 | Refractory solid
tumor malignancies or non-Hodgkin’s lymphoma | I |
| DS-2248 | Advanced solid
tumors | I |
| XL-888 | | |
| XL-888 | Solid tumors | I |
| XL-888 +
AT13387, abiraterone | Prostate
cancer | I |
| XL-888 +
vemurafenib | Unresectable BRAF
mutated stage III/IV melanoma | I |
As illustrated above, the rationale for using HSP90
inhibitors in cancer therapy is well established. Pursuing after
new easily administrable HSP90 inhibitors and their evaluation in
clinical trials is a goal for many pharmaceutical companies.
However, no HSP90 inhibitor has been FDA-approved to date. Lessons
learned in oncology clinical trials give us strategies and future
directions that may enhance therapeutic benefit and accelerate the
drug approval process for safe and efficacious HSP90
inhibitors.
There have been hints that these inhibitors are
minimally effective and with more side-effects as single agents
against various cancer cell line, and that they may show tremendous
promise when used as combination and dual treatment agents
(120).
Inhibition of HSP90 activates the heat shock
response, which compensatorily induces expression of several heat
shock factors, including heat shock factor 1 (HSF1), members of the
HSP70 family and HSP27, which are protective proteins that could
counteract the pro-cell death effects of HSP90 inhibitors (121). Therefore, an interesting approach
for combination studies in the future is to inhibit multiple heat
shock proteins. Silencing HSF1, HSP70 and HSP27, has been shown to
cause a marked increase in the sensitivity of cancer cells to HSP90
inhibition, and induction of apoptosis (112). However, it should be noted that
the toxicity of the combination of HSP70 and HSP90 inhibitors is
unclear, and remains to be understood.
Since targeted therapy was introduced into cancer
treatment, it has brought hope, and it is increasingly believed
that combination therapies targeting parallel signaling pathways
that regulate iconic processes that are absolutely necessary for
cancer cell survival and proliferation may provide better cancer
therapy. As described above, many of HSP90 client proteins are
involved in critical cellular functions, thus, the development of
these HSP90 inhibitors may require close developing client protein
inhibitors, e.g., RAF inhibitors (114).
To enhance the effectiveness of HSP90 inhibitors,
combinatorial targeting of HSP90 cochaperones and/or of
post-translational modifications that influence HSP90’s function is
a potentially attractive approach (112). Then, a greater understanding of
HSP90 cochaperones, and post-translational modifications of HSP90
is needed.
In addition, a better understanding of the HSP90
inhibitors is still the key. For HSP90 has various isoforms and
each has different functions, specific inhibition for HSP90
inhibitors is expected. It has been suggested that HSP90α plays an
essential and unique role in embryo cell differentiation, and its
inhibition blocks macrophagic differentiation in the already formed
animal (122). Furthermore, only
HSP90α was described to emerge in the extracellular (breast cancer
and melanoma) and to play a role in tumor invasion and metastasis
by promoting maturation of extracellular MMP-2 (123). Evidences from that the
concentration of secreted HSP90α positively correlates with tumor
malignancy in liver and breast tumor patients (124). Therefore, future drug discovery
approaches should focus on looking for more specific inhibitors
that only target the HSP90α isoform. Similarly, drugs that
specifically disrupt the interaction of HSP90 with a given
chaperone or client protein without affecting others could be
interesting to avoid the side-effects associated to HSP90
inhibitors (5).
Clinical trial designs may ultimately be critical in
determining if one HSP90 inhibitor has any clear clinical benefit
to exploit. Several strategies can be applied to enhance the
effectiveness of HSP90 inhibitors. Personalizing treatments is
always one of the principles of clinical treatment. Personalizing
treatments to match patients’ genetic profiles and targeting
specific tumors/tumor types should be recognized as ways to
increase the effectiveness of HSP90 inhibitors. As we enter the era
of targeted therapy and personalised medicine, development of
biomarkers to help to stratify patients, ascertain target
inhibition, and monitor or predict response to HSP90 inhibitiors is
of vital importance if HSP90 inhibitors are to succeed. It is also
possible that the therapeutic schedule of HSP90 inhibitors has not
been optimized. Future effective target inhibition would benefit
from a valuable method to optimise drug dosing and scheduling.
Conclusions
Owing to the complicated pathogenesis, poor
prognosis and resistance to treatments, cancer remains a
notoriously unsolved medical issue and desperately requires
efficacious drug candidates. By commanding over the folding and
stabilization of relevant oncoproteins, HSPs are involved in vital
mechanisms of cancerous cells, such as cell proliferation,
differentiation, invasiveness, neoangiogenesis, metastasis and
immune system recognition. Additionally, they have the added
advantage of reducing the likelihood of the tumor acquiring
resistance to any single therapeutic strategy. As a consequence,
HSPs are emerging as interesting targets in cancer therapy,
particularly HSP90, 70 and 27. Owing to the potent anti-apoptotic
function of HSPs 27, 70 and 90 as well as their role in drug
resistance, it is considered that their deletion may increase tumor
cell susceptibility to apoptosis and fight against carcinogenesis
or elicit drug sensitivity (4).
This is one area, which although representing a challenging
endeavor with potential risks, offers very promising alternatives
for the treatment of cancer. Several HSP inhibitors such as 17-AAG,
IPI-504 and BIIB021 are currently in clinical phase trials.
However, specificity is still an important issue for all the tested
HSP inhibitors. HSP90 inhibitors represent the best developed
candidates to treat cancer. However, as we reviewed above, these
inhibitors targeting HSP90 are minimally effective and with more
side-effects as single agents. Consequently, developing drug
candidate stargeting multiple HSPs or the combination of different
HSP inhibitors could be particularly appealing. In addition,
combining HSP inhibitors with other validated drug candidates for
target therapies might provide promising therapeutic benefits.
Although many issues remain unresolved, scientists in the field
still continue to strive toward a better understanding of the
mechanisms of HSPs/HSR and other essential oncogenic pathways,
hoping that this will eventually lead to successful drug candidates
and significantly improve cancer clinical therapeutic index.
Acknowledgements
We acknowledge financial support from
the National Science and Technology Research Supporting Programme
in Chinese Medicine in ‘11th Five-Year Plan’ (2006BAI11B08-01) and
the Project Funded by the Priority Academic Program Development of
Jiangsu Higher Education Institutions (PAPD) and thank for the
assistance from Professor Xu Zhang of the Nanjing University of
Medicine.
References
|
1.
|
Young JC, Agashe VR, Siegers K and Hartl
FU: Pathways of chaperone-mediated protein folding in the cytosol.
Nat Rev Mol Cell Biol. 5:781–791. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Lindquist S and Craig EA: The heat-shock
proteins. Annu Rev Genet. 22:631–77. 1998. View Article : Google Scholar
|
|
3.
|
Lebret T, Watson RW and Fitzpatrick JM:
Heat shock proteins: their role in urological tumours. J Urolog.
169:338–346. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Khalil AA, Kabapy NF, Deraz SF and Smith
C: Heat shock proteins in oncology: diagnostic biomarkers or
therapeutic targets? Biochim Biophys Acta. 1816:89–104.
2011.PubMed/NCBI
|
|
5.
|
Jego G, Hazoumé A, Seigneuric R and
Garrido C: Targeting heat shock proteins in cancer. Cancer Lett.
332:275–285. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Xia Y, Rocchi P, Iovanna JL and Peng L:
Targeting heat shock response pathways to treat pancreatic cancer.
Drug Discov Today. 17:35–43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Garrido C, Brunet M, Didelot C, Zermati Y,
Schmitt E and Kroemer G: Heat shock proteins 27 and 70:
anti-apoptotic proteins with tumorigenic properties. Cell Cycle.
5:2592–2601. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Joly AL, Wettstein G, Mignot G,
Ghiringhelli F and Garrido C: Dual role of heat-shock proteins as
regulator of apoptosis and innate immunity. J Innate Immun.
2:238–247. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Wei MC, Zong WX, Cheng EH, Lindsten T,
Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB and
Korsmeyer SJ: Proapoptotic BAX and BAK: a requisite gateway to
mitochondrial dysfunction and death. Science. 292:727–30. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Zong WX, Lindsten T, Ross AJ, MacGregor GR
and Thompson CB: BH3-only proteins that bind pro-survival Bcl-2
family members fail to induce apoptosis in the absence of Bax and
Bak. Genes Dev. 15:1481–1486. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Garrido C, Galluzzi L, Brunet M, Puig PE,
Didelot C and Kroemer G: Mechanisms of cytochrome c release from
mitochondria. Cell Death Differ. 13:1423–1433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Joza N, Susin SA, Daugas E, Stanford WL,
Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, Ferri KF,
Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW,
Zuniga-Pflucker JC, Kroemer G and Penninger JM: Essential role of
the mitochondrial apoptosis-inducing factor in programmed cell
death. Nature. 410:549–554. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Du C, Fang M, Li Y, Li L and Wang X: Smac,
a mitochondrial protein that promotes cytochrome c-dependent
caspase activation by eliminating IAP inhibition. Cell. 102:33–42.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Luo X, Budihardjo I, Zou H, Slaughter C
and Wang X: Bid, a Bcl2 interacting protein, mediates cytochrome c
release from mitochondria in response to activation of cell surface
death receptors. Cell. 94:481–490. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Shamovsky I and Nudler E: New insights
into the mechanism of heat shock response activation. Cell Mol Life
Sci. 65:855–861. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Akerfelt M, Morimoto RI and Sistonen L:
Heat shock factors: integrators of cell stress, developmentand
lifespan. Nat Rev Mol Cell Biol. 11:545–555. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Green M, Schuetz TJ, Sullivan EK and
Kingston RE: A heat-shock responsive domain of human HSF1 that
regulates transcription activation domain function. Mol Cell Biol.
15:3354–3362. 1995.PubMed/NCBI
|
|
19.
|
Sistonen L, Sarge KD and Morimoto RI:
Human heat shock factors 1 and 2 are differentially activated and
can synergistically induce hsp70 gene transcription. Mol Cell Biol.
14:2087–2099. 1994.PubMed/NCBI
|
|
20.
|
Nakai A, Tanabe M, Kawazoe Y, Inazawa J,
Morimoto RI and Nagata K: HSF4, a new member of the human heat
shock factor family which lacks properties of a transcriptional
activator. Mol Cell Biol. 17:469–481. 1997.PubMed/NCBI
|
|
21.
|
Akerfelt M, Trouillet D, Mezger V and
Sistonen L: Heat shock factors at a cross road between stress and
development. Ann NY Acad Sci. 1113:15–27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Abane R and Mezger V: Roles of heat shock
factors in gameto-genesis and development. FEBS J. 277:4150–4172.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Whitesell L and Lindquist S: Inhibiting
the transcription factor HSF1 as an anticancer strategy. Expert
Opin Ther Targets. 13:469–478. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Westerheide SD, Kawahara TL, Orton K and
Morimoto RI: Triptolide, an inhibitor of the human heat shock
response that enhances stress-induced cell death. J Biol Chem.
281:9616–9622. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Sugiyama Y, Suzuki A, Kishikawa M, Akutsu
R, Hirose T and Waye MM: Muscle develops a specific form of small
heat shock protein complex composed of MKBP/HSPB2 and HSPB3 during
myogenic differentiation. J Biol Chem. 275:1095–1104. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Kostenko S and Moens U: Heat shock protein
27 phosphorylation: kinases, phosphatases, functions and pathology.
Cell Mol Life Sci. 66:3289–3307. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Shin KD, Lee MY, Shin DS, Lee S, Son KH,
Koh S, Paik YK, Kwon BM and Han DC: Blocking tumor cell migration
and invasion with biphenyl isoxazole derivative KRIBB3, a synthetic
molecule that inhibits Hsp27 phosphorylation. J Biol Chem.
280:41439–41448. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Gobbo J, Gaucher-Di-Stasio C, Weidmann S,
Guzzo J and Garrido C: Quantification of HSP27 and HSP70 molecular
chaperone activities. Methods Mol Biol. 787:137–143. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Bruey JM, Paul C, Fromentin A, Hilpert S,
Arrigo AP, Solary E and Garrido C: Differential regulation of HSP27
oligomerization in tumor cells grown in vitro and in vivo.
Oncogene. 19:4855–4863. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Shashidharamurthy R, Koteiche HA, Dong J
and McHaourab HS: Mechanism of chaperone function in small heat
shock proteins: Dissociation of the HSP27 oligomer is required for
recognition and binding of destabilized T4 lysozyme. J Biol Chem.
280:5281–5289. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Charette SJ, Lavoie JN, Lambert H and
Landry J: Inhibition of Daxx-mediated apoptosis by heat shock
protein 27. Mol Cell Biol. 20:7602–7612. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Akbar MT, Lundberg AM, Liu K, Vidyadaran
S, Wells KE, Dolatshad H, Wynn S, Wells DJ, Latchman DS and de
Belleroche J: The neuroprotective effects of heat shock protein 27
overexpression in transgenic animals against kainate-induced
seizures and hippocampal cell death. J Biol Chem. 278:19956–19965.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Straume O, Shimamura T, Lampa MJ,
Carretero J, Øyan AM, Jia D, Borgman CL and Soucheray M:
Suppression of heat shock protein 27 induces long-term dormancy in
human breast cancer. Proc Natl Acad Sci USA. 109:8699–8704. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Bauer K, Nitsche U, Slotta-Huspenina J,
Drecoll E, Von Weyhern CH, Rosenberg R, Höfler H and Langer R: High
HSP27 and HSP70 expression levels are independent adverse
prognostic factors in primary resected colon cancer. Cell Oncol
(Dordr). 35:197–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Chen SF, Nieh S, Jao SW, Liu CL, Wu CH,
Chang YC, Yang CY and Lin YS: Quercetin suppresses drug-resistant
spheres via the p38 MAPK-Hsp27 apoptotic pathway in oral cancer
cells. PLoS One. 7:e492752012. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Ciocca DR, Arrigo AP and Calderwood SK:
Heat shock proteins and heat shock factor 1 in carcinogenesis and
tumor development: an update. Arch Toxicol. 87:19–48. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Pavan S, Musiani D, Torchiaro E, Migliardi
G, Gai M, Di Cunto F, Erriquez J, Olivero M and Di Renzo MF: HSP27
is required for invasion and metastasis triggered by hepatocyte
growth factor. Int J Cancer. 134:1289–1299. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Acunzo J, Katsogiannou M and Rocchi P:
Small heat shock proteins HSP27 (HspB1), αB-crystallin (HspB5) and
HSP22 (HspB8) as regulators of cell death. Int J Biochem Cell Biol.
44:1622–1631. 2012.
|
|
39.
|
Schmitt E, Gehrmann M, Brunet M, Multhoff
G and Garrido C: Intracellular and extracellular functions of heat
shock proteins: repercussions in cancer therapy. J Leukoc Biol.
81:15–27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Voss OH, Batra S, Kolattukudy SJ,
Gonzalez-Mejia ME, Smith JB and Doseff AI: Binding of caspase-3
prodomain to heat shock protein 27 regulates monocyte apoptosis by
inhibiting caspase-3 proteolytic activation. J Biol Chem.
282:25088–25099. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Rocchi P, Jugpal P, So A, Sinneman S,
Ettinger S, Fazli L, Nelson C and Gleave M: Small interference RNA
targeting heat-shock protein 27 inhibits the growth of prostatic
cell lines and induces apoptosis via caspase-3 activation in vitro.
BJU Int. 98:1082–1089. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Paul C, Manero F, Gonin S, Kretz-Remy C,
Virot S and Arrigo AP: Hsp27 as a negative regulator of cytochrome
c release. Mol Cell Biol. 22:816–834. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Chauhan D, Li G, Hideshima T, Podar K,
Mitsiades C, Mitsiades N, Catley L, Tai YT, Hayashi T, Shringarpure
R, Burger R, Munshi N, Ohtake Y, Saxena S and Anderson KC: Hsp27
inhibits release of mitochondrial protein Smac in multiple myeloma
cells and confers dexamethasone resistance. Blood. 102:3379–3386.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Havasi A, Li Z, Wang Z, Martin JL, Botla V
and Ruchalski K: Hsp27 inhibits Bax activation and apoptosis via a
phosphatidylinositol 3-kinase-dependent mechanism. J Biol Chem.
283:12305–12313. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Arrigo AP, Virot S, Chaufour S, Firdaus W,
Kretz-Remy C and Diaz-Latoud C: Hsp27 consolidates intracellular
redox homeostasis by upholding glutathione in its reduced form and
by decreasing iron intracellular levels. Antioxid Redox Signal.
7:414–422. 2005. View Article : Google Scholar
|
|
46.
|
Rogalla T, Ehrnsperger M, Preville X,
Kotlyarov A, Lutsch G, Ducasse C, Paul C, Wieske M, Arrigo AP,
Buchner J and Gaestel M: Regulation of Hsp27 oligomerization,
chaperone function, and protective activity against oxidative
stress/tumor necrosis factor alpha by phosphorylation. J Biol Chem.
274:18947–18956. 1999. View Article : Google Scholar
|
|
47.
|
Sanchez-Niño MD, Sanz AB, Sanchez-Lopez E,
Ruiz-Ortega M, Benito-Martin A, Saleem MA, Mathieson PW, Mezzano S,
Egido J and Ortiz A: HSP27/HSPB1 as an adaptive podocyte
antiapoptotic protein activated by highglucose and angiotensin II.
Lab Invest. 92:32–45. 2012.
|
|
48.
|
Solary E, Droin N, Bettaieb A, Corcos L,
Dimanche-Boitrel MT and Garrido C: Positive and negative regulation
of apoptotic pathways by cytotoxic agents in hematological
malignancies. Leukemia. 14:1833–1849. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Kamada M, So A, Muramaki M, Rocchi P,
Beraldi E and Gleave M: Hsp27 knockdown using nucleotide-based
therapies inhibit tumor growth and enhance chemotherapy in human
bladder cancer cells. Mol Cancer Ther. 6:299–308. 2007. View Article : Google Scholar
|
|
50.
|
Heinrich JC, Tuukkanen A, Schroeder M,
Fahrig T and Fahrig R: RP101 (brivudine) binds to heat shock
protein HSP27 (HSPB1) and enhances survival in animals and
pancreatic cancer patients. J Cancer Res Clin Oncol. 137:1349–1361.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Seigneuric R, Gobbo J, Colas P and Garrido
C: Targeting cancer with peptide aptamers. Oncotarget. 2:557–561.
2011.PubMed/NCBI
|
|
52.
|
Gibert B, Hadchity E, Czekalla A, Aloy MT,
Colas P, Rodriguez-Lafrasse C, Arrigo AP and Diaz-Latoud C:
Inhibition of heat shock protein 27 (HspB1) tumorigenic functions
by peptide aptamers. Oncogene. 30:3672–81. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Kampinga HH, Hageman J, Vos MJ, Kubota H,
Tanguay RM, Bruford EA, Cheetham ME, Chen B and Hightower LE:
Hightower, Guidelines for the nomenclature of the human heat shock
proteins. Cell Stress Chaperones. 14:105–111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Jäättelä M: Heat shock proteins as
cellular lifeguards. Ann Med. 31:261–271. 1999.
|
|
55.
|
Beckmann RP, Mizzen LE and Welch WJ:
Interaction of Hsp 70 with newly synthesized proteins: Implications
for protein folding and assembly. Science. 248:850–854. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Murakami H, Pain D and Blobel G: 70-kD
heat shock-related protein is one of at least two distinct
cytosolic factors stimulating protein import into mitochondria. J
Cell Biol. 107:2051–2057. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Shi Y and Thomas JO: The transport of
proteins into the nucleus requires the 70-kilodalton heat shock
protein or its cytosolic cognate. Mol Cell Biol. 12:2186–2192.
1992.PubMed/NCBI
|
|
58.
|
Nollen EA, Brunsting JF, Roelofsen H,
Weber LA and Kampinga HH: In vivo chaperone activity of heat shock
protein 70 and thermotolerance. Mol Cell Biol. 19:2069–2079.
1999.PubMed/NCBI
|
|
59.
|
Vogel M, Bukau B and Mayer MP: Allosteric
regulation of Hsp70 chaperones by a proline switch. Mol Cell.
21:359–367. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Goloudina AR, Demidov ON and Garrido C:
Inhibition of HSP70: a challenging anti-cancer strategy. Cancer
Lett. 325:117–124. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Bukau B, Weissman J and Horwich A:
Molecular chaperones and protein quality control. Cell.
125:443–451. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Schmitt E, Parcellier A, Gurbuxani S,
Cande C, Hammann A, Morales MC, Hunt CR, Dix DJ, Kroemer RT,
Giordanetto F, Jäättelä M, Penninger JM, Pance A, Kroemer G and
Garrido C: Chemosensitization by a non-apoptogenic heat-shock
protein 70-binding apoptosis-inducing factor mutant. Cancer Res.
63:8233–8240. 2003.PubMed/NCBI
|
|
63.
|
Dix DJ, Allen JW, Collins BW, Mori C,
Nakamura N, Poorman-Allen P, Goulding EH and Eddy EM: Targeted gene
disruption of Hsp70-2 results in failed meiosis, germ cell
apoptosis, and male infertility. Proc Natl Acad Sci USA.
93:3264–3268. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Johnson TR, Stone K and Nikrad M: The
proteasome inhibitor PS-341 overcomes TRAIL resistance in Bax and
caspase 9-negative or Bcl-xL overexpressing cells. Oncogene.
22:4953–4963. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Hui-qing X, Jian-da Z, Xin-min N,
Yan-zhong Z, Cheng-qun L, Quan-yong H, Yi X, Pokharel PB, Shao-hua
W and Dan X: HSP70 inhibits burn serum-induced apoptosis of
cardiomyocytes via mitochondrial and membrane death receptor
pathways. J Burn Care Res. 29:512–518. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Park HS, Cho SG, Kim CK, Hwang HS, Noh KT,
Kim MS, Huh SH, Kim MJ, Ryoo K, Kim EK, Kang WJ, Lee JS, Seo JS, Ko
YG, Kim S and Choi EJ: Heat shock protein hsp72 is a negative
regulator of apoptosis signal-regulating kinase 1. Mol Cell Biol.
22:7721–7730. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
67.
|
Park HS, Lee JS, Huh SH, Seo JS and Choi
EJ: Hsp72 functions as a natural inhibitory protein of c-Jun
N-terminal kinase. EMBO J. 20:446–456. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
68.
|
Lee JS, Lee JJ and Seo JS: HSP70
deficiency results in activation of c-Jun N-terminal kinase,
extracellular signal-regulated kinase, and caspase-3 in
hyperosmolarity-induced apoptosis. J Biol Chem. 280:6634–6641.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
69.
|
Gao T and Newton AC: The turn motif is a
phosphorylation switch that regulates the binding of Hsp70 to
protein kinase C. J Biol Chem. 277:31585–31592. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
70.
|
Zylicz M, King FW and Wawrzynow A: Hsp70
interactions with the p53 tumour suppressor protein. EMBO J.
20:4634–4638. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
71.
|
Yang X, Wang J, Zhou Y, Wang Y, Wang S and
Zhang W: Hsp70 promotes chemoresistance by blocking Bax
mitochondrial translocation in ovariancancer cells. Cancer Lett.
321:137–143. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72.
|
Stankiewicz AR, Lachapelle G, Foo CP,
Radicioni SM and Mosser DD: Hsp70 inhibits heat-induced apoptosis
upstream of mitochondria by preventing Bax translocation. J Biol
Chem. 280:38729–38739. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
73.
|
Li CY, Lee JS, Ko YG, Kim JI and Seo JS:
Heat shock protein 70 inhibits apoptosis down-stream of cytochrome
c release and upstream of caspase-3 activation. J Biol Chem.
275:25665–25671. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
74.
|
Beere HM, Wolf BB, Cain K, Mosser DD,
Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM and Green DR:
Heat-shock protein 70 inhibits apoptosis by preventing recruitment
of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2:469–475.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
75.
|
Creagh EM, Carmody RJ and Cotter TG: Heat
shock protein 70 inhibits caspase-dependent and-independent
apoptosis in Jurkat T cells. Exp Cell Res. 257:58–66. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
76.
|
Ravagnan L, Gurbuxani S, Susin SA, Maisse
C, Daugas E, Zamzami N, Mak T, Jaattela M, Penninger JM, Garrido C
and Kroemer G: Heat-shock protein 70 antagonizes apoptosis-inducing
factor. Nat Cell Biol. 3:839–843. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
77.
|
Gurbuxani S, Schmitt E, Cande C,
Parcellier A, Hammann A, Daugas E, Kouranti I, Spahr C, Pance A,
Kroemer G and Garrido C: Heat shock protein 70 binding inhibits the
nuclear import of apoptosis-inducing factor. Oncogene.
22:6669–6678. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
78.
|
Matsumori Y, Hong SM, Aoyama K, Fan Y,
Kayama T, Sheldon RA, Vexler ZS, Ferriero DM, Weinstein PR and Liu
J: Hsp70 overexpression sequesters AIF and reduces neonatal
hypoxic/ischemic brain injury. J Cereb Blood Flow Metab.
25:899–910. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
79.
|
Kalinowska M, Garncarz W, Pietrowska M,
Garrard WT and Widlak P: Regulation of the human apoptotic
DNase/RNase endonuclease G: Involvement of Hsp70 and ATP.
Apoptosis. 10:821–830. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
80.
|
Sakahira H and Nagata S: Cotranslational
folding of caspase-activated DNase with Hsp70, Hsp40, and inhibitor
of caspase-activated DNase. J Biol Chem. 277:3364–3370. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
81.
|
Liu QL, Kishi H, Ohtsuka K and Muraguchi
A: Heat shock protein 70 binds caspase-activated DNase and enhances
its activity in TCR-stimulated T cells. Blood. 102:1788–1796. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
82.
|
Ribeil JA, Zermati Y, Vandekerckhove J,
Cathelin S, Kersual J, Dussiot M, Coulon S, Moura IC, Zeuner A,
Kirkegaard-Sørensen T, Varet B, Solary E, Garrido C and Hermine O:
Hsp70 regulates erythropoiesis by preventing caspase-3-mediated
cleavage of GATA-1. Nature. 445:102–105. 2007. View Article : Google Scholar
|
|
83.
|
Gyrd-Hansen M, Nylandsted J and Jaattela
M: Heat shock protein 70 promotes cancer cell viability by
safeguarding lysosomal integrity. Cell Cycle. 3:1484–1485. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
84.
|
Nylandsted J, Gyrd-Hansen M, Danielewicz
A, Fehrenbacher N, Lademann U, Hoyer-Hansen M, Weber E, Multhoff G,
Rohde M and Jaattela M: Heat shock protein 70 promotes cell
survival by inhibiting lysosomal membrane permeabilization. J Exp
Med. 200:425–435. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
85.
|
Leu JI, Pimkina J, Frank A, Murphy ME and
George DL: A small molecule inhibitor of inducible heat shock
protein 70. Mol Cell. 36:15–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
86.
|
Schmitt E, Maingret L, Puig PE, Rerole AL,
Ghiringhelli F, Hammann A, Solary E, Kroemer G and Garrido C:
Heat-shock protein 70 neutralization exerts potent antitumor
effects in animal models of colon cancer and melanoma. Cancer Res.
66:4191–4197. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
87.
|
Nadeau K, Nadler SG, Saulnier M, Tepper MA
and Walsh CT: Quantitation of the interaction of the
immunosuppressant deoxyspergualin and analogs with Hsc70 and Hsp90.
Biochemistry. 33:2561–2567. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
88.
|
Fewell SW, Day BW and Brodsky JL:
Identification of an inhibitor of hsc70-mediated protein
translocation and ATP hydrolysis. J Biol Chem. 276:910–914. 2001.
View Article : Google Scholar
|
|
89.
|
Rodina A, Vilenchik M, Moulick K, Aguirre
J, Kim J, Chiang A, Litz J, Clement CC, Kang Y, She Y, Wu N, Felts
S, Wipf P, Massague J, Jiang X, Brodsky JL, Krystal GW and Chiosis
G: Selective compounds define Hsp90 as a major inhibitor of
apoptosis in small-cell lung cancer. Nat Chem Biol. 3:498–507.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
90.
|
Wright CM, Seguin SP, Fewell SW, Zhang H,
Ishwad C, Vats A, Lingwood CA, Wipf P, Fanning E, Pipas JM and
Brodsky JL: Inhibition of SimianVirus 40 replication by targeting
the molecular chaperone function and ATPase activity of T antigen.
Virus Res. 141:71–80. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
91.
|
Massey AJ, Williamson DS, Browne H, Murray
JB, Dokurno P, Shaw T, Macias AT, Daniels Z, Geoffroy S, Dopson M,
Lavan P, Matassova N, Francis GL, Graham CJ, Parsons R, Wang Y,
Padfield A, Comer M, Drysdale MJ and Wood M: A novel, small
molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor
induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother
Pharmacol. 66:535–545. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
92.
|
Jinwal UK, Miyata Y, Koren J III, Jones
JR, Trotter JH, Chang L, O’Leary J, Morgan D, Lee DC, Shults CL,
Rousaki A, Weeber EJ, Zuiderweg ER, Gestwicki JE and Dickey CA:
Chemical manipulation of hsp70 ATPase activity regulates tau
stability. J Neurosci. 29:12079–88. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
93.
|
Wadhwa R, Sugihara T, Yoshida A, Nomura H,
Reddel RR, Simpson R, Maruta H and Kaul SC: Selective toxicity of
MKT-077 to cancer cells is mediated by its binding to the hsp70
family protein mot-2 and reactivation of p53 function. Cancer Res.
60:6818–6821. 2000.PubMed/NCBI
|
|
94.
|
Wadhwa R, Yaguchi T, Hasan MK, Mitsui Y,
Reddel RR and Kaul SC: Hsp70 family member, mot-2/mthsp70/GRP75,
binds to the cytoplasmic sequestration domain of the p53 protein.
Exp Cell Res. 274:246–253. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
95.
|
Britten CD, Rowinsky EK, Baker SD, Weiss
GR, Smith L, Stephenson J, Rothenberg M, Smetzer L, Cramer J,
Collins W, Von Hoff DD and Eckhardt SG: A phase I and
pharmacokinetic study of the mitochondrial-specific rhodacyanine
dye analog MKT 077. Clin Cancer Res. 6:42–49. 2000.PubMed/NCBI
|
|
96.
|
Williams DR, Ko SK, Park S, Lee MR and
Shin I: An apoptosis-inducing small molecule that binds to heat
shock protein 70. Angew Chem Int Ed Engl. 47:7466–7469. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
97.
|
Powers MV, Jones K, Barillari C, Westwood
I, van Montfort RL and Workman P: Targeting HSP70: the second
potentially druggable heat shock protein and molecular chaperone?
Cell Cycle. 9:1542–1550. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
98.
|
Whetstone H and Lingwood C:
3’Sulfogalactolipid binding specifically inhibits Hsp70 ATPase
activity in vitro. Biochemistry. 42:1611–1617. 2003.
|
|
99.
|
Chatterjee M, Andrulis M, Stühmer T,
Müller E, Hofmann C, Steinbrunn T, Heimberger T, Schraud H,
Kressmann S, Einsele H and Bargou RC: The PI3K/Akt signaling
pathway regulates the expression of Hsp70, which critically
contributes to Hsp90-chaperone function and tumor cell survival in
multiple myeloma. Haematologica. 98:1132–1141. 2013. View Article : Google Scholar
|
|
100.
|
Multhoff G and Radons J: Radiation,
inflammation, and immune responses in cancer. Front Oncol.
2:582012. View Article : Google Scholar : PubMed/NCBI
|
|
101.
|
Stangl S, Gehrmann M, Riegger J, Kuhs K,
Riederer I, Sievert W, Hube K, Mocikat R, Dressel R, Kremmer E,
Pockley AG, Friedrich L, Vigh L, Skerra A and Multhoff G: Targeting
membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1
antibody. Proc Nat Acad Sci USA. 108:733–738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
102.
|
Krause SW, Gastpar R, Andreesen R, Gross
C, Ullrich H, Thonigs G, Pfister K and Multhoff G: Treatment of
colon and lung cancer patients with ex vivo heat shock protein
70-peptide-activated, autologous natural killer cells: a clinical
phase I trial. Clin Cancer Res. 10:3699–3707. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
103.
|
Whitesell L and Lindquist SL: HSP90 and
the chaperoning of cancer. Nat Rev Cancer. 5:761–772. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
104.
|
Taipale M, Jarosz DF and Lindquist S:
HSP90 at the hub of protein homeostasis: emerging mechanistic
insights. Nat Rev Mol Cell Biol. 11:515–528. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
105.
|
Sreedhar AS, Kalmár E, Csermely P and Shen
YF: Hsp90 isoforms: functions, expression and clinical importance.
FEBS Lett. 562:11–15. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
106.
|
Hartl FU, Bracher A and Hayer-Hartl M:
Molecular chaperones in protein folding and proteostasis. Nature.
475:324–332. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
107.
|
Onuoha SC, Coulstock ET, Grossmann JG and
Jackson SE: Structural studies on the co-chaperone Hop and its
complexes with Hsp90. J Mol Biol. 379:732–744. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
108.
|
Mayer MP: Gymnastics of molecular
chaperones. Mol Cell. 39:321–331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
109.
|
Krukenberg KA, Street TO, Lavery LA and
Agard DA: Conformational dynamics of the molecular chaperone Hsp90.
Q Rev Biophys. 44:229–255. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
110.
|
Ali MM, Roe SM, Vaughan CK, Meyer P,
Panaretou B and Piper PW: Crysta l structure of an
Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature.
440:1013–1017. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
111.
|
Normant E, Paez G and West KA: The Hsp90
inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor
regression in ALK-driven NSCLC models. Oncogene. 30:2581–2586.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
112.
|
Neckers L and Workman P: HSP90 molecular
chaperone inhibitors: are we there yet? Clin Cancer Res. 18:64–76.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
113.
|
Modi S, Stopeck A and Linden H: HSP90
inhibition is effective in breast cancer: a phase II trial of
tanespimycin (17-AAG) plus trastuzumab in patients with
HER2-positive metastatic breast cancer progressing on trastuzumab.
Clin Cancer Res. 17:5132–5139. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
114.
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
115.
|
Khong T and Spencer A: Targeting HSP 90
induces apoptosis and inhibits critical survival and proliferation
pathways in multiple myeloma. Mol Cancer Ther. 10:1909–1917. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
116.
|
Workman P, Burrows F, Neckers L and Rosen
N: Drugging the cancer chaperone HSP90: combinatorial therapeutic
exploitation of oncogene addiction and tumor stress. Ann NY Acad
Sci. 1113:202–216. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
117.
|
Hartl FU: Chaperone-assisted protein
folding: the path to discovery from a personal perspective. Nat
Med. 17:1206–1210. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
118.
|
Tiroli-Cepeda AO and Ramos CH: An overview
of the role of molecular chaperones in protein homeostasis. Protein
Pept Lett. 18:101–109. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
119.
|
Patel HJ, Modi S, Chiosis G and Taldone T:
Advances in the discovery and development of heat-shock protein 90
inhibitors for cancer treatment. Expert Opin Drug Discov.
6:559–587. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
120.
|
Sequist LV, Gettinger S and Natale R: A
phase II trial of IPI-504 (retaspimycin hydrochloride), a novel
Hsp90 inhibitor, in patients with relapsed and/or refractory stage
IIIb or stage IV non-small cell lung cancer (NSCLC) stratified by
EGFR mutation status. J Clin Oncol. 27:15s2009.
|
|
121.
|
McCollum AK, Teneyck CJ, Sauer BM, Toft DO
and Erlichman C: Up-regulation of heat shock protein 27 induces
resistance to 17-allylamino-demethoxy geldanamycin through a
glutathione-mediated mechanism. Cancer Res. 66:10967–10975. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
122.
|
Didelot C, Lanneau D, Brunet M, Bouchot A,
Cartier J, Jacquel A, Ducoroy P, Cathelin S, Decologne N, Chiosis
G, Dubrez-Daloz L, Solary E and Garrido C: Interaction of
heat-shock protein 90 beta isoform (HSP90 beta) with cellular
inhibitor of apoptosis 1 (c-IAP1) is required for cell
differentiation. Cell Death Differ. 15:859–866. 2008. View Article : Google Scholar
|
|
123.
|
Eustace BK, Sakurai T, Stewart JK,
Yimlamai D, Unger C, Zehetmeier C, Lain B, Torella C, Henning SW,
Beste G, Scroggins BT, Neckers L, Ilag LL and Jay DG: Functional
proteomic screens reveal an essential extracellular role for hsp90
alpha in cancer cell invasiveness. Nat Cell Biol. 6:507–514. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
124.
|
Wang X, Song X, Zhuo W, Fu Y, Shi H, Liang
Y, Tong M, Chang G and Luo Y: The regulatory mechanism of Hsp90
alpha secretion and its function in tumor malignancy. Proc Natl
Acad Sci USA. 106:21288–21293. 2009. View Article : Google Scholar : PubMed/NCBI
|