Introduction
The incidence of cancer is increasing dramatically,
and currently, there is a strong emphasis on identifying
biologically active substances from traditional herbs with
anticancer properties, because these are believed to have fewer
side effects than conventional chemotherapeutic agents (1,2).
Orostachys japonicus, a perennial herbaceous plant belonging
to the family Crassulaceae and referred to as Wa-song in
Korea, has been used traditionally as an anti-inflammatory,
anti-febrile, hemostatic, antidote and anticancer agent (3–7). In
a previous study (8), dried powder
of O. japonicus was extracted and fractionated using a
series of organic solvents, including n-hexane (hexane),
dichloromethane (DCM), ethylacetate (EtOAc), n-butanol (BuOH), and
water (H2O). These extracts were examined for their
anticancer activities on various cancer cell lines, including human
AGS gastric, A549 lung, HepG2 liver, and HT-29 colon cancer cells.
Among the different O. japonicus fractions tested, the
EtOAc-soluble fraction showed the highest anticancer activity
against 4 cancer cell lines, among which, AGS showed the strongest
effect. Furthermore, our previous study identified kaempferol,
quercetin, and gallic acid in the EtOAc fraction (8). However, further research on O.
japonicus is required considering the lack of fundamental data
on the signaling pathways related to its physiological
activity.
Apoptosis, the process of programmed cell death, is
essential to maintain the normal development of organs and for the
persistence of tissues in multi-cellular organisms. Failure of the
apoptosis regulatory machinery is known to induce cancer or other
degenerative diseases (4).
Therefore, the rate of apoptosis in cancer cells is considered a
principal indicator of anticancer activity. In cancer, cells
continue to proliferate in an uncontrolled manner owing to their
inability to sustain normal cellular activity (9). The level of cell cycle arrest has
also been used as an indicator of anticancer activity. The p53 gene
is a well-known transcription factor and a tumor suppressor gene
that participates in several cellular processes, including
apoptosis, cell signaling pathway, and cell cycle control. If cell
DNA is damaged by extracellular stress, p53 is activated and moves
to the nucleus, where it activates target genes. p53 activates
genes involved in apoptosis and genes encoding cell division
suppressors, thereby inducing apoptosis and suppressing cell
division (10,11). During apoptosis, phosphatidylserine
(PS) is exposed to the outer membrane. This results in impeding
anti-apoptosis factors such as B cell lymphoma-2 (bcl-2),
regulation of pro-apoptosis factors such as bcl-2 associated ×
protein (bax), release of mitochondria-mediated apoptosis factors
such as cytochrome c, and activation of apoptosis-induced proteins
such as caspase (12,13). Many therapeutic substances with
anticancer activity arrest the cell cycle and simultaneously induce
apoptotic cell death (14).
Defects in the regulation of the cell cycle are common causes of
abnormal proliferation of cancer cells; therefore, studies of the
cell cycle and cancer have become closely interconnected (15). The eukaryotic cell cycle is
traditionally divided into 4 sequential phases: G1, S,
G2 and M. The cell cycle is regulated by
cyclin/cyclin-dependent kinase (CDK) complexes (16). Mitogen activated protein kinases
(MAPKs) are important mediators involved in the intracellular
network of interacting proteins that transduce extracellular
signals to intracellular responses. When MAPKs such as p38, c-Jun
N-terminal kinase (JNK), and extracellular regulated protein kinase
1/2 (ERK1/2) are activated, their signals lead to the activation of
diverse molecules that regulate proliferation, gene expression,
differentiation, mitosis, cell survival and apoptosis (17,18).
In this study, the effect of the EtOAc fraction
derived from O. japonicus on the activation of signaling
pathways involving p53 and its downstream and upstream effectors
was investigated.
Materials and methods
Cell line and reagents
The EtOAc fraction was prepared from O.
japonicas in our laboratory using methods described previously
(5,8,19,20).
After analyzing the EtOAc fraction by GC-MS system, 12 peaks were
identified. Nine peaks were unknown, and 3 peaks were identified as
gallic acid (4.24%), kaempferol (6.81%) and quercetin (5.08%)
(8). AGS cells were obtained from
the Korean cell line bank (KCLB, Seoul, Korea). Dulbecco’s modified
Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin and
streptomycin were purchased from Hyclone (Thermo Scientific, Logan,
UT, USA). Monoclonal antibodies against p53; bcl-2; bax; cytochrome
c; caspase-3, -8 and -9; cleaved caspase-3, -8 and -9; phospho-p38
(p-p38), -JNK (p-JNK), -ERK1/2 (p-ERK1/2); p38; JNK; ERK1/2; and
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were obtained from
Cell Signaling Technology (Berverly, MA, USA). Goat anti-rabbit
IgG-horseradish peroxidase (HRP) secondary antibody was obtained
from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Z-VAD-FMK (a
pancaspase inhibitor) was purchased from BD Pharmingen™ (BD
Biosciences, Franklin Lakes, NJ, USA). All other reagents used were
of the highest available grade.
Cell culture
The AGS cells were cultured in DMEM containing 10%
FBS and 1% penicillin/streptomycin and incubated at 37°C in a 5%
CO2 incubator. The cells were subcultured every 5–7 days
at 1:5 split ratios, and the culture medium was changed every 2
days. The cells at ∼80–90% confluency were used in the experiments.
Cells were treated either with 0.1% dimethyl sulfoxide (DMSO) or
with various concentrations of the EtOAc fraction for 12 h.
Flow cytometric analysis of
apoptosis
Apoptosis in the AGS cells was evaluated by Annexin
V-fluorescein isothiocyanate (Annexin V-FITC) and propidium iodide
(PI) staining by using the Annexin V-FITC apoptosis detection kit
(BD Biosciences), according to the manufacturer’s instructions. AGS
cells (4×105 cells/ml in a 24-well plate) were added to
different concentrations of the EtOAc fraction for 12 h and then
harvested by centrifugation at 300 × g. After centrifugation, the
pellets were washed twice with cold phosphate-buffered saline (PBS;
137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, pH
7.4) and suspended in 100 μl of 1X binding buffer (10 mM
HEPES/NaOH, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4). The
cells were incubated with 5 μl of Annexin V-FITC and 5
μl of PI at the room temperature for 15 min in the dark.
After incubation, 400 μl of 1X binding buffer was added to
each tube and the cells were analyzed immediately by FACSCalibur
flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA).
Flow cytometric analysis of the cell
cycle
The cell cycle phase was assayed by DNA fragment
staining with PI using the Cell Cycle Phase determination kit
(Cayman Chemical, MI, USA), according to the manufacturer’s
instructions. AGS cells (4×105 cells/ml in a 24-well
plate) were added to different concentrations of the EtOAc fraction
for 12 h and then harvested. After centrifugation, the pellets were
washed and suspended in cell-based assay buffer. The cells were
fixed and permeabilized by adding 1 ml of a fixative to each tube
for more than 2 h. After centrifugation, the fixatives were
decanted and the cell pellets were suspended in 500 μl of a
staining solution (200 μl of RNase and 200 μl of PI),
followed by incubation for 30 min at room temperature in the dark.
Then, the cells were analyzed immediately by FACSCalibur flow
cytometry.
RT-PCR analysis
Total RNA was isolated using TRI reagent
(Sigma-Aldrich, St. Louis, MO, USA). The cells were lysed using the
TRI reagent, followed by the addition of chloroform
(Sigma-Aldrich). RNA precipitated by 2-propanol (Sigma-Aldrich).
The purity and concentration of the total RNA was checked by
Optizen 2120 UV plus spectrophotometer (Mecasys Co., Ltd., Daejeon,
Korea). Equal amounts of RNA were reverse-transcribed into cDNA
using the DiaStar™ RT kit (Solgent Co., Ltd., Daejeon, Korea) by
incubation at 65°C for 5 min, 50°C for 60 min, and 95°C for 5 min.
PCR was performed using a 50 μl reaction mixture containing
cDNA, 10X h-Taq buffer, 10 mM dNTP mix, 10 pM of each primer, h-Taq
polymerase (Solgent Co., Ltd.), and RNase-free water. Primers used
for amplification were as follows: i) CDK1: forward
5′-TTTTCAGAGCTTTGGGCACT-3′ and reverse 5′-AAACATGGCAGTGACACCAA-3′,
ii) cyclin B1: forward 5′-CGGGAAGTCACTGGAAACAT-3′ and reverse
5′-AAACA TGGCAGTGACACCAA-3′, iii) GAPDH: forward 5′-GAGTC
AACGGATTTGGTCGT-3′ and reverse 5′-TTGATTTTGG AGGGAGCTCG-3′. The
samples were subjected to 25 cycles of denaturation for 20 sec at
95°C, annealing for 40 sec at 56°C (cyclin B1 and GAPDH) and 54°C
(CDK1), extension for 1 min at 72°C, and a final extension step at
72°C for 5 min. The housekeeping gene GAPDH served as the control.
The PCR products were electrophoresed in 2% agarose gel, followed
by staining with 0.5 μg/ml ethidium bromide (EtBr) at 100 V
for 30 min and visualization under a UV transilluminator.
Western blotting
The AGS cells were treated with the EtOAc fraction,
washed twice with ice-cold PBS, and harvested using a cell scraper.
The cells were then pelleted by centrifugation, the pellets were
resuspended in lysis buffer on ice for 1 h, and the cell debris was
removed by centrifugation at 10,000 x g for 10 min. Protein
concentrations were determined using the Bicinchoninic acid (BCA)
Protein Assay kit (Thermo Scientific, Rockford, IL, USA). Equal
amounts of protein were mixed with 2X Laemmli loading buffer and
preheated at 95°C for 5 min. The samples were electrophoresed on
10–15% sodium dodecyl sulfate (SDS)-polyacrylamide gels and
transferred onto a polyvinylidene fluoride (PVDF) membrane for 1 h
by using a semi-dry transfer system (Bio-Rad Laboratories,
Hercules, CA, USA). The membrane was blocked with 5% non-fat milk
in PBS containing 0.1% Tween-20 (PBST) for 2 h at 4°C and then
incubated overnight with primary antibodies. After hybridization
with primary antibodies, the membranes were washed for 5 min with
PBST, 3 times. The membranes were subsequently incubated with
HRP-secondary antibody for 2 h at 4°C and washed for 5 min with
PBST, 3 times. The signal of the membranes was developed by using a
western blotting luminal reagent (Santa Cruz Biotechnology).
Statistical analysis
All experiments were performed at least 3 times. The
data are expressed as the mean ± standard deviation (SD). The
statistical differences were calculated using the Student’s t-test
using the SigmaStat software (SigmaStat for Windows version 3.0.).
A p-value of <0.05 was considered statistically significant.
Results
Induction of apoptosis
At the initial stages of apoptosis, the PS is
exposed, which is considered an early marker of apoptosis. In
addition, Annexin V, a Ca2+-dependent
phospholipid-binding protein, can now bind to the exposed PS
(21). The exposed PS could be
detected based on this characteristic. At the same time, the
damaged DNA is stained using PI. Thus, from the amount of Annexin V
binding and the intensity of PI staining, apoptosis can be
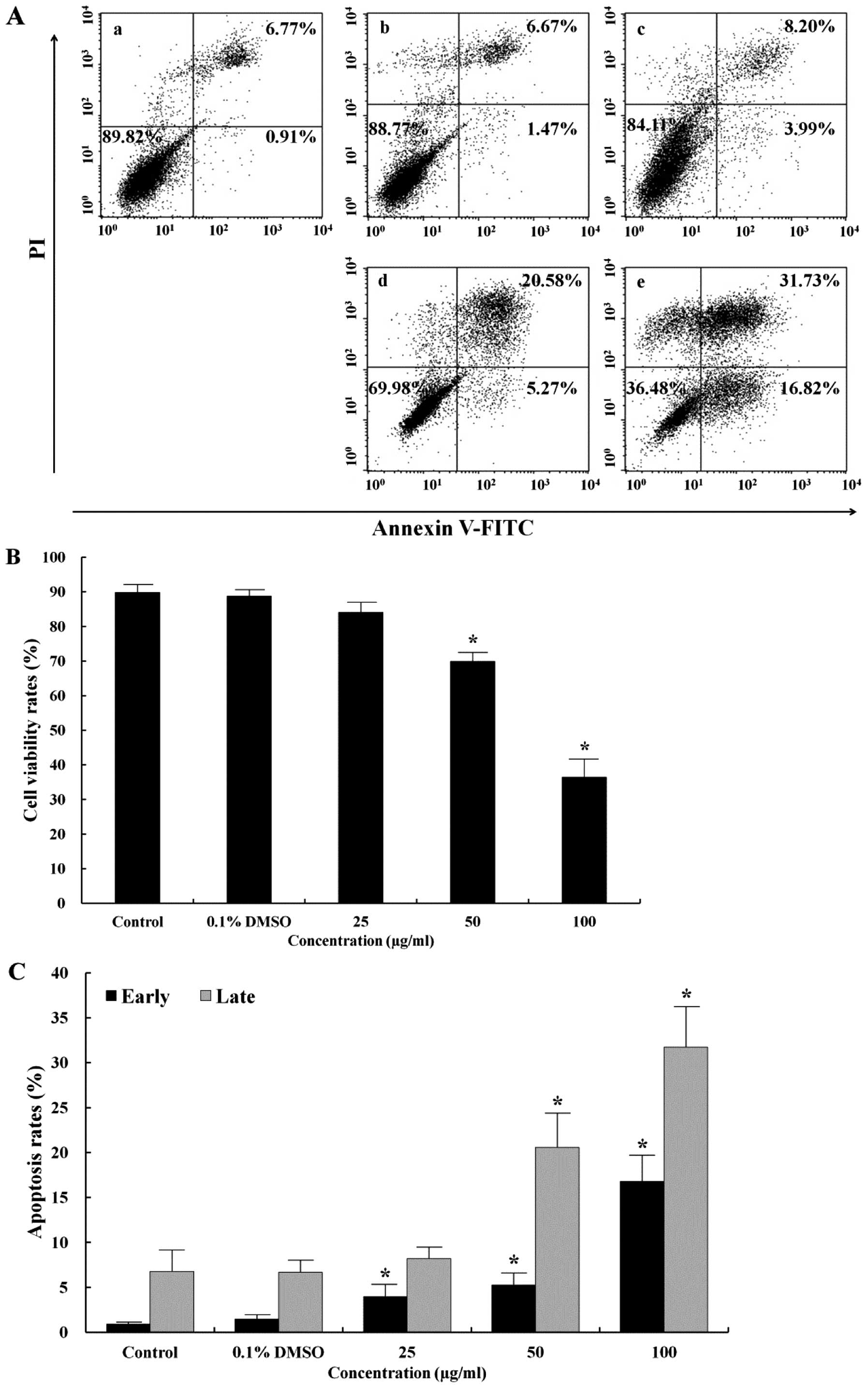
detected. As shown in Fig. 1A,
stained cell populations were defined as follows: lower left (LL),
viable cells (Annexin V−/PI−); lower right
(LR), cells undergoing early apoptosis (Annexin
V+/PI−); upper right (UR), and late apoptotic
or necrotic cells (Annexin V+/PI+). Apoptosis
induction in AGS cells treated with the EtOAc fraction was
evaluated by flow cytometry. The EtOAc fraction increased the
number of early (LR) and late (UR) apoptotic cells in a
dose-dependent manner (Fig. 1C).
In addition, the total apoptosis rate was more than 6-fold higher
than that in the control (48.55 vs. 7.68%) (Fig. 1A). Under control conditions, 89.82%
of cells were viable (LL). In contrast, the survival rate induced
by the 100 μg/ml EtOAc fraction was 36.48% (Fig. 1B).
Induction of cell cycle arrest
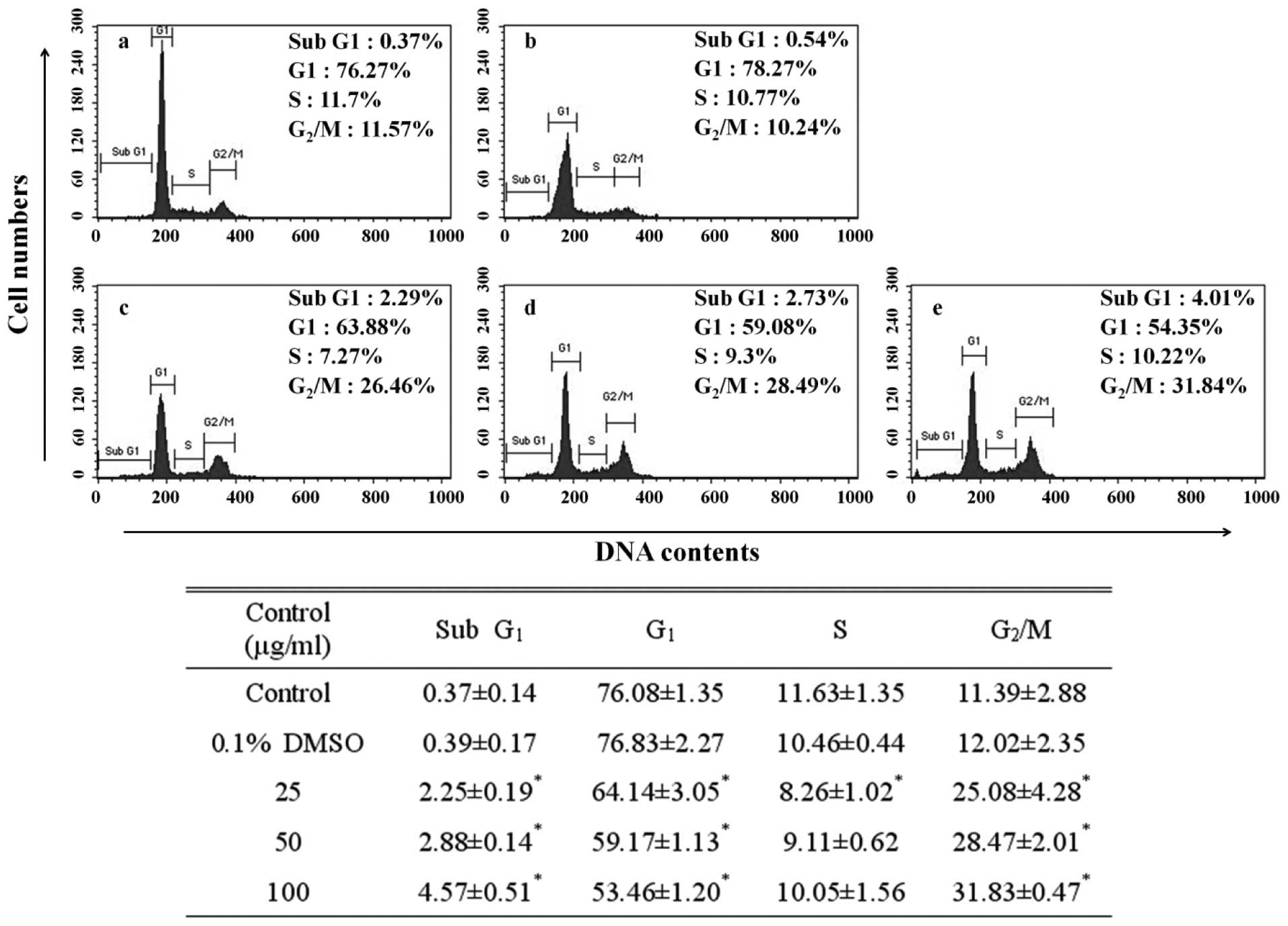
The DNA content of the PI-stained AGS cells was
determined by flow cytometry. The fragmented DNA that serves as
evidence of apoptosis is manifested on the left side rather than at
the G1 peak of a cell cycle (22).
Apoptosis can be observed by detecting the presence of this sub-G1
peak. To probe the relationship between the inhibitory effect of
the EtOAc fraction on AGS cell growth and the apoptotic effect, we
investigated the alteration of the cell cycle using flow cytometry.
In the control cells, this sub-G1 peak was negligible (0.37%), but
accounted for 4.57% of the cells treated with the 100 μg/ml
EtOAc fraction. The sub-G1 phase cell population of the EtOAc
fraction-treated cells increased in a dose-dependent manner
(Fig. 2). Further, apart from the
sub-G1 peak as evidence of apoptosis, the G2/M peak was
31.83%, simultaneously being measured at a significantly higher
rate than that of the control (11.39%). The G2/M phase
cell population of the EtOAc fraction-treated cells increased in a
dose-dependent manner. Depending on the control period of DNA
synthesis, the cell cycle is divided into four phases: G1, S,
G2 and M. The cell cycle has a checkpoint that
determines whether the general progress of cell division is
implemented from one phase to another without disorder. The
G2/M transition is a point at which the completion of
DNA replication is confirmed prior to cell division (23). An increase in G2/M peak
refers to the G2/M cell cycle arrest shows inhibited
cell division, incapacitation by further cell division due to the
damage in the DNA of the AGS cells.
Effect of the EtOAc fraction on the
expression level of the cell cycle-related genes
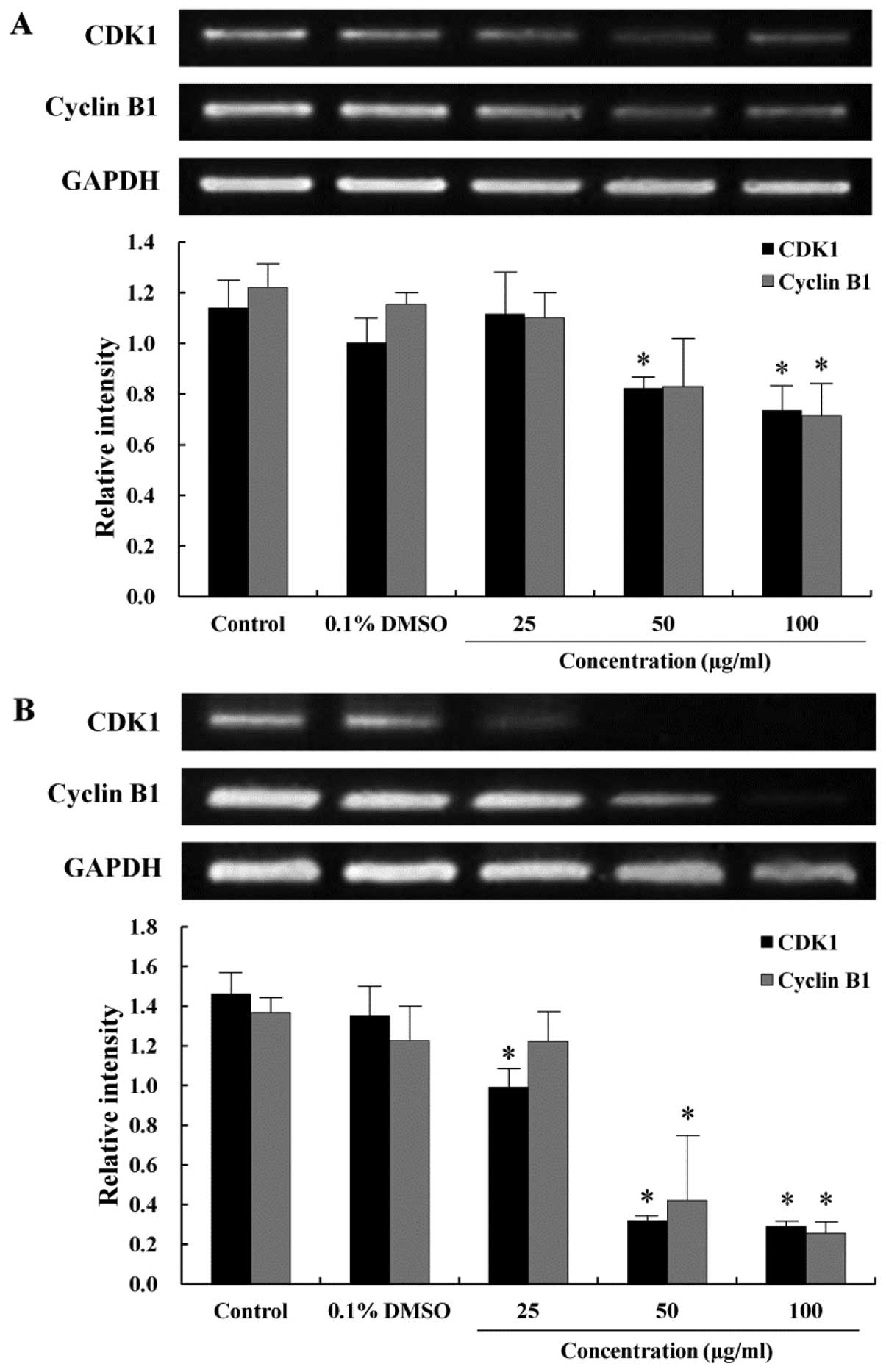
As the EtOAc fraction arrested the AGS cells in the
G2/M phase of the cell cycle, RT-PCR was performed to
study the correlation between G2/M arrest and the
transcription of the cell cycle-related genes. The CDK1 is
primarily activated in association with cyclin B1 during the
progression of the G2/M phase (24). To examine the expression of mRNA
levels regulating the cell cycle progression at the G2/M
phase, which was remarkably arrested as shown in Fig. 2, CDK1 and cyclin B1 expression were
measured by RT-PCR. The expression of CDK1 decreased when the cells
were treated with the EtOAc fraction in a time- and dose-dependent
manner (Fig. 3). Cyclin B1 was
also expressed at a lower level in the EtOAc fraction-treated cells
than in the control cells. The decrease in the expression of CDK1
and cyclin B1 correlated to the increase in distribution of the
G2/M peak, which was consistent with the G2/M
arrest observed from the cell cycle analysis.
Induction of mitochondrial-mediated
apoptosis
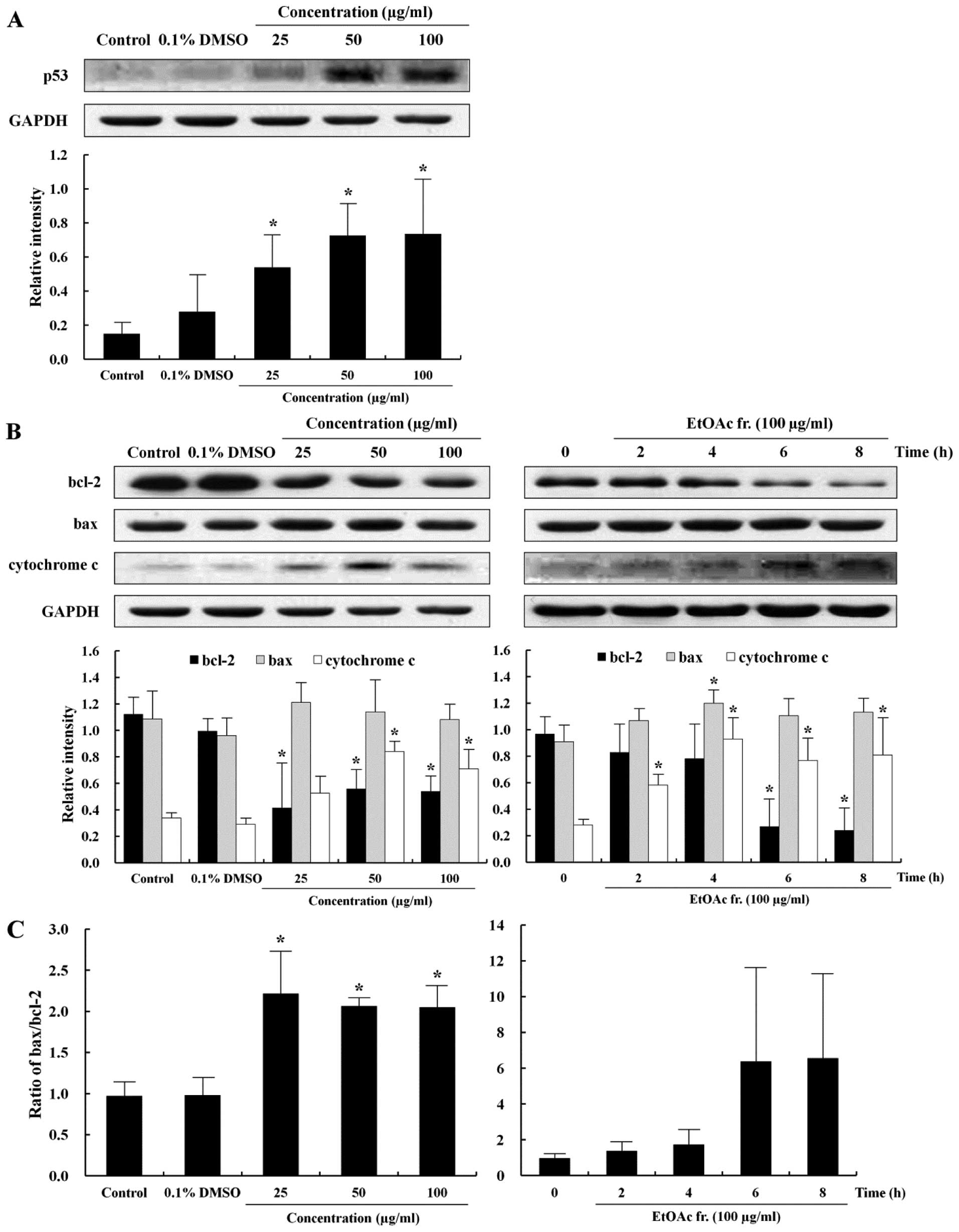
To further provide insight into the apoptotic effect
of the EtOAc fraction, this study measured the expression level of
p53, bcl-2, bax and cytochrome c proteins, which are relevant to
the understanding of apoptotic signaling pathways after exposure to
the EtOAc fraction for 12 h in the AGS cells. p53 is a tumor
suppressor gene and transcription factor that induces either
apoptosis or cell cycle arrest in response to extracellular stress
signals and DNA damage (25). In
addition, the imbalance of bcl-2 and bax protein expression could
be influenced by p53 (26). The
level of p53 protein expression increased in a dose-dependent
manner (Fig. 4A). Major proteins
involved in apoptosis are bcl-2 and bax. The bcl-2 protein inhibits
the emission of cytochrome c from the mitochondria to defend the
cell from apoptosis signals. In contrast, bax protein induces
apoptosis by inducing stress in the mitochondria (27). The release of cytochrome c from the
mitochondria is promoted by bax and blocked by bcl-2 (28). The levels of anti-apoptotic and
apoptotic proteins were examined by western blotting to determine
whether the EtOAc fraction induced the apoptosis of AGS cells
through the regulation of bcl-2, bax and cytochrome c. The
expression of bcl-2, bax and cytochrome c proteins in the AGS cells
with the EtOAc fraction is shown in Fig. 4B. The level of bcl-2 protein
expression decreased and that of cytochrome c increased in a time-
and dose-dependent manner, while that of bax was not significantly
different between the EtOAc fraction-treated cells and the control
cells. However, the bax/bcl-2 ratios increased significantly in a
dose-dependent manner (Fig. 4C).
These results suggest that the EtOAc fraction induced apoptosis of
the AGS cells via the mitochondrial-mediated pathway.
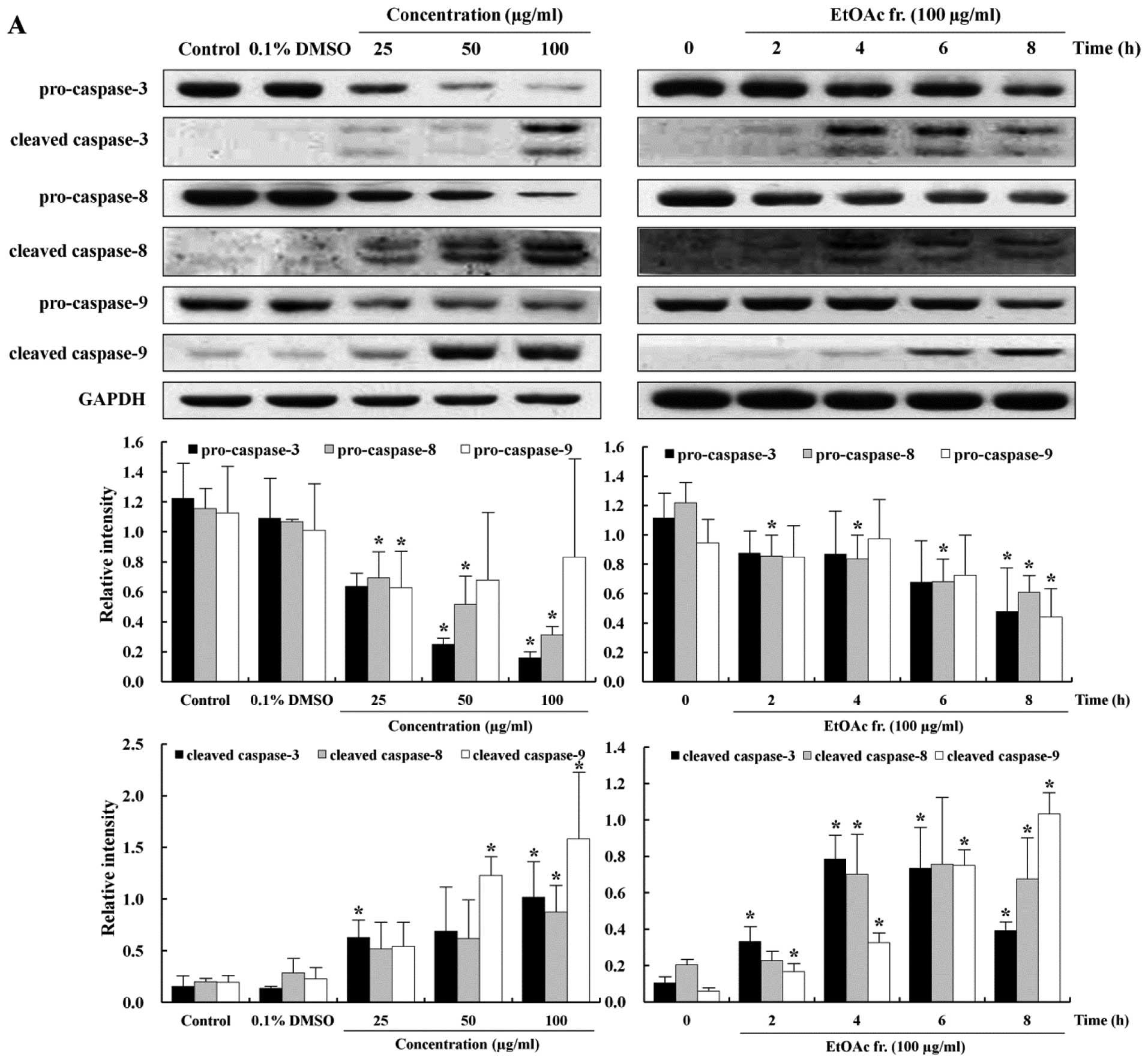
Induction of caspase-mediated
apoptosis
The apoptotic pathway is regulated by caspases that
are responsible, either directly or indirectly, for the cleavage of
other protein substrates within the cell, which is a characteristic
of apoptosis (29). Caspase-3 is
an effector caspase that can trigger the apoptotic process by
activation of initiator caspases such as caspase-8 and -9 (30). In order to understand the
mechanisms involved in the caspase-mediated apoptosis, we measured
the expression levels of pro-caspase-3, -8, -9 and cleaved
caspase-3, -8, -9 proteins. The treatment of the AGS cell with the
EtOAc fraction caused decreases in the levels of pro-caspase-3, -8
and -9, inactive forms of caspase, in a time- and dose-dependent
manner (Fig. 5A). The cleavage of
caspases is directly related to the activation of the apoptotic
process. Cleaved caspase-3, -8 and -9, the active forms of caspase,
increased substantially in the EtOAc fraction-treated AGS cells in
a time- and dose-dependent manner (Fig. 5A). To investigate whether apoptosis
inductions was caspase-dependent, we used the general caspase
inhibitor Z-VAD-FMK. As shown in Fig.
5B, the addition of 20 μM Z-VAD-FMK significantly
reduced the induction of apoptosis in response to the EtOAc
fraction in a dose-dependent manner. These results show that
caspase-3, -8 and -9 act as fundamental factors in the apoptotic
effect of the EtOAc fraction in the AGS cells.
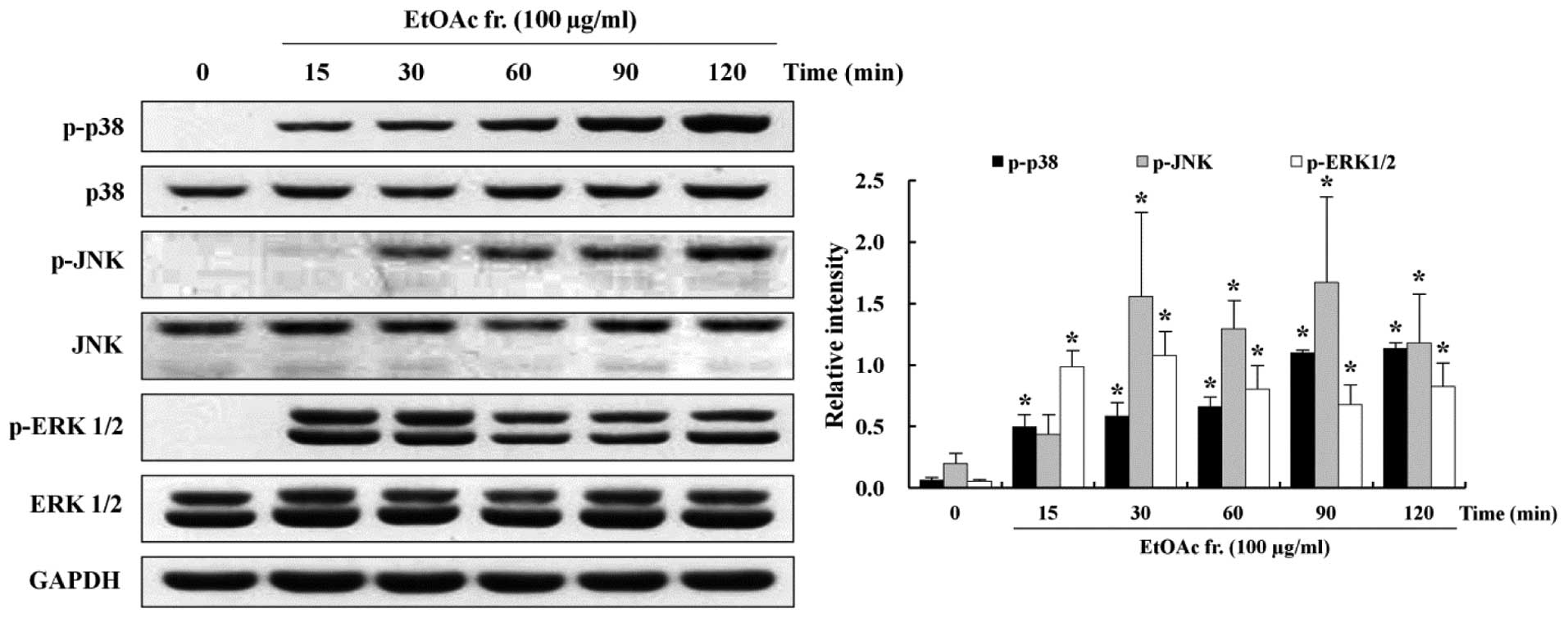
Involvement of MAPKs in the induction of
apoptosis
MAPKs, such as p38, JNK and ERK1/2, activated by
extracellular signals are involved in pro-survival and
pro-apoptotic activity (31). To
understand the role of pro-survival- and proapoptotic-signaling
pathways in the induction of apoptosis, this study assessed 100
μg/ml of the EtOAc fraction-treated AGS cells by western
blotting using antibodies against the phosphorylated and total
forms of MAPKs. Western blotting showed that the phosphorylation of
p38 and JNK increased in the EtOAc fraction-treated AGS cells in a
time-dependent manner. Activation of p38 and JNK was increased at
15 min and 30 min, respectively, and continued to increase until
120 min. On the other hand, activation of ERK1/2 increased markedly
at 15 min, but thereafter, it decreased until 120 min in a
time-dependent manner (Fig. 6).
The expression of the total forms of p38, JNK and ERK1/2 did not
change.
Discussion
Our results showed that the EtOAc fraction induced
apoptosis and cell cycle arrest in the AGS cells. The apoptosis of
the AGS cells with the EtOAc fraction was induced by p53 and
mitochondrial-mediated apoptotic proteins through the MAPK
signaling pathway. In addition, the EtOAc fraction arrested the AGS
cells in the G2/M phase of the cell cycle by regulating
CDK1 and cyclin B1. In our previous study (8), we demonstrated that the EtOAc
fraction, from among the 5 other fractions tested, possessed the
highest anticancer activity in the AGS cells, as evidenced by
4’-6-diamidino-2-phenylindole (DAPI) staining, DNA fragmentation
assay and flow cytometry analysis. The present study is the first
to investigate the signaling pathway at the molecular biological
level in the context of exposure time and dose.
Apoptosis is the process of programmed cell death
that is characterized by cell changes, including exposure of PS
from the cell membrane and DNA fragmentation (32). To detect the characteristics of
apoptosis in the AGS cells treated with the EtOAc fraction, flow
cytometry analysis was performed following Annexin V/PI staining
for detection of early or late apoptosis, and PI labeling assay was
performed to detect DNA damage. As shown in Fig. 1, the early apoptosis rate (16.82%)
in the AGS cells treated with the 100 μg/ml EtOAc fraction
was higher than that of the control (0.91%), and the late apoptosis
rate (31.73%) was higher than that of the control (6.77%). The
EtOAc fraction increased the total apoptosis rate and decreased the
survival rate in a dose-dependent manner. The fragmented DNA that
indicated the induction of apoptosis observed in the sub-G1 peak of
the cell cycle. The EtOAc fraction increased the sub-G1 phase cell
population in a dose-dependent manner (Fig. 2). These results suggest that the
EtOAc fraction may not only induce apoptosis, but also inhibit the
growth of AGS cells.
The normal cell growth is regulated through cell
cycle progression, and uncontrolled cell growth is a characteristic
of cancer. Several anticancer agents have been reported to arrest
the cell cycle and simultaneously induce apoptosis. Cell cycle
arrest has been used as an indicator of anticancer activity
(33). The eukaryotic cell cycle
is divided into the G1 phase for DNA duplication, the synthetic S
phase for effective DNA duplication, the G2 phase for
cell growth ready for mitosis, and the M phase for mitosis
(34). There are 3 checkpoints
that check the general progress for problem-free cell division with
regular transition between the phases, i.e., in the late G1 phase,
G2/M transition, and metaphase-to-anaphase transition
(24). The CDKs are protein
kinases involved in regulating the cell cycle and binding a
regulatory protein called cyclin. The cyclin-CDK complexes regulate
the general progress for a cell cycle. The cyclin D-CDK4 (and/or 6)
complex is necessary for initiating the G1 phase, while the cyclin
E-CDK2 complex is needed to progress to the S phase. The cyclin
A-CDK2 complex is involved from S to G2 phase, and the
cyclin B1-CDK1 complex is required for progression to the M phase
(22,35). The effect of the EtOAc fraction on
the cell cycle progression of the AGS cells is shown in Figs. 2 and 3. The G2/M phase cell
population of the EtOAc fraction-treated cells increased in a
dose-dependent manner. In the present study, we investigated the
correlation between G2/M arrest and the regulating genes
of the cell cycle. Expression of CDK1 and cyclin B1 decreased
comparison with the control in a time- and dose-dependent manner
(Fig. 3). These results revealed
that the EtOAc fraction act on the G2/M transition
checkpoint of the cell cycle.
p53 is a tumor suppressor gene, a transcription
factor, which becomes activated in the cytoplasm and accumulated in
the nucleus when the DNA is damaged by extracellular stress. p53
activates the target genes, including those encoding apoptotic
proteins and cell division suppressor genes to induce apoptosis and
arrest the cell cycle (36). The
basal level of p53 was low in the AGS cells, and the EtOAc fraction
upregulated its expression in a time- and dose-dependent manner
(Fig. 4A). Previous studies
investigating the involvement of p53 in the cellular response to
active substances from natural plants have shown that it induces
apoptosis and upregulates p53 in various cancer cells (37). The p53 activation causes induction
of apoptotic cell death, which leads to the release of cytochrome c
from the mitochondria (38). There
are 2 major apoptotic pathways: the intrinsic mitochondrial pathway
and the extrinsic death receptor pathway (39,40).
Both these pathways are regulated by caspases for the cleavage of
cellular proteins. The mitochondrial pathway induces apoptosis by
releasing pro-apoptotic protein such as cytochrome c. The
mitochondrial-mediated apoptosis is regulated by apoptotic proteins
that suppress the anti-apoptotic protein bcl-2 and promote the
pro-apoptotic protein bax (41).
We showed that the EtOAc fraction decreased the expression of bcl-2
and increased the expression levels of cytochrome c and bax/bcl-2
ratio in a time- and dose-dependent manner (Fig. 4B and C). Cytochrome c is released
from the mitochondria into the cytosol during apoptosis. The
released cytochrome c forms an apoptosome composed of apoptotic
protease activating factor-1 (Apaf1), cytochrome c, and caspase-9,
which subsequently activates caspase-3 (29,42).
As one of the primary pathways of apoptosis, the mitochondrial
pathway is regulated by bcl-2 family proteins and is finalized by
DNA fragmentation, and the apoptotic bodies are regulated through
the activation of caspase-3, a final executing factor. The death
receptor pathway is initiated by Fas receptor that transduces
apoptotic signals into caspase-8-dependent cascade (43). During this cascade, caspase-8
induces the release of cytochrome c and activates caspase-3.
Several studies have provided evidence that caspase
plays a crucial role in the induction of apoptosis (44,45).
Caspases are grouped into initiator caspases such as caspase-8 and
-9 and effector caspases such as caspase-3. Caspase-3 is an
executioner caspase that is activated by a mitochondrial-mediated
apoptosis involving caspase-9 or a death receptor-mediated
apoptosis involving caspase-8 (46). In the present study, procaspase-3,
-8 and -9, inactive forms of caspase, decreased in the cells
treated with the EtOAc fraction in a time- and dose-dependent
manner (Fig. 5A). In contrast,
cleaved caspase-3, -8 and -9, active forms of caspase, increased
substantially in the cells treated with the EtOAc fraction in a
time- and dose-dependent manner (Fig.
5A). In addition, pretreatment with the general caspase
inhibitor Z-VAD-FMK significantly inhibited the EtOAc
fraction-induced apoptosis, indicating that the EtOAc fraction
induced caspase-dependent apoptosis in the AGS cells. The
expression levels of caspase-3 and -8 were significantly blocked in
the presence of Z-VAD-FMK, but the inhibition of caspase-9 had a
mild inhibitory effect (Fig. 5B).
Caspase-9 was activated at ∼6 h, while caspase-3 was activated at 4
h. These findings indicate that caspase-3 has a feedback action on
caspase-9. These results suggested that p53 is a key regulator of
apoptosis and cell cycle arrest in the EtOAc fraction-treated AGS
cells. Furthermore, bcl-2 and anti-apoptotic protein was impeded by
the release of cytochrome c from the mitochondria, and caspases
were sequentially activated to induce apoptosis of the EtOAc
fraction-treated AGS cells.
The MAPK signaling pathways regulate intra cellular
functions such as apoptosis and cell proliferation (47). Major members in the MAPK signaling
pathway belong to 3 subgroups of the MAPK family: p38, JNK and
ERK1/2 (48). Several studies have
reported that the activation of MAPKs is linked to cell growth and
induction of apoptosis in various cancer cells (49–51).
It has been shown that activation of p38 and JNK are involved in
mitochondrial-mediated apoptosis and caspase activation and that
ERK1/2 is associated with cell proliferation and survival. This
study hypothesized that the anticancer activity of the EtOAc
fraction was related to the activation of the MAPK signaling
pathway. To screen the MAPK-signaling pathway underlying the
anticancer effects of the EtOAc fraction, the phosphorylation of
p38, JNK and ERK 1/2 was determined. We observed that the EtOAc
fraction increased phosphorylation of p38 and JNK in a
time-dependent manner (Fig.
6).
In conclusion, we showed that the EtOAc fraction
could effectively suppress the growth of AGS cells by the induction
of apoptosis and cell cycle arrest. The expression levels of p53,
cytochrome c, cleaved caspase-3, -8 and -9 were upregulated, and
the expression levels of CDK1, cyclin B1, bcl-2, pro-caspase-3, -8
and -9 were downregulated through phosphorylation of p38 and JNK in
the AGS cells treated with the EtOAc fraction. These results
provide, for the first time, a useful foundation for understanding
the anticancer effects of O. japonicus at the molecular
level and show that O. japonicus could be used for the
development of novel anticancer agents for the treatment of gastric
cancers.
Acknowledgements
This study was supported by the
Post-doctoral Research Program of Inje University 2012.
References
|
1.
|
Kelloff GJ: Perspectives on cancer
chemoprevention research and drug development. Adv in Cancer Res.
78:199–334. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Ryu DS, Baek GO, Kim EY, Kim KH and Lee
DS: Effects of polysaccharides derived from Orostachys
japonicus on induction of cell cycle arrest and apoptotic cell
death in human colon cancer cells. BMB Rep. 43:750–755.
2010.PubMed/NCBI
|
|
3.
|
Choi SY, Chung MJ, Seo WD, Shin JH, Shon
MY and Sung NJ: Inhibitory effects of Orostachys japonicus
extracts on the formation of N-nitrosodimethylamine. J Agric Food
Chem. 54:6075–6078. 2006.
|
|
4.
|
Jung HJ, Choi J, Nam JH and Park HJ:
Anti-ulcerogenic effects of the flavonoid-rich fraction from the
extract of Orostachys japonicus in mice. J Med Food.
10:702–706. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Lee HS, Ryu DS, Lee GS and Lee DS:
Anti-inflammatory effects of dichloromethane fraction from
Orostachys japonicus in RAW 264.7 cells: Suppression of
NF-κB activation and MAPK signaling. J Ethnopharmacol. 140:271–276.
2012.PubMed/NCBI
|
|
6.
|
Je Ma C, Jung WJ, Lee KY, Kim YC and Sung
SH: Calpain inhibitory flavonoids isolated from Orostachys
japonicus. J Enzyme Inhib Med Chem. 24:676–679. 2009.PubMed/NCBI
|
|
7.
|
Park HJ, Young HS, Park KY, Rhee SH, Chung
HY and Choi JS: Flavonoids from the whole plants of Orostachys
japonicus. Arch Pharm Res. 14:167–171. 1991. View Article : Google Scholar
|
|
8.
|
Ryu DS, Lee HS, Lee GS and Lee DS: Effect
of the ethylacetate extract of Orostachys japonicus on
induction of apoptosis through the p53-mediated signaling pathway
in human gastric cancer cells. Biol Pharm Bull. 35:660–665.
2012.
|
|
9.
|
Singh SV, Herman-Antosiewicz A, Singh AV,
Lew KL, Srivastava SK, Kamath R, Brown KD, Zhang L and Baskarah R:
Sulforaphane-induced G2/M phase cell cycle arrest involves
checkpoint kinase 2-mediated phosphorylation of cell division cycle
25C. J Biol Chem. 279:25813–25822. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Harris SL and Levin AJ: The p53 pathway:
positive and negative feedback loops. Oncogene. 24:2899–2908. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Vazquez A, Bond EE, Levine AJ and Bond GL:
The genetics of the p53 pathway, apoptosis and cancer therapy. Nat
Rev Drug Discov. 7:979–987. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Shiozaki EN and Shi Y: Caspases, IAPs and
Smac/DIABLO: mechanisms from structural biology. Trends Biochem
Sci. 29:486–494. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Tsujimoto Y: Bcl-2 family of proteins:
life-or-death switch in mitochondria. Biosci Rep. 22:47–58. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Dhanalakshmi S, Agarwal P, Globe L and
Agarwal R: Silibinin sensitizes human prostate carcinoma DU145
cells to cisplatinand carboplatin-induced growth inhibition and
apoptotic death. Int J Cancer. 106:699–705. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Brooks G: Cyclin, cyclin-dependent
kinases, and cyclin-dependent kinase inhibitors: detection methods
and activity measurements. Methods Mol Biol. 296:291–298.
2005.PubMed/NCBI
|
|
16.
|
Vermeulen K, Berneman ZN and Van
Bockstaele DR: Cell cycle and apoptosis. Cell Prolif. 36:165–175.
2003. View Article : Google Scholar
|
|
17.
|
Sebolt-Leopold JS: Development of
anticancer drugs targeting the MAP kinase pathway. Oncogene.
19:6594–6599. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Sebolt-Leopold JS and Herrera R: Targeting
the mitogen-activated protein kinase cascade to treat cancer. Nat
Rev Cancer. 4:937–947. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Jeong JH, Ryu DS, Suk DH and Lee DS:
Anti-inflammatory effects of ethanol extract from Orostachys
japonicus on modulation of signal pathways in LPS-stimulated
RAW 264.7 cells. BMB Rep. 44:399–404. 2011.PubMed/NCBI
|
|
20.
|
Lee HS, Bilehal D, Lee GS, Ryu DS, Kim HK,
Suk DH and Lee DS: Anti-inflammatory effect of the hexane fraction
from Orostachys japonicus in RAW 264.7 cells by suppression
of NF-κB and PI3K-Akt signaling. J Funct Foods. 5:1217–1225.
2013.
|
|
21.
|
Kaufmann SH and Henqartner MO: Programmed
cell death: alive and well in the new millennium. Trends Cell Biol.
11:526–534. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Ryu DS, Kim SH and Lee DS:
Anti-proliferative effect of polysaccharides from Salicornia
herbacea on induction of G2/M arrest and apoptosis in human
colon cancer cells. J Microbiol Biotechnol. 19:1482–1489.
2009.PubMed/NCBI
|
|
23.
|
Nakanishi M, Shimada N and Niida H:
Genetic instability in cancer cells by impaired cell cycle
checkpoints. Cancer Sci. 97:984–989. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Elledge SJ and Harpae JW: CDK inhibitors;
on the threshold of checkpoints and development. Curr Opin Cell
Biol. 6:847–852. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Vousden KH: Apoptosis. p53 and PUMA: a
deadly duo. Science. 309:1685–1686. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Lavin MF and Gueven N: The complexity of
p53 stabilization and activation. Cell Death Differ. 13:941–950.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Ai Z, Lu W and Qin X: Arsenic trioxide
induces gallbladder carcinoma cell apoptosis via down regulation of
bcl-2. Biochem Biophys Res Commun. 348:1075–1081. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Wang JB, QI LL, Zheng SD and Wu TX:
Curcumin induces apoptosis through the mitochondria-mediated
apoptotic pathway in HT-29 cells. J Zhejizng Univ Sci B. 10:93–102.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Lavrik I, Golks A and Krammer PH: Death
receptor signaling. J Cell Sci. 118:265–267. 2005. View Article : Google Scholar
|
|
30.
|
Fujita E, Egashira J, Urase K, Kuida K and
Momoi T: Caspase-9 processing by caspase-3 via a feedback
amplification loop in vivo. Cell Death Differ. 8:335–344. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Seger R and Krebs EG: The MAPK signaling
cascade. FASEB J. 9:726–735. 1995.PubMed/NCBI
|
|
32.
|
Meier P, Finch A and Evan G: Apoptosis in
development. Nature. 407:796–801. 2000. View Article : Google Scholar
|
|
33.
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Schwartz GK and Shah MA: Targeting the
cell cycle: a new approach to cancer therapy. J Clin Oncol.
23:9408–9421. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Koff A, Giordano A, Desai D, Yamashita K,
Harper JW, Elledge S, Nishimoto T, Morgan DO, Franza BR and Roberts
JM: Formation and activation of a cyclin E-cdk2 complex during the
G1 phase of the human cell cycle. Science. 257:1689–1694. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Fisher DE: The p53 tumor suppressor:
critical regulator of life & death in cancer. Apoptosis.
6:7–15. 2001. View Article : Google Scholar
|
|
37.
|
Moll UM and Zaika A: Nuclear and
mitochondrial apoptotic pathways of p53. FEBS Lett. 493:65–69.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Erster S, Mihara M, Kim RH, Petrenko O and
Moll UM: In vivo mitochondrial p53 translocation triggers a rapid
first wave of cell death in response to DNA damage that can precede
p53 target gene activation. Mol Cell Biol. 24:6728–6741. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Jiang X and Wang X: Cytochrome c-mediated
apoptosis. Annu Rev Biochem. 73:87–106. 2004. View Article : Google Scholar
|
|
40.
|
Nagata S: Fas ligand-induced apoptosis.
Annu Rev Genet. 33:29–55. 1999. View Article : Google Scholar
|
|
41.
|
Willis SN and Adams JM: Life in the
balance: how BH3-only proteins induce apoptosis. Curr Opin Cell
Biol. 17:617–625. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Pop C, Timmer J, Sperandio S and Salvesen
GS: The apoptosome activates caspase-9 by dimerization. Mol Cell.
22:269–275. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Zhou Z, Sun X and Kang YJ: Ethanol-induced
apoptosis in mouse liver: Fas- and cytochrome c-mediated caspase-3
activation pathway. Am J Pathol. 159:329–338. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Jin UH, Song KH, Motomura M, Suzuki I, Gu
YH, Kang YJ, Moon TC and Kim CH: Caffeic acid phenethyl ester
induces mitochondria-mediated apoptosis in human myeloid leukemia
U937 cells. Mol Cell Biochem. 310:43–48. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Zheng Y, Zhou M, Ye A, Li Q, Bai Y and
Zhang Q: The conformation change of Bcl-2 is involved in arsenic
trioxide-induced apoptosis and inhibition of proliferation in
SGC7901 human gastric cancer cells. World J Sur Oncol. 8:312010.
View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Cho SH, Chung KS, Choi JH, Kim DH and Lee
KT: Compound K, a metabolite of ginseng saponin, induces apoptosis
via caspase-8-dependent pathway in HL-60 human leukemia cells. BMC
Cancer. 9:4492009. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Martin GS: Cell signaling and cancer.
Cancer Cell. 4:167–174. 2003. View Article : Google Scholar
|
|
48.
|
Freeman SM and Whartenby KA: The role of
the mitogen-activated protein kinase cellular signaling pathway in
tumor cell survival and apoptosis. Drug News Perspect. 17:237–242.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Lin A and Dibling B: The true face of JNK
activation in apoptosis. Aging Cell. 1:112–116. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Lunghi P, Giuliani N, Mazzera L, Lombardi
G, Ricca M, Corradi A, Cantoni AM, Salvatore L, Riccioni R and
Costanzo A: Targeting MEK/MAPK signal transduction module
potentiates ATO-induced apoptosis in multiple myeloma cells through
multiple signaling pathways. Blood. 112:2450–2462. 2008. View Article : Google Scholar
|
|
51.
|
Mansouri A, Ridgway LD, Korapati AL, Zhang
Q, Tian L, Wang Y, Siddik ZH, Mills GB and Claret FX: Sustained
activation of JNK/p38 MAPK pathways in response to cisplatin leads
to Fas ligand induction and cell death in ovarian carcinoma cells.
J Biol Chem. 278:19245–19256. 2003. View Article : Google Scholar : PubMed/NCBI
|