1. Introduction
Acute lymphoblastic leukemia (ALL) is caused by the
uncontrolled clonal proliferation of immature lymphoid cells which
accumulate in the bone marrow (BM) and other body sites. The
neoplastic lymphoblasts display an impaired differentiation
program, are blocked at various maturation steps and are resistant
to apoptotic stimuli and cell death. ALL accounts for approximately
20% of acute leukemias in the adult, however it is the most common
malignant disease in the childhood (1). The clinical management of ALL is
challenging, especially in the adults, even though current
therapies can induce a complete remission in 65–90% of patients.
Nevertheless, patients who are refractory to induction therapy or
relapse after induction face a poor prognosis (2). ALL can be classified in two main
subgroups, namely B-cell and T-cell ALL (B-ALL and T-ALL,
respectively) (3).
T-ALL is an aggressive form of leukemia which arises
in the thymus from T-cell progenitors expressing immature T-cell
immunophenotypic markers (4,5).
T-ALL accounts for 10–15% and 25% of pediatric and adult ALL,
respectively. In the childhood, cure rate for T-ALL patients
reaches 70–75%. In the adults, the cure rate remains low: 30–40%
for adults below 60 years of age and 10% above this age (6,7). By
immunophenotyping, it is possible to distinguish three subtypes of
T-ALL, i.e., early, cortical and mature, which reflect different
stages of healthy thymocyte differentiation. This classification is
prognostically relevant, as early and mature T-ALLs have a poorer
outcome than cortical T-ALL (8).
Recent findings have documented that T-ALL is an
extremely heterogeneous disease, characterized by chromosomal
rearrangements causing aberrant expression of transcription factors
(Myb; TAL/SCL; HOX) (9,10), altered expression of oncogenes
(10), somatic gene mutations
(11,12), multiple signal transduction pathway
impairment (13–16) and microRNA dysregulation (17–19).
Activating mutations in Notch-1, the master
regulator of T-cell development, are found in more than 60% of
T-ALL patients, independently of the subtype (20). All of these alterations impact on
T-ALL cell proliferation, differentiation, survival and
drug-resistance (21).
In general, leukemia pathogenesis, treatment
resistance and relapse are thought to be caused by leukemic stem
cells or leukemia initiating cells (LICs) (22). LICs are characterized by asymmetric
cell division and self-renewal capacity, unlimited repopulating
potential and production of partially differentiated cells. Whereas
the bulk of leukemic cells rapidly proliferate, LICs are mainly
quiescent (23). This feature is
associated with chemoresistance, as conventional chemotherapy
strategies mainly target rapidly dividing cells (24).
The phenotype of T-ALL LICs is still under
discussion. Cox et al (25)
reported that either CD34+/CD4− or
CD34+/CD7− cells were capable of serial
engraftment in NOD/SCID mice (25). Afterwards, the leukemia initiating
potential in xenografts of the CD7+/CD1a−
subset of primary T-ALL samples was found to be superior to other
subsets (26). The importance of
CD34 as a marker of LIC activity in T-ALL patients has nevertheless
been documented by independent groups (27,28).
However, it has been shown that also
CD34−/CD7+ T-ALL cells displayed LIC
proprieties, although at lower levels than CD34+ cells
(28). The above-outlined
discrepancies could well reflect differences among distinct
molecular T-ALL subtypes. Nevertheless, these studies have
disclosed the complexity of LICs in human T-ALL.
Among the deregulated signaling pathways that have
been identified in T-ALL, the phosphoinositide 3-kinase
(PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling network
has been reported to be active in a high percentage (75–80%) of
patients, where it portends a poorer prognosis (29).
Over the last 10 years, mTOR has become an
attractive therapeutic target in cancer patients, as several small
molecule mTOR inhibitors have been developed and are being tested
as monotherapy in clinical trials (30–32).
Moreover, the use of targeted drugs combined with traditional
anticancer agents could increase treatment efficacy, by lowering
the required dosage of chemotherapeutic drugs and limiting their
systemic side effects (33). In
this review, we will describe the potential of several strategies
for mTOR inhibition to improve the outcome of T-ALL patients.
2. The PI3K/Akt/mTOR pathway
mTOR is a 289-kDa serine/threonine (Ser/Thr) kinase
which belongs to the phosphoinositide kinase-related family of
protein kinases (PIKK) (34). The
PIKK family includes ataxia telangiectasia mutated (ATM), ataxia
telangiectasia- and RAD3-related (ATR), human suppressor of
morphogenesis in genitalia-1 (hSMG-1) and the catalytic subunit of
DNA-dependent protein kinase (DNA-PK) (35).
mTOR collects input from several signal transduction
networks, such as the PI3K/Akt, the Ras/Raf/mitogen-activated
protein kinase (MEK)/extracellular signal-regulated kinase (ERK)
and the AMP-activated protein kinase (AMPK) pathways, for
regulating several physiological events. Indeed, mTOR is involved
in cell cycle progression, cell survival, translation, metabolism,
motility, autophagy and ageing (36). mTOR is the catalytic subunit of two
distinct multi-protein complexes known as mTOR complex 1 (mTORC1)
and mTOR complex 2 (mTORC2), both of which are characterized by
their different partner proteins and their substrate specificity
(36) (Fig. 1).
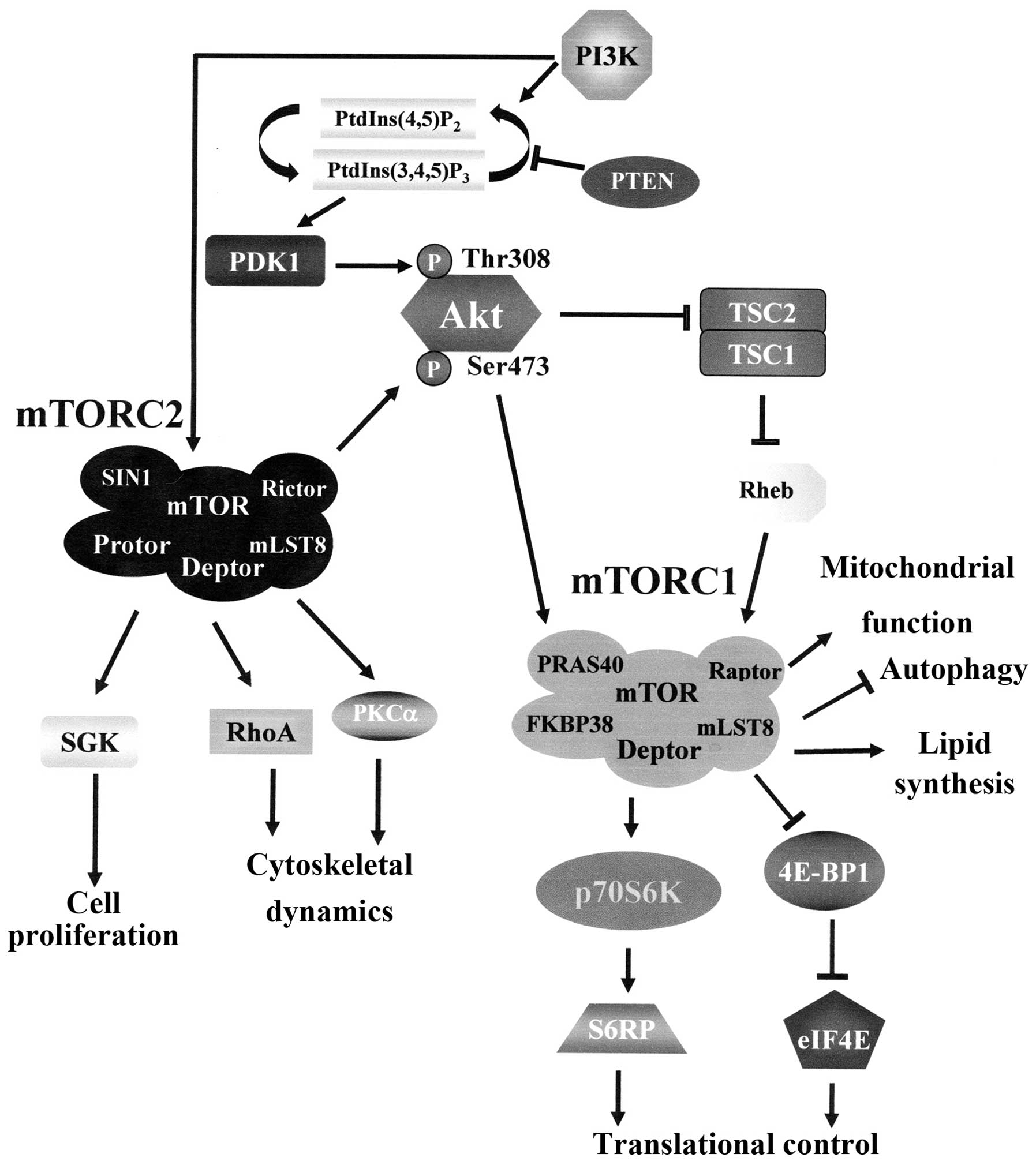 | Figure 1The PI3K/Akt/mTOR pathway. PI3K
generates PtdIns(3,4,5)P3 from PtdIns(4,5)P2. PtdIns(3,4,5)P3 attracts to the plasma
membrane PDK1 which phosphorylates Akt at Thr 308. Full Akt
activation requires Ser 473 phosphorylation by mTORC2. Active Akt
inhibits TSC2 activity through direct phosphorylation. TSC2 is a
GTP-ase activating protein (GAP) that functions in association with
TSC1 to inactivate the small G protein Rheb. Akt-driven TSC1/TSC2
complex inactivation allows Rheb to accumulate in a GTP-bound
state. Rheb-GTP then upregulates mTORC1 activity. However, mTORC1
is controlled by Akt also through PRAS40 phosphorylation. The
activation mechanisms of mTORC2 are not fully understood yet, but
they require PI3K activity. Arrows indicate activating events,
while perpendicular lines indicate inhibitory events. 4E-BP1,
eukaryotic initiation factor 4E-binding protein 1; Deptor,
DEP-domain-containing mTOR interacting protein; eIF4E, eukaryotic
initiation factor 4E; FKBP38, FK-506 binding protein 38; mLST8,
mammalian lethal-with-sec-thirteen 8; mTOR, mammalian target of
rapamycin; mTORC1, mTOR complex 1; mTORC2, mTOR complex 2; PDK1,
phosphoinositide-dependent kinase 1; PI3K, phosphoinositide
3-kinase; PKCα, protein kinase C α; PRAS40, proline-rich Akt
substrate of 40-kDa; Protor, protein observed with Rictor;
PtdIns(4,5)P2, phosphoinositide
(4,5) bisphosphate; PtdIns(3,4,5)P3, phosphoinositide
(3,4,5)
trisphosphate; PTEN, phosphatase and tensin homolog deleted on
chromosome ten; p70S6K, p70S6 kinase; Raptor, regulatory associated
protein of mTOR; Rheb, Ras homolog enriched in brain; Rictor,
rapamycin insensitive companion of mTOR; S6RP, S6 ribosomal
protein; SGK, serum- and glucocorticoid-stimulated kinase; SIN1,
stress-activated protein kinase-interacting protein 1; TSC1,
tuberous sclerosis 1; TSC2, tuberous sclerosis 2. |
mTORC1 is composed of the regulatory associated
protein of mTOR (Raptor, a scaffolding protein), mammalian
Lethal-with-Sec-Thirteen 8 (mLST8), proline-rich Akt substrate of
40-kDa (PRAS40), FK-506 binding protein 38 (FKBP38) and
DEP-domain-containing mTOR interacting protein (Deptor). mTORC1 is
sensitive to rapamycin and its derivatives (rapalogs) (37). Multiple exogenous stimuli regulate
mTORC1 activity, including growth factors such as insulin and
insulin-like growth factor-1 (IGF-1), stress signals, cellular
energy status and amino acids (38).
mTORC1 activation is mainly regulated by PI3K/Akt
signaling. Akt phosphorylates 200-kDa tuberous sclerosis 2 (TSC2 or
hamartin). TSC2 is a GTPase-activating protein (GAP) that
associates with TSC1 (tuberous sclerosis 1 or tuberin) for
inactivating the small G protein Rheb (Ras homolog enriched in
brain). Once TSC2 is phosphorylated by Akt, the GAP activity of the
TSC1/TSC2 complex is repressed, allowing Rheb to accumulate in a
GTP-bound state. As a consequence, Rheb-GTP upregulates the protein
kinase activity of mTORC1 (39).
Furthermore, Akt phosphorylates PRAS40 at Thr246. Phosphorylated
PRAS40 dissociates from mTORC1 in response to growth factors, as
well as glucose and nutrients, and thereby releases the inhibitory
function of PRAS40 on mTORC1 (40).
mTORC1 positively regulates cell growth and
proliferation by promoting many anabolic processes and by limiting
catabolic processes such as autophagy (41) (Fig.
1). Regarding protein translation, mTORC1 phosphorylates
components of the protein synthesis machinery, such as p70 S6
kinase (p70S6K) and eukaryotic translation initiation factor
4E-binding protein 1 (4E-BP1). In turn, p70S6K phosphorylates the
40S ribosomal protein S6 (S6RP), leading to active translation of
mRNA involved in ribosome biogenesis (42), while 4E-BP1 interacts with the
eukaryotic initiation factor 4E (eIF4E), which critically regulates
cap-dependent mRNA translation (43). Once phosphorylated by mTORC1,
4E-BP1 releases eIF4E, which then associates with eIF4G to
stimulate translation initiation (44,45).
In addition to its role in protein translation,
activation of mTORC1 triggers metabolic changes that are critically
important in carcinogenesis, such as mitochondrial biogenesis and
oxidative metabolism, aerobic glycolysis and de novo
lipogenesis (41). mTORC1 controls
mitochondrial biogenesis and oxidative metabolism by regulating the
interactions between the transcription factor yin-yang 1 (YY1) and
the peroxisomal proliferator-activated receptor γ (PPARγ)
coactivator 1 (PGC-1), thereby preventing the coactivation of YY1
(46).
As far as aerobic glycolysis is concerned, mTORC1
promotes it through induction of a transcriptional program
affecting metabolic glycolytic gene targets of hypoxia-inducible
factor 1α (HIF1α) (47,48).
Regarding lipid synthesis, mTORC1 activates the
transcription factors sterol regulatory element binding protein 1
(SREBP1) and PPARγ which are necessary and sufficient for the
differentiation of preadipocytes and lipid accumulation (41).
mTORC1 negatively regulates autophagy, a complex
catabolic process that sustains cellular metabolism through
recycling of cellular components during growth unfavorable
conditions. Nevertheless, autophagy has also been associated with
promoting cell survival during nutrient or hypoxic stress and may
promote cancer cell survival (49). mTORC1 suppresses the kinase
activity of unc-51-like kinase 1 (ULK1), thus preventing the
ULK1/autophagy-related gene 13 (Atg13)/FIP200 complex formation
(50) that plays an essential role
at the early stages of autophagosome formation (51).
mTORC2 comprises rapamycin-insensitive companion of
mTOR (Rictor), mLST8, stress-activated protein kinase-interacting
protein 1 (SIN1), protein observed with Rictor (Protor), and
Deptor, and is generally described as being insensitive to
rapamycin/rapalogs. Nevertheless, it has been demonstrated that
long-term rapamycin treatment leads to dissociation of mTORC2 with
resulting inhibition of Akt feedback phosphorylation at Ser 473, in
primary leukemic cells both in vitro and in vivo
(52). mTORC2 is mainly activated
by growth factors through PI3K/Akt, and controls several downstream
AGC kinases such as Akt itself, serum- and glucocorticoid
stimulated kinase (SGK) and protein kinases Cα (PKCα) (53–55)
(Fig. 1). Therefore, mTORC2
regulates cell proliferation, but it is also involved in the
spatial control of cell growth via cytoskeleton regulation, through
actin fibers, paxillin, RhoA, Rac1 and PKCα (56).
The regulation of PI3K/Akt/mTOR axis is extremely
complex, and this is due mainly to the existence of multiple
feedback loops and direct activation mechanisms that place mTOR
both upstream and downstream of several oncogenic pathways.
Importantly, these regulation loops are relevant in vivo and
influence therapeutic responses based on mTOR inhibition,
contributing to the drug-resistance that can occur in mTOR-targeted
therapies using rapamycin or rapalogs (45). When Akt activates mTORC1, a
negative feedback circuit antagonizes the formation of mTORC2 and
reduces Akt activity (57).
Moreover, when activated, mTORC1 phosphorylates p70S6K, which in
turn inhibits insulin receptor substrate 1 (IRS-1) by
phosphorylating it at multiple sites (Ser 307 and Ser 636/639),
inducing its degradation and altering its localization, all of
which ultimately dampen PI3K/Akt/mTORC1 activation (58–61).
mTORC1 is also capable of downregulating IRS-2 expression by
enhancing its proteosomal degradation (62). Recent findings have also
highlighted the existence of a rapamycin-sensitive,
mTORC1/p70S6K-mediated phosphorylation of Rictor on Thr1135. This
phosphorylative event exerts a negative regulatory effect on the
mTORC2-dependent phosphorylation of Akt at Ser 473 in vivo
(63).
PI3K/Akt/mTOR signaling is antagonized by
phosphatases. Phosphatase and tensin homolog deleted on chromosome
10 (PTEN) is a potent repressor of this pathway that removes
3′-phosphate from phosphoinositide (3,4,5)
trisphosphate [PtdIns(3,4,5)P3] to yield PtdIns
(4,5) bisphosphate [PtdIns(4,5)P2]
(64), thus counterbalancing the
action of PI3K (Fig. 1). Loss of
PTEN, due to inactivating mutations or silencing, has been
reported in a wide range of sporadic human cancers, including
leukemias, and it has been correlated to cellular proliferation,
cancer susceptibility and tumor progression (65). PTEN plays an important role
in T-ALL pathophysiology (see below).
The lipid phosphatases, Src homology
domain-containing inositol phosphatase (SHIP) 1 and 2, remove
5′-phosphate from PtdIns(3,4,5)P3 to yield
PtdIns(3,4)P2 (66),
and play a fundamental role in the inhibition of proliferation and
survival of hematopoietic cells (67). Mutations of SHIP1, that is
predominantly expressed in hematopoietic cells, have been
implicated in the development of different blood disorders,
including T-ALL (68). Also
protein phosphatases, such as protein phosphatase 2A (PP2A), impact
on PI3K/Akt/mTOR signaling, as PP2A dephosphorylates Akt at Thr308
(69).
3. Disregulated mTOR activity and T-ALL
development
It is established that PTEN deletion led to
T-ALL development in mice (70)
and that rapamycin treatment of preleukemic mice prevented LIC
formation and halted T-ALL development (71). Both mTORC1 and mTORC2 have been
implicated in T-ALL pathophysiology. Regarding mTORC1, it has been
documented that loss of mTORC1 activity caused by Raptor
deficiency, eradicated T-ALL in a murine model of disease,
suggesting that mTORC1 played a key role in T-ALL LIC survival
(72). However, rapamycin was not
sufficient for T-ALL eradication. This could be due to the fact
that rapamycin is an incomplete blocker of mTORC1 outputs (73). Therefore, dual PI3K/mTOR inhibitors
or ATP-competitive mTORC1/mTORC2 inhibitors (see below) could be
more effective agents against T-ALL, as they efficiently targeted
rapamycin-resistant mTORC1 activity in T-ALL cells (74–76).
An important role for mTORC2 in T-ALL development is
suggested by the findings of another group (77). It was documented that deletion of
the mTORC2 component, Rictor, prevented leukemogenesis and
hematopoietic stem cell (HSC) depletion after PTEN deletion
in adult mice. These observations implicated an important role for
mTORC2 activation in these processes. However, Rictor
deletion (and hence mTORC2 function inactivation) had little effect
on the physiology of healthy (i.e., non-PTEN-deleted) HSCs.
Moreover, PTEN deletion from neonatal HSCs did not activate
PI3K/Akt signaling or promote HSC proliferation/depletion or
leukemogenesis. Therefore, it was concluded that PTEN is required
in adult, but not neonatal, HSCs for inhibiting mTORC2 signaling
downstream of PI3K/Akt (77).
These findings could explain why B-ALL, where PTEN deletions
are very uncommon (78), is a
disease of the early childhood with a peak incidence at 2–5 years
of age (79), whereas pediatric
T-ALL, in which PTEN deletion/inactivation is quite
frequently observed (80),
displays an older mean age of presentation (approximately 9–10
years) (81).
4. Causes for PI3K/Akt/mTOR pathway
activation in T-ALL
PI3K/Akt/mTOR pathway aberrant activation is a
common feature in T-ALL, being detectable in 70–85% of the patients
(82) and is associated with a
poorer outcome (80,83).
Mutations in PI3K, Akt and PTEN have been described
in T-ALL patients. Collectively, they were found in about 50% of 44
T-ALL samples (84). However,
while PI3K or Akt mutations are extremely rare (two and one case,
respectively, in the above mentioned study), PTEN mutations
occur more frequently in both adult and pediatric T-ALL (85,86).
In adults, PTEN mutations have been identified in 10% of
patients in a study in which 90 T-ALL cases were analyzed (87), whereas in children, PTEN was
found mutated in 52 out of 301 (17.3%) patients (85). However, some PTEN mutations
affected exon 7, and were predicted to truncate the C2 domain
without disrupting the lipid phosphatase domain of PTEN (84). Therefore, these mutations should
not impact on PI3K/Akt/mTOR signaling, even though this has never
been documented.
Moreover, PTEN could be either deleted
(84) or repressed due to several
mechanisms. In T-ALLs displaying Notch-1 activation (50–60% of
cases), PTEN could be repressed through the hairy enhancer
of Split-1 (HES-1), a downstream target of Notch-1 signaling
(88). Another Notch-1 target gene
which negatively impacts on PTEN expression is c-Myc
(89,90). Overexpression of miR-19 has also
been documented in T-ALL patients and resulted in lower expression
of several genes controlling the PI3K/Akt/mTOR cascade, including
PTEN (91).
However, in most pediatric T-ALL clinical samples,
PTEN is expressed, but displays elevated phosphorylation at the
C-terminal Ser/Thr cluster, due to phosphorylation by casein kinase
2 (CK2), and/or oxidation by reactive oxygen species (ROS).
Phosphorylation and/or oxidation resulted in PTEN stabilization and
functional inactivation, with ensuing overactivation of
PI3K/Akt/mTOR signaling (92).
Decreased activity of PP2A on Thr308 p-Akt could also account for
PI3K/Akt/mTOR upregulation in PTEN-null T-ALL cells
(93).
IGF-1/IGF-1R signaling plays an important role in
the activation of the PI3K/Akt/mTOR cascade in T-ALL cells. Indeed,
pharmacologic inhibition or genetic deletion of IGF-1R negatively
affected T-ALL cell proliferation and survival (94). Interestingly, IGF-1R is a Notch-1
target gene and Notch-1 was required to maintain IGF-1R expression
at high levels in T-ALL cells. Furthermore, a moderate decrease in
IGF1-R signaling compromised T-ALL LIC activity (94).
Cytokines produced by the thymic/BM
microenvironment, including interleukin (IL)-4 (95) and IL-7 (96), could be involved in upregulation of
PI3K/Akt/mTOR signaling in T-ALL. An important source for IL-7
could be represented by thymic epithelial cells (97). In this connection, it has been
recently reported that ROS, produced through IL-7 signaling, are
critical for activating PI3K/Akt/mTOR which in turn mediates
proliferation and survival of T-ALL cells (29). However, in T-ALL patients,
increased signaling downstream of the IL-7 receptor α chain
(IL-7Rα) could also be a consequence of gain-of-function IL-7Rα
mutations, which were detected in about 9% of pediatric T-ALL
patients (98).
CXC chemokine ligand 12 (CXCL12), referred to as
SDF-1α (stromal cell-derived factor 1α), the ligand for the CXC
chemokine receptor 4 (CXCR4), is another cytokine with the
potential for activating PI3K/Akt/mTOR signaling (99). CXCL12 is produced by BM stromal
cells in T-ALL patients (100)
and it has been recently demonstrated to be involved in
PI3K/Akt/mTOR activation and drug-resistance in T-ALL cells
(101).
5. mTOR inhibitors
We will summarize the three main classes of mTOR
inhibitors that have been tested in pre-clinical models and/or
entered clinical trials for treatment of T-ALL: rapamycin/rapalogs
that are allosteric mTORC1 inhibitors; dual PI3K/mTOR inhibitors
that target both PI3K and mTORC1/mTORC2; ATP-competitive,
‘active-site’ mTORC1/mTORC2 inhibitors that target the catalytic
site of mTOR (Fig. 2).
 | Figure 2Targets of mTOR inhibitors.
Allosteric mTOR inhibitors (rapamycin and rapalogs) associate with
FKBP12 leading to dissociation of Raptor from mTORC1 complex and
loss of contact between mTORC1 and its substrates. Dual PI3K/mTOR
inhibitors target both PI3K and mTORC1/mTORC2. ATP-competitive
mTORC1/mTORC2 inhibitors target the catalytic site of the enzyme,
thus acting on both mTORC1 and mTORC2. FKBP12, FK506 binding
protein 12; mTOR, mammalian target of rapamycin; mTORC1, mTOR
complex 1; mTORC2, mTOR complex 2; PI3K, phosphoinositide 3-kinase;
PtdIns(4,5)P2, phosphoinositide
(4,5) bisphosphate; PtdIns(3,4,5)P3, phosphoinositide
(3,4,5)
trisphosphate; TSC1, tuberous sclerosis 1; TSC2, tuberous sclerosis
2. |
Rapamycin/rapalogs
Rapamycin (sirolimus), a natural compound discovered
from the bacterium Streptomyces hygroscopicus in the Easter
Island more than 30 years ago, is an allosteric mTORC1 inhibitor
that at first interacts with the intracellular protein, FK506
binding protein 12 (FKBP12) (102). The rapamycin/FKBP12 complex
results in the dissociation of Raptor from mTORC1 and loss of
contact between mTORC1 and its substrates (103). Therefore, rapamycin does not
directly target the mTOR catalytic site and does not affect mTORC2
activity, except in some cell types after prolonged exposure
(52). Since no other cellular
protein has been identified as rapamycin targets and a cofactor
(i.e., FKBP12) is required, rapamycin is a very selective mTORC1
inhibitor. Rapamycin is now FDA-approved as an immunosuppressive
agent in solid organ transplantation.
Rapamycin derivatives (rapalogs) display an improved
bioavailability when compared to rapamycin, and include CCI-779
(temsirolimus), RAD001 (everolimus) and AP23573 (ridaforolimus).
The orally available RAD001 is more efficacious than rapamycin, as
it has a higher affinity to FKBP12 (104).
Rapamycin has been tested in vitro in
pre-clinical models of T-ALL, where it induced apoptosis and/or
cell cycle arrest and synergized with chemotherapeutic drugs
(doxorubicin, idarubicin, dexamethasone) (105–107). Interestingly, rapamycin
synergized with the glycolysis inhibitor, 3-BrOP in T-ALL cell
lines, where the combined treatment induced apoptosis (108). It was concluded that when ATP is
depleted by glycolysis inhibition, blocking mTORC1 may further
limit nutrient uptake, which resulted in additional
cytotoxicity.
CCI-779 was able to block in vitro
IL-7-induced proliferation, survival and cell cycle progression of
primary T-ALL cells, and synergized with both doxorubicin and
dexamethasone (109).
The Pediatric Preclinical Testing Program (PPTP)
evaluated rapamycin against T-ALL cell lines and xenografts.
Rapamycin induced regression in the two T-cell ALL xenografts
studied during PPTP (110).
However, the efficacy of rapamycin/rapalogs as
broad-based monotherapies for acute leukemia treatment has not been
as promising as initially expected. Indeed, several mechanisms
emerged as barriers to anti-leukemic activity of this class of
mTORC1 inhibitors which could explain the mostly disappointing
results of clinical trials (111,112).
The rapamycin/rapalog modest effects on leukemic
cells, could be due to several reasons. Firstly, these drugs have
only a poor pro-apoptotic activity, being mainly cytostatic.
Secondly, they do not target all the mTORC1 outputs. In particular,
phosphorylation of 4E-BP1 is usually resistant to
rapamycin/rapalogs (73,113,114). This is a very critical issue, as
4E-BP1 controls the cap-dependent translation of mRNAs coding for
critical factors which regulates cell survival and proliferation in
cancer cells. These include c-Myc, cyclin-dependent kinase-2
(CDK-2), cyclin D1, signal activator and transducer of
transcription-3 (STAT-3), B-cell lymphoma 2 (Bcl-2), Bcl-xL,
survivin, myeloid cell leukemia-1 (Mcl-1), ornithine decarboxylase
(45,82). Thirdly, upregulation of PIM protein
kinase activity has been shown to contribute to resistance to
rapamycin (115). Indeed, PIM1
protein kinase phosphorylates PRAS40 at the same amino acidic
residue (Thr246) as Akt and, by doing so, activates mTORC1
(116). Moreover, PIM2 protein
kinase phosphorylated 4E-BP1 at Ser 65 residue and this
phosphorylative event was documented to be essential for oncogenic
protein translation independent of mTORC1 activity in acute
myelogenous leukemia cells (113). Interestingly, a small molecule
inhibitor of PIM protein kinases (SMI-4a) was cytotoxic to T-ALL
cell lines through the induction of a G1 phase cell
cycle arrest, and apoptosis (117). SMI-4a treatment reduced mTORC1,
but not mTORC2, activity. However, it upregulated MEK/ERK
signaling, possibly due to mTORC1/p70S6K inhibition (117).
In this connection, the disappointing performances
of rapamycin/rapalogs have been also ascribed to the feedback loops
that lead to re-activation of either PI3K/Akt and/or MEK/ERK
signaling upon mTORC1 inhibition (118–121). However, it should be pointed out
that the existence of these feedback loops has never been
documented in T-ALL cells treated with rapamycin/rapalogs.
In agreement with pre-clinical studies, clinical
trials with rapalogs combined with chemotherapy have provided more
encouraging clinical results (122,123). Phase I/II clinical trials are
ongoing in which CCI-779 is being tested in combination with
intensive re-induction therapy (dexamethasone, mitoxantrone,
vincristine and PEG-asparaginase) in children with relapsed T-ALL
(ClinicalTrials.gov: NCT01403415). Also RAD001 has entered phase
I/II clinical trials for T-ALL, in combination with standard
chemotherapy regimens (ClinicalTrials.gov: NCT00968253;
NCT01523977; NCT01403415).
Dual PI3K/mTOR inhibitors
PI3K and mTOR belong to the PIKK family of kinases,
and share high sequence homology in their catalytic domains. Dual
PI3K/mTOR inhibitors are ATP-competitive inhibitors that target the
active sites of both the holoenzymes. The first compound of this
class to be disclosed was the morpholinoquinazoline derivative,
PI-103 (124). Dual PI3K/mTOR
inhibitors downregulate signaling both upstream and downstream of
Akt, thus avoiding the issue of Akt re-activation which follows
mTORC1 inhibition. These compounds are more powerful apoptotic
inducers than rapamycin/rapalogs and inhibit rapamycin-resistant
mTORC1 outputs (125,126). They also target mTORC2 activity
(127). PI-103 was cytotoxic to
T-ALL cell lines and patient samples, where it inhibited 4E-BP1
phosphorylation, as well as oncogenic protein translation, more
efficiently than rapamycin (74,75).
Interestingly, Shepherd and coworkers have documented that PI-103
treatment of T-ALL cell lines with activating Notch-1 mutations,
caused a compensatory upregulation of Notch-1 signaling, as
demonstrated by increased levels of c-Myc (128). PI-103 and a γ-secretase inhibitor
(compound E, which targets Notch-1 signaling) synergized in
inducing T-ALL cell death, thus providing a rational basis for the
use of drug combinations that target both the signaling networks
(128). Although PI-103 displayed
low toxicity and was well tolerated in mouse xenografts (124), it did not enter clinical trials,
mainly because of its rapid in vivo metabolism (129).
NVP-BEZ235 is an orally bioavailable
imidazoquinoline dual PI3K/mTOR inhibitor (130) that has entered a phase I clinical
trial for relapsed/refractory ALL patients
(ClinicalTrials.gov:NCT01756118). NVP-BEZ235 inhibited the
proliferation and induced apoptosis in T-ALL cell lines and primary
lymphoblasts (114). The drug
synergized with several chemotherapeutic agents (cyclophosphamide,
Ara-C, dexamethasone) currently used for treating T-ALL patients
(114,131). In this connection, it is very
important to emphasize that NVP-BEZ235 also inhibited DNA-PK and
ATM/ATR kinases, that are key players of DNA damage response (DDR)
(132). Chemotherapeutic drugs,
such as Ara-C and doxorubicin, induce DDR and activate ATR
(17,133). Therefore, abrogation of DNA
repair by NVP-BEZ235 could potentiate the effects of traditional
chemotherapeutic drugs.
NVP-BGT226 is another dual PI3K/mTOR inhibitor which
has been tested in vitro against T-ALL cell lines and
primary lymphoblasts (134).
NVP-BGT226 was more powerful in inducing apoptosis than NVP-BEZ235.
Nevertheless, a phase I clinical study of NVP-BTG226 in patients
with advanced solid tumors, revealed that the drug displayed only a
limited anti-neoplastic activity and inconsistent target
inhibition. This was probably due to the fact that efficacious
plasma concentrations were not achieved at the maximum safety dose
(135).
The main limit of dual PI3K/mTOR inhibitors is that
these drugs, by inhibiting PIKK family of kinases, could also
result in more toxic side effects than rapamycin/rapalogs (136), even if they seem to be well
tolerated when administered orally (137,138).
ATP-competitive mTORC1/mTORC2
inhibitors
Due to the limited success of rapalogs in the
treatment of leukemia, a new generation of mTOR inhibitors, which
target the ATP-binding site of mTOR and inhibit the catalytic
activity of both mTORC1 and mTORC2, were developed. Acting on both
mTOR complexes, these compounds display stronger effects on cell
growth, proliferation and survival than rapalogs, and they offer a
more efficient alternative to rapalogs in the treatment of T-ALL.
Their use also minimize the re-activation feedback loops of Akt
seen with rapamycin/rapalogs (139). This class of inhibitors
displayed, in pre-clinical evaluations, more potent anti-leukemic
effects when compared with rapamycin/rapalogs. In particular, they
strongly suppressed both mTORC1-dependent phosphorylation of p70S6K
and 4E-BP1 (140,141) and mTORC2-dependent
phosphorylation of Akt at Ser 473, without affecting PI3K (136,142).
PP-242 was one of the first mTORC1/mTORC2 ATP-
competitive inhibitors to be disclosed (143). PP-242 displayed cytotoxic
activity against T-ALL and was a potent repressor of cap-dependent
mRNA translation in T-ALL cells, most likely via inhibition of the
rapamycin-resistant phosphorylation of 4E-BP1 (75). PP-242 has not been developed into
the clinic, however its derivative, MLN0128 (formerly INK128), has
entered phase I/II clinical trials for cancer patients, including
hematological malignancies (e.g., ClinicalTrials.gov: NCT01058707;
NCT01351350). MLN0128 displayed potent anti-leukemic activity in
pre-clinical models of B-ALL (144).
Other mTORC1/mTORC2 ATP-competitive inhibitors which
have been successfully tested in vitro against T-ALL cells
include AZD-8055 and OSI-027 (75,76).
Both of these drugs are being evaluated in clinical trials for
individuals with lymphomas (ClinicalTrials.gov: NCT01194193;
NCT00698243).
6. Conclusion
mTOR is activated in most T-ALL cell lines and
primary samples, due to several mechanisms, which include
PTEN gene deletion/suppression or PTEN protein
phosphorylation/oxidation. mTOR activation confers a poorer
prognosis to T-ALL patients. Both mTORC1 and mTORC2 play an
important role in the pathophysiology of T-ALL, as they are
involved in the proliferation/survival of T-ALL LICs.
Three main classes of mTOR inhibitors have been
tested both in vitro and in vivo in pre-clinical
settings of T-ALL: allosteric mTORC1 inhibitors
(rapamycin/rapalogs), dual PI3K/mTOR inhibitors and ATP-competitive
mTORC1/mTORC2 inhibitors. Some of these are now being tested, alone
or in combination with chemotherapeutic drugs, in T-ALL patients.
Therefore, in the future also T-ALL could be added to the growing
list of disorders where mTOR inhibition is beneficial to patient
outcome.
7. Perspectives
A growing body of evidence has documented that mTOR
is a key node of the PI3K/Akt/mTOR signaling pathway, which is by
far one of the most commonly upregulated signal transduction
cascades in human cancer (32).
The literature reviewed in this article suggests that there is a
strong rationale for targeting mTOR in T-ALL, including the fact
that both mTORC1 and mTORC2 are important for T-ALL LIC survival
(72). These findings suggest that
mTOR inhibition, by targeting LICs, has the potential for
eradicating T-ALL.
Could it be possible to specifically target mTOR
signaling in T-ALL LICs, without affecting the functions of healthy
HSCs? Indeed, evidence suggests that mTOR is important for the
biology of normal HSCs (145).
However, preliminary findings have indicated that there are subtle
differences in how HSCs and LICs utilize the same signaling
pathways. This has been demonstrated in murine LICs treated with
rapamycin (146), where the drug
did not affect HSCs, while it was cytotoxic to LICs. Some of the
side effects of rapamycin/rapalogs (anemia, leukopenia,
thrombocytopenia) seem to indicate that this class of drugs does
indeed affect normal hematopoiesis. However, these side effects are
usually quite mild (147). The
side effects of dual PI3K/mTOR inhibitors and of ATP-competitive
mTORC1/mTORC2 inhibitors on healthy HSCs are at present not well
known, although the only hematological toxicity which emerged from
a phase I study of BGT-226 was anemia (148).
A major challenge in the clinical use of mTOR
inhibitors remains the identification of patients who will likely
respond to the treatment. For example, it has been recently
documented that B-lymphoma cell lines which did not express 4E-BP1,
were resistant to ATP-competitive mTORC1/mTORC2 inhibitors
(149).
Additional work is therefore required to identify
and confirm predictive biomarkers of constitutive/acquired
resistance and sensitivity to each drug in large scale clinical
trials using homogeneous patient populations (32). Future studies could also benefit
from a more thorough analysis of the entire PI3K/Akt/mTOR pathway
and of its cross-talk with other signal transduction networks
aberrantly activated in T-ALL, including the Notch-1 pathway
(85,86). All of these studies could provide
the rationale for developing personalized pharmacological
treatments, based on mTOR inhibitors, with or without
chemotherapeutics or other targeted agents, aimed at T-ALL
eradication.
Acknowledgements
This study was supported by a grant from MIUR FIRB
2010 (RBAP10447J_003) to A.M.M.
References
|
1
|
Farhi DC and Rosenthal NS: Acute
lymphoblastic leukemia. Clin Lab Med. 20:17–28. 2000.
|
|
2
|
Mullighan CG: Molecular genetics of
B-precursor acute lymphoblastic leukemia. J Clin Invest.
122:3407–3415. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brown C: The genomics revolution:
relevance in healthcare today and tomorrow. J R Coll Physicians
Edinb. 42:248–250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao WL: Targeted therapy in T-cell
malignancies: dysregulation of the cellular signaling pathways.
Leukemia. 24:13–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kox C, Zimmermann M, Stanulla M, et al:
The favorable effect of activating NOTCH1 receptor mutations on
long-term outcome in T-ALL patients treated on the ALL-BFM 2000
protocol can be separated from FBXW7 loss of function. Leukemia.
24:2005–2013. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pui CH, Robison LL and Look AT: Acute
lymphoblastic leukaemia. Lancet. 371:1030–1043. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Koch U and Radtke F: Notch in T-ALL: new
players in a complex disease. Trends Immunol. 32:434–442. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hoelzer D and Gokbuget N: T-cell
lymphoblastic lymphoma and T-cell acute lymphoblastic leukemia: a
separate entity? Clin Lymphoma Myeloma. 9:S214–S221. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alharbi RA, Pettengell R, Pandha HS and
Morgan R: The role of HOX genes in normal hematopoiesis and acute
leukemia. Leukemia. 27:1000–1008. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Iacobucci I, Papayannidis C, Lonetti A,
Ferrari A, Baccarani M and Martinelli G: Cytogenetic and molecular
predictors of outcome in acute lymphocytic leukemia: recent
developments. Curr Hematol Malig Rep. 7:133–143. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bains T, Heinrich MC, Loriaux MM, et al:
Newly described activating JAK3 mutations in T-cell acute
lymphoblastic leukemia. Leukemia. 26:2144–2146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jenkinson S, Koo K, Mansour MR, et al:
Impact of NOTCH1/FBXW7 mutations on outcome in pediatric T-cell
acute lymphoblastic leukemia patients treated on the MRC UKALL 2003
trial. Leukemia. 27:41–47. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Blackburn JS, Liu S, Raiser DM, et al:
Notch signaling expands a pre-malignant pool of T-cell acute
lymphoblastic leukemia clones without affecting
leukemia-propagating cell frequency. Leukemia. 26:2069–2078. 2012.
View Article : Google Scholar
|
|
14
|
Lhermitte L, Ben Abdelali R, Villarese P,
et al: Receptor kinase profiles identify a rationale for
multitarget kinase inhibition in immature T-ALL. Leukemia.
27:305–314. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cialfi S, Palermo R, Manca S, et al:
Glucocorticoid sensitivity of T-cell lymphoblastic
leukemia/lymphoma is associated with glucocorticoid
receptor-mediated inhibition of Notch1 expression. Leukemia.
27:485–488. 2013. View Article : Google Scholar
|
|
16
|
Malyukova A, Brown S, Papa R, et al: FBXW7
regulates glucocorticoid response in T-cell acute lymphoblastic
leukaemia by targeting the glucocorticoid receptor for degradation.
Leukemia. 27:1053–1062. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Correia NC, Durinck K, Leite AP, et al:
Novel TAL1 targets beyond protein-coding genes: identification of
TAL1-regulated microRNAs in T-cell acute lymphoblastic leukemia.
Leukemia. 27:1603–1606. 2013. View Article : Google Scholar
|
|
18
|
Lv M, Zhang X, Jia H, et al: An oncogenic
role of miR-142-3p in human T-cell acute lymphoblastic leukemia
(T-ALL) by targeting glucocorticoid receptor-a and cAMP/PKA
pathways. Leukemia. 26:769–777. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schotte D, Pieters R and Den Boer ML:
MicroRNAs in acute leukemia: from biological players to clinical
contributors. Leukemia. 26:1–12. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tosello V and Ferrando AA: The NOTCH
signaling pathway: role in the pathogenesis of T-cell acute
lymphoblastic leukemia and implication for therapy. Ther Adv
Hematol. 4:199–210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Van Vlierberghe P and Ferrando A: The
molecular basis of T cell acute lymphoblastic leukemia. J Clin
Invest. 122:3398–3406. 2012.PubMed/NCBI
|
|
22
|
Buss EC and Ho AD: Leukemia stem cells.
Int J Cancer. 129:2328–2336. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Clevers H: The cancer stem cell: premises,
promises and challenges. Nat Med. 17:313–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kreso A and Dick JE: Evolution of the
cancer stem cell model. Cell Stem Cell. 14:275–291. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cox CV, Martin HM, Kearns PR, Virgo P,
Evely RS and Blair A: Characterization of a progenitor cell
population in childhood T-cell acute lymphoblastic leukemia. Blood.
109:674–682. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chiu PP, Jiang H and Dick JE:
Leukemia-initiating cells in human T-lymphoblastic leukemia exhibit
glucocorticoid resistance. Blood. 116:5268–5279. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ma W, Gutierrez A, Goff DJ, et al: NOTCH1
signaling promotes human T-cell acute lymphoblastic leukemia
initiating cell regeneration in supportive niches. PloS One.
7:e397252012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gerby B, Clappier E, Armstrong F, et al:
Expression of CD34 and CD7 on human T-cell acute lymphoblastic
leukemia discriminates functionally heterogeneous cell populations.
Leukemia. 25:1249–1258. 2011. View Article : Google Scholar
|
|
29
|
Silva A, Girio A, Cebola I, Santos CI,
Antunes F and Barata JT: Intracellular reactive oxygen species are
essential for PI3K/Akt/mTOR-dependent IL-7-mediated viability of
T-cell acute lymphoblastic leukemia cells. Leukemia. 25:960–967.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Benjamin D, Colombi M, Moroni C and Hall
MN: Rapamycin passes the torch: a new generation of mTOR
inhibitors. Nat Rev Drug Discov. 10:868–880. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lv X, Ma X and Hu Y: Furthering the design
and the discovery of small molecule ATP-competitive mTOR inhibitors
as an effective cancer treatment. Expert Opin Drug Discov.
8:991–1012. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dienstmann R, Rodon J, Serra V and
Tabernero J: Picking the point of inhibition: a comparative review
of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 13:1021–1031.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Steelman LS, Franklin RA, Abrams SL, et
al: Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy.
Leukemia. 25:1080–1094. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Memmott RM and Dennis PA: Akt-dependent
and -independent mechanisms of mTOR regulation in cancer. Cell
Signal. 21:656–664. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Finlay MR and Griffin RJ: Modulation of
DNA repair by pharmacological inhibitors of the PIKK protein kinase
family. Bioorg Med Chem Lett. 22:5352–5359. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zoncu R, Efeyan A and Sabatini DM: mTOR:
from growth signal integration to cancer, diabetes and ageing. Nat
Rev Mol Cell Biol. 12:21–35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fingar DC and Blenis J: Target of
rapamycin (TOR): an integrator of nutrient and growth factor
signals and coordinator of cell growth and cell cycle progression.
Oncogene. 23:3151–3171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Inoki K, Li Y, Zhu T, Wu J and Guan KL:
TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR
signalling. Nat Cell Biol. 4:648–657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Volkers M and Sussman M: mTOR/PRAS40
interaction: hypertrophy or proliferation. Cell Cycle.
12:3579–3580. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Laplante M and Sabatini DM: mTOR signaling
at a glance. J Cell Sci. 122:3589–3594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Browne GJ and Proud CG: A novel
mTOR-regulated phosphorylation site in elongation f actor 2 kinase
modulates the activity of the kinase and its binding to calmodulin.
Mol Cell Biol. 24:2986–2997. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ma XM and Blenis J: Molecular mechanisms
of mTOR-mediated translational control. Nat Rev Mol Cell Biol.
10:307–318. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Mutations and deregulation of Ras/Raf/MEK/ERK and
PI3K/PTEN/Akt/mTOR cascades which alter therapy response.
Oncotarget. 3:954–987. 2012.PubMed/NCBI
|
|
45
|
Martelli AM, Evangelisti C, Chappell W, et
al: Targeting the translational apparatus to improve leukemia
therapy: roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia.
25:1064–1079. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cunningham JT, Rodgers JT, Arlow DH,
Vazquez F, Mootha VK and Puigserver P: mTOR controls mitochondrial
oxidative function through a YY1-PGC-1a transcriptional complex.
Nature. 450:736–740. 2007. View Article : Google Scholar
|
|
47
|
Majumder PK, Febbo PG, Bikoff R, et al:
mTOR inhibition reverses Akt-dependent prostate intraepithelial
neoplasia through regulation of apoptotic and HIF-1-dependent
pathways. Nat Med. 10:594–601. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yecies JL and Manning BD: Transcriptional
control of cellular metabolism by mTOR signaling. Cancer Res.
71:2815–2820. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hosokawa N, Hara T, Kaizuka T, et al:
Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200
complex required for autophagy. Mol Biol Cell. 20:1981–1991. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mizushima N: The role of the Atg1/ULK1
complex in autophagy regulation. Curr Opin Cell Biol. 22:132–139.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zeng Z, Sarbassov dos D, Samudio IJ, et
al: Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT
activation in AML. Blood. 109:3509–3512. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Garcia-Martinez JM and Alessi DR: mTOR
complex 2 (mTORC2) controls hydrophobic motif phosphorylation and
activation of serum- and glucocorticoid-induced protein kinase 1
(SGK1). Biochem J. 416:375–385. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ikenoue T, Inoki K, Yang Q, Zhou X and
Guan KL: Essential function of TORC2 in PKC and Akt turn motif
phosphorylation, maturation and signalling. EMBO J. 27:1919–1931.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Oh WJ and Jacinto E: mTOR complex 2
signaling and functions. Cell Cycle. 10:2305–2316. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Tamburini J, Green AS, Chapuis N, et al:
Targeting translation in acute myeloid leukemia: a new paradigm for
therapy? Cell Cycle. 8:3893–3899. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Shah OJ, Wang Z and Hunter T:
Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces
IRS1/2 depletion, insulin resistance, and cell survival
deficiencies. Curr Biol. 14:1650–1656. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Bhaskar PT and Hay N: The two TORCs and
Akt. Dev Cell. 12:487–502. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lang SA, Hackl C, Moser C, et al:
Implication of RICTOR in the mTOR inhibitor-mediated induction of
insulin-like growth factor-I receptor (IGF-IR) and human epidermal
growth factor receptor-2 (Her2) expression in gastrointestinal
cancer cells. Biochim Biophys Acta. 1803:435–442. 2010. View Article : Google Scholar
|
|
61
|
Xu X, Sarikas A, Dias-Santagata DC, et al:
The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1
for ubiquitin-dependent degradation. Mol Cell. 30:403–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sriburi R, Jackowski S, Mori K and Brewer
JW: XBP1: a link between the unfolded protein response, lipid
biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell
Biol. 167:35–41. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Boulbes D, Chen CH, Shaikenov T, et al:
Rictor phosphorylation on the Thr-1135 site does not require
mammalian target of rapamycin complex 2. Mol Cancer Res. 8:896–906.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Maehama T and Dixon JE: The tumor
suppressor, PTEN/MMAC1, dephosphorylates the lipid second
messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem.
273:13375–13378. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sansal I and Sellers WR: The biology and
clinical relevance of the PTEN tumor suppressor pathway. J Clin
Oncol. 22:2954–2963. 2004. View Article : Google Scholar
|
|
66
|
Kalesnikoff J, Sly LM, Hughes MR, et al:
The role of SHIP in cytokine-induced signaling. Rev Physiol Biochem
Pharmacol. 149:87–103. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Liu Q, Sasaki T, Kozieradzki I, et al:
SHIP is a negative regulator of growth factor receptor-mediated
PKB/Akt activation and myeloid cell survival. Genes Dev.
13:786–791. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Bunney TD and Katan M: Phosphoinositide
signalling in cancer: beyond PI3K and PTEN. Nat Rev Cancer.
10:342–352. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Seshacharyulu P, Pandey P, Datta K and
Batra SK: Phosphatase: PP2A structural importance, regulation and
its aberrant expression in cancer. Cancer Lett. 335:9–18. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Guo W, Lasky JL, Chang CJ, et al:
Multi-genetic events collaboratively contribute to Pten-null
leukaemia stem-cell formation. Nature. 453:529–533. 2008.
View Article : Google Scholar
|
|
71
|
Guo W, Schubbert S, Chen JY, et al:
Suppression of leukemia development caused by PTEN loss. Proc Natl
Acad Sci USA. 108:1409–1414. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hoshii T, Kasada A, Hatakeyama T, et al:
Loss of mTOR complex 1 induces developmental blockage in early
T-lymphopoiesis and eradicates T-cell acute lymphoblastic leukemia
cells. Proc Natl Acad Sci USA. 111:3805–3810. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kang SA, Pacold ME, Cervantes CL, et al:
mTORC1 phosphorylation sites encode their sensitivity to starvation
and rapamycin. Science. 341:12365662013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Chiarini F, Fala F, Tazzari PL, et al:
Dual inhibition of class IA phosphatidylinositol 3-kinase and
mammalian target of rapamycin as a new therapeutic option for
T-cell acute lymphoblastic leukemia. Cancer Res. 69:3520–3528.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Evangelisti C, Ricci F, Tazzari P, et al:
Targeted inhibition of mTORC1 and mTORC2 by active-site mTOR
inhibitors has cytotoxic effects in T-cell acute lymphoblastic
leukemia. Leukemia. 25:781–791. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bressanin D, Evangelisti C, Ricci F, et
al: Harnessing the PI3K/Akt/mTOR pathway in T-cell acute
lymphoblastic leukemia: eliminating activity by targeting at
different levels. Oncotarget. 3:811–823. 2012.PubMed/NCBI
|
|
77
|
Magee JA, Ikenoue T, Nakada D, Lee JY,
Guan KL and Morrison SJ: Temporal changes in PTEN and mTORC2
regulation of hematopoietic stem cell self-renewal and leukemia
suppression. Cell Stem Cell. 11:415–428. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Mullighan CG: Genomic profiling of
B-progenitor acute lymphoblastic leukemia. Best Pract Res Clin
Haematol. 24:489–503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Inaba H, Greaves M and Mullighan CG: Acute
lymphoblastic leukaemia. Lancet. 381:1943–1955. 2013. View Article : Google Scholar
|
|
80
|
Jotta PY, Ganazza MA, Silva A, et al:
Negative prognostic impact of PTEN mutation in pediatric T-cell
acute lymphoblastic leukemia. Leukemia. 24:239–242. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Karrman K, Forestier E, Heyman M, et al:
Clinical and cytogenetic features of a population-based consecutive
series of 285 pediatric T-cell acute lymphoblastic leukemias: rare
T-cell receptor gene rearrangements are associated with poor
outcome. Genes Chromosomes Cancer. 48:795–805. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Martelli AM, Chiarini F, Evangelisti C, et
al: Two hits are better than one: targeting both
phosphatidylinositol 3-kinase and mammalian target of rapamycin as
a therapeutic strategy for acute leukemia treatment. Oncotarget.
3:371–394. 2012.PubMed/NCBI
|
|
83
|
Nemes K, Sebestyen A, Mark A, et al:
Mammalian target of rapamycin (mTOR) activity dependent
phospho-protein expression in childhood acute lymphoblastic
leukemia (ALL). PLoS One. 8:e593352013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gutierrez A, Sanda T, Grebliunaite R, et
al: High frequency of PTEN, PI3K, and AKT abnormalities in T-cell
acute lymphoblastic leukemia. Blood. 114:647–650. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Bandapalli OR, Zimmermann M, Kox C, et al:
NOTCH1 activation clinically antagonizes the unfavorable effect of
PTEN inactivation in BFM-treated children with precursor T-cell
acute lymphoblastic leukemia. Haematologica. 98:928–936. 2013.
View Article : Google Scholar
|
|
86
|
Trinquand A, Tanguy-Schmidt A, Ben
Abdelali R, et al: Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic
risk classification of adult T-cell acute lymphoblastic leukemia: a
Group for Research in Adult Acute Lymphoblastic Leukemia study. J
Clin Oncol. 31:4333–4342. 2013. View Article : Google Scholar
|
|
87
|
Grossmann V, Haferlach C, Weissmann S, et
al: The molecular profile of adult T-cell acute lymphoblastic
leukemia: mutations in RUNX1 and DNMT3A are associated with poor
prognosis in T-ALL. Genes Chromosomes Cancer. 52:410–422. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Palomero T, Sulis ML, Cortina M, et al:
Mutational loss of PTEN induces resistance to NOTCH1 inhibition in
T-cell leukemia. Nat Med. 13:1203–1210. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Palomero T, Lim WK, Odom DT, et al: NOTCH1
directly regulates c-MYC and activates a feed-forward-loop
transcriptional network promoting leukemic cell growth. Proc Natl
Acad Sci USA. 103:18261–18266. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Gutierrez A, Grebliunaite R, Feng H, et
al: Pten mediates Myc oncogene dependence in a conditional
zebrafish model of T cell acute lymphoblastic leukemia. J Exp Med.
208:1595–1603. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Mavrakis KJ, Wolfe AL, Oricchio E, et al:
Genome-wide RNA-mediated interference screen identifies miR-19
targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat
Cell Biol. 12:372–379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Silva A, Yunes JA, Cardoso BA, et al: PTEN
posttranslational inactivation and hyperactivation of the PI3K/Akt
pathway sustain primary T cell leukemia viability. J Clin Invest.
118:3762–3774. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Hales EC, Orr SM, Larson Gedman A, Taub JW
and Matherly LH: Notch1 receptor regulates AKT protein activation
loop (Thr308) dephosphorylation through modulation of the PP2A
phosphatase in phosphatase and tensin homolog (PTEN)-null T-cell
acute lymphoblastic leukemia cells. J Biol Chem. 288:22836–22848.
2013. View Article : Google Scholar
|
|
94
|
Medyouf H, Gusscott S, Wang H, et al:
High-level IGF1R expression is required for leukemia-initiating
cell activity in T-ALL and is supported by Notch signaling. J Exp
Med. 208:1809–1822. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Cardoso BA, Martins LR, Santos CI, et al:
Interleukin-4 stimulates proliferation and growth of T-cell acute
lymphoblastic leukemia cells by activating mTOR signaling.
Leukemia. 23:206–208. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Barata JT, Silva A, Brandao JG, Nadler LM,
Cardoso AA and Boussiotis VA: Activation of PI3K is indispensable
for interleukin 7-mediated viability, proliferation, glucose use,
and growth of T cell acute lymphoblastic leukemia cells. J Exp Med.
200:659–669. 2004. View Article : Google Scholar
|
|
97
|
Scupoli MT, Vinante F, Krampera M, et al:
Thymic epithelial cells promote survival of human T-cell acute
lymphoblastic leukemia blasts: the role of interleukin-7.
Haematologica. 88:1229–1237. 2003.PubMed/NCBI
|
|
98
|
Zenatti PP, Ribeiro D, Li W, et al:
Oncogenic IL7R gain-of-function mutations in childhood T-cell acute
lymphoblastic leukemia. Nat Genet. 43:932–939. 2011. View Article : Google Scholar
|
|
99
|
Wong D and Korz W: Translating an
antagonist of chemokine receptor CXCR4: from bench to bedside. Clin
Cancer Res. 14:7975–7980. 2008. View Article : Google Scholar
|
|
100
|
Scupoli MT, Donadelli M, Cioffi F, et al:
Bone marrow stromal cells and the upregulation of interleukin-8
production in human T-cell acute lymphoblastic leukemia through the
CXCL12/CXCR4 axis and the NF-κB and JNK/AP-1 pathways.
Haematologica. 93:524–532. 2008.PubMed/NCBI
|
|
101
|
Pillozzi S, Masselli M, De Lorenzo E, et
al: Chemotherapy resistance in acute lymphoblastic leukemia
requires hERG1 channels and is overcome by hERG1 blockers. Blood.
117:902–914. 2011. View Article : Google Scholar
|
|
102
|
Heitman J, Movva NR and Hall MN: Targets
for cell cycle arrest by the immunosuppressant rapamycin in yeast.
Science. 253:905–909. 1991. View Article : Google Scholar
|
|
103
|
Zhou H, Luo Y and Huang S: Updates of mTOR
inhibitors. Anticancer Agents Med Chem. 10:571–581. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Schuler W, Sedrani R, Cottens S, et al:
SDZ RAD, a new rapamycin derivative: pharmacological properties in
vitro and in vivo. Transplantation. 64:36–42. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Avellino R, Romano S, Parasole R, et al:
Rapamycin stimulates apoptosis of childhood acute lymphoblastic
leukemia cells. Blood. 106:1400–1406. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Chan SM, Weng AP, Tibshirani R, Aster JC
and Utz PJ: Notch signals positively regulate activity of the mTOR
pathway in T-cell acute lymphoblastic leukemia. Blood. 110:278–286.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wu KN, Zhao YM, He Y, et al: Rapamycin
interacts synergistically with idarubicin to induce T-leukemia cell
apoptosis in vitro and in a mesenchymal stem cell simulated
drug-resistant microenvironment via Akt/mammalian target of
rapamycin and extracellular signal-related kinase signaling
pathways. Leuk Lymphoma. 55:668–676. 2014.
|
|
108
|
Akers LJ, Fang W, Levy AG, Franklin AR,
Huang P and Zweidler-McKay PA: Targeting glycolysis in leukemia: a
novel inhibitor 3-BrOP in combination with rapamycin. Leukemia Res.
35:814–820. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Batista A, Barata JT, Raderschall E, et
al: Targeting of active mTOR inhibits primary leukemia T cells and
synergizes with cytotoxic drugs and signaling inhibitors. Exp
Hematol. 39:457–472. e4532011. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Houghton PJ, Morton CL, Kolb EA, et al:
Initial testing (stage 1) of the mTOR inhibitor rapamycin by the
pediatric preclinical testing program. Pediatr Blood Cancer.
50:799–805. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Yee KW, Zeng Z, Konopleva M, et al: Phase
I/II study of the mammalian target of rapamycin inhibitor
everolimus (RAD001) in patients with relapsed or refractory
hematologic malignancies. Clin Cancer Res. 12:5165–5173. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Rizzieri DA, Feldman E, Dipersio JF, et
al: A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a
novel mammalian target of rapamycin inhibitor, in patients with
relapsed or refractory hematologic malignancies. Clin Cancer Res.
14:2756–2762. 2008. View Article : Google Scholar
|
|
113
|
Tamburini J, Green AS, Bardet V, et al:
Protein synthesis is resistant to rapamycin and constitutes a
promising therapeutic target in acute myeloid leukemia. Blood.
114:1618–1627. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Chiarini F, Grimaldi C, Ricci F, et al:
Activity of the novel dual phosphatidylinositol 3-kinase/mammalian
target of rapamycin inhibitor NVP-BEZ235 against T-cell acute
lymphoblastic leukemia. Cancer Res. 70:8097–8107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Fox CJ, Hammerman PS and Thompson CB: The
Pim kinases control rapamycin-resistant T cell survival and
activation. J Exp Med. 201:259–266. 2005. View Article : Google Scholar
|
|
116
|
Zhang F, Beharry ZM, Harris TE, et al:
PIM1 protein kinase regulates PRAS40 phosphorylation and mTOR
activity in FDCP1 cells. Cancer Biol Ther. 8:846–853. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lin YW, Beharry ZM, Hill EG, et al: A
small molecule inhibitor of Pim protein kinases blocks the growth
of precursor T-cell lymphoblastic leukemia/lymphoma. Blood.
115:824–833. 2010. View Article : Google Scholar
|
|
118
|
Tamburini J, Chapuis N, Bardet V, et al:
Mammalian target of rapamycin (mTOR) inhibition activates
phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like
growth factor-1 receptor signaling in acute myeloid leukemia:
rationale for therapeutic inhibition of both pathways. Blood.
111:379–382. 2008. View Article : Google Scholar
|
|
119
|
Carracedo A, Ma L, Teruya-Feldstein J, et
al: Inhibition of mTORC1 leads to MAPK pathway activation through a
PI3K-dependent feedback loop in human cancer. J Clin Invest.
118:3065–3074. 2008.PubMed/NCBI
|
|
120
|
Efeyan A and Sabatini DM: mTOR and cancer:
many loops in one pathway. Curr Opin Cell Biol. 22:169–176. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Bertacchini J, Guida M, Accordi B, et al:
Feedbacks and adaptive capabilities of the PI3K/Akt/mTOR axis in
acute myeloid leukemia revealed by pathway selective inhibition and
phosphoproteome analysis. Leukemia. Apr 4–2014.(E-pub ahead of
print).
|
|
122
|
Park S, Chapuis N, Saint Marcoux F, et al:
A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in
association with chemotherapy for AML patients in first relapse.
Leukemia. 27:1479–1486. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Daver N, Kantarjian H, Thomas D, et al: A
phase I/II study of hyper-CVAD plus everolimus in patients with
relapsed/refractory acute lymphoblastic leukemia. In: 55th ASH
Annual Meeting; Blood. 122. abs. 3916. 2013
|
|
124
|
Fan QW, Knight ZA, Goldenberg DD, et al: A
dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma.
Cancer Cell. 9:341–349. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Cho DC, Cohen MB, Panka DJ, et al: The
efficacy of the novel dual PI3-kinase/mTOR inhibitor NVP-BEZ235
compared with rapamycin in renal cell carcinoma. Clin Cancer Res.
16:3628–3638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Karar J, Cerniglia GJ, Lindsten T,
Koumenis C and Maity A: Dual PI3K/mTOR inhibitor NVP-BEZ235
suppresses hypoxia-inducible factor (HIF)-1α expression by blocking
protein translation and increases cell death under hypoxia. Cancer
Biol Ther. 13:1102–1111. 2012.PubMed/NCBI
|
|
127
|
Schenone S, Brullo C, Musumeci F, Radi M
and Botta M: ATP-competitive inhibitors of mTOR: an update. Curr
Med Chem. 18:2995–3014. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Shepherd C, Banerjee L, Cheung CW, et al:
PI3K/mTOR inhibition upregulates NOTCH-MYC signalling leading to an
impaired cytotoxic response. Leukemia. 27:650–660. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Raynaud FI, Eccles S, Clarke PA, et al:
Pharmacologic characterization of a potent inhibitor of class I
phosphatidylinositide 3-kinases. Cancer Res. 67:5840–5850. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Maira SM, Stauffer F, Brueggen J, et al:
Identification and characterization of NVP-BEZ235, a new orally
available dual phosphatidylinositol 3-kinase/mammalian target of
rapamycin inhibitor with potent in vivo antitumor activity. Mol
Cancer Ther. 7:1851–1863. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Schult C, Dahlhaus M, Glass A, et al: The
dual kinase inhibitor NVP-BEZ235 in combination with cytotoxic
drugs exerts anti-proliferative activity towards acute
lymphoblastic leukemia cells. Anticancer Res. 32:463–474.
2012.PubMed/NCBI
|
|
132
|
Shortt J, Martin BP, Newbold A, et al:
Combined inhibition of PI3K-related DNA damage response kinases and
mTORC1 induces apoptosis in MYC-driven B-cell lymphomas. Blood.
121:2964–2974. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Woods D and Turchi JJ: Chemotherapy
induced DNA damage response: convergence of drugs and pathways.
Cancer Biol Ther. 14:379–389. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Kampa-Schittenhelm KM, Heinrich MC, Akmut
F, et al: Cell cycle-dependent activity of the novel dual
PI3K-mTORC1/2 inhibitor NVP-BGT226 in acute leukemia. Mol Cancer.
12:462013. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Soria JC, Cortes J, Massard C, et al:
Phase I safety, pharmacokinetic and pharmacodynamic trial of
BMS-599626 (AC480), an oral pan-HER receptor tyrosine kinase
inhibitor, in patients with advanced solid tumors. Ann Oncol.
23:463–471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Janes MR, Limon JJ, So L, et al: Effective
and selective targeting of leukemia cells using a TORC1/2 kinase
inhibitor. Nat Med. 16:205–213. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Garcia-Echeverria C and Sellers WR: Drug
discovery approaches targeting the PI3K/Akt pathway in cancer.
Oncogene. 27:5511–5526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Garcia-Echeverria C: Allosteric and
ATP-competitive kinase inhibitors of mTOR for cancer treatment.
Bioorg Med Chem Lett. 20:4308–4312. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Gentzler RD, Altman JK and Platanias LC:
An overview of the mTOR pathway as a target in cancer therapy.
Expert Opin Ther Targets. 16:481–489. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Altman JK, Sassano A, Kaur S, et al: Dual
mTORC2/mTORC1 targeting results in potent suppressive effects on
acute myeloid leukemia (AML) progenitors. Clin Cancer Res.
17:4378–4388. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Willems L, Chapuis N, Puissant A, et al:
The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor
activity in acute myeloid leukemia. Leukemia. 26:1195–1202. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Gupta M, Hendrickson AE, Yun SS, et al:
Dual mTORC1/mTORC2 inhibition diminishes Akt activation and induces
Puma-dependent apoptosis in lymphoid malignancies. Blood.
119:476–487. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Feldman ME, Apsel B, Uotila A, et al:
Active-site inhibitors of mTOR target rapamycin-resistant outputs
of mTORC1 and mTORC2. PLoS Biol. 7:e382009. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Janes MR, Vu C, Mallya S, et al: Efficacy
of the investigational mTOR kinase inhibitor MLN0128/INK128 in
models of B-cell acute lymphoblastic leukemia. Leukemia.
27:586–594. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Peng C, Chen Y, Li D and Li S: Role of
Pten in leukemia stem cells. Oncotarget. 1:156–160. 2010.PubMed/NCBI
|
|
146
|
Yilmaz OH, Valdez R, Theisen BK, et al:
Pten dependence distinguishes haematopoietic stem cells from
leukaemia-initiating cells. Nature. 441:475–482. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Kaplan B, Qazi Y and Wellen JR: Strategies
for the management of adverse events associated with mTOR
inhibitors. Transplant Rev. Mar 12–2014.(Epub ahead of print).
|
|
148
|
Markman B, Tabernero J, Krop I, et al:
Phase I safety, pharmacokinetic, and pharmacodynamic study of the
oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in
patients with advanced solid tumors. Ann Oncol. 23:2399–2408. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Mallya S, Fitch BA, Lee JS, So L, Janes MR
and Fruman DA: Resistance to mTOR kinase inhibitors in lymphoma
cells lacking 4EBP1. PloS One. 9:e888652014. View Article : Google Scholar
|














