Introduction
Heat shock protein 90 (Hsp90) is a molecular
chaperone that contributes to the maintenance of the correct
folding of critical protein effectors involved in cell survival,
growth and differentiation (1).
Hsp90, in association with other cochaperone proteins, catalyzes
via its ATPase activity the conformational changes of client
proteins, that are mainly involved in cell signalling,
proliferation and survival (2). In
addition, Hsp90 ensures the stability of a number of mutated client
proteins required for tumor growth and resistance (3). In recent years this molecular target
has gained considerable interest for the discovery and development
of novel anticancer drugs (4,5),
because of their putative therapeutic use in multiple cancer
indications.
Thus, a number of novel synthetic Hsp90 inhibitors,
such as NVP-AUY922 (Novartis/Vernalis) (6), STA-9090 (Synta) (7), AT13387 (Astex) delivered
intravenously and XL-888 (Exelixis), DS-2248 and Debio 0932 orally
administered (8), are currently
under oncology clinical investigations. Besides having the unusual
ability of disrupting the activity of many receptors, kinases and
transcription factors, all these drugs overcome the potential
limitations of the ansamycin-derived Hsp90 inhibitors including
complex formulations, poor solubility and hepatotoxicity (8).
Preclinical data in human tumor xenograft models
show that these Hsp90 inhibitors are efficacious in a wide variety
of tumor types, through activity against different oncoproteins
(9). Antitumor efficacy ranges
from minimal effects to tumor growth stasis, but rarely leads to
tumor regression (10–15). The different efficacy of antitumor
response in various xenograft models may be attributable to
differences in client protein dependence on Hsp90, tumor dependence
on the client protein, kinetics of client protein degradation and
turnover, as well as drug pharmacokinetic and pharmacologic
properties. This complexity makes it difficult to predict antitumor
response in xenograft models and renders patient stratification in
the clinic challenging (3). Since
Hsp90 also plays a key role in regulating protein function and
stability in normal cells (16),
the balancing of efficacy and toxicity is important to achieve as
suitable therapeutic index in patients.
We have previously described the discovery of a
novel series of 3,4-isoxazolediamides based Hsp90 inhibitors which
were shown to have a very interesting activity both in vitro
and in vivo (17). In
particular, within this group, we selected the compound SST0116CL1
as a synthetic, new chemical entity designed to potently inhibit
Hsp90. SST0116CL1 binds to the ATP binding pocket of Hsp90, and
interferes with Hsp90 chaperone function thus resulting in client
protein degradation and tumor growth inhibition.
We report on the in vitro activity and in
vivo pharmacokinetic and efficacy profiles of SST0116CL1 in
human cancer cell lines from different etiology. These results
support the selection of SST0116CL1 for clinical development.
Materials and methods
Compound preparation
For in vitro experiments, stock solutions of
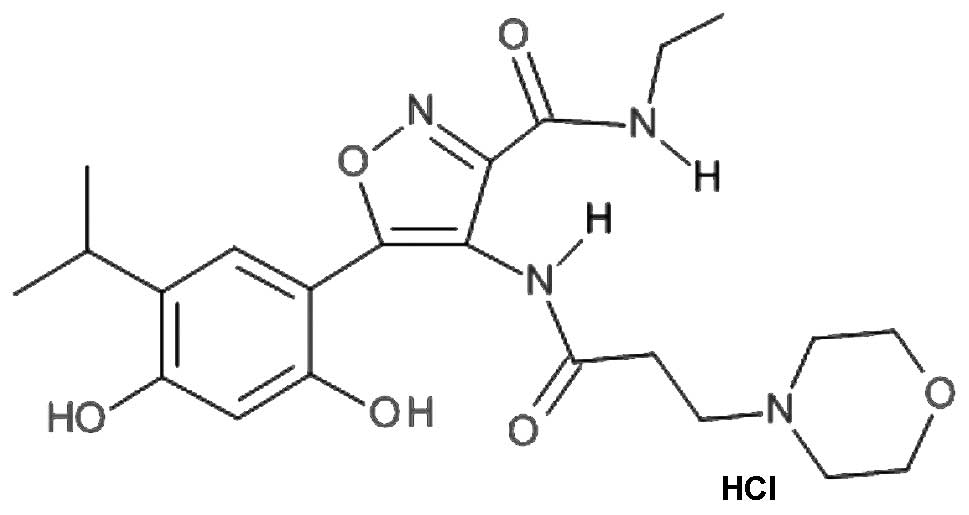
SST0116CL1 (property of Sigma-Tau Research Switzerland S.A) (see
Fig. 1) were prepared in 100%
dimethyl sulfoxide (DMSO) at 10 mM and stored at −20°C. For
intraperitoneal or intravenous administration, SST0116CL1 was
formulated in 2.5% ethanol, 20% 50 mM tartaric acid, 77.5% (5%
glucose in water containing 1% Tween-80) vol/vol and delivered in a
volume of 10 ml/kg.
Binding on Hsp90 by a fluorescence
polarization assay
GM-FITC, supplied by Invivogen (cat. no. 06C23-MT,
San Diego, CA, USA), was previously dissolved in DMSO to obtain 10
mM stock solutions and kept at −20°C until use. Recombinant human
Hsp90, purchased from Stressgen (cat. no. SPP-776, Victoria, BC,
Canada), was previously dissolved in assay buffer (HFB) to form 2.2
μM stock solutions and kept at −80°C until use. On the day of the
experiment, compound solutions at various concentrations were
prepared by serial dilutions in assay buffer (HFB) containing 20 mM
HEPES (K+), pH 7.3, 50 mM KCl, 5 mM MgCl2, 20
mM Na2MoO4, and 0.01% NP-40. Before each use,
0.1 mg/ml bovine γ-globulin and 2 mM DTT were added. Fluorescence
polarization (FP) was performed in Opti-Plate-96F well plates
(Perkin-Elmer, Zaventem, Belgium) using the Wallac Envision 2101
multilabel plate reader (Perkin-Elmer). To evaluate the binding
affinity of the molecule, 50 μl of the GM-FTC solution (5 nM) were
added to 30 nM Hsp90 in the presence of 5 μl of the test compounds
at increasing concentrations. The plates were shaken at 4°C for 4
h, and the FP values in mP (millipolarization units) were then
recorded. The IC50 value was calculated as the inhibitor
concentration that displaced 50% of the tracer, each data point
being the result of the average of triplicate wells, and was
determined from a plot using non-linear least-squares analysis.
Curve fitting was performed using Prism GraphPad software program
(GraphPad Software, Inc., San Diego, CA, USA).
Cell lines and cell sensitivity to
drug
A non-small cell lung carcinoma (NSCLC, NCI-H460)
cell line, a breast carcinoma (BT-474) cell line, a fibrosarcoma
(HT-1080) cell line and an acute monocytic leukemia (MV4;11) cell
line were purchased from the American Type Culture Collection
(Manassas, VA, USA). The sensitive ovarian carcinoma cell line
(A2780) was from European Collection of Animal Cell Cultures
(ECACC). The gastric carcinoma (GTL-16) and the epidermoid
carcinoma (A431) cell lines were kindly provided by Metheresis and
by Istituto Tumori di Milano, respectively. The NSCLC, the breast
carcinoma and the epidermoid carcinoma cells, as well as the acute
monocytic leukaemia and the ovarian carcinoma cells were grown in
RPMI-1640 (Lonza, Verviers, Belgium) supplemented with 10% fetal
bovine serum (FBS, Invitrogen, Geithersburg, MD, USA). The gastric
carcinoma GTL-16 cells were grown in DMEM (Lonza) supplemented with
10% fetal bovine serum (FBS, Invitrogen). The fibrosarcoma cells
were grown in EMEM (Lonza) supplemented with 10% fetal bovine serum
(FBS, Invitrogen). Cells were routinely maintained in a humidified
atmosphere with 5% CO2 at 37°C. All experiments were
performed starting from frozen cell stocks of each cell line. When
thawed, such cells were characterized in house, by assessing cell
morphology, cell growth kinetics curve and absence of mycoplasma.
The cell sensitivity to the drug was measured in vitro by
assessing the inhibition of proliferation by sulphorodamine B (SRB)
assay. Briefly, cells were seeded in 96-well tissue culture plates
in complete medium (10% FBS), and 24 h after seeding were treated
for 72 h with various concentrations of SST0116CL1. The drug
cytotoxic potency was evaluated by means of the ‘ALLFIT’ computer
program and defined as IC50 (drug concentration required
for 50% inhibition of cell survival).
Her2 degradation assay
BT-474 cells were seeded into Viewplates-384TC (cat.
6007480, Perkin-Elmer Inc., Waltham, MA, USA) at the density of
2×104 cells/well in 100 μl of culture medium, and were
then incubated at 37°C, in the presence of 5% CO2 for 24
h. Different concentrations of the drug or vehicle (DMSO) were then
added to each well, and cells were cultured for further 24 h. After
washing the cells with PBS, 30 μl of Her2 AlphaLISA Immunoassay
buffer were added to wells and mixed on a shaker at 22°C for 30
min. Then 15 μl of a solution (10 μg/ml) consisting in Her2
AlphaELISA Anti-ERBB2/Her2 Acceptor beads and (1 nM) Biotinylated
Anti-ERBB2/Her2 Antibody were added to wells, and incubated at 22°C
for 2 h. Finally, 5 μl of (40 μg/ml) Streptavidin Donor beads were
added, and the plate was incubated at 22°C for 1 h, in the dark,
and measurements were done through the multilabel reader Envision
(Wallac Envision 2101 multilabel reader, Perkin-Elmer, Zaventem,
Belgium). To rule out that the declining signal in drug-treated
cells was not the result of reduced cell number caused by
unspecific cell death but due to decreased Her2 content, suitable
viability studies were performed by means of the luminescence ATP
detection assay system (ATPlite 1 Step cat. 6016941, Perkin-Elmer,
Zaventem, Belgium). IC50 was calculated as the drug
concentration needed to degrade 50% of the total Her2; each data
point was the result of the average of triplicate wells, and
determined from a plot using nonlinear least-squares analysis.
Curve fitting was performed using the Prism GraphPad software
program (GraphPad software, Inc., San Diego, CA, USA).
Western blot analysis
For the in vitro experiments, A431 (human
epidermoid carcinoma) cells were seeded at 1×106
cells/100 mm dish in complete culture medium, and allowed to grow
overnight at 37°C with 95% air and 5% CO2. The day
after, the medium was renewed and cells were treated, for 24 h,
with various concentrations of SST0116CL1. 17-DMAG (at the
concentration of 0.2 μM) was used as internal reference inhibitor.
Following treatments, cells were rinsed twice with ice-cold PBS and
then lysed in RIPA buffer supplemented with protease and
phosphatase inhibitors. After determination of the protein
concentration by Bradford Protein Assay (Thermo Scientific,
Rockford, IL, USA), equal amounts of cellular extracts were
separated by SDS-PAGE and then transferred onto nitrocellulose
membranes. Non-specific binding sites were blocked by incubation of
the membranes with 5% non-fat dry milk in TBS, overnight at 4°C.
Membranes were finally probed with the following primary
antibodies: anti-EGFR (Upstate Biotechnology, Millipore Corporate,
Billerica, MA, USA); anti-Cdk4 (Santa Cruz Biotechnology Inc.,
Santa Cruz, CA, USA); anti-Akt (Cell Signaling Technology, Inc.,
MA, USA); anti-HSP70 (BRM-22) and anti-Actin (Sigma Chemical Co.,
St. Louis, MO, USA). After extensive washings in TBS,
immunoreactive bands were revealed by horseradish
peroxidase-conjugated secondary antibodies, using an enhanced
chemiluminescence detection reagent (ECL Plus, GE Healthcare
Bio-Sciences, Uppsala, Sweden), and acquired by a phosphoimaging
system (STORM 860; Molecular Dynamics, Sunnyvale, CA, USA). Protein
loading equivalence was corrected in relation to the expression of
actin. For quantification of signals, blots were subjected to
densitometry analysis.
Murine xenograft model
All experiments were carried out at Sigma-Tau (Rome,
Italy) using 5–6 week-old female athymic nude mice (Harlan, Italy).
Mice were maintained in laminar flow rooms with constant
temperature and humidity. Experimental protocols were approved by
the Ethics Committee for Animal Experimentation of Sigma-Tau
according to the United Kingdom Coordinating Committee on Cancer
Research Guidelines. The following human tumor xenograft models
were used for antitumor activity studies: GTL-16 gastric carcinoma,
MV4;11 AML, A2780/ADR multi-drug resistant ovarian carcinoma.
Exponentially growing tumor cells (5×106/mouse) were
s.c. inoculated in the right flank of nude mice. Groups of eight
mice were employed to assess antitumor activity. Drug treatments
were started 3 or 4 days after tumor injection. SST0116CL1 was
delivered intraperitoneally or intravenously in a volume of 10
ml/kg according to different schedules (qdx5/w: daily from Monday
to Friday; q2d/w: Monday, Wednesday, Friday; q4d/w: Monday and
Friday). Tumor growth was followed by measurement of tumor
diameters with a Vernier caliper. Tumor volume (TV) was calculated
using the formula: TV (mm3) = [d2 × D]/2,
where d and D are the shortest and the longest diameter,
respectively. The efficacy of the drug treatment was assessed as:
TV inhibition percentage (TVI%) in treated vs. control mice,
calculated as: TVI% = 100 − [(mean TV treated/mean TV control)
×100)]. CR (complete response) was defined as no evidence of tumor
at the end of the drug-treatment. When tumors reached a maximum
volume of 2,000 mm3, mice were sacrificed by cervical
dislocation. To examine the possible toxicity of treatment, body
weight was recorded throughout the study. BWL% (body weight loss)
was calculated as 100 − [(mean BWdayx/mean
BWday1)] ×100), where day 1 is the first day of
treatment and day × is any day after (maximum BWL%). In order to
assess the in vivo effect of SST0116CL1 on the expression of
typical HSP90 client proteins, GTL-16 tumor xenografts (4
samples/group) were excised at different times after the last
treatment, and then total protein lysates were prepared through the
homogenization of tumor samples in T-PER (Tissue Protein Extraction
Reagent, Pierce, Rockland, IL, USA), supplemented with 10 μg/ml of
protease inhibitor cocktail (Sigma Chemical Co., St. Louis, MO,
USA). Determination of the protein concentration and western blot
analysis were finally performed as previously described for the
in vitro experiments.
PK sampling and analysis
CD1 nude mice bearing A431 epidermoid carcinoma
xenografts were used. Mice were treated with a single
intraperitoneal dose of SST0116CL1 at 80 mg/10 ml/kg. Levels of
SST0116CL1 were determined in blood, lung and tumor samples
collected at 1, 2, 4, 8 and 24 h post-treatment. Bioanalysis was
conducted by quantitative HPLC-MS/MS and PK analysis was carried
out according to a non-compartmental approach for sparse data
sampling (WinNonLin, Pharsight). The maximum plasma concentration
(Cmax) of SST0116, and the corresponding times of
occurrence (Tmax) were obtained directly from the mean
plasma or tissue concentration data. The slope of the terminal
disposition phase (k) was determined by logarithmic-linear
regression and used to calculate the terminal half-life
(T1/2) as 0.693/k. The linear trapezoidal rule was used
to estimate the area under the plasma concentration versus time
curve from zero to the last time point with a measurable drug
concentration (Clast) for AUC0-last, and
extrapolating to infinity by addition of the quantity
Clast/k for AUC0-inf. The apparent total
clearance, (CL/F) and the apparent terminal volume of distribution
(Vz/F) referenced to plasma were calculated as
Dose/AUC0-inf, and Dose/(k*AUC0-inf),
respectively.
Statistical analysis
The data shown represent mean values ± standard
error of mean (SEM) and standard deviation (SD). For comparison
between a control and a treatment group, an unpaired Mann-Whitney
test was used. A P-value ≤0.05 was considered significant.
Results
SST0116CL1 inhibits cell growth, and
recombinant HSP90α
In a competitive binding fluorescence polarization
assay, SST0116CL1 was shown to inhibit recombinant Hsp90α, with an
IC50 value of 0.2 μM (Table
I). The ability of SST0116CL1 to induce degradation of Her2 in
BT-474 human breast carcinoma cells, was also measured by means of
a specific Her2 AlphaELISA immunoassay. As shown in Table I, exposure of BT-474 cells for 24 h
to increasing concentrations of SST0116CL1 was able to cause a
significant reduction in cellular levels of Her2, with an
IC50 value of 0.2 μM. The ability of SST0116CL1 to
target Hsp90 directly on human tumor cells was evaluated by a tumor
cell proliferation assay against a representative panel of human
tumor cell lines, including fibrosarcoma (HT-1080), acute monocytic
leukemia (MV4;11), NSCLC (NCI-H460), ovarian (A2780), epidermoid
(A431), gastric (GTL-16), breast (BT-474) carcinoma cell lines,
characterized by constitutively activated oncogenic pathways
(Table II). SST0116CL1 was
effective in inhibiting cell growth in all the different tumor cell
lines, following 72-h exposure, with IC50 values ranging
from 0.1 to 0.8 μM (Table II).
Because the broad mechanism of action of SST0116CL1, such as for
other Hsp90 inhibitors, it was not possible to associate the
sensitivity observed in the cell panel with the mutation of a
single gene. These results correlated well with those obtained from
the Her2 degradation assay as well as with the Hsp90 competitive
binding assay.
 | Table IBinding affinity to recombinant human
HSP90α and Her2 degradation on BT474 breast carcinoma cells. |
Table I
Binding affinity to recombinant human
HSP90α and Her2 degradation on BT474 breast carcinoma cells.
| Compound | HSP90α
(IC50 ± SD, μM) | Her2 (IC50
± SD, μM) |
|---|
| SST0116CL1 | 0.21±0.03 | 0.20±0.02 |
| Table IIAntiproliferative activity of
SST0116CL1 on different tumor cell lines. |
Table II
Antiproliferative activity of
SST0116CL1 on different tumor cell lines.
| Cell line | Tissue | SST0116CL1
IC50 ± SD (μM) | Alterated oncogenic
pathway |
|---|
| A431 | Epidermoid
carcinoma | 0.81±0.02 | EGFR |
| NCI-H460 | NSCLC | 0.11±0.1 | KRAS |
| A2780 | Ovarian
carcinoma | 0.81±0.1 | PTEN |
| MV4;11 | Acute monocytic
leukemia | 0.40±0.07 | FLT3 |
| GTL-16 | Gastric
carcinoma | 0.23±0.04 | MET |
| BT474 | Breast
carcinoma | 0.62±0.1 | HER2 |
| HT-1080 | Fibrosarcoma | 0.34±0.09 | NRAS |
SST0116CL1 inhibits the expression of
specific Hsp90 client proteins
The ability of SST0116CL1 to downregulate the
expression of a few representative Hsp90 protein clients and to
induce the expression of Hsp70 protein was assessed, by western
blot analysis, in the A431 human squamous carcinoma cell line,
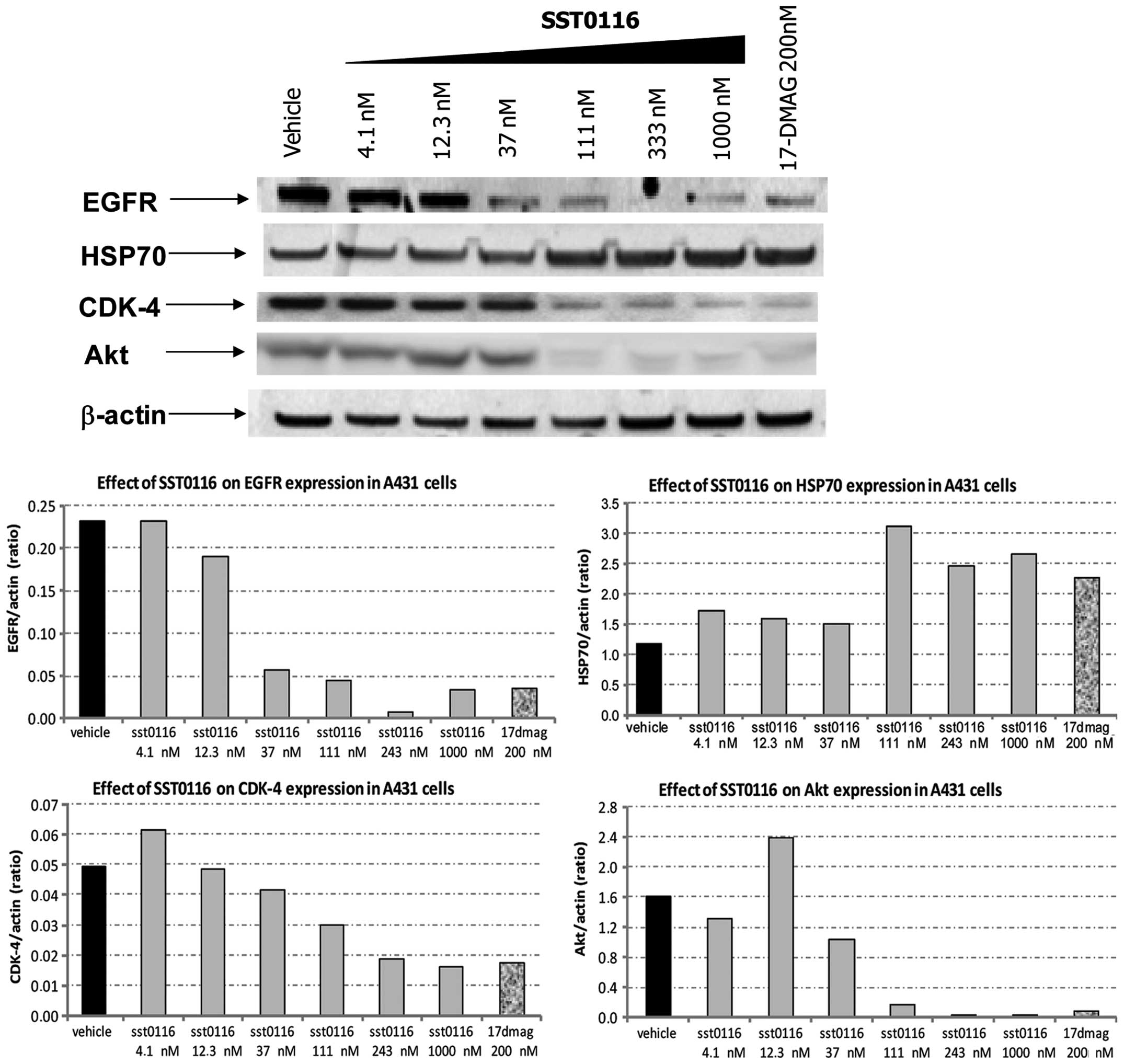
characterized by the constitutive overexpression of EGFR. As shown
in Fig. 2, a dramatic depletion of
selected Hsp90 client proteins (EGFR, CDK4 and AKT), associated to
a very strong increase in the expression levels of Hsp70, was
achieved in A431 cells after a 24-h exposure of cells even to
concentrations of SST0116 almost 10-fold lower than the cytotoxic
doses of the drug. Thus, the modulation of client proteins and the
upregulation of Hsp70 cochaperone were consistent with inhibition
of Hsp90 function, and confirmed that target modulation of Hsp90
was achieved.
SST0116CL1 inhibits tumor growth in
different cancer cell xenografts
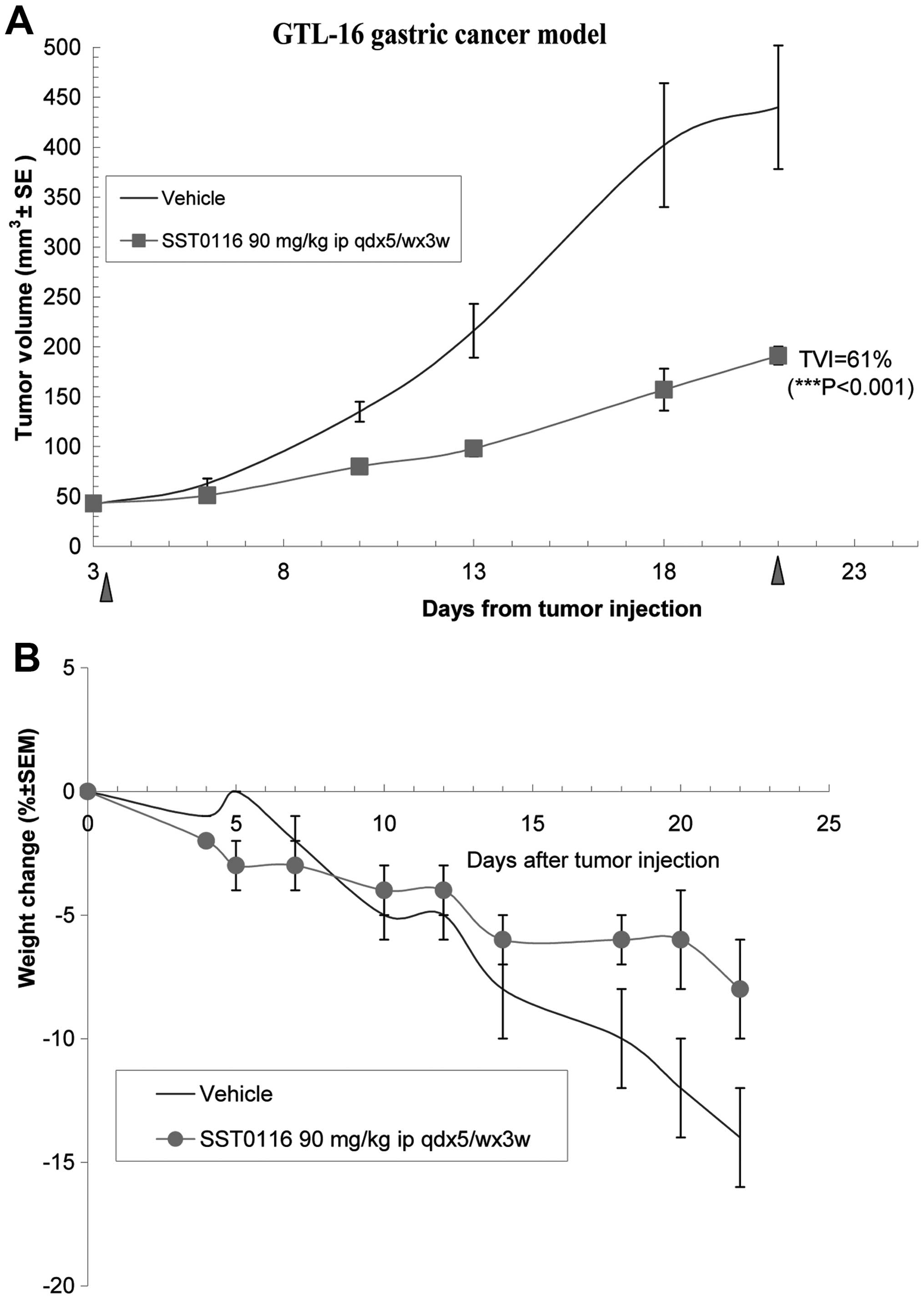
SST0116CL1 significantly reduced solid tumor growth
in different xenograft models. Against the gastric carcinoma
(GTL-16), SST0116CL1, administered at 90 mg/10 ml/kg i.p. according
to the schedule qdx5/wx3w, was able to induce a significantly Tumor
Volume Inhibition of 61% (P<0.001; Fig. 3A). Interestingly, the compound
revealed to be well-tolerated and to protect mice from cachexia
induced by this tumor xenograft (see the body weight of treated
group in comparison with the drug-treated group) (Fig. 3B). To validate that the in
vivo antitumor effect of SST0116CL1 was effectively related to
the inhibition of Hsp90, the modulation of selected Hsp90 client
proteins was assessed by western blot in tumor xenografts a few
hours after the last treatment. As shown in Fig. 3C, SST0116CL1 induced a relevant
decrease of the protein levels of three typical client proteins
(c-MET, AKT and CDK4) in GTL-16 tumor lysates and, at the same
time, significantly increased the expression levels of the
chaperone Hsp70, thus confirming that inhibition of Hsp90 function
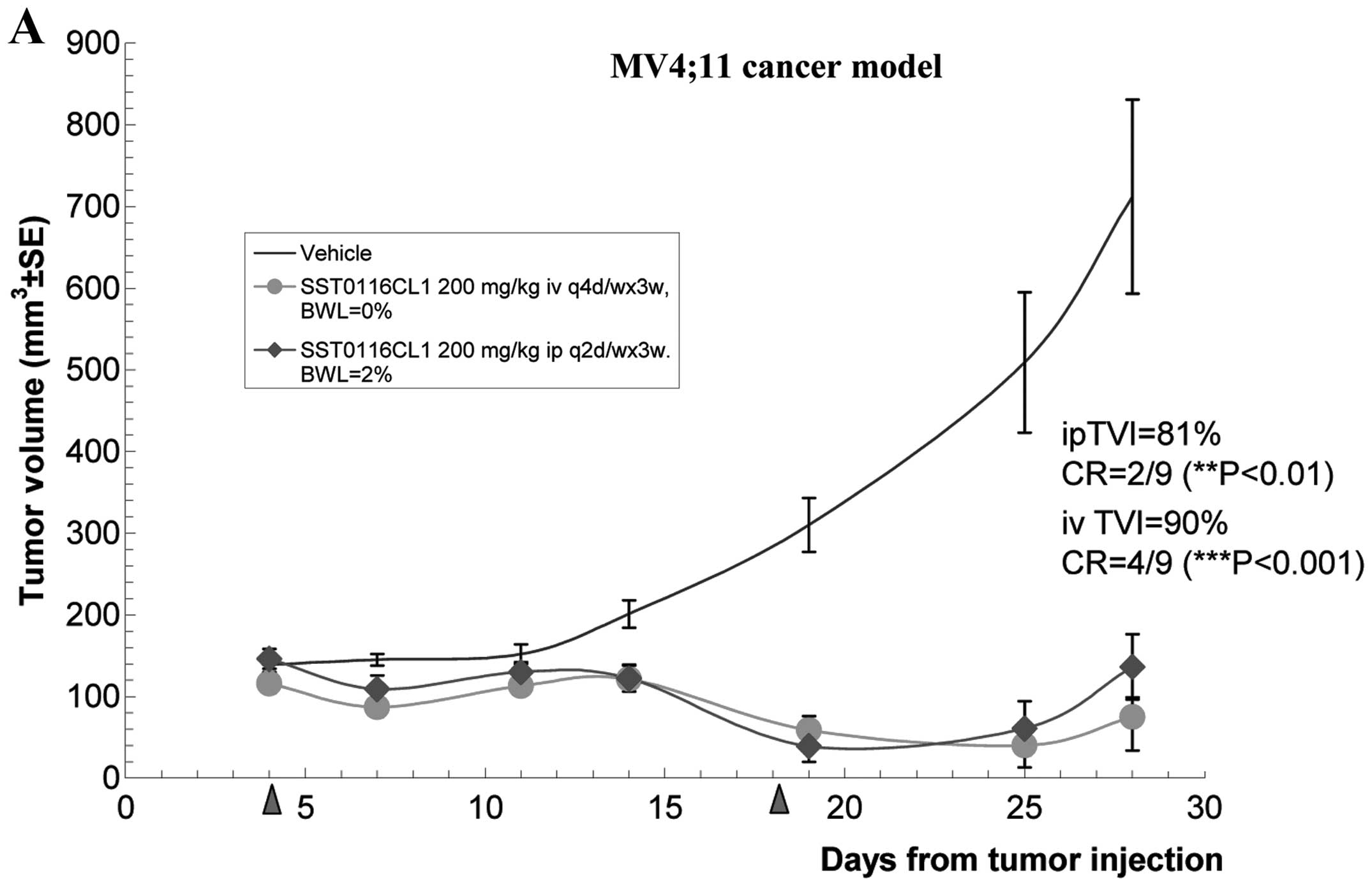
was achieved. The in vivo antitumor efficacy of SST0116CL1
against MV4;11 AML cell line was also investigated. Administration
of SST0116CL1 (200 mg/10 ml/kg) intravenously (q4d/wx2w) and (200
mg/10 ml/kg) intraperitoneally (q2d/wx2w) showed a very potent
antitumor effect (TVI=90%, P<0.001, with 50% of complete
response, and TVI=81%, P<0.01, with 22% of complete response,
respectively) (Fig. 4A). Thus, the
antitumor efficacy was comparable irrespectively of the routes and
schedules. In both treatments, SST0116CL1 was revealed to be well
tolerated. The A2780/Dx ovarian tumor cell line, overexpressing
P-glycoprotein, also showed a significant responsiveness to the
compound (TVI=71%, P<0.01). Against this tumor model, SST0116CL1
was delivered at 200 mg/10 ml/kg i.p. according to the schedule
q4d/wx3w, and again it did not show any toxicity (Fig. 4B).
SST0116CL1 shows a preferential
distribution in tumors
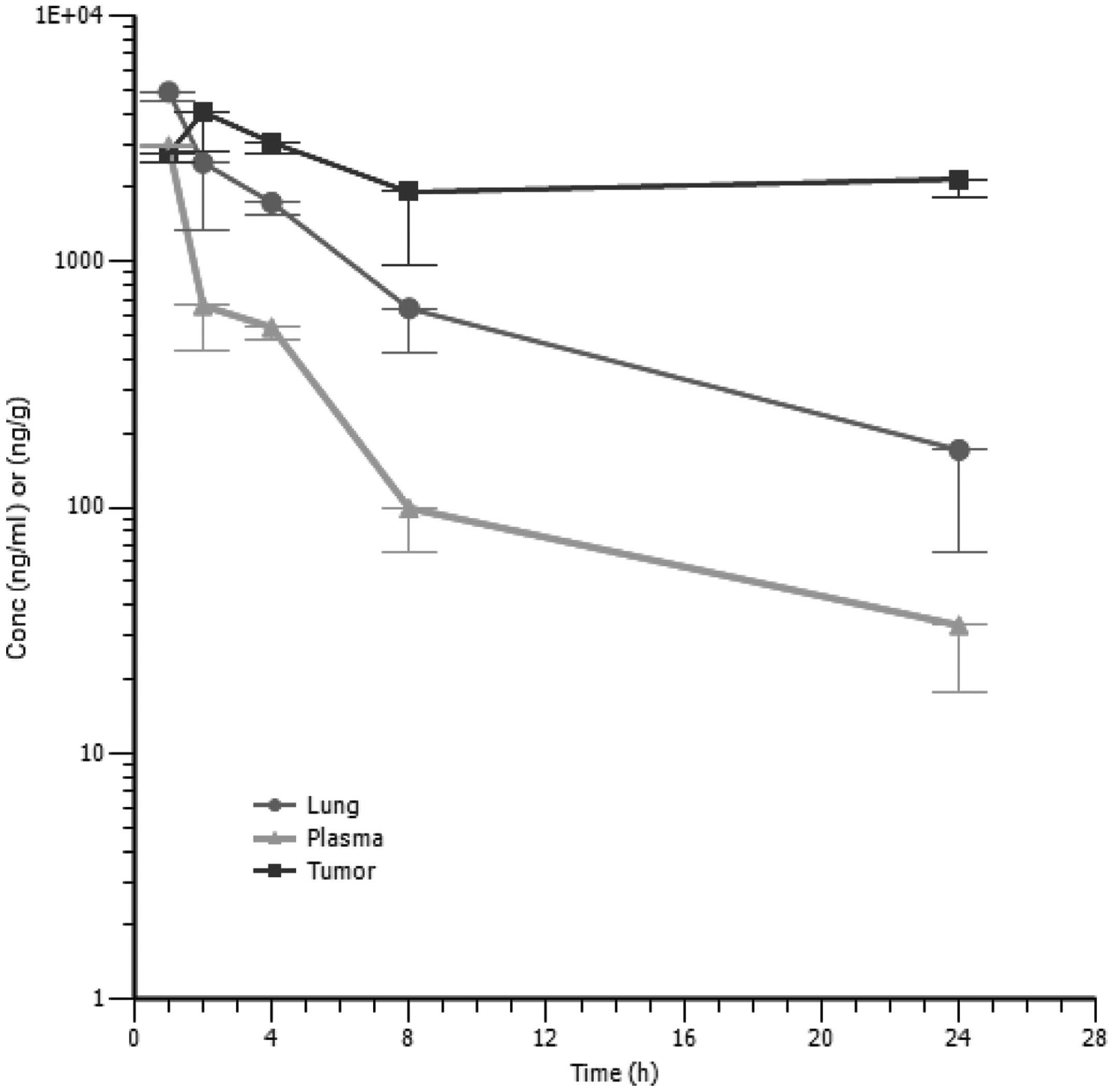
The PK profile of SST0116CL1 in tumor bearing
animals was determined after a single dose of 80 mg/10 ml/kg of
compound administered intraperitoneally. The concentrations of
SST0116CL1 were assessed in both blood and tissue samples (tumor
and lung). A Cmax of 2,953 ng/ml was observed in plasma
1 h after dosing, then plasma concentrations decreased according to
a bi-exponential profile, having a terminal half-life of 5.8 h. As
suggested by its plasma concentration versus time profile,
SST0116CL1 distributed outside the systemic circulation, reaching
Cmax of 4,890 ng/ml in lung and 4,046 ng/ml in tumor
within 1 and 2 h, respectively (Table III). The terminal elimination
phase in lung paralleled that of plasma (T1/2 of 6.5 h);
conversely SST0116CL1 seemed to accumulate in tumor; in fact, in
this tissue its concentration declined much more slowly than in
plasma and lung (Fig. 5). The
tissue to plasma AUC0-last ratios turned out to be about
3 for lung and 8 for tumor; thus these data reveal enhanced tissue
distribution. The CL/F and Vz/F evaluated from plasma data were
11,268 ml/h/kg and 94,551 ml/kg, respectively. The high values of
CL/F and Vz/F might also reflect an incomplete bioavailability of
the tested compound in nude mouse.
| Table IIIModel independent pharmacokinetic
analysis of SST0116CL1 in the plasma and tissues of tumor-bearing
mice (A431 epidermoid carcinoma). |
Table III
Model independent pharmacokinetic
analysis of SST0116CL1 in the plasma and tissues of tumor-bearing
mice (A431 epidermoid carcinoma).
| Tissue | Tmax
(h) | Cmax
(ng/ml) | AUClast
(h*ng/ml) | HL_Lambda_z
(h) | CL_F_obs
(ml/h/kg) | AUCinf_obs
(h*ng/ml) | Vz_F_obs
(ml/kg) |
|---|
| Plasma | 1.00 | 2,953 | 6,822 | 5.8 | 11,268 | 7,100 | 94,551 |
| Lung | 1.00 | 4,890 | 21,676 | 6.5 | | 23,287 | |
| Tumor | 2.00 | 4,046 | 54,387 | NE | | 267,987 | |
Discussion
Hsp90 is a component of a molecular chaperone
complex that plays critical roles in regulating the folding,
maturation and stabilisation of key signalling molecules which
control cell proliferation, survival and transformation. Hsp90
works in association with other cochaperones and catalyzes, via its
ATPase, the conformational changes of a set of cancer-associated
proteins, collectively referred to as ‘clients’. Inhibition of
Hsp90 causes simultaneous destabilization and eventual degradation
of client proteins that in turn result in suppression of tumor
growth. A number of novel synthetic Hsp90 inhibitors are currently
under oncology clinical investigations for the treatment of a wide
variety of tumor types (8,18,19).
The earlier geldanamycin analogues (i.e., 17-AAG or 17-DMAG),
despite potent in vitro and in vivo preclinical
activity, have not shown clear clinical benefit (5,20).
The disappointing clinical activity was due to their poor
selectivity, pharmaceutical properties and toxicity profiles in
patients (21,22). Given this precedent, we planned to
identify novel Hsp90 inhibitors with a superior pharmacological and
tolerability profile. We previously identified a new class of
3,4-isoxazolediamides (17), where
the compound SST0116CL1 was selected as a potential new drug
candidate, unrelated to the ansamycin class of natural products. We
chose to investigate molecules with innovative changes and to
assess the benefits of such changes through well-focused in
vivo studies, based on suitable tumor models.
SST0116CL1 in the studies in vitro was shown
to inhibit recombinant Hsp90α and to induce the degradation of the
oncogenic Her2 tyrosine kinase in BT-474 human breast cancer cells.
The degradation of the oncogenic Her2 tyrosine kinase represents a
biological effect observed upon addition of known Hsp90 inhibitors
to cancer cells (22).
Overexpression of Her2 in cancer cells, such as breast and ovarian
carcinoma cells, usually results in Akt activation which in turn
promotes cell survival. Hsp90 inhibitors induce Her2 degradation
through disruption of the Her2/Hsp90 association, and this effect
is detrimental to the cell leading to its death (23). Moreover, SST0116CL1 was able to
induce the destabilization and depletion of different client
proteins, often overexpressed and constitutively activated in
numerous types of hematological or solid human tumors. These
results well correlated with those obtained from the cell
proliferation assay as well as with the Hsp90 competitive binding
assay, and clearly confirmed that target modulation of Hsp90 was
achieved. We also showed a putative broad spectrum of
antiproliferative activity on a panel of tumor cell lines harboring
different gene mutations: K-Ras mutations (H460), EGFR and Her2
amplification (A431 and BT474, respectively), N-Ras mutation
(HT1080), PTEN loss (A2780), FLT3 mutation (MV4;11) and c-met
amplification (GTL-16).
Although the in vitro activity of SST0116CL1
was not particularly potent with respect to analogues Hsp90
inhibitors currently undergoing clinical trials, it showed,
however, a great versatility and a good pharmacological profile
when assessed in vivo for tolerability, pharmacokinetics,
pharmacodynamics and antitumor activity. In some solid and
haematological tumor xenograft models, SST0116CL1 delivered
intravenously or intraperitoneally at different schedules, from
once a day to once every two or four days (qdx5/w; q2d/w; q4d/w),
in both sensitive and doxorubicin-resistant tumor models, showed a
statistically significant tumor growth inhibition with a TVI
ranging from 61 to 90%, associated to a slight body weight loss
(BWL) during the drug-treatment. Three tumor cell lines were
selected as tumor xenograft models to represent a diversity of
tumor types and oncogenic drivers. The GTL-16 tumor cell line was
chosen because of amplification of the receptor tyrosine kinase
c-Met, a client protein of Hsp90, and for its dependency on c-Met
for growth and survival (24),
MV4; 11 leukemia driven by the tyrosine kinase receptor FLT3ITD
mutation, was also analyzed. The activating internal tandem
duplications (ITD) in the juxtamembrane domain of FLT3 have been
identified in 35% AML patients (25). MV4;11 has been shown to be
dependent on FLT3-ITD by its sensitivity to selective FLT3 kinase
inhibitors (26). The best
approach to the treatment of FLT3-ITD AML is currently undefined
and multiple clinical trials are investigating inhibitors of the
FLT3 kinase (27). Their action is
very often transient, possibly due to inadequate dosing or
insufficient selectivity of these drugs. SST0116CL1 treatment of
the MV4;11 AML resulted in eradication of a good percentage of
tumors after subcutaneous tumor cell implantation.
Since many types of cancer express relatively high
levels of P-glycoprotein, a major type of MDR protein (28), we show that, unlike Hsp90
inhibitors such as 17-AAG and its derivatives (29), SST0116CL1 exhibited no MDR
dependency in A2780/ADR tumor xenograft model. This characteristic
makes SST0116CL1 a potentially superior Hsp90 inhibitor in the
situation where P-gp is expressed, enabling it, to overcome the MDR
barrier that commonly undermines cancer therapy. Differently,
17-AAG could itself induce P-gp expression, rendering the drug less
effective during treatment (29).
A modulation of PD biomarkers in terms of
downregulation of EGFR, AKT and CDK4 client proteins was achieved
either in vitro, on A431 tumor cells treated with
SST0116CL1, and in terms of down-modulation of c-Met, AKT and CDK4
ex vivo in tumor lesions collected from GTL-16 tumor-bearing
mice. The molecular signature of Hsp90 inhibition represents a
fundamental pharmacodynamic biomarker of efficacy in cancer cell
lines, and has been well validated in human tumor xenografts as
well as to measure target inhibition in cancer patients receiving
treatments with selective Hsp90 inhibitors (30–32).
It has been extensively demonstrated that targeting cellular Hsp90
protein function with pharmacological doses of Hsp90 inhibitors
results in the depletion of the well-established client proteins,
mainly represented by oncogenic proteins or, more generally,
cellular proteins involved in the modulation of critical cell
pathways and mechanisms, such as cell cycle, proliferation and
survival, and in the upregulation of other members of the Hsp90
family. An advantage of Hsp90 inhibitors is their ability to affect
multiple oncoproteins simultaneously. This is relevant given the
emerging data showing resistant phenotypes arising from mutation,
activation of alternative signaling pathways or feedback loops,
frequently seen with therapeutics targeting a single oncogene or
pathway (33). The preclinical
data of tolerability, manageability and activity profile make
SST0116CL1 a very attractive antitumor therapeutic agent ready to
undergo clinical studies.
Acknowledgements
The authors wish to thank Dr M.B. Guglielmi, Dr M.
Barbarino, Dr R. Foderà, Dr M. Castorina, Dr M.L. Cervoni, Mrs P.
Tobia and Mr. A. Marconi for their excellent technical assistance.
The authors declare no financial interest.
References
|
1
|
Li J and Buchner J: Structure, function,
and regulation of the Hsp90 machinery. Biomed J. 36:106–117. 2013.
View Article : Google Scholar
|
|
2
|
Hao H, Naomoto Y, Bao X, Watanabe N,
Sakurama K, Noma K, Motoki T, Tomono Y, Fukazawa T, Shirakawa Y,
Yamatsuji T, Matsuoka J and Takaoka M: Hsp90 and its inhibitors.
Oncol Rep. 23:1483–1492. 2010.PubMed/NCBI
|
|
3
|
Whitesell L and Lindquist SL: HSP90 and
the chaperoning of cancer. Nat Rev Cancer. 5:761–772. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mahalingam D, Swords R, Carew JS, Nawrocki
ST, Bhalla K and Gilles FJ: Targeting HSP90 for cancer therapy. Br
J Cancer. 100:1523–1529. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim YS, Alarcon SV, Lee S, Lee MJ,
Giaccone G, Neckers L and Trepel JB: Update on Hsp90 inhibitors in
clinical trial. Curr Top Med Chem. 9:1479–1492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sessa C, Shapiro GI, Bhalla KN, Britten C,
Jacks KS, Mita M, Papadimitrakopoulou V, Pluard T, Samuel TA,
Akimov M, Quadt C, Fernandez-Ibarra C, Lu H, Bailey S, Chica S and
Banerji U: First-in-human phase I dose-escalation study of the
Hsp90 inhibitor AUY922 in patients with advanced solid tumors. Clin
Cancer Res. 19:3671–3680. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goldman JW, Raju RN, Gordon GA, El-Hariry
I, Teofilivici F, Vukovic VM, Bradley R, Karol MD, Chen Y, Guo W,
Inoue T and Rosen LS: A first in human, safety, pharmacokinetics,
and clinical activity phase I study of once weekly administration
of the Hsp90 inhibitor ganetespib (STA-9090) in patients with solid
malignancies. Biomed Central Cancer. 13:152–161. 2013.
|
|
8
|
Soga S, Akinaga S and Shiotsu Y: Hsp90
inhibitors as anti-cancer agents, from basic discoveries to
clinical development. Curr Pharm Des. 19:366–376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li J, Soroka J and Buchner J: The Hsp90
chaperone machinery: conformational dynamics and regulation by
co-chaperons. Biochim Biophys Acta. 1823.624–635. 2012.PubMed/NCBI
|
|
10
|
Eccles SA, Massey A, Raynaud FI, Sharp SY,
Box G, Valenti M, Patterson L, de Haven Brandon A, Gowan S, Boxall
F, Aherne W, Rowlands M, Hayes A, Martins V, Urban F, Boxall K,
Prodromou C, Pearl L, James K, Matthews TP, Cheung KM, Kalusa A,
Jones K, McDonald E, Barril X, Brough PA, Cansfield JE, Dymock B,
Drysdale MJ, Finch H, Howes R, Hubbard RE, Surgenor A, Webb P, Wood
M, Wright L and Workman P: NVP-AUY922: a novel heat shock protein
90 inhibitor active against xenograft tumor growth, angiogenesis,
and metastasis. Cancer Res. 68:2850–2860. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jensen MR, Schoepfer J, Radimerski T,
Massey A, Guy CT, Brueggen J, Quadt C, Buckler A, Cozens R,
Drysdale MJ, Garcia-Echeverria C and Chène P: NVP-AUY922: a small
molecule HSP90 inhibitor with potent antitumor activity in
preclinical breast cancer models. Breast Cancer Res. 10:R332008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Trepel JB, Neckers LM and Giaccone
G: STA-9090, a small-molecule Hsp90 inhibitor for the potential
treatment of cancer. Curr Opin Investig Drugs. 11:1466–1476.
2010.PubMed/NCBI
|
|
13
|
Okawa Y, Hideshima T, Steed P, Vallet S,
Hall S, Huang K, Rice J, Barabasz A, Foley B, Ikeda H, Raje N,
Kiziltepe T, Yasui H, Enatsu S and Anderson KC: SNX-2112, a
selective Hsp90 inhibitor, potently inhibits tumor cell growth,
angiogenesis, and osteoclastogenesis in multiple myeloma and other
hematologic tumors by abrogating signaling via Akt and ERK. Blood.
113:846–855. 2009. View Article : Google Scholar
|
|
14
|
Lin TY, Bear M, Du Z, Foley KP, Ying W,
Barsoum J and London C: The novel HSP90 inhibitor STA-9090 exhibits
activity against Kit-dependent and -independent malignant mast cell
tumors. Exp Hematol. 36:1266–1277. 2008. View Article : Google Scholar
|
|
15
|
Lundgren K, Zhang H, Brekken J, Huser N,
Powell RE, Timple N, Busch DJ, Neely L, Sensintaffar JL, Yang YC,
McKenzie A, Friedman J, Scannevin R, Kamal A, Hong K, Kasibhatla
SR, Boehm MF and Burrows FJ: BIIB021, an orally available, fully
synthetic small-molecule inhibitor of the heat shock protein Hsp90.
Mol Cancer Ther. 8:921–929. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Picard D: Heat shock protein 90, a
chaperone for folding and regulation. Cell Mol Life Sci.
59:1640–1648. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baruchello R, Simoni D, Grisolia G,
Barbato G, Marchetti P, Rondanin R, Mangiola S, Giannini G,
Brunetti T, Alloatti D, Gallo G, Ciacci A, Vesci L, Castorina M,
Milazzo FM, Cervoni ML, Guglielmi MB, Barbarino M, Foderà R, Pisano
C and Cabri W: Novel 3.4-isoxazolediamides as potent inhibitors of
chaperone heat shock protein 90. J Med Chem. 54:8592–8604. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jhaveri K and Modi S: Hsp90 inhibitors for
cancer therapy and overcoming drug resistance. Adv Pharmacol.
65:471–517. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Patel HJ, Modi S, Chiosis G and Taldone T:
Advances in the discovery and development of heat-shock protein 90
inhibitors for cancer treatment. Expert Opin Drug Discov.
6:559–587. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Trepel J, Mollapour M, Giaccone G and
Neckers L: Targeting the dynamic Hsp90 complex in cancer. Nat Rev
Cancer. 10:537–549. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ramanathan RK and Egorin MJ, Eiseman JL,
Ramalingam S, Friedland D, Agarwala SS, Zuhowski EG, Lan J, Potter
DM, Ivy SP, Ramalingam S, Brufsky AM, Wong MK, Tutchko S and Egorin
MJ: Phase I and pharmacodynamic study of
17-(allylamino)-17-demethoxygenldanamycin in adult patients with
refractory advanced cancers. Clin Cancer Res. 13:1769–1774. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ramanathan RK, Egorin MJ, Erlichman C,
Remick SC, Ramalingam SS, Naret C, Holleran JL, TenEyck CJ, Ivy SP
and Belani CP: Phase I pharmacokinetic and pharmacodynamic study of
17-dimethylaminoethylamino-17-demethoxygeldanamycin, an inhibitor
of heat-shock protein 90, in patients with advanced solid tumors. J
Clin Oncol. 28:1520–1526. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mimnaugh EG, Chavany C and Neckers L:
Polyubiquitination and proteasomal degradation of the p185c-erbB-2
receptor protein-tyrosine kinase induced by geldanamycin. J Biol
Chem. 271:22796–22801. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Christensen JG, Schreck R, Burrows J,
Kuruganti P, Chan E, Le P, Chen J, Wang X, Ruslim L, Blake R,
Lipson KE, Ramphal J, Do S, Cui JJ, Cherrington JM and Mendel DB: A
selective small molecule inhibitor of c-Met kinase inhibits
c-Met-dependent phenotypes in vitro and exhibits cytoreductive
antitumor activity in vivo. Cancer Res. 63:7345–7355.
2003.PubMed/NCBI
|
|
25
|
Gilliland DG and Griffin JD: Role of FLT3
in leukemia. Curr Opin Hematol. 9:274–281. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lopes de Menezes DE, Peng J, Garrett EN,
Louie SG, Lee SH, Wiesmann M, Tang Y, Shephard L, Goldbeck C, Oei
Y, Ye H, Aukerman SL and Heise C: CHIR-258: a potent inhibitor of
FLT3 kinase in experimental tumor xenograft model of human acute
myelogenous leukemia. Clin Cancer Res. 11:5281–5291.
2005.PubMed/NCBI
|
|
27
|
Fathi AT and Chabner BA: FLT3 inhibition
as therapy in acute myeloid leukemia: a record of trials and
tribulations. Oncologist. 16:1162–1174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gottesman MM, Bates SE and Fojo T:
Multidrug resistance in cancer: role of ATP-dependent transporters.
Nat Rev Cancer. 2:48–58. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
McCollum AK, TenEyck CJ, Stensgard B,
Morlan BW, Ballman KV, Jenkins RB, Toft DO and Erlichman C:
P-glycoprotein mediated resistance to Hsp90-directed therapy is
eclipsed by the heat shock response. Cancer Res. 68:7419–7427.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Banerji U, O’Donnell A, Scurr M, Pacey S,
Stapleton S, Asad Y, Simmons L, Maloney A, Raynaud F, Campbell M,
Walton M, Lakhani S, Kaye S, Workman P and Judson I: Phase I
pharmacokinetic and pharmacodynamic study of 17-allylamino,
17-demethoxygeldanamycin in patients with advanced malignancies. J
Clin Oncol. 23:4152–4161. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kelland LR, Sharp SY, Rogers PM, Myers TG
and Workman P: DT Diaphorase expression and tumor cell sensitivity
to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat
shock protein 90. J Natl Cancer Inst. 91:1940–1949. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hostein I, Robertson D, DiStefano F,
Workman P and Clarke PA: Inhibition of signal transduction by the
Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in
cytostasis and apoptosis. Cancer Res. 61:4003–4009. 2001.PubMed/NCBI
|
|
33
|
Ellis LM and Hicklin DJ: Resistance to
targeted therapies: refining anticancer therapy in the era of
molecular oncology. Clin Cancer Res. 15:7471–7478. 2009. View Article : Google Scholar : PubMed/NCBI
|