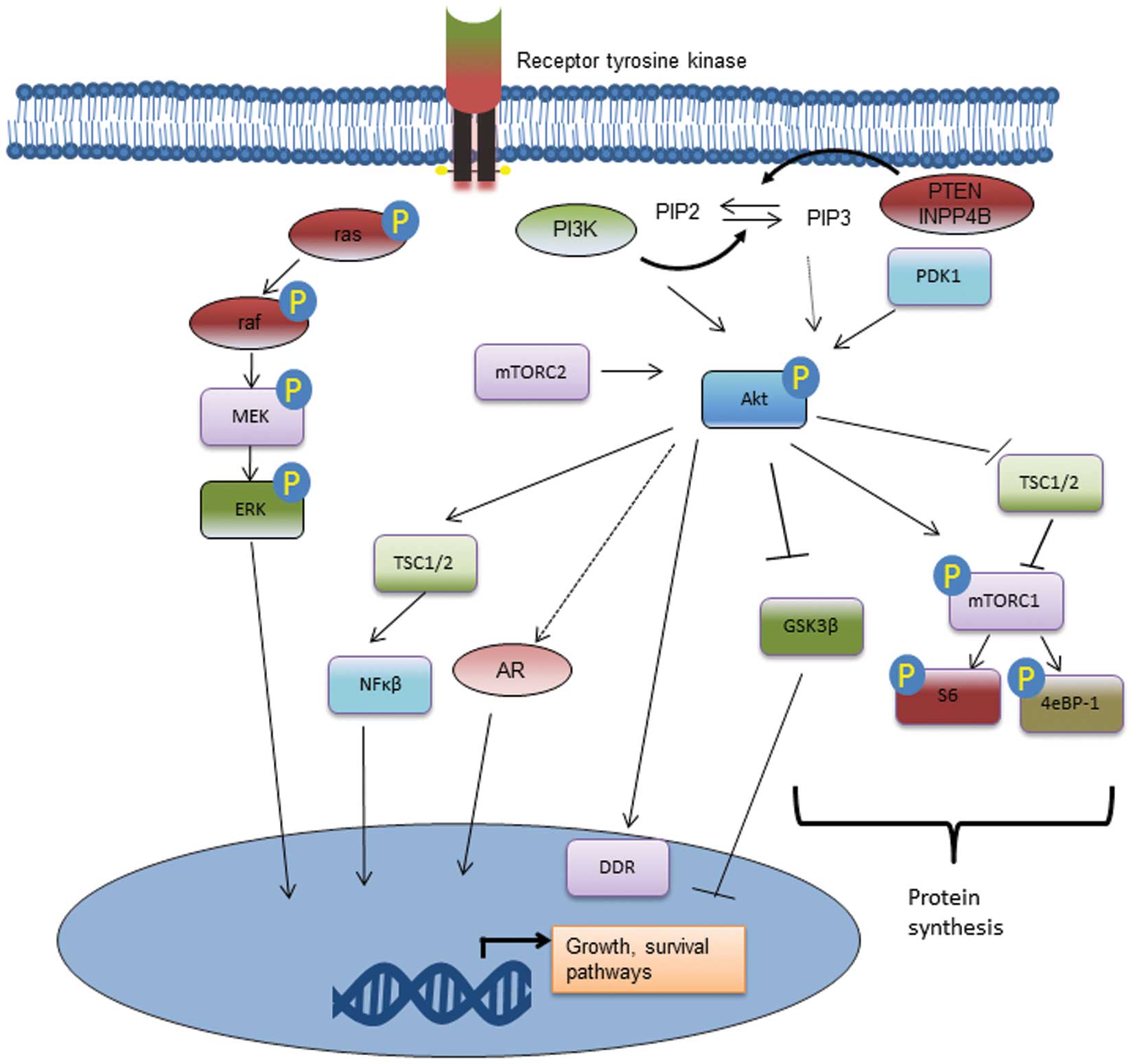
1. Introduction
Prostate cancer is the second most frequent cause of
cancer death in American men, with approximately 1 in 36 men dying
of prostate cancer (1). Over the
last several years, new therapies have emerged for treatment of
castrate resistant prostate cancer (CRPC) which have improved the
survival of patients with this disease (2,3).
Nonetheless, cure remains elusive with resistance developing over
time. With the increasing use of potent anti-androgens such as
abiraterone and enzalutamide, there is renewed interest in
targeting non-AR pathways in prostate cancer progression.
One of the most prominent alternate pathways in
prostate cancer is the PI3K/Akt signalling pathway. Activation of
this pathway is implicated in many aggressive human cancers
(4). Accordingly, there has been
significant investment toward developing targeted inhibitors of
this pathway in various hematologic and solid cancers. In this
review, we will discuss the relevance of the PI3K/Akt pathway in
prostate cancer, highlighting both basic science and clinical
aspects.
2. PI3K/Akt signalling pathway
The PI3K/Akt signalling pathway regulates cellular
metabolism, tumour development, growth, proliferation, metastases
and cytoskeletal reorganization. It is part of a complex
intracellular cell signalling cascade (Fig. 1). PI3K is a plasma
membrane-associated protein kinase consisting of three subunits:
the regulatory subunits p85 and p55; referred by convention as
collectively p85 and a catalytic subunit, p110 (5). There are three classes; it is the
class IA PI3Ks which are the most clearly implicated in human
cancer, including prostate cancer. Moreover, this class is usually
downstream of receptor tyrosine kinases. Activation of receptor
tyrosine kinases at the cell membrane results in conformational
changes which removes auto-inhibition of the catalytic domain of
PI3K. Three catalytic isoforms p110α, p110β and p110δ are,
respectively, the product of the genes PIK3CA, PIK3CB and PIK3CD.
Once activated, PI3K catalyzes the phosphorylation of PIP2 to
produce PIP3. PIP3 then activates intracellular signalling through
its binding to pleckstrin homology (PH) domains of many signalling
proteins, including Akt. In prostate cancer, it appears that the
p110β isoform is most relevant to prostate cancer progression and
resistance (6). It has been
associated with basal activation of Akt in prostate cancer models
(7).
The PI3K/Akt pathway functions downstream of
receptor tyrosine kinases (RTKs) as well as independently of RTKs.
Non-RTK activation of this pathway may be from other intracellular
signalling pathways or from other membrane receptors including
G-protein coupled receptors. The main upstream activators likely
are context specific. In autopsy specimens of metastatic prostate
lesions, various RTKs were associated with Akt activation (8). Of note, many of the RTKs which
activated the PI3K/Akt pathway, including EGFR, IGF-IR, FGFR and
c-MET receptors, are actively researched as targets in CRPC.
Nonetheless, some in vitro studies in prostate cancer cells
suggests that basal activation of this pathway occurs independently
of RTKs (7). Notably,
phospho-proteomic analysis of metastatic tumour samples collected
on rapid autopsy found that Akt was the tyrosine kinase most
commonly found to be active in metastastic prostate cancer
(8). Activated Akt is a kinase
which in turn phosphorylates and activates many oncogenic features
within cancer cells. Upon recruitment to the cell membrane, it is
phosphorylated by phosphoinositide-dependent kinase 1 (PDK1), a
reaction catalyzed by PIP3 binding to the PH domains of both
molecules (Fig. 1). Once
phosphorylated at both Ser473 and Thr308 phosphosites, the
activated Akt can activate many downstream functions via its kinase
activity.
Mammalian target of rapamycin (mTOR) is a major
downstream signalling protein involved in protein translation via
the eIF4E complex and S6K which is activated by Akt. Both mTOR and
S6K are found in higher levels in prostate cancer compared to
benign controls (9). The proteins
differentially associated with mTOR defined the TORC1 andTORC2
complexes. These have overlapping, but different functions, with
TORC2 providing negative feedback regulation on the PI3K/Akt
pathway via S6K (10,11). There are many other downstream
oncogenic effects of Akt phosphorylation. Cell survival is promoted
through anti-apoptotic effects, particularly inhibition of the
pro-apoptotic Bcl-2 family members BAD and BAX (12). Transcription factor FOX01 acts as a
tumour suppressor and its phosphorylation by Akt induces its
ubiquitation and degradation by the proteasome. Further, inhibition
of glycogen synthase kinase 3 (GSK-3) increases cellular
translation of proteins as does phosphorylation of 4eBP-1.
Regulation of cell growth and survival by Akt also occurs by the
NF-κB pathway via activation of IκB kinase (IKK) (Fig. 1). Further, the PI3k/Akt pathway in
prostate cancer appears to be involved with modulation of DNA
damage repair pathway (13). More
recently, the PI3K/Akt pathway has been implicated in modulating a
more aggressive phenotype through modulation of cholesterol ester
formation in prostate cancer cells (14). This suggests a possible
relationship with metabolic pathway disturbances and the
development of aggressive prostate cancer. Overall, there are a
plethora of downstream cellular functions of the Akt pathway which
correspond to a clinically aggressive phenotype.
3. Regulation by phosphatases
The PI3K/Akt pathway is antagonized by several
phosphatases, including phosphatase and tensin homolog gene (PTEN),
PH and leucine-rich repeat protein phosphatase (PHLPP), cellular
prostatic acid phosphatase, PP2A and INPP4B (15–17)
(Table I). Genetic loss or other
inactivation of these phosphatases results in greater amounts of
phospho-Akt and subsequent increased or sustained oncogenic
signaling. Notably, the PTEN gene on chromosome 10q23.3 is the
most-commonly deleted gene in prostate cancer (18). However, genomic loss of PTEN does
not always correlate with activation of the PI3K/Akt pathway
(19). Pre-clinical models and
patient samples also show that loss of PTEN results in a
particularly aggressive phenotype when found in combination with
activation of receptor tyrosine kinases (20,21).
PHLPP is regulated by the AR via FKBP5 and explains in part the
upregulation of the PI3K/Akt pathway seen following androgen
deprivation (17). INPP4B is
decreased following androgen deprivation and may be another
mechanism through which the Akt pathway is activated resulting in
earlier disease recurrence (16).
 | Table ICommon genomic alterations
potentially involved in activation of the PI3K/Akt/mTOR pathway
from the MSKCC dataset (47). |
Table I
Common genomic alterations
potentially involved in activation of the PI3K/Akt/mTOR pathway
from the MSKCC dataset (47).
| Gene | Type of
alteration | Prevalence in
metastatic disease (%) | Prevalence in
localized disease (%) |
|---|
| PTEN | Loss or
inactivation | 4 | 42 |
| INPP4B | Loss or
inactivation | 8 | 47 |
| PIK3R1 | Loss or
inactivation | 22 | 58 |
| PIK3R3 | Loss or
inactivation | 2 | 16 |
| PIK3CA | Activating
mutation | 6 | 16 |
| PHLPP | Loss or
inactivation | 11 | 37 |
4. Role of PI3K/Akt in prostate
carcinogenesis and progression
The mechanisms through which the PI3K pathway may
induce carcinogenesis include the activation of growth and survival
pathways. Further, activation of this pathway may also alter
epigenetic regulators, such as BIM1 (22). The PI3K/Akt pathway has also been
shown to be important to the survival and proliferation of prostate
cancer stem cells (23).
PTEN deletion is commonly used to model prostate
cancer progression in mice (24,25).
PTEN loss in mice has been shown to suppress androgen-responsive
genes and promote cell autonomous growth (26). Activation of the P3K/Akt pathway in
mice may also occur using myristolated Akt or constitutive
activation of p110β. In a murine subrenal xenograft model,
activation of both AR and Akt has been noted to synergize to
increase prostate tumour growth (27). Nonetheless, the exact role of this
pathway in carcinogenesis in humans is uncertain. On the contrary,
a recent genome wide sequencing analysis suggests that PTEN loss is
a late-stage feature in the progression of prostate cancer
(28).
Pre-clinical studies suggest that concomitant loss
of certain proteins together with PTEN loss appear to accelerate
prostate cancer progression. This has been demonstrated in mice and
correlated with features of aggressiveness, such as Gleason score,
in patient samples for the tumour suppressors NKX3.1, EAF2/U19,
Gata3 and Sox9 (29–33). B-raf and Stat3 activation and loss
of SMAD4 and p53 signalling have also been shown in murine models
to cooperate with PTEN loss to enhance prostate cancer progression
(34–37). This complex network with other
pathways highlights why monotherapy against the PI3K/Akt pathway
may not be an optimal strategy. Table
II lists different combination strategies which have been
explored targeting the PI3K/Akt/mTOR pathway in pre-clinical models
of prostate cancer.
| Table IISelected pre-clinical combination
studies of PI3K/Akt/mTOR inhibitors. |
Table II
Selected pre-clinical combination
studies of PI3K/Akt/mTOR inhibitors.
| PI3K/Akt/mTOR
pathway inhibitor | Additional pathway
target | Selected outcomes
assessed | Author (Refs.) |
|---|
| ZSTK474 (Pan-PI3K
inhibitor) | PSMA | In vitro and
in vivo (C4-2luc) tumour growth | Baiz et al
(83) |
| BEZ235 (PI3K-mTOR
inhibitor) | HDAC | Attenuation of DNA
damage repair protein ATM | Ellis et al
(13) |
| AZD5363 (Akt
inhibitor) | AR | Apoptosis,
proliferation, LNCaP tumour growth | Thomas et al
(38) |
| BEZ235 (PI3K/mTOR
inhibitor) | Microtubules | In vitro and
in vivo tumour growth | Yasumizu et
al (84) |
| AZD5363 (Akt
inhibitor) | Autophagy | Apoptosis, tumour
growth | Lamoureux et
al (85) |
| Akt inhibitors | Pim-1 | Apoptosis, tumour
growth | Cen et al
(86) |
| Rapamycin (mTOR
inhibitor) | MEK | In vitro and
in vivo tumour growth | Kinkade et
al (87) |
| Everolimus (mTOR
inhibitor) | Propachlor (from
drug screen panel) | Authophagic cell
death | Tai et al
(88) |
| Perifosine (Akt
inhibitor) | EGFR | Apoptosis | Festuccia et
al (89) |
5. PI3K/Akt and the AR pathway
The relationship between the PI3K/Akt and AR
pathways is of significant interest as a co-targeting strategy in
prostate cancer (17,38). Reciprocal interactions between
these pathways have been demonstrated in several pre-clinical
studies (17,39,40).
Blockade of the AR pathway results in PHLPP-mediated Akt
inactivation via a decrease in androgen regulated FKBP5 (17,26).
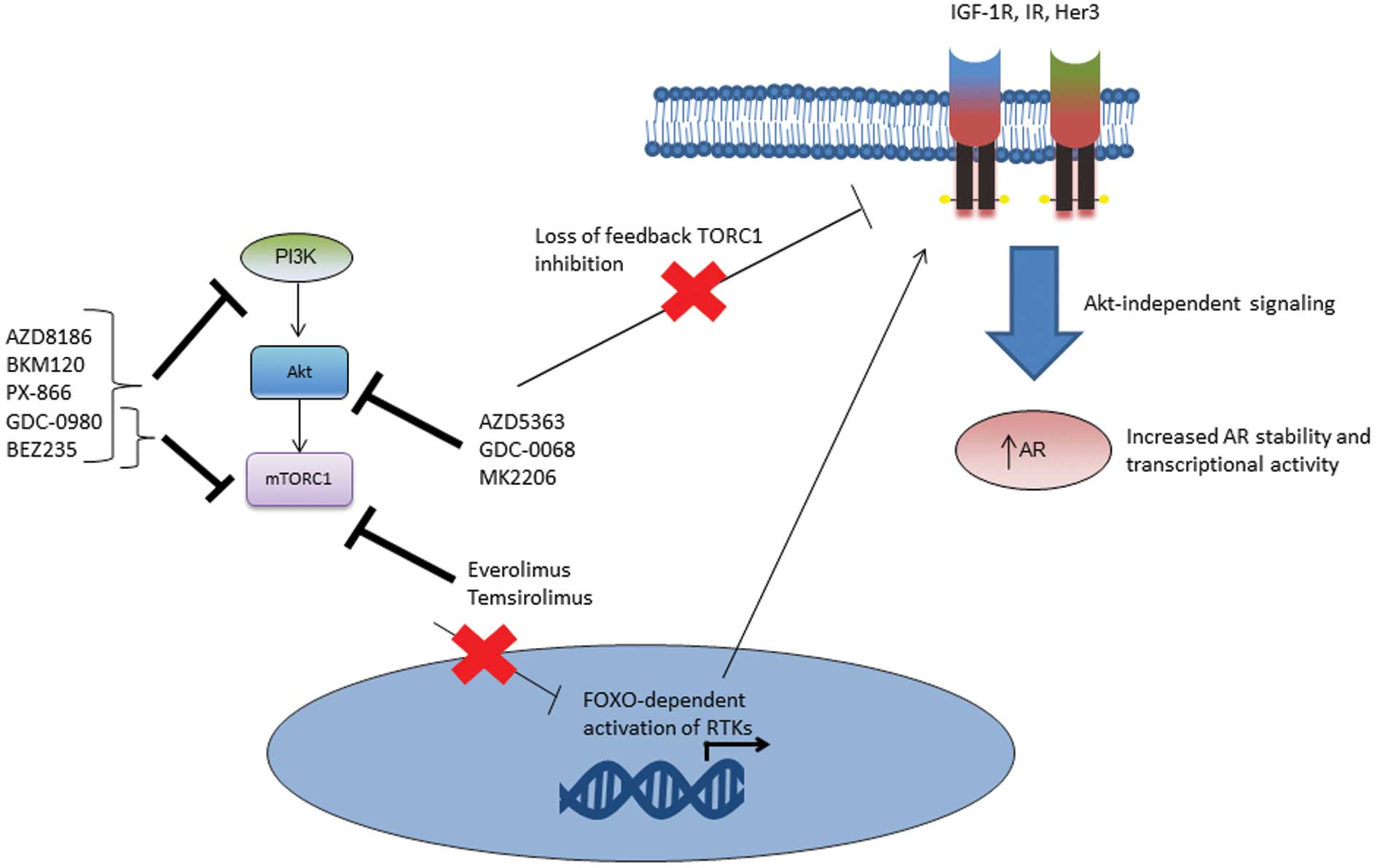
Inhibition of the PI3K/Akt pathway may result in upregulation of AR
transcriptional activity via activation of membrane signalling
proteins such as HER3 (Fig. 2)
(17,41). Direct AR phosphorylation by Akt
appears to predominantly be relevant in the low-testosterone state
(i.e., during androgen deprivation) (27,42,43).
Akt has been shown to phosphorylate the AR at Ser-213 and Ser-791,
but the significance of these phosphosites is unclear (27). In vitro models suggest that
Akt may regulate AR transcription (27), but that this is not by direct
phosphorylation of the AR (27,44).
The combination of bicalutamide and a pan-Akt
inhibitor, AZD5363 have been noted in vitro and in
vivo in LNCaP models to synergize to decreased tumour growth
(38). Early results suggest
markedly decreased in vivo tumour volumes with the use of
potent pan-Akt or PI3K isoform specific inhibitors in combination
with potent AR inhibitors (45,46).
These pre-clinical findings have supported the development of
clinical trials of combination blockade of both the AR and the
PI3k/Akt/mTOR pathways (Table
III). Further understanding of the interactions between these
pathways in pre-clinical models may aid in design of future
clinical trials.
| Table IIIPI3K/Akt inhibitors in current
clinical evaluation for prostate cancer. |
Table III
PI3K/Akt inhibitors in current
clinical evaluation for prostate cancer.
| Drug | Inhibitors | Phase | Regimen | Population | Trial registry
no.b |
|---|
| AZD8186
(AstraZeneca) | PI3Kβ and δ
inhibitor | I | Monotherapy | CRPC | NCT01884285 |
| BKM120
(Novartis) | Pan-PI3K
inhibitor | I | + abiraterone
acetate | Post-abiraterone
CRPC | NCT01634061 |
| I | + abiraterone
acetate | mCRPC | NCT01741753 |
| II | Monotherapy | Post-chemo | NCT01385293 |
| PX-866
(Oncothyreon) | Pan-PI3K
inhibitor | II | Monotherapy | mCRPC post ADT | NCT01331083 |
| BEZ235
(Novartis) | PI3K/mTOR
inhibitor | I | + abiraterone
acetate | Post-abiraterone
CRPC | NCT01634061 |
| GDC-0980
(Genetech) | PI3K/mTOR
inhibitor | I/II | + abiraterone
acetate | Post-docetaxel | NCT01485861 |
| | | 2011-004126-10 |
| AZD5363
(AstraZeneca) | Akt inhibitor | I | Monotherapy | mCRPC | NCT01692262 |
| II | + enzalutamide | mCRPC | 2013-004091-34 |
| I/II | + docetaxel | mCRPC | NCT02121639 |
| GDC-0068
(Genetech) | Akt inhibitor | I/II | + abiraterone
acetate | Post-docetaxel | NCT01485861 |
| | | 2011-004126-10 |
| MK2206 (Merck) | Akt inhibitor | II | + bicaluatmide | Biochemical failure
after primary therapy | NCT01251861 |
| Everolimus
(Novartis) | mTORC1
inhibitor | II | + pasireotide
(somatostatin) | Chemo-naïve
CRPC | NCT01313559 |
| I/II | + docetaxel,
bevacizumab (VEGF inhibitor) | mCRPC | NCT00574769 |
| I/II | + carboplatin,
everolimus, and prednisone | Post-docetaxel
mCRPC | NCT01051570 |
| II | + bicalutamide | Recurrent or mCRPC
post ADT | NCT00814788 |
| Temsirolimus
(Wyeth) | mTORC1
inhibitor | I/II | + bevacizumab | Post-docetaxel
mCRPC | NCT01083368 |
| I | + vorinostat | Post-docetaxel
mCRPC | NCT01174199 |
| II | Monotherapy | CRPC | 2011-002087-24 |
| I/II | + docetaxel | CRPC | NCT01206036 |
| | | 2010-018370-21 |
| I/II | + cixutumumab | mCRPC | NCT01026623 |
6. PI3K/Akt pathway as a biomarker in
prostate cancer
With the aggressive oncogenic characteristics of the
PI3K/Akt pathway, there has been significant interest to use this
pathway as a biomarker to differentiate more significant, lethal
prostate cancer from more indolent disease. While it does appear
from the current data that this pathway does provide prognostic
information, it is unclear that it provides any significant
improvement over currently used clinical and pathologic markers.
However, there is ongoing interest to use this pathway as a
predictive biomarker for newer targeted agents of this pathway.
The challenges of using this pathway as a biomarker
relate in large part to the complexity of the biology in advanced
prostate cancer and tumour heterogeneity. Firstly, there is a large
diversity of mutations and genomic alterations which may activate
this pathway, making any single marker less sensitive. Further, the
context of this pathway in prostate cancer differs from other
malignancies where this pathway also plays an important role. For
example, while activating PI3KCA mutations are relatively common
among advanced malignancies, it does not appear to be as common in
prostate cancer (47). Factors in
the tumour microenvironment can influence signalling of this
pathway, which is downstream of various cell surface receptors.
Therefore, both tumour and patient heterogeneity contribute to a
complexity making biomarker evaluation and validation more
challenging. For example, in circulating tumour cells PTEN allelic
loss has significant heterogeneity when analyzed by fluorescent
in situ hybridization (48). Further, on immunohistochemistry,
analysis of downstream targets does not clearly correlate in
patient samples with the phosphorylation of Akt. In one study,
phosphorylation of downstream GSK3β and a forkhead transcription
factor was noted in only 29 and 40% of cases, respectively, in
localized prostate cancer samples with phospho-Akt (49). Finally, technical variation,
antibody limitations, tissue acquisition and processing also
present challenges to use of this pathway as a clinical
biomarker.
It is estimated that genomic PTEN alterations are
found in 9–45% of high grade prostate intra-epithelial neoplasia
(HG-PIN), increase to 20–60% in localized prostate cancer, and are
altered in up to 100% of cases of metastatic prostate cancer
(47,50). Homozygous deletion of PTEN is
linked with CRPC (51). Further,
PTEN is the most common gene with loss of heterozygosity in
circulating tumour cells CTCs (52). Similarly, mutations in the PI3K
pathway occur more frequently in metastatic tissue compared to
primary tumours. In the Taylor et al dataset, mutations in
the PI3K regulatory genes PIK3R1, PIK3R3 and PIK3CS occur at a
frequency of 22, 2 and 6% in primary tumours, respectively. In
metastatic tissues, the frequency increases to 58, 16 and 16%,
respectively (47). Mutations in
PIK3CA catalytic gene of PI3K are known to be activating to the
pathway and also may predict response to therapy with PI3K
inhibitors, though they are not highly prevalent in prostate cancer
(53). Mutations in the regulatory
phosphatase PHLPP gene can also result in activation of the Akt
pathway, occurring at 11 and 37% in primary and metastatic tumours,
respectively (47).
On immunohistochemistry, both PTEN status and
phospho-Akt are the most commonly investigated biomarkers. Akt can
be phosphorylated at Thr308 and Ser473; it appears that both sites
are usually phosphorylated in the active state. It does appear that
Akt staining is specific to tumour cells, without any staining in
adjacent stromal tissue (49). Not
surprisingly given the molecular features of the activated PI3K/Akt
pathway, the loss of PTEN staining in localized prostate cancer
samples correlates with higher Gleason score and pathologic stage
(54,55) as well as an increased risk of
positive lymph nodes (56). PTEN
protein loss on immunohistochemistry of the primary tumour has also
been associated with shorter time to biochemical recurrence post
radical prostatectomy, but not consistently (57). Levels of phospho-Akt increase with
higher Gleason grade (58,59) and are associated with poorer
survival in CRPC (60). However,
it is unclear whether they hold any prognostic significance in low
and intermediate grade disease (Gleason score 6–7) (59). Levels of phospho-Akt also predict
for biochemical recurrence post radical prostatectomy, with
improved prediction when used in combination with PTEN protein loss
(61). Loss of INPP4B has been
noted to be a good marker of aggressive breast cancer (62), but has not been explored in
prostate cancer. Another area which remains to be more fully
explored is the cellular localization of phospho-Akt. It is unclear
whether increased nuclear staining improves prognostication, as
suggested by one study which found that greater nuclear phospho-Akt
staining was associated with higher Gleason grade (63).
In addition to the above mentioned studies on the
use of PTEN, phospho-Akt or other related proteins as prognostic
biomarkers, there is significant interest into the use of
predictive biomarkers for PI3K/Akt targeting agents. Only a few
studies to date have investigated predictive biomakers, but this
area is expected to increase as more inhibitors of this pathway
enter clinical evaluation. In unselected men with CRPC, PTEN status
on immunohistochemistry did not predict response to everolimus
(64). Activating mutations in
mTOR were found in one patient with an excellent response to
combination everolimus and pazopanib (65). Similar anecdotal responses to
AZD5363, a pan-Akt inhibitor has been reported to be associated
with genetic alterations, but not yet in prostate cancer patients
(66). Prospected clinical
investigation and validation is ongoing to identify and evaluate
appropriate predictive biomarkers in patients for response to
PI3K/Akt inhibitor therapy in prostate cancer patients.
7. Clinical studies of PI3K/Akt/mTOR
inhibitors in prostate cancer
Several novel PI3K/Akt/mTOR pathway inhibitors are
in clinical development (Table
III) for advanced prostate cancer. Two studies have evaluated
monotherapy with the Akt inhibitor perifosine in prostate cancer.
Perifosine is an alkylphospholid with Akt inhibitor properties.
Akylphospholipids are known to accumulate in cell membranes, but
the exact reason for the anticancer activity is unclear, but this
is presumed due to its capacity to inhibit the Akt pathway. A phase
II trial of perifosine monotherapy in 25 patients with biochemical
recurrence after primary therapy did not alter PSA doubling time
and was ended early for lack of response (67). No significant toxicities were
reported in this relatively healthy population. A second trial in
19 patients with metastatic prostate cancer also demonstrated
minimal benefit (68). The median
time to progression was 4 weeks, with only 4 patients having stable
disease beyond 12 weeks. A recent phase III trial with perifosine
in colon cancer has also found no therapeutic benefit (69).
The novel agents now in development (Table III) differ significantly from
perifosine’s mechanism of action and limited side effects. Most are
small molecule reversible inhibitors of the catalytic function of
PI3K, Akt, and/or mTOR. The common adverse effects of PI3k/Akt
inhibitors reported to date include insulin resistance,
hyperglycemia, nausea and mood alterations. PI3K inhibitors include
non-specific and isoform-specific inhibitors. The three isoforms
(p110α, β, and δ) of class IA PI3K may play relatively different
roles in the progression of prostate cancer (7). Isoform-specific inhibitors in
prostate cancer aim to inhibit only the PI3Kβ and δ isoforms to
decrease insulin resistance and hyperglycemia associated with PI3Kα
inhibition (70).
BKM120 is an oral pan-PI3K inhibitor with reported
clinical data in advanced solid tumours, including prostate cancer
(71). Among 31 patients in the
phase I trial, one had a partial response and 12 (52%) had stable
disease. Treatment-related adverse events include rash,
hyperglycemia, diarrhea, anorexia, mood alteration, nausea and
fatigue. Reversible neuropsychiatric adverse events may be due to
BKM120 crossing the blood-brain barrier and inhibiting
PI3K/Akt/mTOR signalling modulating neurotransmitter
concentrations. Similarly, results with the oral pan-Akt inhibitor
AZD5363 showed two partial responses out of 92 patients in two
phase I trials (66). Notably,
both patients had mutations in Akt1 or PI3KCA, suggesting mutations
in these genes may be predictive of response.
Results to date with inhibition of downstream TORC1
or TORC2 in prostate cancer have been disappointing. Rapalogs such
as everolimus, temsirolimus, and ridaforolimus have had poor
results as single agents in prostate cancer clinical trials
(64,72). This may be due in part to TORC2
mediated feedback on Akt (10).
Dual TORC1/TORC2 inhibition has demonstrated improved inhibition of
downstream effectors such as the eIF-4E protein translation complex
not seen with mTORC1 inhibition (73). Dual mTORC1/mTORC2 inhibitors have
entered clinical testing for advanced solid tumours, including
prostate cancer (74).
8. Combination targeting in prostate
cancer
From pre-clinical data to date, as well as the
failure of prior monotherapy trials, it appears that targeting of
the PI3K/Akt pathway in prostate cancer is optimally done in
combination with other agents. While monotherapy with newer agents
may have some activity, the abundance of cross-talk with other
pathways seen in pre-clinical studies suggests that resistance will
develop as other pathways are reciprocally upregulated. In breast
cancer, the combination of the aromatase inhibitor targeting the
estrogen receptor with everolimus targeting mTOR demonstrated
significant synergy (75). In an
analogous manner in prostate cancer, rationale combination therapy
may significantly improve clinical response rates and reduce the
development of resistance.
Combination therapy with PI3K/Akt/mTOR inhibitors in
prostate cancer can be conceptualized as vertical blockade, with
inhibition of multiple nodes in the PI3K/Akt pathway or horizontal
blockade, with inhibition of the PI3K/Akt pathway together with
other parallel pathways. For example, pre-clinical work in
transgenic mice suggests that dual targeting of the Akt and mTOR
signalling (i.e., vertical blockade) has significantly more
activity compared to either monotherapy (76). Dual PI3K/mTOR blockade with one
molecule is facilitated by similarities of the PI3K and mTOR
catalytic sites. Similarly, effective combinations of Akt or mTOR
inhibition with AR blockade has been noted in other pre-clinical
studies (17,38,77).
Combination of Akt inhibition with receptor tyrosine kinases is
another horizontal blockade strategy with good pre-clinical data
(10).
Many of the ongoing clinical trials are evaluating a
PI3K/Akt inhibitor in combination with an AR pathway inhibitor
(Table III). A limited number of
trials targeting both pathways have been reported in prostate
cancer. A study in 36 patients with CRPC of the mTOR inhibitor
everolimus in combination with bicalutamide has been reported
(78). This study did not show any
benefit for the combination, though it was well tolerated. Reasons
for the lack of benefit could include a possible partial agonist
effect of bicalutamide, lack of effective AR inhibition with
bicalutamide as well as the activation of upstream Akt as a result
of mTOR inhibition. However, another phase I/II trial found that of
13 patients treated with bicalutamide and everolimus, 9 had a
partial response, one an unconfirmed partial response and 3 had
stable disease. Of the 5 patients treated with placebo +
bicalutamide, 1 had partial response, one unconfirmed partial
response, 2 stable disease and 1 had disease progression. The mean
time to relapse was 220 days for the everolimus + bicalutamide vs
109 days for placebo + bicalutamide (79). Very early results of the
combination of BKM120 with abiraterone and prednisone are also
promising (80).
One putative benefit of combination therapy is
decreased toxicity while maintaining therapeutic efficacy. However,
this has yet to be consistently observed in clinical trials in
patients. Increased toxicity has been noted in some early studies
to date when using combination therapy (81,82).
In a clinical trial of 11 patients with CRPC treated with
ridaforolimus and bicalutamide, 3 had dose-limiting toxicity,
including hyperglycemia and stomatitis. It is anticipated that
continued experience with these newer PI3K inhibitors will result
in better dosing schedules to minimize adverse effects, while
maintaining therapeutic efficacy.
9. Conclusion
Activation of the PI3K/Akt pathway clearly plays a
major role in the aggressive nature of many prostate cancers. With
the use of newer AR pathway inhibitors and combination therapy,
this non-androgen receptor pathway may become increasingly relevant
as more patients develop non-AR driven tumours. Clinical trials are
now assessing the efficacy of targeting this pathway in CRPC. An
improved understanding of the biology and relevant biomarkers of
this pathway in prostate cancer will be important to understand
which patients will benefit from PI3K/Akt/mTOR inhibitors and at
what point in the disease course they should be given. The use of
combination therapy has potential to substantially improve the
outcome of patients, but needs to be balanced against toxicities,
particularly if combination therapies are utilized earlier in the
course of disease.
References
|
1
|
What are the key statistics about prostate
cancer? American Cancer Society. http://www.cancer.org/cancer/prostatecancer/detailedguide/prostate-cancer-key-statistics.
Accessed April 27, 2014
|
|
2
|
Scher HI, Fizazi K, Saad F, et al:
Increased survival with enzalutamide in prostate cancer after
chemotherapy. N Engl J Med. 367:1187–1197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ryan CJ, Smith MR, de Bono JS, et al:
Abiraterone in metastatic prostate cancer without previous
chemotherapy. N Engl J Med. 368:138–148. 2013. View Article : Google Scholar
|
|
4
|
Donahue TR, Tran LM, Hill R, et al:
Integrative survival-based molecular profiling of human pancreatic
cancer. Clin Cancer Res. 18:1352–1363. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martini M, Ciraolo E, Gulluni F and Hirsch
E: Targeting PI3K in cancer: any good news? Front Oncol. 3:1082013.
View Article : Google Scholar
|
|
6
|
Jia S, Liu Z, Zhang S, et al: Essential
roles of PI(3)K-p110beta in cell growth, metabolism and
tumorigenesis. Nature. 454:776–779. 2008.PubMed/NCBI
|
|
7
|
Jiang X, Chen S, Asara JM and Balk SP:
Phosphoinositide 3-kinase pathway activation in phosphate and
tensin homolog (PTEN)-deficient prostate cancer cells is
independent of receptor tyrosine kinases and mediated by the
p110beta and p110delta catalytic subunits. J Biol Chem.
285:14980–14989. 2010. View Article : Google Scholar
|
|
8
|
Drake JM, Graham NA, Lee JK, et al:
Metastatic castration-resistant prostate cancer reveals
intrapatient similarity and interpatient heterogeneity of
therapeutic kinase targets. Proc Natl Acad Sci USA.
110:E4762–E4769. 2013. View Article : Google Scholar
|
|
9
|
Kremer CL, Klein RR, Mendelson J, et al:
Expression of mTOR signaling pathway markers in prostate cancer
progression. Prostate. 66:1203–1212. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bitting RL and Armstrong AJ: Targeting the
PI3K/Akt/mTOR pathway in castration-resistant prostate cancer.
Endocr Relat Cancer. 20:R83–R99. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dibble CC, Asara JM and Manning BD:
Characterization of Rictor phosphorylation sites reveals direct
regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 29:5657–5670.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ellis L, Ku SY, Ramakrishnan S, et al:
Combinatorial antitumor effect of HDAC and the PI3K-Akt-mTOR
pathway inhibition in a Pten defecient model of prostate cancer.
Oncotarget. 4:2225–2236. 2013.PubMed/NCBI
|
|
14
|
Yue S, Li J, Lee SY, et al: Cholesteryl
ester accumulation induced by PTEN loss and PI3K/AKT activation
underlies human prostate cancer aggressiveness. Cell Metab.
19:393–406. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Muniyan S, Ingersoll MA, Batra SK and Lin
MF: Cellular prostatic acid phosphatase, a PTEN-functional
homologue in prostate epithelia, functions as a prostate-specific
tumor suppressor. Biochim Biophys Acta. pii: S0304-419X(14)00042-0.
Apr 18–2014.(Epub ahead of print). View Article : Google Scholar
|
|
16
|
Hodgson MC, Shao LJ, Frolov A, et al:
Decreased expression and androgen regulation of the tumor
suppressor gene INPP4B in prostate cancer. Cancer Res. 71:572–582.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Carver BS, Chapinski C, Wongvipat J, et
al: Reciprocal feedback regulation of PI3K and androgen receptor
signaling in PTEN-deficient prostate cancer. Cancer Cell.
19:575–586. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Phin S, Moore MW and Cotter PD: Genomic
rearrangements of in prostate cancer. Front Oncol. 3:2402013.
View Article : Google Scholar
|
|
19
|
Fata JE, Debnath S, Jenkins EC Jr and
Fournier MV: Nongenomic mechanisms of PTEN regulation. Int J Cell
Biol. 2012:3796852012.PubMed/NCBI
|
|
20
|
Ahmad I, Patel R, Singh LB, et al: HER2
overcomes PTEN (loss)-induced senescence to cause aggressive
prostate cancer. Proc Natl Acad Sci USA. 108:16392–16397. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zu K, Martin NE, Fiorentino M, et al:
Protein expression of PTEN, insulin-like growth factor I receptor
(IGF-IR), and lethal prostate cancer: a prospective study. Cancer
Epidemiol Biomarkers Prev. 22:1984–1993. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nacerddine K, Beaudry JB, Ginjala V, et
al: Akt-mediated phosphorylation of Bmi1 modulates its oncogenic
potential, E3 ligase activity, and DNA damage repair activity in
mouse prostate cancer. J Clin Invest. 122:1920–1932. 2012.
View Article : Google Scholar
|
|
23
|
Dubrovska A, Kim S, Salamone RJ, et al:
The role of PTEN/Akt/PI3K signaling in the maintenance and
viability of prostate cancer stem-like cell populations. Proc Natl
Acad Sci USA. 106:268–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang S, Gao J, Lei Q, et al:
Prostate-specific deletion of the murine Pten tumor suppressor gene
leads to metastatic prostate cancer. Cancer Cell. 4:209–221. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Blando J, Portis M, Benavides F, et al:
PTEN deficiency is fully penetrant for prostate adenocarcinoma in
C57BL/6 mice via mTOR-dependent growth. Am J Pathol. 174:1869–1879.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mulholland DJ, Tran LM, Li Y, et al: Cell
autonomous role of PTEN in regulating castration-resistant prostate
cancer growth. Cancer Cell. 19:792–804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xin L, Teitell MA, Lawson DA, Kwon A,
Mellinghoff IK and Witte ON: Progression of prostate cancer by
synergy of AKT with genotropic and nongenotropic actions of the
androgen receptor. Proc Natl Acad Sci USA. 103:7789–7794. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Baca Sylvan C, Prandi D, Lawrence Michael
S, et al: Punctuated evolution of prostate cancer genomes. Cell.
153:666–677. 2013.PubMed/NCBI
|
|
29
|
Nguyen AH, Tremblay M, Haigh K, et al:
Gata3 antagonizes cancer progression in Pten-deficient prostates.
Hum Mol Genet. 22:2400–2410. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ai J, Pascal LE, O’Malley KJ, et al:
Concomitant loss of EAF2/U19 and Pten synergistically promotes
prostate carcinogenesis in the mouse model. Oncogene. 33:2286–2294.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thomsen MK, Ambroisine L, Wynn S, et al:
SOX9 elevation in the prostate promotes proliferation and
cooperates with PTEN loss to drive tumor formation. Cancer Res.
70:979–987. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim MJ, Cardiff RD, Desai N, et al:
Cooperativity of Nkx3.1 and Pten loss of function in a mouse model
of prostate carcinogenesis. Proc Natl Acad Sci USA. 99:2884–2889.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song H, Zhang B, Watson MA, Humphrey PA,
Lim H and Milbrandt J: Loss of Nkx3.1 leads to the activation of
discrete downstream target genes during prostate tumorigenesis.
Oncogene. 28:3307–3319. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen Z, Trotman LC, Shaffer D, et al:
Crucial role of p53-dependent cellular senescence in suppression of
Pten-deficient tumorigenesis. Nature. 436:725–730. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Blando JM, Carbajal S, Abel E, et al:
Cooperation between Stat3 and Akt signaling leads to prostate tumor
development in transgenic mice. Neoplasia. 13:254–265.
2011.PubMed/NCBI
|
|
36
|
Ding Z, Wu CJ, Chu GC, et al:
SMAD4-dependent barrier constrains prostate cancer growth and
metastatic progression. Nature. 470:269–273. 2011. View Article : Google Scholar
|
|
37
|
Wang J, Kobayashi T, Floc’h N, et al:
B-Raf activation cooperates with PTEN loss to drive c-Myc
expression in advanced prostate cancer. Cancer Res. 72:4765–4776.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Thomas C, Lamoureux F, Crafter C, et al:
Synergistic targeting of PI3K/AKT pathway and androgen receptor
axis significantly delays castration-resistant prostate cancer
progression in vivo. Mol Cancer Ther. 12:2342–2355. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang Y, Kreisberg JI and Ghosh PM:
Cross-talk between the androgen receptor and the
phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr
Cancer Drug Targets. 7:591–604. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kaarbo M, Mikkelsen OL, Malerod L, et al:
PI3K-AKT-mTOR pathway is dominant over androgen receptor signaling
in prostate cancer cells. Cell Oncol. 32:11–27. 2010.PubMed/NCBI
|
|
41
|
Chandarlapaty S, Sawai A, Scaltriti M, et
al: AKT inhibition relieves feedback suppression of receptor
tyrosine kinase expression and activity. Cancer Cell. 19:58–71.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wen Y, Hu MCT, Makino K, et al: HER-2/neu
promotes androgen-independent survival and growth of prostate
cancer cells through the Akt pathway. Cancer Res. 60:6841–6845.
2000.PubMed/NCBI
|
|
43
|
Manin M, Baron S, Goossens K, et al:
Androgen receptor expression is regulated by the phosphoinositide
3-kinase/Akt pathway in normal and tumoral epithelial cells.
Biochem J. 366:729–736. 2002.PubMed/NCBI
|
|
44
|
Nan B, Snabboon T, Unni E, X-J Y, Whang Y
and Marcelli M: The PTEN tumor suppressor is a negative modulator
of androgen receptor transcriptional activity. J Mol Endocrinol.
31:169–183. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schwartz S, Carver B, Wongvipat J, et al:
The antitumor effects of PI3K beta inhibitors in PTEN negative
prostate cancer are enhanced by inhibition of reactivated PI3K
alpha signaling. In: Proc 105th Annual Meeting Amer Assoc Cancer
Res; abs. 4774. 2014
|
|
46
|
Toren P, Kim S, Gleave M and Zoubeidi A:
Combined targeting of PI3K/Akt and AR pathway with AZD5363 and
enzalutamide induces anticancer activity in preclinical models of
prostate cancer. J Urol. 189(Suppl l4): e4032013. View Article : Google Scholar
|
|
47
|
Taylor BS, Schultz N, Hieronymus H, et al:
Integrative genomic profiling of human prostate cancer. Cancer
Cell. 18:11–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Attard G, Swennenhuis JF, Olmos D, et al:
Characterization of ERG, AR and PTEN gene status in circulating
tumor cells from patients with castration-resistant prostate
cancer. Cancer Res. 69:2912–2918. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jendrossek V, Henkel M, Hennenlotter J, et
al: Analysis of complex protein kinase B signalling pathways in
human prostate cancer samples. BJU Int. 102:371–382. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jia S, Gao X, Lee SH, et al: Opposing
effects of androgen deprivation and targeted therapy on prostate
cancer prevention. Cancer Discov. 3:44–51. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sircar K, Yoshimoto M, Monzon FA, et al:
PTEN genomic deletion is associated with p-Akt and AR signalling in
poorer outcome, hormone refractory prostate cancer. J Pathol.
218:505–513. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Schmidt H, DeAngelis G, Eltze E, Gockel I,
Semjonow A and Brandt B: Asynchronous growth of prostate cancer is
reflected by circulating tumor cells delivered from distinct, even
small foci, harboring loss of heterozygosity of the PTEN gene.
Cancer Res. 66:8959–8965. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Janku F, Tsimberidou AM, Garrido-Laguna I,
et al: PIK3CA mutations in patients with advanced cancers treated
with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther. 10:558–565.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
McMenamin ME, Soung P, Perera S, Kaplan I,
Loda M and Sellers WR: Loss of PTEN expression in paraffin-embedded
primary prostate cancer correlates with high Gleason score and
advanced stage. Cancer Res. 59:4291–4296. 1999.PubMed/NCBI
|
|
55
|
Dreher T, Zentgraf H, Abel U, et al:
Reduction of PTEN and p27kip1 expression correlates with tumor
grade in prostate cancer. Analysis in radical prostatectomy
specimens and needle biopsies. Virchows Arch. 444:509–517. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Schmitz M, Grignard G, Margue C, et al:
Complete loss of PTEN expression as a possible early prognostic
marker for prostate cancer metastasis. Int J Cancer. 120:1284–1292.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Koumakpayi IH, Le Page C, Mes-Masson AM
and Saad F: Hierarchical clustering of immunohistochemical analysis
of the activated ErbB/PI3K/Akt/NF-kappaB signalling pathway and
prognostic significance in prostate cancer. Br J Cancer.
102:1163–1173. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Malik SN, Brattain M, Ghosh PM, et al:
Immunohistochemical demonstration of phospho-Akt in high Gleason
grade prostate cancer. Clin Cancer Res. 8:1168–1171.
2002.PubMed/NCBI
|
|
59
|
Hammarsten P, Cipriano M, Josefsson A, et
al: Phospho-Akt immunoreactivity in prostate cancer: relationship
to disease severity and outcome, Ki67 and phosphorylated EGFR
expression. PLoS One. 7:e479942012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
McCall P, Gemmell LK, Mukherjee R,
Bartlett JM and Edwards J: Phosphorylation of the androgen receptor
is associated with reduced survival in hormone-refractory prostate
cancer patients. Br J Cancer. 98:1094–1101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Bedolla R, Prihoda TJ, Kreisberg JI, et
al: Determining risk of biochemical recurrence in prostate cancer
by immunohistochemical detection of PTEN expression and Akt
activation. Clin Cancer Res. 13:3860–3867. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Won JR, Gao D, Chow C, et al: A survey of
immunohistochemical biomarkers for basal-like breast cancer against
a gene expression profile gold standard. Mod Pathol. 26:1438–1450.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Van de Sande T, Roskams T, Lerut E, et al:
High-level expression of fatty acid synthase in human prostate
cancer tissues is linked to activation and nuclear localization of
Akt/PKB. J Pathol. 206:214–219. 2005.PubMed/NCBI
|
|
64
|
Templeton AJ, Dutoit V, Cathomas R, et al:
Phase 2 trial of single-agent everolimus in chemotherapy-naive
patients with castration-resistant prostate cancer (SAKK 08/08).
Eur Urol. 64:150–158. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wagle N, Grabiner BC, Van Allen EM, et al:
Activating mTOR mutations in a patient with an extraordinary
response on a phase I trial of everolimus and pazopanib. Cancer
Discov. 4:546–553. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Banerji U: Results of two phase 1
multicenter trials of AZD5363, an inhibitor of AKT1, 2 and 3:
biomarker and early clinical evaluation in Western and Japanese
patients with advanced solid tumors. In: 2013, Proc Annual Meeting
Amer Assoc Cancer Res (abstract LB-66); 2013;
|
|
67
|
Chee KG, Longmate J, Quinn DI, et al: The
AKT inhibitor perifosine in biochemically recurrent prostate
cancer: a phase II California/Pittsburgh cancer consortium trial.
Clin Genitourin Cancer. 5:433–437. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Posadas EM, Gulley J, Arlen PM, et al: A
phase II study of perifosine in androgen independent prostate
cancer. Cancer Biol Ther. 4:1133–1137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bendell JC, Ervin TE, Senzer N, et al:
Results of the X-PECT study: a phase III randomized double-blind,
placebo-controlled study of perifosine plus capecitabine (P-CAP)
versus placebo plus capecitabine (CAP) in patients (pts) with
refractory metastatic colorectal cancer (mCRC). J Clin Oncol.
30:LBA35012012.
|
|
70
|
Busaidy NL, Farooki A, Dowlati A, et al:
Management of metabolic effects associated with anticancer agents
targeting the PI3K-Akt-mTOR pathway. J Clin Oncol. 30:2919–2928.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Bendell JC, Rodon J, Burris HA, et al:
Phase I dose-escalation and -expansion study of buparlisib
(BKM120), an oral pan-class I PI3K inhibitor, in patients with
advanced solid tumors. J Clin Oncol. 30:282–290. 2012. View Article : Google Scholar
|
|
72
|
Amato RJ, Wilding G, Bubley G, Loewy J,
Haluska F and Gross ME: Safety and preliminary efficacy analysis of
the mTOR inhibitor ridaforolimus in patients with taxane-treated,
castration-resistant prostate cancer. Clin Genitourin Cancer.
10:232–238. 2012. View Article : Google Scholar
|
|
73
|
Hsieh AC, Liu Y, Edlind MP, et al: The
translational landscape of mTOR signalling steers cancer initiation
and metastasis. Nature. 485:55–61. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Mateo J, Schoffski P, Olmos D, et al:
Abstract B187: Pharmacodynamics of OSI-027, a dual mTORC1/mTORC2
inhibitor, in tumor and surrogate tissues: Results from the
expansion phase of a first-in-man study. Mol Cancer Ther.
12:B1872013. View Article : Google Scholar
|
|
75
|
Baselga J, Campone M, Piccart M, et al:
Everolimus in post-menopausal hormone-receptor-positive advanced
breast cancer. N Engl J Med. 366:520–529. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Floc’h N, Kinkade CW, Kobayashi T, et al:
Dual targeting of the Akt/mTOR signaling pathway inhibits
castration-resistant prostate cancer in a genetically engineered
mouse model. Cancer Res. 72:4483–4493. 2012.PubMed/NCBI
|
|
77
|
Squillace RM, Miller D, Wardwell SD, Wang
F, Clackson T and Rivera VM: Synergistic activity of the mTOR
inhibitor ridaforolimus and the antiandrogen bicalutamide in
prostate cancer models. Int J Oncol. 41:425–432. 2012.PubMed/NCBI
|
|
78
|
Nakabayashi M, Werner L, Courtney KD, et
al: Phase II trial of RAD001 and bicalutamide for
castration-resistant prostate cancer. BJU Int. 110:1729–1735. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Pan C, Robles D, D’Abronzo L, et al:
Synergistic effects of everolimus and bicalutamide in
castration-resistant prostate cancer: results from a phase I/II
clinical trial. Cancer Res. 72:57502012. View Article : Google Scholar
|
|
80
|
Patnaik A, Loda M, Kung J, et al: A phase
Ib study of BKM120 combined with abiraterone acetate for
castrate-resistant, metastatic prostate cancer. In: Proc 105th
Annual Meeting Amer Assoc Cancer Res; abs. CT418. 2014
|
|
81
|
Janne PA, Cohen RB, Laird AD, et al: Phase
I safety and pharmacokinetic study of the PI3K/mTOR inhibitor
SAR245409 (XL765) in combination with erlotinib in patients with
advanced solid tumors. J Thorac Oncol. 9:316–323. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Meulenbeld HJ, de Bono JS, Tagawa ST, et
al: Tolerability, safety and pharmacokinetics of ridaforolimus in
combination with bicalutamide in patients with asymptomatic,
metastatic castration-resistant prostate cancer (CRPC). Cancer
Chemother Pharmacol. 72:909–916. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Baiz D, Hassan S, Choi YA, et al:
Combination of the PI3K inhibitor ZSTK474 with a PSMA-targeted
immunotoxin accelerates apoptosis and regression of prostate
cancer. Neoplasia. 15:1172–1183. 2013.PubMed/NCBI
|
|
84
|
Yasumizu Y, Miyajima A, Kosaka T, Miyazaki
Y, Kikuchi E and Oya M: Dual PI3K/mTOR inhibitor NVP-BEZ235
sensitizes docetaxel in castration resistant prostate cancer. J
Urol. 191:227–234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Lamoureux F, Thomas C, Crafter C, et al:
Blocked autophagy using lysosomotropic agents sensitizes resistant
prostate tumor cells to the novel Akt inhibitor AZD5363. Clin
Cancer Res. 19:833–844. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Cen B, Mahajan S, Wang W and Kraft AS:
Elevation of receptor tyrosine kinases by small molecule AKT
inhibitors in prostate cancer is mediated by Pim-1. Cancer Res.
73:3402–3411. 2013. View Article : Google Scholar
|
|
87
|
Kinkade CW, Castillo-Martin M, Puzio-Kuter
A, et al: Targeting AKT/mTOR and ERK MAPK signaling inhibits
hormone-refractory prostate cancer in a preclinical mouse model. J
Clin Invest. 118:3051–3064. 2008.PubMed/NCBI
|
|
88
|
Tai S, Sun Y, Liu N, et al: Combination of
Rad001 (everolimus) and propachlor synergistically induces
apoptosis through enhanced autophagy in prostate cancer cells. Mol
Cancer Ther. 11:1320–1331. 2012. View Article : Google Scholar
|
|
89
|
Festuccia C, Gravina GL, Muzi P, et al:
Akt down-modulation induces apoptosis of human prostate cancer
cells and synergizes with EGFR tyrosine kinase inhibitors.
Prostate. 68:965–974. 2008. View Article : Google Scholar : PubMed/NCBI
|