Introduction
Leiomyosarcoma is a highly malignant neoplasm that
shows a high rate of local recurrence and distant metastasis,
associated with aggressive growth and poor prognosis (1). The standard multimodal treatment
strategies are surgery, radiation and conventional chemotherapy.
Due to the insufficient effectiveness of the current treatment
options, novel therapy options for treatment with target-specific
drugs are urgently needed (2,3).
Inhibition of tumor-caused angiogenesis has emerged
as a new therapeutic tool for therapy of diverse cancers. Growth of
new blood vessels via release of angiogenic factors such as
vascular endothelial growth factors (VEGFs) has been shown to be
essential for tumor growth, nutrient supply and migration in
metastasis (4). VEGFs signal
through their cognate receptor tyrosine kinases (RTKs)
(VEGFR-1/Flt-1, VEGFR-2/Flk-1/KDR and VEGFR-3/Flt-4). Importantly,
it has been shown that VEGF receptor (VEGFR) family members are
expressed not only in cells of the tumor cell microenvironment
(vascular, lymphatic, endothelial and non-endothelial cells)
(5) but on various cancer cells
such as multiple myeloma, leukaemia, breast, colon, pancreatic
(5–8) and leiomyosarcoma cells (2). VEGF-A has been demonstrated to have a
stimulatory effect on prolife ration and migration of diverse
VEGF-A-expressing carcinoma cells in vitro and in
vivo (6,7,9–13).
Thus, anti-angiogenic drugs for inhibition of angiogenesis and
tumor cell growth are considered as promising alternative or
supportive tools to conventional tumor therapy (4). Different strategies are available to
repress tumor angiogenesis and have been approved for clinical use
in diverse cancers, e.g., ligand-specific antibodies (bevacizumab)
(14) or small molecule inhibitors
(e.g., pazopanib, sorafenib, sunitinib) (4,15).
Inhibition of VEGFR family members has been proven
to be effective in several malignancies (16–18).
In particular, simultaneous inhibition of multiple, related RTK
families was suggested to be a more efficient strategy for
antitumor treatment compared to single receptor targeting (19). The multi-targeted tyrosine kinase
inhibitor PTK787/ZK222584 (PTK787) (Vatalanib) has been shown to
inhibit not only VEGFR-1, -2 and -3 but also platelet-derived
growth factor receptor (PDGFR)-α and -β kinase activity (20). VEGF-induced phosphorylation of
VEGFR-1, -2 and -3 in vitro is specifically blocked by
PTK787, which leads to inhibition of endothelial cell
proliferation, differentiation, tumor cell migration and VEGF- and
platelet-derived growth factor (PDGF)-induced angiogenesis
(6,20–25).
Additional activity of PTK787 in vivo (26) has led to
clinical trials in different malignant diseases. In a phase II and
III trial, PTK787 treatment showed promising results in relapsed or
progressing non-small cell lung cancer (27) and in a subgroup of metastatic
colorectal cancer patients, respectively (14,18,28).
PTK787 exerts an antitumor activity on the tumor
endothelium via reduction of vessel density in tumor tissues of
many different entities (6,24).
However, in order to understand the mechanism of action of the drug
it is essential not only to focus studies on the effects of PTK787
on the tumor cell environment/vasculature but also on the tumor
cells themselves, which were also shown to express VEGFR family
members (2). In this study we
evaluated the rationale for using the VEGFR tyrosine kinase
inhibitor PTK787 in leiomyosarcoma cells. We found high expression
of VEGFR family members and PDGFR-β in leiomyosarcoma tissue
specimens and in the leiomyosarcoma cell lines SK-LMS-1 and SK-UT-1
in addition to ligand secretion. Intracellular signalling pathways
were partially inhibited by PTK787. Leiomyosarcoma cell growth
remained unchanged upon PTK787 treatment alone or in combination
with VEGF-A or PDGF-BB. However, PTK787 treatment affected cell
migration and cell death.
The expression of angiogenic growth factors, their
corresponding receptors and functional responsiveness to inhibition
of VEGFR/PDGFR signalling provides strong evidence that
leiomyosarcoma patients with VEGFR- and/or PDGFR-positive tumor
samples might benefit from anti-angiogenic treatment by inhibition
of both autocrine stimulation of tumor cell growth and paracrine
stimulation of angiogenesis.
Materials and methods
Cell cultures and reagents
Human umbilical cord vein endothelial cells (HUVECs)
were isolated from human umbilical chords with a standardized
protocol as described (29). Human
leiomyosarcoma cell lines SK-UT-1 and SK-LMS-1, and human
promyelocytic leukemia cells (HL-60) were obtained from the
American Type Culture Collection (ATCC) (Manassas, VA, USA).
Leiomyosarcoma cell lines were cultured under standard conditions
in Dulbecco’s modified Eagle’s medium (DMEM) with high glucose
content (PAA Laboratories GmbH, Pasching, Austria) and supplemented
with 10% fetal calf serum (FCS) (CCPro, Oberdorla, Germany), 2 mM
glutamine and penicillin/streptomycin (both from PAA Laboratories
GmbH, Cölbe, Germany). HUVEC cells were isolated and cultured under
standard conditions in MCD131 medium as previously described
(29,30). The tyrosine kinase inhibitor PTK787
was provided by Novartis AG (Dr J. Wood, Oncology Research Group,
Basel, Switzerland) and was developed as a joint venture of
Novartis AG and Schering AG (Berlin, Germany). A 100 mM stock
solution was prepared in DMSO and stored at −20°C. For all assays
the inhibitor was diluted in culture medium to a final
concentration as indicated. The concentration of DMSO was diluted
to 0.1% for all assays. Recombinant VEGF165 (cat. no. 300-076;
ReliaTech GmbH, Braunschweig, Germany) and recombinant PDGF-BB
(cat. no. GF018; Chemicon International, Temecula, CA, USA) were
applied at final concentrations of 100 or 50 ng/ml,
respectively.
Analysis of mRNA expression with
RT-PCR
VEGFR-1, -2 and -3, and PDGFR-β mRNA expression was
assessed in leiomyosarcoma cell lines with RT-PCR. Briefly, RNA was
extracted using the RNeasy mini kit (Qiagen, Hilden, Germany)
following the manufacturer’s instructions. RNA concentration was
quantified by UV spectrophotometry. RT reaction was performed with
Superscript® Reverse Transcriptase (Invitrogen Life
Technologies, Darmstadt, Germany) according to the manufacturer’s
protocol. Subsequently, cDNA was amplified in a
RoboCycler® (Stratagene, San Francisco, CA, USA) using
sequence specific primers (Table
I) (Eurofins MWG Operon, Ebersberg, Germany) and Taq polymerase
(cat. no. M1245; Promega GmbH, Mannheim, Germany) with a precycle
of 4 min at 94°C and an amplification reaction of 35 cycles (94°C
for 1 min, 58°C for 1 min and 72°C for 2 min). The reaction was
terminated by 7 min at 72°C. Expression of GAPDH was used as a
control to measure the integrity of the RNA samples. To exclude DNA
contamination, purified RNA was incubated with the appropriate
primers and Taq polymerase, but without reverse transcriptase. cDNA
isolated from HUVEC and HL-60 cells was used as positive control
for all five sets of primers.
 | Table IGene specific primers for RT-PCR. |
Table I
Gene specific primers for RT-PCR.
| Gene name | Sequence
(5′→3′) | Size (bp) |
|---|
| VEGFR-1 | F: ATT TGT GAT TTT
GGC CTT GC
R: CAG GCT CAT GAA CTT GAA AGC | 555 |
| VEGFR-2 | F: GTG ACC AAC ATG
GAG TCG TG
R: CCA GAG ATT CCA TGC CAC TT | 630 |
| VEGFR-3 | F: TCC TTG TCG GTA
CCG GCG TC
R: GAG GAT CTT GAG CTC CGA CA | 368 |
| PDGFR-β | F: TGA CCA CCC AGC
CAT CCT TC
R: GAG GAG GTG TTG ACT TCA TTC | 228 |
| GAPDH | F: GCG GGG CTC TCC
AGA ACA TCA T
R: CCA GCC CCA GCG TCA AAG GTG | 301 |
Flow cytometric analysis of protein
expression
For analysis of VEGFR-1, -2 and -3, and PDGFR-β
protein expression in leiomyosarcoma, 1×105 cells were
seeded in 100 mm plates and cultured in DMEM/0.1% FCS for 12 h.
Cells were then washed twice with phosphate buffered saline (PBS)
and removed from the plate with HEPES/EDTA buffer after 20 min
incubation at 37°C. Cells were washed in PBS/3% BSA and fixed in 1%
paraformaldehyde (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany)
for 20 min at room temperature. After washing with PBS/3% BSA and
permeabilization buffer (PBS/0.5% saponin/3% BSA) cells were
incubated with the following primary antibodies diluted 1:100 in
PBS/3% BSA for 30 min at 4°C on a shaking device: mouse monoclonal
anti-VEGFR-1 antibody [Flt-1 (C-17), cat. no. sc-316], rabbit
polyclonal anti-VEGFR-3 antibody [Flt-4 (C-20), cat. no. sc-321],
rabbit anti-PDGFR-β antibody (P-20, cat. no. sc-339; all from Santa
Cruz Biotechnology, Inc., Heidelberg, Germany) or mouse monoclonal
anti-VEGFR-2 antibody (clone KDR-1, cat. no. V9134; Sigma-Aldrich
Chemie GmbH). After two washing steps with permeabilization buffer
cells were incubated with secondary phycoerythrin (PE)-coupled goat
anti-mouse or anti-rabbit Fab fragment (dilution 1:200 in
permeabilization buffer; both from Dianova GmbH, Hamburg, Germany)
for 1 h at 4°C. Control cells were stained with mouse IgG1, κ
(MOPC-21, cat. no. M5284; Sigma-Aldrich Chemie GmbH) or rabbit
(Clone DA1E; Cell Signalling Technology, Inc., Danvers, MA, USA)
isotype control antibodies for the primary antibody in combination
with the respective PE-Fab fragment. After a final washing step in
PBS cells were resuspended in 2 ml PBS and analyzed flow
cytometrically. Cells were kept in the dark during preparation.
Flow cytometric assessment of cell cycle
and events with lower than G1 DNA content
The extent of cell death was quantified by staining
of the cellular DNA content and determination of the fraction of
events with lower DNA content than G1-phase cells (sub G1
fraction). Cells (1×106) were seeded in DMEM/10% FCS and
grown for 24 h. Samples were supplied with fresh medium and then
pre-exposed to 1 μM PTK787 or DMSO (control) and subsequently
treated with VEGF or PDGF for 24, 48, and 72 h. After harvesting
with 0.05% Trypsin/0.02% EDTA (PAN-Biotech GmbH, Aidenbach,
Germany) cells were washed with PBS and 1×106 cells per
sample were fixed with 70% ethanol for 24 h at 4°C. Cells were then
washed in PBS and treated with 10 U/ml RNase (Sigma-Aldrich Chemie
GmbH) for 20 min at 37°C. Cells were next stained with propidium
iodide (PI) (final concentration: 25 μg/ml), and incubated for 15
min. The sub G1 fraction and the fraction of live cells (cells in
G1-, S-, and G2/M-phase) were determined flow cytometrically with a
flow rate of 300 events/sec.
Flow cytometric data acquisition and
analysis
Flow cytometric measurements were done with a
FACScan flow cytometer using CellQuest software (BD Biosciences,
San Jose, CA, USA). PE and PI fluorescence were exited with a 488
nm argon laser. Fluorescence emission was measured with a 585/42 nm
band pass filter (PE) or a >670 nm long pass filter (PI) and
visualized on a logarithmic scale. Data were stored as list mode
FCS2.0 files.
Ligand quantification in cell culture
supernatants
The amount of secreted human VEGF-A, PDGF-BB was
quantified in cell culture supernatants by specific ELISA kits
(R&D Systems, Wiesbaden, Germany). Leiomyosarcoma cells were
counted, plated at a 40–50% density and cultured in DMEM with
either 10, 1 or 0,1% FCS. After an incubation interval of 24 or 48
h, 1 ml cell culture supernatant was removed and analyzed according
to the manufacturer’s protocol. Protein levels are expressed as
pg/ml.
SDS-PAGE and western blotting
Tumor cells were starved for 24 h in DMEM/0.1% FCS
and then pre-incubated with 0.1, 1 and 10 μM PTK787 or DMSO to
serve as control. Subsequently, cells were stimulated with either
VEGF or PDGF for 5, 10, 20, 30, 60 min as described above. Cells
were then lysed in Laemmli buffer (Rotiphorese® 10X
SDS-PAGE cat. no. 3060.1; Carl Roth GmbH & Co. KG, Karlsruhe,
Germany) and denatured for 5 min at 95°C. Protein concentrations
were measured with the BCA Protein Assay kit (Thermo Fisher
Scientific, Bonn, Germany). Protein (20 μg) of each sample was
separated by SDS-PAGE on a 10% polyacrylamide gel in a mini gel
chamber (Peqlab Biotechnologie GmbH, Erlangen, Germany). Proteins
were transferred onto Protran nitrocellulose membranes (Schleicher
& Schuell, Dassel, Germany), probed with an antibody cocktail
(PathScan® Multiplex Western Cocktail ; Cell Signaling
Technology, Inc.) containing the following antibodies:
phospho-p90RSK (Ser380) (9D9) rabbit mAb, phospho-S6 ribosomal
protein (Ser235/236) (D57.2.2E) rabbit mAb, phospho-p44/42 MAPK
(ERK1/2) (Thr202/Tyr204) (D13.14.4W) XP® rabbit mAb,
phospho-AKT (Ser473) (D9E) XP® rabbit mAb as well as
eIF4E as protein loading control. Blots were washed, incubated with
horseradish peroxidase (HRP)-conjugated secondary antibody
(anti-rabbit IgG HRP-linked antibody, dilution 1:2,000; Cell
Signaling Technology, Inc.) for 2 h and washed again.
ECL-chemiluminescence substrate (ECL Plus Western Blotting
Detection System; GE Healthcare GmbH, Freiburg, Germany) was used
for detection. Membranes were stripped with Restore Western Blot
Stripping Buffer (Pierce/Thermo Fisher Scientific) and re-analysed
with either phospho-p38 (Thr180/Tyr182) (Clone D3F9) or rabbit mAb,
phospho-FAK (Tyr925) or phospho-paxillin (Tyr118) antibody
(dilution 1:1,000; all from Cell Signaling Technology, Inc.) using
the same protocol as described above.
MTT assay
To evaluate the effect of PTK787 on cell growth,
leiomyosarcoma cells were seeded into 96-well plates
(1×103 cells/well) on day 0 in DMEM/10% FCS. On day 1
medium was changed and cells were exposed to 0.1, 1 or 10 μM
PTK787. After incubation for 24, 48 or 72 h at 37°C cell growth was
assessed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) (Sigma-Aldrich Chemie GmbH) at a final concentration
of 0.2 mg/ml. After an incubation of 2 h, medium was removed and
cells were dissolved in acidic isopropanol (90% isopropanol, 0.5%
sodium dodecyl sulphate, 40 mM HCl). The absorbance of the coloured
solution was quantified in a spectrophotometer at 490 nm with
isopropanol as reference.
Tumor cell migration assay
To determine the inhibitory effect of 0.1, 1 and 10
μM PTK787 on leiomyosarcoma cell motility in vitro,
migration assays were performed using a modified Boyden chamber as
previously described (31).
Briefly, 1×105 cells were suspended in DMEM/1% FCS and
seeded into inserts with 8 μm filter pores (BD Biosciences,
Heidelberg, Germany). As chemoattractant 50 ng/ml VEGF-A or 10
ng/ml PDGF-BB diluted in DMEM/10% FCS were used. Control samples
were incubated in medium without growth factor addition. After 48 h
cells were fixed, migrated cells were stained (Diff-Quick reagent;
Dade Behring, Inc., Newark, DE, USA), counted under microscope in
four random fields and average cell numbers were calculated.
Immunohistochemistry of patient
samples
Leiomyosarcoma tissue samples were culled from the
tissue archives of the Institute of Pathology and Neuropathology,
University Mainz, Germany. Immediately after surgery, tissue
samples were fixed in buffered formaldehyde (4%; SG Planung,
Holzkirchen, Germany) and embedded in paraffin (Sigma-Aldrich
Chemie GmbH). The specimens were diagnosed by at least two
experienced pathologists as leiomyosarcomas and graded after the
FNCLCC grading scheme.
For immunohistochemical staining 5 μm sections were
prepared. Stains were performed using the ready-to-use,
peroxidase-based EnVision® kit (Dako, Hamburg, Germany)
according to the manufacturer’s protocol and developed with the
Avidin-Biotin Complex (ABC) method with 3-amino-9-ethylcarbazole
(AEC) or 3,3′-diaminobenzidine (DAB) staining solution,
respectively. The antibodies used are listed in Table II. The sections were
counterstained with haematoxylin and mounted with
Aquatex® (Merck, Darmstadt, Germany). In control
sections, the primary antibody was either omitted or substituted
with non-specific rabbit or mouse immunoglobulins. The specimens
were analysed by light microscopy (Zeiss Axiophot; Carl Zeiss
Microscopy GmbH, Göttingen, Germany).
| Table IIAntibody types and source used in
this study. |
Table II
Antibody types and source used in
this study.
| Antibody | Antigen | Provider | Dilution | Epitope
retrieval | Incubation | Control |
|---|
| Mouse IgG1 | VEGFR-1
Clone C-17 | Santa Cruz
Biotechnology, Inc., Heidelberg, Germany | 1:100 | 6×5 min in CB, pH
6.0 at 500 W | Overnight RT | Umbilical vein |
| Mouse IgG1 | VEGFR-2
Clone A-3 | Santa Cruz
Biotechnology, Inc., Heidelberg, Germany | 1:50 | 40 min in CB, pH
6.0 at 240 W | Overnight RT | Colon
carcinoma |
| Rabbit polyclonal
IgG | VEGFR-3
Clone C-20 | Santa Cruz
Biotechnology, Inc., Heidelberg, Germany | 1:50 | 6×5 min in CB, pH
6.0 at 500 W | Overnight RT | Colon
carcinoma
Umbilical vein |
| Rabbit monoclonal
IgG | PDGFR-β Clone
28E1 | Cell Signaling
Technology, Inc., Frankfurt, Germany | 1:50 | 6×5 min in CB, pH
6.0 at 500 W | 1 h at RT | Ovarian
carcinoma |
Results
Expression of VEGFRs and PDGFR-β in
leiomyosarcoma tissue specimens
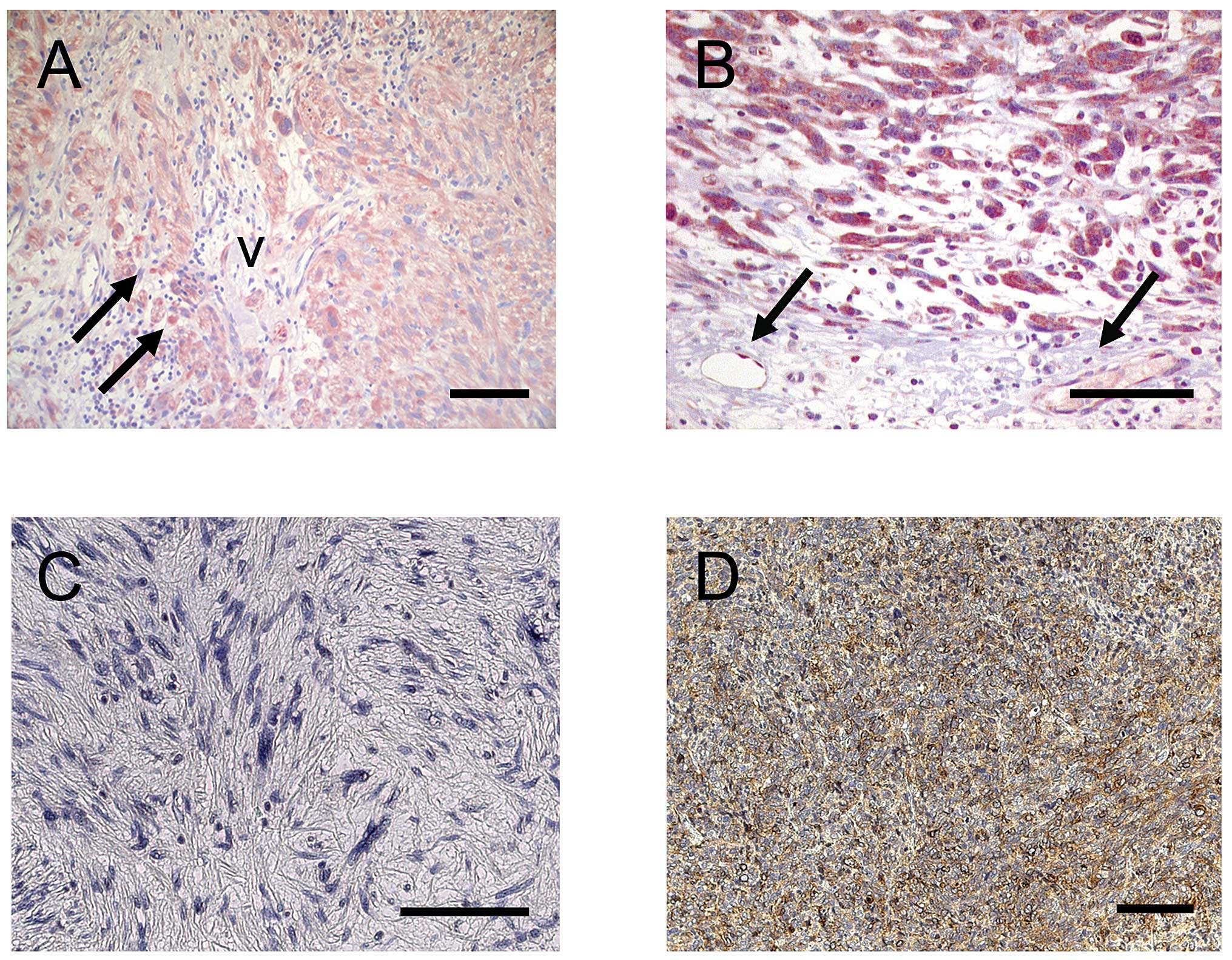
Immunohistochemical investigations showed a strong
expression for VEGFR-1/2 in the cytoplasm as well as the cell
membrane indicating a prominent protein expression in tumor cells
(Fig. 1A and B). Also vascular
endothelial cells expressed VEGFR-2 (Fig. 1B) and to a lesser extent VEGFR-1
(Fig. 1A). In addition, VEGFR-1
was present in tumor-associated macrophages (TAM) (Fig. 1A). On the other hand VEGFR-3 was
not present in sarcoma cells and could not be detected in lymphatic
vessels in our series (Fig. 1C).
In contrast to VEGFR-3, PDGFR-β was prominently expressed in the
cytoplasm as well as in the cell membrane of sarcoma cells
(Fig. 1D) emphasizing that
VEGFR/PDGFR family members play an important role in sarcoma cells.
Also PDGFR-β was expressed in perivascular cells as reported
previously (19,21,22).
Expression of VEGFR family members,
PDGFR-β and corresponding ligands in leiomyosarcoma cell lines
Positive expression of VEGFR family members and
PDGFR-β in leiomyosarcoma tissue specimen (Fig. 1) suggested further functional
studies on their potential as therapeutic targets for specific
tyrosine kinase inhibition. Since it is known that the small
molecule inhibitor PTK787 is able to sufficiently block several
RTKs (20) we first examined the
expression of VEGFR-1, -2 and -3, and PDGFR-β in the two
leiomyosarcoma cell lines SK-LMS-1 and SK-UT-1. Analysis of PCR
products revealed that VEGFR-1, -2 and -3 were detectable in both
sarcoma cell lines as well as in control HUVEC and HL-60 cells. In
addition, PDGFR-β mRNA was strongly expressed in both sarcoma cell
lines (data not shown). Since the detection of mRNA does not
necessarily predict the functional expression of the receptors, we
further assessed the protein expression at the cellular surface of
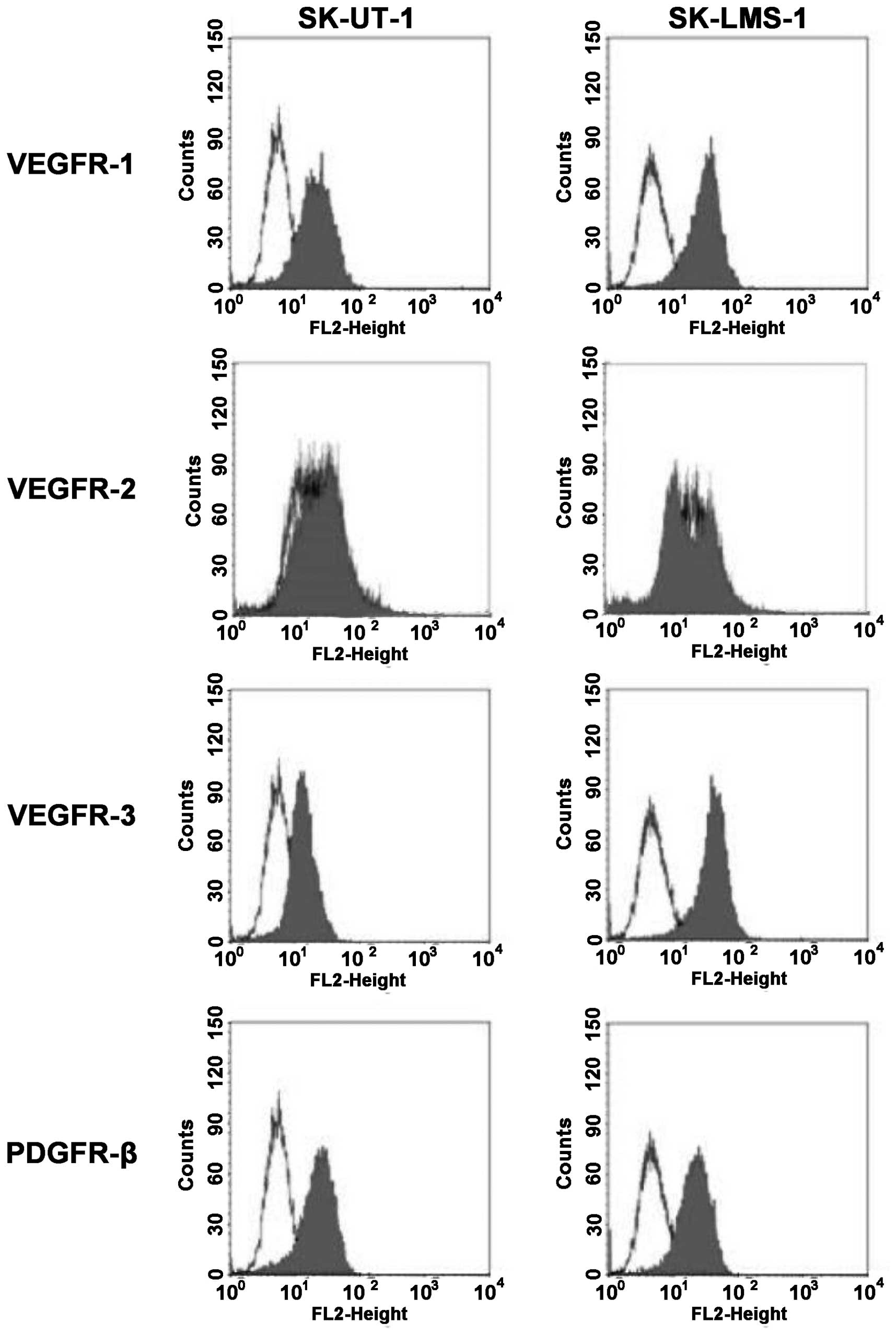
the tumor cell lines by flow cytometry (Fig. 2). We were able to show that VEGFR-1
and -3 are strongly expressed in both leiomyosarcoma cell lines
whereas VEGFR-2 staining was slightly positive in SK-UT-1 but
ambiguous in SK-LMS-1. In addition, PDGFR-β was detectable at the
cellular surface of both cell lines.
Expression of VEGF and PDGF receptor
ligands in leiomyosarcoma cell lines
Binding of the corresponding ligands causes receptor
activation and subsequent intracellular signalling. Therefore,
expression and secretion of corresponding growth factors for the
RTKs are essential for their functional activity. Thus, we
investigated the secretion of VEGF-A and PDGF-BB for both
leiomyosarcoma cell lines with specific ELISA assays. We could
detect high amounts of VEGF-A in SK-LMS-1 (1278±148.4 pg/ml after
24 h) which exceeded the detection limit after 48 h. VEGF-A
secretion in SK-UT-1 cells was 10.9-fold lower (117±6.9 pg/ml)
compared to SK-LMS-1 after 24 h and increased to 371.1±5.2 pg/ml
after 48 h. When assessing secreted PDGF-BB levels both cell lines
showed comparable amounts after 24 h (SK-UT-1: 36.97±3.6 pg/ml;
SK-LMS-1: 26.35±1.04 pg/ml) and 48 h (SK-UT-1: 38.81±12.4 pg/ml;
SK-LMS-1: 28.91±1.5 pg/ml) of cell culture.
The effect of PTK787 on RTK
signalling
We investigated whether PTK787 could interfere with
VEGF-A- or PDGF-BB-caused activation of intracellular signalling
intermediates, which are known to be involved in VEGFR and
PDGFR-signalling and regulate cell proliferation and migration. In
Fig. 3 we show representative data
gained with SK-UT-1. Results were similar for SK-LMS-1. Cells were
incubated with either 0.1, 1 or 10 μM PTK787 or DMSO and
subsequently stimulated with growth factors for different time
intervals.
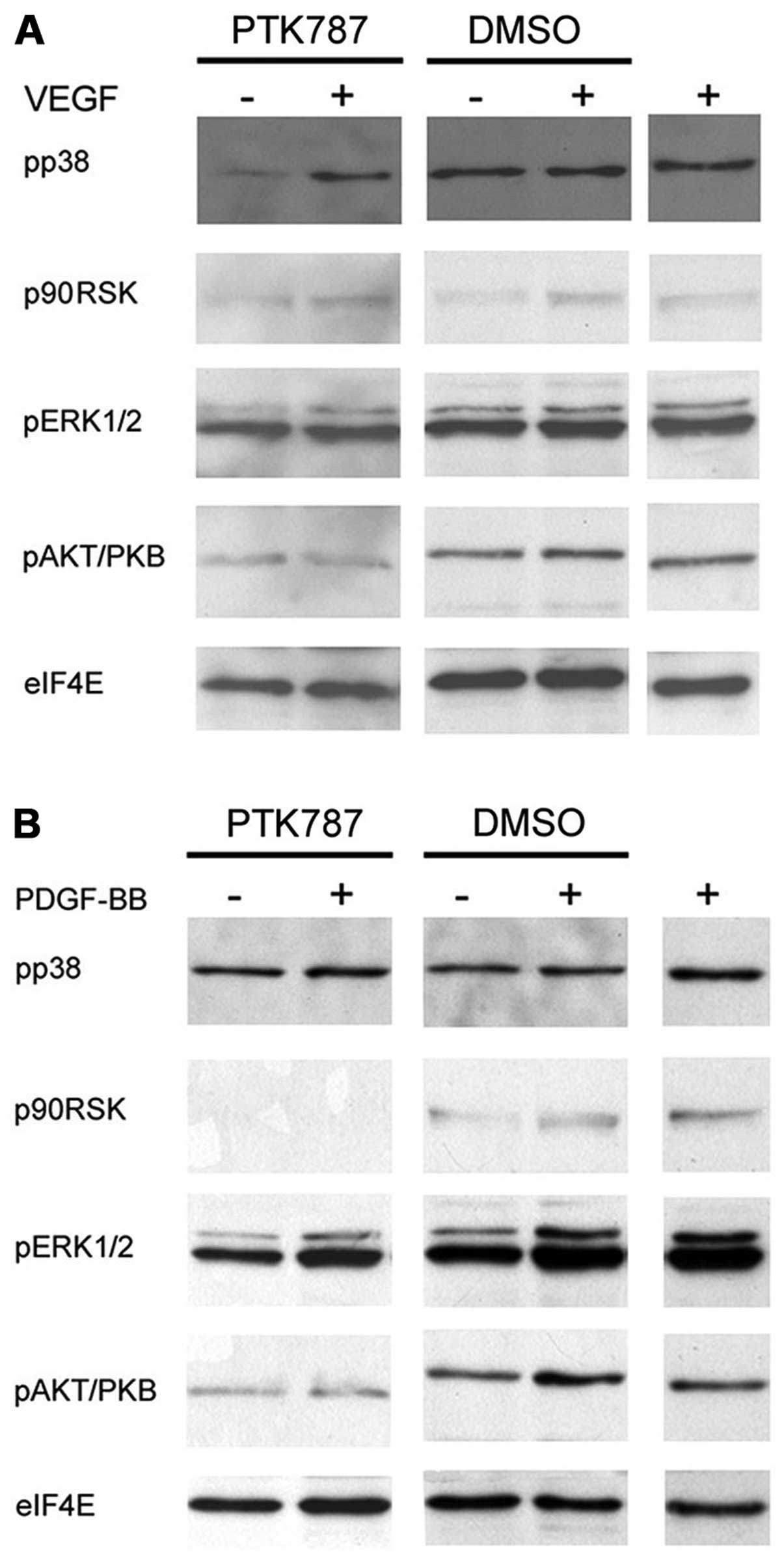
VEGF-A stimulation alone or in combination with
PTK787 did not affect phosphorylation of ERK1/2 (Fig. 3A). However, both with and without
VEGF-A treatment PTK787 seemed to slightly reduce the level of
AKT/PKB phosphorylation compared to DMSO control samples. In
addition, p38 activation seemed to be reduced upon PTK787 treatment
but was brought back to basal levels upon concomitant VEGF-A
treatment. Comparable results were obtained for 1 and 10 μM PTK787
and different incubation intervals with growth factors (data not
shown). PDGF-BB stimulation (Fig.
3B) increased the phosphorylation of AKT/PKB and ERK1/2 in DMSO
control samples. Additional PTK787 treatment reduced the level of
ERK1/2 phosphorylation in the presence and absence of PDGF-BB, but
a PDGF-BB-caused increase in ERK1/2 phosphorylation remained
stable. In line, compared to the DMSO control samples AKT/PKB
phosphorylation levels were reduced both for PTK787 treatment alone
and for combination treatment of PTK787 and PDGF-BB. Strikingly,
PTK787 seemed to completely compensate the stimulatory potential of
PDGF-BB on AKT/PKB phosphorylation. Furthermore, p90RSK
phosphorylation was abrogated by PTK787 treatment independent of
PDGF-BB stimulation.
However, concerning the phosphorylation of p38 no
difference could be seen between PTK787 treatment and DMSO alone.
Since p38 is a key regulator of cellular migration we also
investigated whether alternative signalling pathways for regulation
of migration are activated in these cell lines. However, no
suppression of either FAK or paxillin phosphorylation, two key
regulators of tumor cell migration (32,33),
could be observed for both leiomyosarcoma cell lines (data not
shown).
The effect of PTK787 on growth and
migration in leiomyosarcoma cell lines
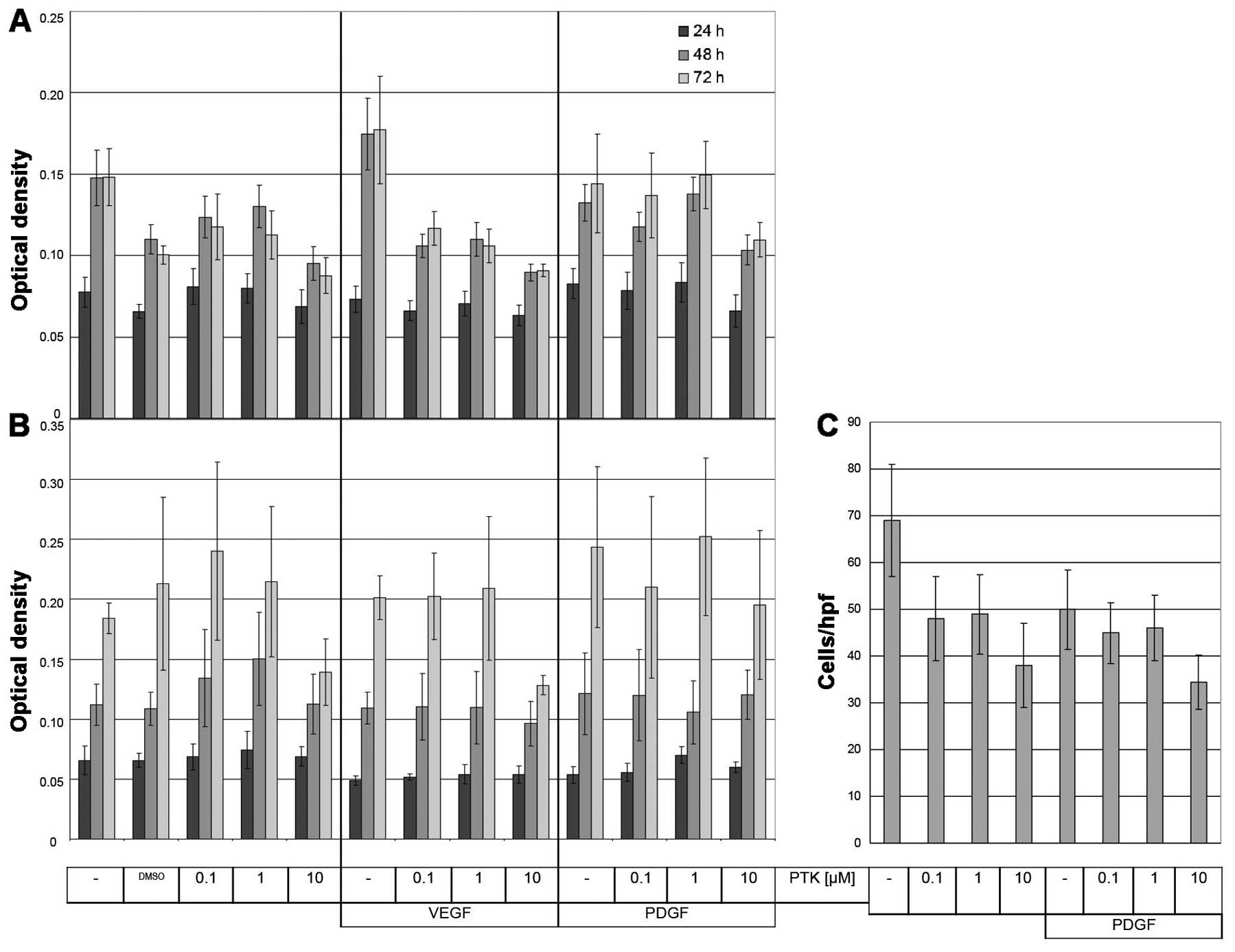
MTT assay was used to assess the cellular growth.
Different concentrations of PTK787 were applied and cells were
subsequently stimulated with VEGF-A or PDGF-BB. Even with 10 μM
PTK787 treatment SK-UT-1 and SK-LMS-1 cells did not show a
significant decrease in optical density (OD) compared to control
samples (Fig. 4A and B). A
positive effect on SK-UT-1 cell growth by VEGF-A treatment was
completely compensated by 10 μM PTK787 treatment reaching OD levels
similar to samples without VEGF-A addition (Fig. 4A). In SK-LMS-1 VEGF-A treatment had
no effect on cell growth. The presence of PDGF-BB alone or in
combination with PTK787 treatment caused no difference in OD
compared to the respective control samples (Fig. 4A and B).
The modified Boyden chamber assay was applied to
assess the effect of PTK787 on cell migration. In Fig. 4C a representative example is shown.
Although PTK787 treatment reduced SK-UT-1 cell migration when
applied alone, we could not observe a further effect upon PDGF-BB
(Fig. 4C) or VEGF-A stimulation
(data not shown). Similar results were obtained for SK-LMS-1 (data
not shown). Therefore, VEGF-A or PDGF-BB treatment does not affect
PTK787-reduced migration of SK-UT-1 and SK-LMS-1 leiomyosarcoma
cell lines.
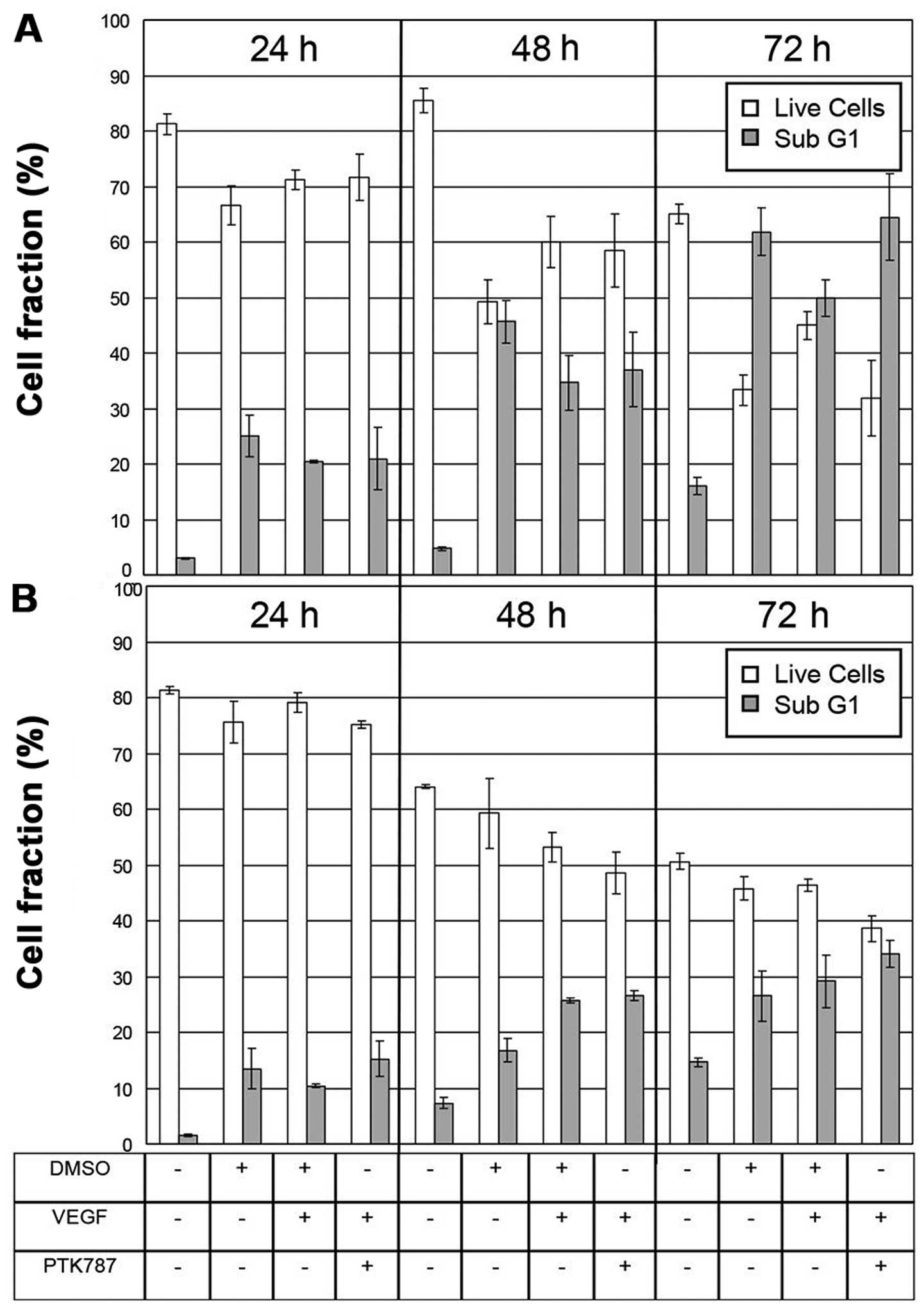
Increase in sub G1 fraction upon
PTK787/growth factor treatment
Compared to VEGF-A stimulated cells additional
PTK787 treatment increased the sub G1 fraction from 49.9±3.3% to
64.5±7.8% in SK-UT-1 (Fig. 5A) and
from 29.2±4.8% to 34.1±2.4% in SK-LMS-1 cells (Fig. 5B) after 72 h of incubation. The
fraction of live cells (defined as the sum of G1-, S- and G2/M-cell
fractions) decreased in SK-UT-1 from 45±2.5% to 31.9±6.9% and in
SK-LMS-1 from 46.4±1% to 38.6±2.4%. However, both cell lines were
insensitive to PDGF-BB treatment alone and in combination with
PTK787 (data not shown). The 48 and 72 h VEGF-A treatment seemed to
increase the fraction of live cells and decrease the number of
events with lower than G1 cell content only in SK-UT-1 (Fig. 5A) but not in SK-LMS-1 (Fig. 5B).
Discussion
Conventional therapy of leiomyosarcomas is of
limited effect (3). Therefore it
is essential to identify new potential therapeutic approaches to
improve patient outcome. Targeting the tumor and its vasculature by
specific anti-angiogenic drugs has emerged as promising tool to
disrupt the outgrowth of new blood vessels, and subsequently the
nutrient supply of tumor cells and to directly inhibit tumor growth
(4). However, in anti-angiogenic
therapy biomarkers to select responders are not available (34).
In the present study, we have demonstrated
expression of key proteins for angiogenesis in leiomyosarcoma
cells. Strong expression of VEGFR-1, -2 and PDGFR-β in tumor and
endothelial cells (Fig. 1) may
thus represent the prerequisite for response to inhibition with the
multi-targeting anti-angiogenic small molecule inhibitor PTK787.
Furthermore, prominent expression of PDGFR-β in perivascular
cells/pericytes (Fig. 1D) may
represent a complimentary target for efficacious anti-angiogenic
therapy by causing pericyte detachment, resulting in immature
vessels that are prone to regression (19). The availability of PTK787 target
proteins in patient tissue led us to investigate the role and
function of PTK787 in a leiomyosarcoma cell culture model to
outline the potential of PTK787 for therapy of leiomyosarcoma
patients.
We confirmed concomitant expression of angiogenic
receptors (VEGFR-1, -2, -3, PDGFR-β, data not shown) and the
corresponding ligands (VEGF-A, PDGF-BB) in leiomyosarcoma cell
lines SK-UT-1 and SK-LMS-1. In other tumor cell lines it was
previously shown that the VEGF/VEGFR system represents an autocrine
stimulatory unit (7). Therefore,
we investigated the cellular effects of inhibition of VEGFRs with
PTK787 upon stimulation with VEGF-A and PDGF-BB.
Our data indicate that upon VEGFR stimulation with
VEGF-A the growth-inhibitory effects of PTK787 are predominantly
achieved through induction of cell death. This observation is in
agreement with other studies that showed an increase in apoptotic
cell death upon PTK787 treatment in chronic lymphocytic leukemia
(5) and upon PTK787 addition to
IFN/5-FU therapy or hypoxia in hepatocellular carcinoma cell lines
(35,36).
PTK787 does not display absolute selectivity for the
VEGFRs but also blocks the activity of, e.g., PDGFR-β at higher
concentrations (20). Despite
prominent expression of PDGFRs in SK-UT-1 and SK-LMS-1 (data not
shown) cell death was not affected by PTK787 treatment in PDGF-BB
activated cells (Fig. 5). However,
this finding was accompanied by PDGF-BB-caused phosphorylation of
AKT/PKB (cell survival pathway) and ERK1/2 (cell proliferation
pathway) and a reversion of these phosphorylation events by PTK787
treatment (Fig. 3). Therefore, we
provide evidence that the antitumor efficiency of PTK787 may not
only be mediated by AKT-related pathways regulating cell survival
(36) but also by affecting cell
proliferation (35) via ERK1/2
signalling. In addition, our study emphasizes that PTK787
effectively counteracts PDGF-BB-induced signalling in tumor cells
despite a relatively low inhibitory effect for PDGFRs
[IC50=580 nM vs. VEGFR-1 IC50, 77 nM; VEGFR-2
IC50, 37 nM (24)].
Therefore, the expression of PDGFRs in leiomyosarcoma cells is
likely to significantly participate in tumorigenesis.
However, the lack of induction of cell death upon
PDGF-BB/PTK787 treatment raises the intriguing possibilities: I)
that the level of PTK787-caused inhibition of cell signalling is
not sufficient to result in a significant cellular response; and/or
II) that further PDGF-BB-activated signalling cascades are involved
in compensation pathways. Such a compensation mechanism or switch
may significantly contribute to therapy resistance, which might be
counteracted by a combination of anti-angiogenic drugs with
conventional or further target-specific treatment (20,35,37).
The lack of VEGF-A-caused effects on phosphorylation
of signalling proteins (Fig. 3A)
may at least in part be due to a high level of VEGF-A secretion
particularly in SK-LMS-1 cells, which probably results in autocrine
activation of VEGFR kinase activity and thereby interferes with an
effective exogenous supplementation with VEGF-A. Only in VEGF-A low
expressing SK-UT-1 cells, VEGF-A treatment resulted in an increase
in cell growth (Fig. 4A) and in
the number of live cells and in decreased cell death (Fig. 5A). Similarly, a mitogenic response
to exogenous VEGF has been shown in different tumor entities, e.g.,
pancreatic carcinoma, chorioncarcinoma and melanoma (11–13).
The VEGF-A-caused increase in cell growth was
reversed by PTK787 to basal levels (Fig. 4A). Furthermore, other studies
showed that exogenous VEGF-A compensates a reduction in cell growth
caused by VEGFR inhibition with neutralizing antibodies or VEGF
ablation with oligonucleotides (7). However, PTK787 treatment of
VEGF-A-stimulated SK-UT-1 and SK-LMS-1 did not affect signalling
proteins studied herein. Our observations contrast in part with
those of previous publications, where PTK787 inhibited VEGF-induced
ERK-phosphorylation and cell proliferation of multiple myeloma cell
lines (6) and in Chinese hamster
ovary cells (25). Other studies
showed in hepatocellular carcinoma cell lines that PTK787 treatment
alone reduced AKT-phosphorylation, Cyclin D1 and anti-apoptotic
Bcl-2 protein expression, which correlated with cell cycle
retardation/arrest and reduced cell growth (36). Further studies have to reveal the
signalling pathways involved in reverting the VEGF-A-caused
increase in cell growth by PTK787 in leiomyosarcoma cell lines.
Similarly, the compensating mechanisms that prevent PDGF-BB-treated
cells from PTK787-caused cell death despite an efficient inhibition
of key proteins in survival pathways (AKT/PKB and p90RSK) have to
be further investigated.
Signalling pathways responsible for cell migration
were impaired by PTK787 (p38) but not in combination with growth
factors (p38 and FAK/paxillin), which correlated with the lack of
PTK787 activity on cell migration of VEGF-A- or PDGF-BB-treated
cells in Boyden chamber assays. However, in multiple myeloma cell
lines PTK787 blocks VEGF-caused cell migration at a concentration
of 1 μM (6). In hepatocellular
carcinoma cell lines PTK787 treatment reduced expression of
migration-related proteins Rac1 and Rho and significantly inhibited
cell migration at higher concentrations than studied herein (>20
μM). In addition, cell migration of human leukemic cells was
inhibited by anti-VEGFR-1 antibody and VEGFR-2 neutralizing
antibody IMC-1C11 suggesting that both VEGFR-1 and -2 take part in
regulation of migration (8).
However, other authors provide evidence that mainly VEGFR-1 is
responsible for regulation of cell migration (38) whereas VEGFR-2 mediates mitogenic
signalling, growth and survival. In the present study we found
prominent expression of VEGFR-1/-2 in SK-UT-1 and of VEGFR-1 in
SK-LMS-1 cell lines, which seemed to be sufficient for inhibition
of cell migration by PTK787 (Fig.
4C). However, activation of cell signalling via the
VEGFR/VEGF-A or PDGFR-β/PDGF-BB system as well as concomitant
PTK787-treatement was shown to be insufficient in effectively
reduce migration of leiomyosarcoma cell lines.
In summary, we have shown that both leiomyosarcoma
cell lines and patient leiomyosarcoma specimens express members of
the VEGFR and PDGFR tyrosine kinase family and their cognate
ligands VEGF-A and PDGF-BB that are the key players in angiogenesis
for providing tumor nutrient supply. The VEGFR low-molecular weight
inhibitor PTK787 has limited impact on leiomyosarcoma cell lines in
terms of inhibition of signalling pathways responsible for cell
proliferation and cell survival resulting in an induction of cell
death. These observations support the notion that anti-angiogenic
therapy with PTK787 may be a new therapeutic option for
leiomyosarcoma patients with positive expression of PTK787 target
molecules. However, compared to blocking angiogenesis by other
anti-angiogenic drugs, e.g., bevacizumab, the addition of PTK787 to
chemotherapy was less effective in clinical trials (14). On the other hand, two recent phase
III clinical studies suggested that a high serum lactate
dehydrogenase (LH) level might be useful as a predictive marker for
response to PTK787 treatment (18,28).
Further in vitro, in vivo and clinical studies are
needed to reveal the involvement of PTK787 target proteins and
potential predictive markers for response to treatment. The
expression level and interplay of angiogenic growth factor
receptors and their cognate ligands in tumor cells, the surrounding
endothelial cells and perivascular cells/pericytes have to be taken
into consideration offering new strategies to overcome drug
resistance by target-specific anticancer therapy.
Acknowledgements
The authors wish to acknowledge the excellent
technical support of Ursula Hofmann, Cornelia Michel, and Martina
Waeber.
References
|
1
|
García-Martínez E, Egea Prefasi L,
García-Donas J, Escolar-Pérez PP, Pastor F and González-Martín A:
Current management of uterine sarcomas. Clin Transl Oncol.
13:307–314. 2011.
|
|
2
|
Gaumann AK, Schermutzki G, Mentzel T,
Kirkpatrick CJ, Kriegsmann JB and Konerding MA: Microvessel density
and VEGF/VEGF receptor status and their role in sarcomas of the
pulmonary artery. Oncol Rep. 19:309–318. 2008.PubMed/NCBI
|
|
3
|
Maki RG: Gemcitabine and docetaxel in
metastatic sarcoma: past, present, and future. Oncologist.
12:999–1006. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Paesler J, Gehrke I, Gandhirajan RK, et
al: The vascular endothelial growth factor receptor tyrosine kinase
inhibitors vatalanib and pazopanib potently induce apoptosis in
chronic lymphocytic leukemia cells in vitro and in vivo. Clin
Cancer Res. 16:3390–3398. 2010. View Article : Google Scholar
|
|
6
|
Lin B, Podar K, Gupta D, et al: The
vascular endothelial growth factor receptor tyrosine kinase
inhibitor PTK787/ZK222584 inhibits growth and migration of multiple
myeloma cells in the bone marrow microenvironment. Cancer Res.
62:5019–5026. 2002.PubMed/NCBI
|
|
7
|
Masood R, Cai J, Zheng T, Smith DL, Hinton
DR and Gill PS: Vascular endothelial growth factor VEGF is an
autocrine growth factor for VEGF receptor-positive human tumors.
Blood. 98:1904–1913. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dias S, Hattori K, Zhu Z, et al: Autocrine
stimulation of VEGFR-2 activates human leukemic cell growth and
migration. J Clin Invest. 106:511–521. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Podar K, Tai YT, Davies FE, et al:
Vascular endothelial growth factor triggers signalling cascades
mediating multiple myeloma cell growth and migration. Blood.
98:428–435. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Volm M, Koomagi R and Mattern J: Vascular
endothelial growth factor and basic fibroblast growth factor in
primary lung carcinomas and the incidence of metastases. Int J
Oncol. 9:711–714. 1996.PubMed/NCBI
|
|
11
|
Itakura J, Ishiwata T, Shen B, Kornmann M
and Korc M: Concomitant over-expression of vascular endothelial
growth factor and its receptors in pancreatic cancer. Int J Cancer.
85:27–34. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Charnock-Jones DS, Sharkey AM, Boocock CA,
Ahmed A, Plevin R, Ferrara N and Smith SK: Vascular endothelial
growth factor receptor localization and activation in human
trophoblast and choriocarcinoma cells. Biol Reprod. 51:524–530.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu B, Earl HM, Baban D, Shoaibi M, Fabra
A, Kerr DJ and Seymour LW: Melanoma cell lines express VEGF
receptor KDR and respond to exogenously added VEGF. Biochem Biophys
Res Commun. 217:721–727. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Los M, Roodhart JM and Voest EE: Target
practice: lessons from phase III trials with bevacizumab and
vatalanib in the treatment of advanced colorectal cancer.
Oncologist. 12:443–450. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Escudier B, Eisen T, Stadler WM, et al:
Sorafenib for treatment of renal cell carcinoma: Final efficacy and
safety results of the phase III treatment approaches in renal
cancer global evaluation trial. J Clin Oncol. 27:3312–3318. 2009.
View Article : Google Scholar
|
|
16
|
Rini BI, Escudier B, Tomczak P, et al:
Comparative effectiveness of axitinib versus sorafenib in advanced
renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet.
378:1931–1939. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Llovet JM, Ricci S, Mazzaferro V, et al:
Sorafenib in advanced hepatocellular carcinoma. N Engl J Med.
359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hecht JR, Trarbach T, Hainsworth JD, et
al: Randomized, placebo-controlled, phase III study of first-line
oxaliplatin-based chemotherapy plus PTK787/ZK 222584, an oral
vascular endothelial growth factor receptor inhibitor, in patients
with metastatic colorectal adenocarcinoma. J Clin Oncol.
29:1997–2003. 2011. View Article : Google Scholar
|
|
19
|
Bergers G, Song S, Meyer-Morse N,
Bergsland E and Hanahan D: Benefits of targeting both pericytes and
endothelial cells in the tumor vasculature with kinase inhibitors.
J Clin Invest. 111:1287–1295. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wood JM, Bold G, Buchdunger E, et al:
PTK787/ZK 222584, a novel and potent inhibitor of vascular
endothelial growth factor receptor tyrosine kinases, impairs
vascular endothelial growth factor-induced responses and tumor
growth after oral administration. Cancer Res. 60:2178–2189.
2000.
|
|
21
|
Hasumi Y, Kłosowska-Wardega A, Furuhashi
M, Ostman A, Heldin CH and Hellberg C: Identification of a subset
of pericytes that respond to combination therapy targeting PDGF and
VEGF signalling. Int J Cancer. 121:2606–2614. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Erber R, Thurnher A, Katsen AD, et al:
Combined inhibition of VEGF and PDGF signalling enforces tumor
vessel regression by interfering with pericyte-mediated endothelial
cell survival mechanisms. FASEB J. 18:338–340. 2004.
|
|
23
|
De Bock K, Mazzone M and Carmeliet P:
Antiangiogenic therapy, hypoxia, and metastasis: risky liaisons, or
not? Nat Rev Clin Oncol. 8:393–404. 2011.
|
|
24
|
Drevs J, Müller-Driver R, Wittig C, et al:
PTK787/ZK 222584, a specific vascular endothelial growth
factor-receptor tyrosine kinase inhibitor, affects the anatomy of
the tumor vascular bed and the functional vascular properties as
detected by dynamic enhanced magnetic resonance imaging. Cancer
Res. 62:4015–4022. 2002.
|
|
25
|
Drevs J, Hofmann I, Hugenschmidt H, et al:
Effects of PTK787/ZK 222584, a specific inhibitor of vascular
endothelial growth factor receptor tyrosine kinases, on primary
tumor, metastasis, vessel density, and blood flow in a murine renal
cell carcinoma model. Cancer Res. 60:4819–4824. 2000.
|
|
26
|
Schomber T, Zumsteg A, Strittmatter K, et
al: Differential effects of the vascular endothelial growth factor
receptor inhibitor PTK787/ZK222584 on tumor angiogenesis and tumor
lymphangiogenesis. Mol Cancer Ther. 8:55–63. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gauler TC, Besse B, Mauguen A, et al:
Phase II trial of PTK787/ZK 222584 vatalanib administered orally
once-daily or in two divided daily doses as second-line monotherapy
in relapsed or progressing patients with stage IIIB/IV
non-small-cell lung cancer (NSCLC). Ann Oncol. 23:678–687. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Van Cutsem E, Bajetta E, Valle J, et al:
Randomized, placebo-controlled, phase III study of oxaliplatin,
fluorouracil, and leucovorin with or without PTK787/ZK 222584 in
patients with previously treated metastatic colorectal
adenocarcinoma. J Clin Oncol. 29:2004–2010. 2011.
|
|
29
|
Hewett PW and Murray JC: Isolation of
microvascular endothelial cells using magnetic beads coated with
anti-PECAM-1 antibodies. In Vitro Cell Dev Biol Anim. 32:4621996.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jaffe EA, Nachman RL, Becker CG and Minick
CR: Culture of human endothelial cells derived from umbilical
veins. Identification by morphologic and immunologic criteria. J
Clin Invest. 52:2745–2756. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bauer TW, Fan F, Liu W, et al: Targeting
of insulin-like growth factor-I receptor with a monoclonal antibody
inhibits growth of hepatic metastases from human colon carcinoma in
mice. Ann Surg Oncol. 14:2838–2846. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao X and Guan JL: Focal adhesion kinase
and its signalling pathways in cell migration and angiogenesis. Adv
Drug Deliv Rev. 63:610–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Deakin NO and Turner CE: Paxillin comes of
age. J Cell Sci. 121:2435–2444. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jain RK, Duda DG, Willett CG, et al:
Biomarkers of response and resistance to antiangiogenic therapy.
Nat Rev Clin Oncol. 6:327–338. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Murakami M, Kobayashi S, Marubashi S, et
al: Tyrosine kinase inhibitor PTK/ZK enhances the antitumor effects
of interferon-α/5-fluorouracil therapy for hepatocellular carcinoma
cells. Ann Surg Oncol. 18:589–596. 2011.PubMed/NCBI
|
|
36
|
Yang ZF, Poon RT, Liu Y, et al: High doses
of tyrosine kinase inhibitor PTK787 enhance the efficacy of
ischemic hypoxia for the treatment of hepatocellular carcinoma:
dual effects on cancer cell and angiogenesis. Mol Cancer Ther.
5:2261–2270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Riesterer O, Oehler-Jänne C, Jochum W,
Broggini-Tenzer A, Vuong V and Pruschy M: Ionizing radiation and
inhibition of angiogenesis in a spontaneous mammary carcinoma and
in a syngenic heterotopic allograft tumor model: a comparative
study. Radiat Oncol. 6:662011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Barleon B, Sozzani S, Zhou D, Weich HA,
Mantovani A and Marmé D: Migration of human monocytes in response
to vascular endothelial growth factor (VEGF) is mediated via the
VEGF receptor flt-1. Blood. 87:3336–3343. 1996.PubMed/NCBI
|