Introduction
The tumor suppressor p53 plays a key role in
regulation of the cell cycle, apoptosis, DNA repair, and senescence
in response to various acute stresses, such as DNA damage, hypoxia,
changes in redox potential and abnormal expression of oncogenes
(1,2). Activation of p53 after DNA damage
affects genes associated with cell cycle arrest, DNA repair and
apoptosis, including p21, Bax, PUMA and murine double minute 2
(MDM2) (3,4). In addition to its role as a
transcriptional activating protein, p53 also serves as a negative
transcription factor, downregulating the expression of genes, such
as the anti-apoptotic gene bcl-2 (5).
The regulation of p53 activity is mostly through
post-translational modification. Stabilization is an essential step
for efficient functioning of p53 in response to cellular stresses.
Under physiological conditions, p53, in most cells, is expressed at
a low or undetectable level, with a half-life of a few minutes.
This rapid degradation is, at least in part, mediated by the
ubiquitination pathway following the interaction of MDM2 with the
N-terminus of p53. Under a condition of stress, such as DNA damage
caused by ionizing radiation or cytotoxic agents, endogenous p53 is
stabilized through a series of physiological responses, including
ataxia telangiectasia mutated (ATM)/ATM and Rad3-related (ATR)
activation (6,7), phosphorylation and acetylation of
p53, and weakening of the binding of MDM2 to p53.
Because of the strong possibility of p53 eliciting
apoptosis or growth arrest in cells, pharmacological reactivation
of the p53 tumor suppressor is a promising strategy for anticancer
therapy. Previous studies showed that some small compounds induce
cancer cell cycle arrest and apoptosis through restoration of the
p53 pathway (8,9). Some other molecules were also
identified that induce the activity of p53, which effectively
represses angiogenesis (10) and
the growth of xenograft tumors (11).
Pyrimidine derivatives are well known for their
pharmacological activities. Various drugs containing a pyrimidine
nucleus are synthesized and used as anticancer drugs; these include
5-fluorouracil (5-FU), tegafur and thioguanine (12). Other pyrimidine derivatives, such
as 2-cyanopyrimidines (13),
hydrazinopyrimidine-5-carbonitriles (14), and some series of
N-(2-(trifluoromethyl)pyridin-4-yl) anthranilic acids, show
antiproliferative activity against human cancer cells (15). 5-FU is the optimal choice of all
chemotherapeutic agents when treating patients with colorectal
cancer. Intracellular metabolites of 5-FU can exert cytotoxic
effects via inhibition of thymidylate synthetase or incorporation
into DNA or RNA, events that ultimately activate apoptosis
(16).
We sought to develop a new agent capable of
activating p53 that could be used as an anticancer agent (17). In our process, we screened a
chemical-compound library for p53 activators and identified
N-[2-(dimethylamino)ethyl]-2,3-dimethyl-4-oxo-4H-pyrido[1,2-a]thieno[2,3-d]pyrimidine-9-carboxamide
(PTP), which significantly increased p53 reporter activity. In the
present study, we identified PTP as a novel activator of the p53
response and characterized its antitumor mechanism in human
colorectal cancer cells.
Materials and methods
Cells and reagents
The human colorectal adenocarcinoma cancer cell
lines HCT116 and HT-29 were obtained from the Korean Cell Line Bank
(KCLB, Seoul, Korea). All cells were maintained in RPMI-1640 medium
(PAA Laboratories, Pasching, Austria) supplemented with 10%
heat-inactivated fetal bovine serum (PAA Laboratories) and 100 U/ml
penicillin/streptomycin. PTP was obtained from ChemBridge (San
Diego, CA, USA). U-0126 was purchased from Assay Designs (Ann
Arbor, MI, USA); anacardic acid was from Santa Cruz Biotechnology
(Santa Cruz, CA, USA).
Luciferase assay
HCT116 cells were transfected with the p53-Luc
reporter vector using Lipofectamine 2000 (Invitrogen, Carlsbad, CA,
USA) according to the manufacturer’s instructions, and stably
transfected cell lines were established by selection in medium
containing G418 (1 mg/ml). The cells were inoculated into duplicate
96-well plates at 15,000 cells/well, incubated for 24 h, and then
treated for an additional 24 h with each of the 7920 small-molecule
compounds at a final concentration of 10 μM. The cells were then
washed with phosphate-buffered saline (PBS) and lysed in 100 μl
cell lysis buffer (Promega, Madison, WI, USA), after which
p53-dependent luciferase activity was determined using a Luciferase
Assay kit (Promega).
RNA isolation and real-time PCR
Total RNA was isolated using the Hybrid-R Total RNA
purification kit (GeneAll, Seoul, Korea). One microgram of total
RNA was reverse transcribed with PrimeScript RT Master Mix (Takara
Bio, Shiga, Japan). After 1:10 dilution, 2 μl cDNA was used as
template in a 20-μl PCR reaction mixture. Real-time PCR was
performed using qPCR SYBR-Green Master Mix (MBiotech, Seoul, Korea)
on a CFX96 Real-Time PCR Detection system (Bio-Rad Laboratories,
Hercules, CA, USA). The following primers were used for real-time
PCR: p21 (5′-CTGCGCCAGCTGAGGTGTGAG-3′ and
5′-GCCGCATGGGTTCTGACGGA-3′); PUMA (5′-CCTGGAGGGTCCTGTACAATCT-3′ and
5′-GCACCTAATTGGGCTCCATCT-3′); CYPA (5′-CCCACCGTGTTCTTCGACAT-3′ and
5′-CCAGTGCTCAGAGCACGAAA-3′); RPL13A (5′-CCTGGAGGAGAAGAGGAAAGAGA-3′
and 5′-TTGAGGACCTCTGTGTATTTGTCAA-3′). The relative quantification
of gene expression was carried out using the ΔΔCt method and CFX
Manager Software (Bio-Rad Laboratories) with multiple reference
genes (CYPA and RPL13A). The PCR program was as follows; initial
denaturation at 95°C for 10 min followed by 45 cycles, of 95°C for
5 sec and 60°C for 30 sec. The last amplification cycle was
followed by a melt curve analysis to confirm the specificity of the
PCR amplification.
Fluorescence-activated cell sorting
(FACS) analysis
The cells were harvested by trypsinization, fixed by
overnight incubation in 70% ethanol at −20°C, stained with
propidium iodide (50 μg/ml) containing 50 μg/ml RNase A
(Sigma-Aldrich, St. Louis, MO, USA), and the cell death and cell
cycle profile was analyzed by flow cytometry. Cell death was
measured as the percentage of cells in the sub-G1 population.
Senescence-associated (SA)
β-galactosidase assay
HCT116 cells (5×104) were plated in 35-mm
culture dishes and treated with 0, 5 and 10 μM PTP for 6 days.
Cells were fixed in 2% formaldehyde/0.2% glutaraldehyde for 5 min
at room temperature, and SA β-galactosidase staining was performed
according to the manufacturer’s instructions (Cell Signaling
Technology, Danvers, MA, USA).
Cell viability and proliferation
assay
HCT116 and HT-29 cells (2×103) were
plated in 96-well plates and allowed to attach for 24 h prior to
treatment. The cells were exposed to various concentrations of PTP
for 48 h, and cell viability was evaluated using the Ez-Cytox Cell
viability, Proliferation & Cytotoxicity Assay kit (Daeil Lab
Service, Seoul, Korea) according to the manufacturer’s
instructions.
Immunofluorescence
Cells cultured on 18×18-mm cover glasses were fixed
in 4% paraformaldehyde for 10 min, permeabilized with 1% Triton
X-100 for 5 min, and then blocked in PBS containing 10% FBS for 30
min. Cells were incubated with anti-γ-H2AX antibody (Millipore,
Billerica, MA, USA) overnight at 4°C and then with FITC-conjugated
secondary antibodies (Invitrogen) for 1 h at room temperature.
After counterstaining with 4′,6-diamidino-2-phenylindole (DAPI),
immunofluorescence images were captured with a laser-scanning
confocal microscope (LSM710; Carl Zeiss MicroImaging, Jena,
Germany).
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay
(RIPA) buffer [50 mM Tris-Cl (pH 8.0), 150 mM NaCl, 0.1% SDS, 0.5%
deoxycholic acid, 1% NP-40], and protease inhibitor and phosphatase
inhibitor cocktail and briefly sonicated. Protein content was
measured using the Coomassie (Bradford) Protein Assay kit (Thermo
Fisher Scientific, Rockford, IL, USA) and equal amounts of cell
lysates were separated on SDS-polyacrylamide gels and transferred
to nitrocellulose membranes (Bio-Rad Laboratories). Membranes were
immunoblotted with antibodies against p53, p21, cyclin D1, cyclin
E, cyclin A, p300, β-actin (Santa Cruz Biotechnology), phospho-p53
(Ser15), acetyl-p53 (Lys382), phospho-Chk2 (Thr68), Erk1/2,
phospho-Erk1/2, phospho-JNK, phospho-p38 (Cell Signaling
Technology), phospho-ATM (Ser1981), Chk2, γ-H2AX (Ser139)
(Millipore), ATM (Epitomics, Burlingame, CA, USA) and
chemiluminescence was detected using ECL detection reagents.
Small interfering RNAs (siRNAs)
The siRNA duplexes with the following sequences were
synthesized by Genolution Pharmaceuticals, Inc. (Seoul, Korea):
Erk1 (5′-UUAGAGAGCAUCUCAGCCAGAAUGC-3′), Erk2
(5′-AAGAGGAUUGAAGUAGAACAGdTdT-3′), and p300
(5′-AACCCCUCCUCUUCAGCACCAdTdT-3′). Cells in 60-mm culture plates
were transfected with 20 nM siRNA oligonucleotides using
Lipofectamine RNAiMAX reagent (Invitrogen) according to the
manufacturer’s instructions.
In situ proximity ligation assay
(PLA)
In situ PLA was performed according to the
manufacturer’s instructions (Olink Bioscience, Uppsala, Sweden).
Briefly, cells cultured on 18 × 18-mm cover glasses were fixed in
4% paraformaldehyde for 10 min, permeabilized with 1% Triton X-100
for 5 min, and then blocked for 30 min at 37°C. Cells were
incubated with anti-p53 antibody (Santa Cruz Biotechnology)
together with anti-MDM2, anti-p300, anti-histone deacetylase
(HDAC)1 (Santa Cruz Biotechnology), or anti-sirtuin (SIRT)1
antibody (Millipore) overnight at 4°C. After removal of unbound
primary antibodies, cells were incubated with proximity probes
(anti-rabbit PLUS and anti-mouse MINUS) (Olink Bioscience) for 1 h
at 37°C. The ligation reaction was performed for 30 min at 37°C
followed by polymerization reaction for 2 h at 37°C. The cells were
counterstained with DAPI, and immunofluorescence images were
captured with a laser-scanning confocal microscope.
Results
PTP increases p53 luciferase
activity
The tumor suppressor p53 is involved in multiple
cellular processes and is a good molecular target for cancer
therapy. To identify novel chemotherapeutic agents that activate
the p53 response, a p53-dependent promoter-luciferase reporter
system was developed. Small-molecule compounds in a chemically
diverse library, dissolved in DMSO at 10 μM, were assayed in a
96-well format in duplicate in order to measure p53-dependent
transactivation. In an initial screen using a luciferase assay, we
selected 26 of the 7920 tested molecules on the basis of a
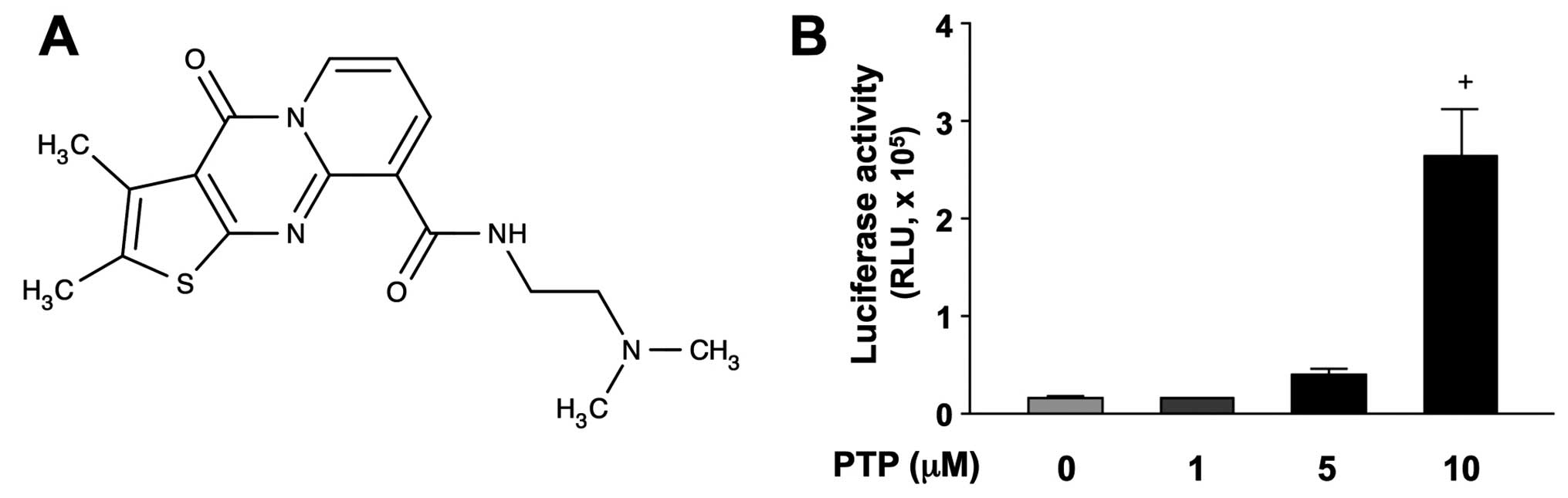
>2-fold increase in activity. One of the positive compounds,
N-[2-(dimethylamino)ethyl]-2,3-dimethyl-4-oxo-4H-pyrido[
1,2-a]thieno[2,3-d]pyrimidine-9-carboxamide (PTP), structure of
which is shown in Fig. 1A,
significantly increased p53-luciferase activity in HCT116 cells in
a concentration-dependent manner (Fig.
1B).
PTP increases wild-type p53 protein and
induces p53 target gene expression
To verify the effect of PTP on p53 activation in
vivo, two colon cancer cell lines were treated with PTP. As
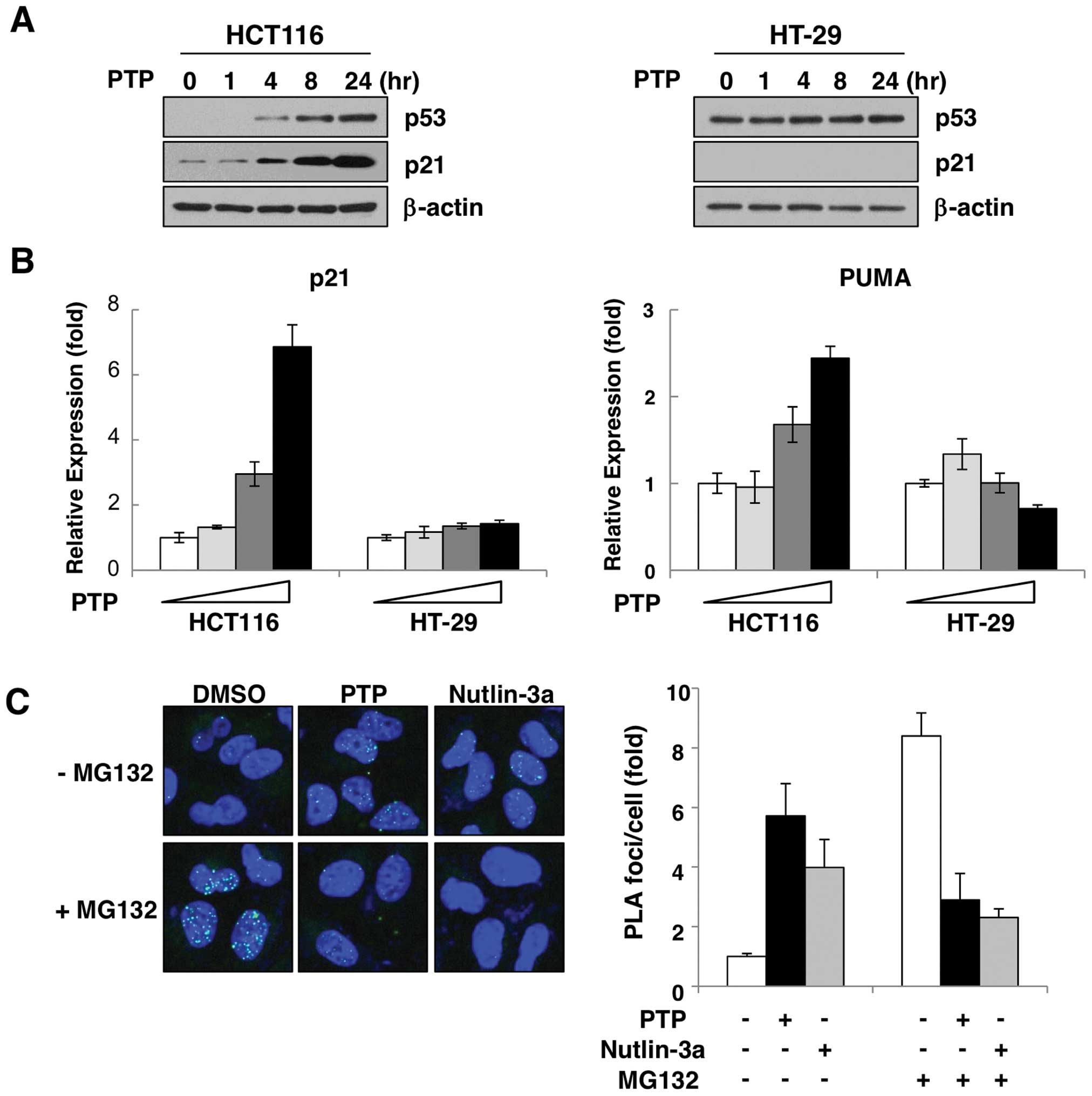
shown in Fig. 2A, treatment with
10 μM PTP increased the p53 protein level in a time-dependent
manner in HCT116 cells expressing wild-type p53. To confirm the
activity of induced p53, we examined the expression of the
p53-target genes, p21 and PUMA. Both transcript and protein levels
of p21 were increased by PTP in a time- (Fig. 2A) and dose-dependent manner
(Fig. 2B). Also, the level of PUMA
mRNA was increased dose-dependently (Fig. 2B). However, in HT-29 cells that
harbor p53 mutation, PTP did not affect either the protein level of
p53 or the induction of p21 or PUMA (Fig. 2A and B). The p53 protein is
maintained at a very low level in unstressed cells, and this level
is determined by its rate of degradation rather than transcription
or translation. This degradation is ensured by negative feedback
regulation due to an ubiquitin ligase, MDM2 (18,19).
To determine the cause of the PTP-mediated p53 accumulation, we
investigated the interaction of p53 and MDM2 in PTP-treated HCT116
cells by in situ PLA. As Fig.
2C shows, in the presence of MG132, an inhibitor of proteasomal
degradation, interaction between p53 and MDM2 was increased by
~8.2–11.9-fold compared with untreated cells. However, this
increase of p53-MDM2 interaction was suppressed in cells treated
with PTP or the MDM2 antagonist nutlin-3a. These results suggest
that PTP-induced p53 accumulation is due to suppression of p53-MDM2
interaction.
PTP induces p53-dependent G1-phase cell
cycle arrest and senescence
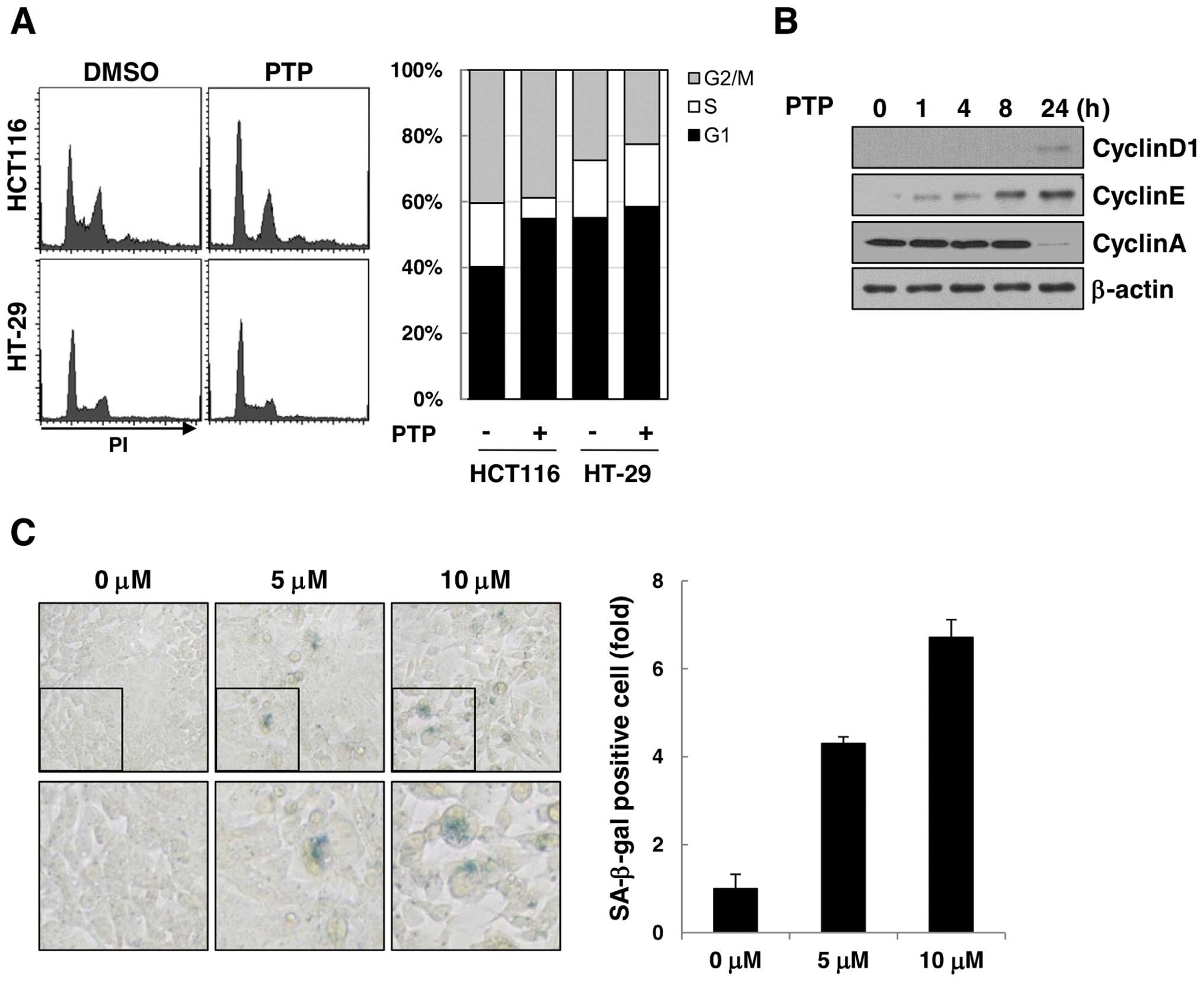
The effect of PTP on cell cycle progression was
examined by flow cytometric analysis of PTP-treated HCT116 and
HT-29 cells (Fig. 3A). The cell
cycle analysis revealed that HCT116 cells were arrested at the G1
phase of the cell cycle after 24 h of exposure to 10 μM PTP, but
the cell cycle distribution of HT-29 cells was unchanged. The
levels of cyclin D1 and cyclin E (markers of G1 phase) increased
time-dependently, and cyclin A (marker of S phase) decreased after
24 h (Fig. 3B). As cell cycle
arrest in G1-phase led to premature senescence, we carried out SA
β-galactosidase assay to determine if long-term exposure to PTP
could induce senescence of HCT116 cells. As shown in Fig. 3C, SA β-galactosidase-stained HCT116
cells increased by 4.3- and 6.7-fold after treatment with 5 and 10
μM PTP, respectively, for 6 days. These results suggest that PTP
induces G1 phase cell cycle arrest and leads to premature
senescence in cancer cells through a p53-dependent mechanism.
PTP suppresses proliferation and induces
cell death of HCT116 cells
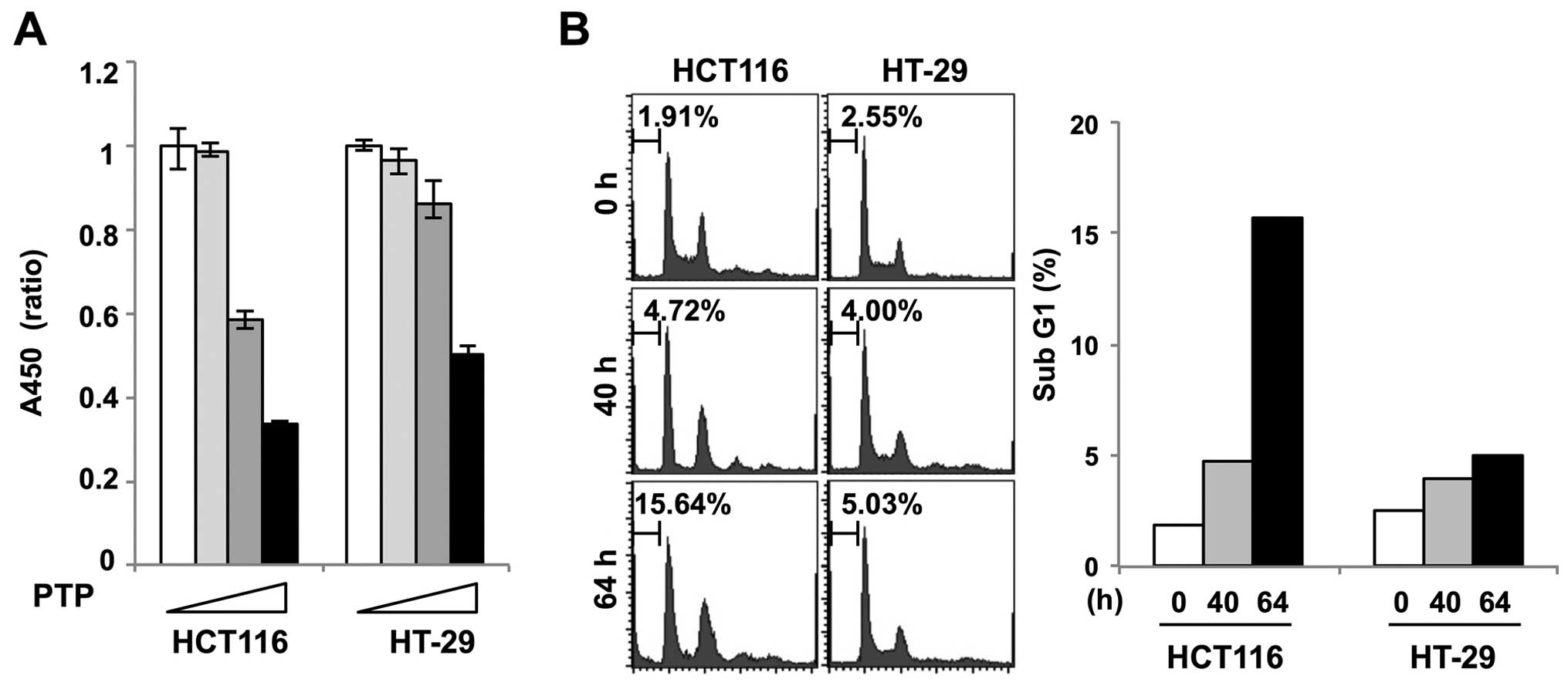
Cell viability assay was performed in PTP-treated
HCT116 and HT-29 cells to determine the effect of PTP on
proliferation of colon cancer cells. Fig. 4A shows that the growth of HCT116
cells treated with 5 and 10 μM PTP was markedly inhibited. However,
there was no significant growth inhibition of HT-29 cells treated
with 5 μM PTP. To examine if PTP caused cell death, we performed
flow cytometric analysis of PTP-treated HCT116 and HT-29 cells. The
cell cycle analysis showed that HCT116 cells appeared to undergo
cell death after 64 h of exposure to PTP, as evidenced by the
accumulation of sub-G1 cells. In contrast, no sub-G1 accumulation
was observed in PTP-treated HT-29 cells (Fig. 4B).
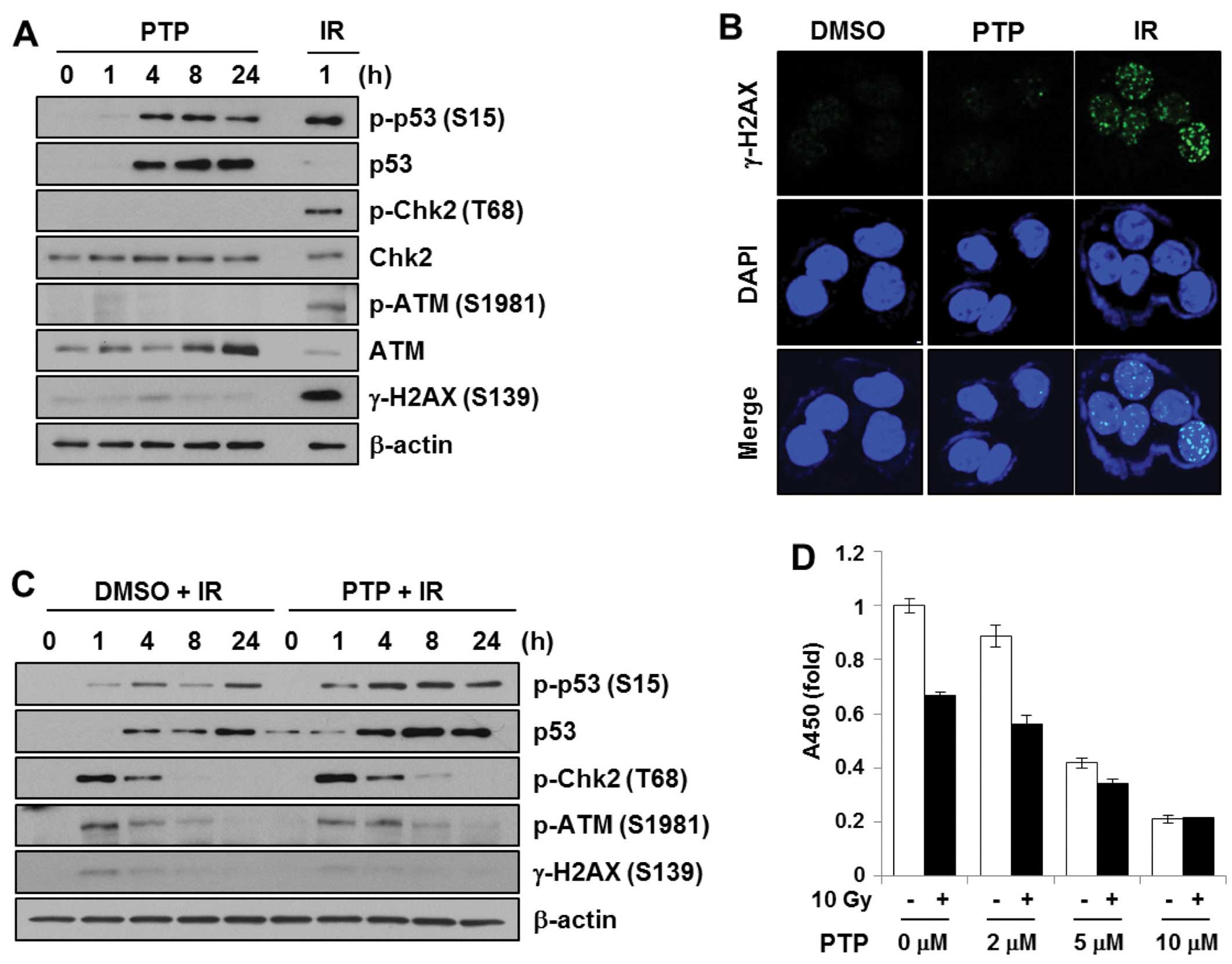
PTP induces phosphorylation of p53
irrespective of DNA damage
We originally speculated that PTP might act as a
DNA-damaging agent. The DNA damage signal cascade was confirmed
after treatment of HCT116 cells with PTP. HCT116 cells were treated
with 10 μM PTP for 0, 1, 4, 8 or 24 h. Cell lysates were subjected
to SDS-PAGE and probed with antibodies against proteins in the DNA
damage signal cascade. To prepare a DNA damage control sample,
HCT116 cells were irradiated at 5 Gy and harvested after 1 h. As
shown in Fig. 5A, PTP exposure and
γ-irradiation induced phosphorylation of p53 at Ser15. However, PTP
treatment did not lead to phosphorylation of ATM (Ser1981), Chk2
(Thr68), or H2AX (Ser139), whereas γ-irradiation did. Together with
western blot analysis, immunofluorescence detection of γ-H2AX foci
formation was used to confirm the generation of DNA double-strand
breaks (Fig. 5B). The γ-H2AX foci
formation was observed in γ-irradiated samples (5 Gy, 30 min).
However, γ-H2AX foci were not detected in PTP-treated cells. This
suggests that PTP does not induce DNA damage and that p53
phosphorylation by PTP is mediated by protein kinases other than
ATM or Chk2. Then, to determine whether PTP enhanced the
radiation-induced DNA damage signal, HCT116 cells were pretreated
with PTP for 1 h followed by γ-irradiation. The levels of p53 and
phospho-p53 (Ser15) were increased in a time-dependent manner after
irradiation. However, pre-treatment with PTP did not cause any
significant change in DNA damage signaling (Fig. 5C). Although PTP pre-treatment
followed by irradiation induced a higher level of p53
phosphorylation, this might be due to the increased level of p53
protein by PTP. The effect of PTP pre-treatment on cell
proliferation and survival after γ-irradiation was also
investigated. HCT116 cells were pretreated for 16 h with various
concentrations of PTP. Afterwards, 10 Gy of γ-irradiation was
applied. Although the proliferation of HCT116 cells was inhibited
by PTP in a dose-dependent manner and by irradiation, cells
pretreated with >5 μM PTP did not show further growth inhibition
with irradiation (Fig. 5D). This
suggests that PTP and γ-irradiation do not have a synergistic
effect on DNA damage signaling or DNA damage-mediated cell
death.
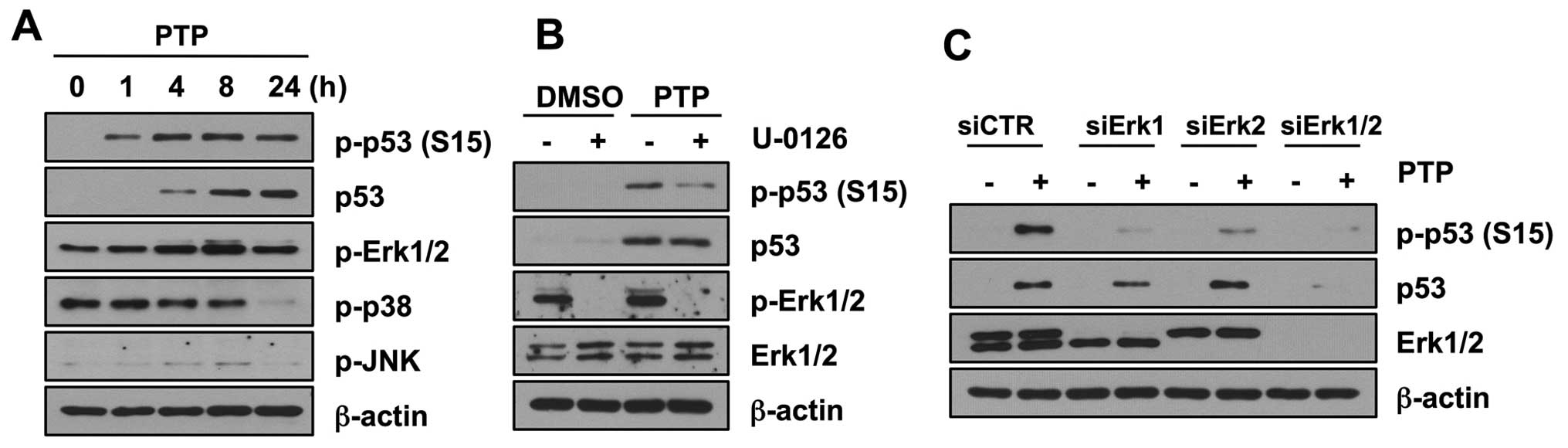
PTP induces p53 phosphorylation through
activation of Erk1/2
MAP kinases have been shown to phosphorylate
wild-type p53 under stress conditions. Therefore, to explore the
mechanism of p53 phosphorylation, we first determined whether PTP
activates mitogen-activated protein kinases (MAPKs). Of three
MAPKs, phosphorylation of Erk1/2 increased in 8 h (Fig. 6A). However, phosphorylation of
other members of the MAPK family, JNK and p38, did not increase. To
confirm that Erk1/2 mediates p53 phosphorylation, we investigated
the levels of phospho-p53 (Ser15) after PTP treatment in the
presence and absence of U-0126, an inhibitor of Erk1/2.
Phosphorylation of p53 was inhibited in HCT116 cells treated with
10 μM U-0126. However, the total level of p53 protein was not
affected by this U-0126 treatment (Fig. 6B). To clarify the contribution of
Erk MAPKs on the phosphorylation of p53 by PTP, Erk MAPKs were
depleted by siRNA transfection, and then cells were treated with 10
μM PTP for 8 h. Both Erk1 and Erk2 were involved in phosphorylation
of p53 at Ser15 (Fig. 6C). Similar
to the case with U-0126 treatment, Erk1/2 knockdown affected only
the phosphorylation of p53 and not the total level of p53 protein.
All these results suggest that PTP induces the phosphorylation of
p53 mediated by Erk1/2, but the accumulation of p53 is not caused
by modification by Erk1/2.
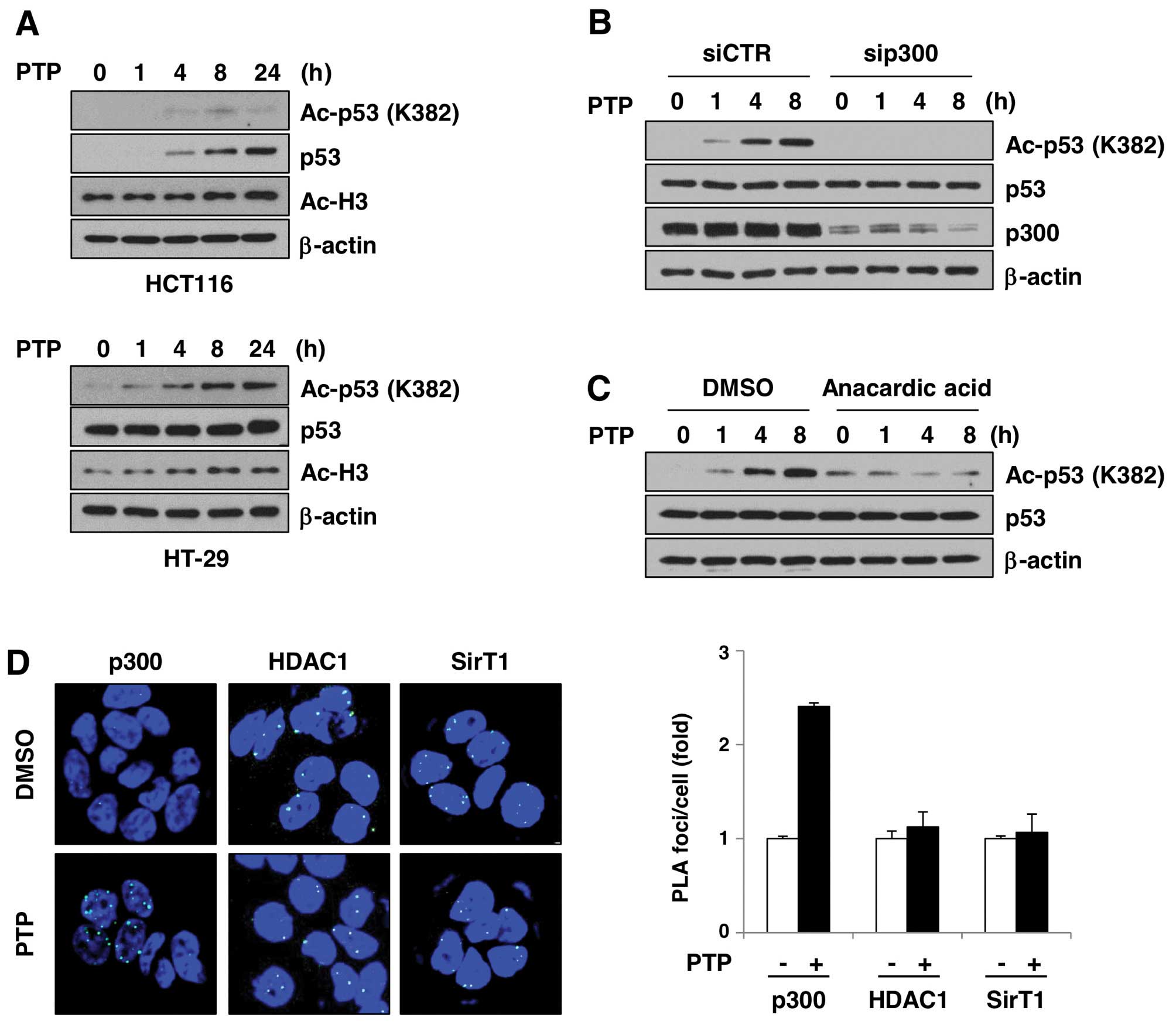
PTP induces p300-dependent acetylation of
p53
A number of groups showed that p53 can be acetylated
in vivo in response to a variety of cellular stress signals.
p300/CBP and PCAF acetylate p53 at different sites and increase its
stability. To test whether increased p53 level is related to
acetylation, we investigated the levels of acetylated p53 in
PTP-treated HCT116 and HT-29 cells (Fig. 7A). In the HCT116 and HT-29 cells,
acetylation of p53 at Lys382 increased in a time-dependent manner.
Interestingly, acetylation of histone H3 was also increased by PTP.
This suggests that PTP affects the activity of histone
acetyltransferases and/or HDACs in cells, which leads to
acetylation of proteins, including p53. In order to test whether
p300 is involved in PTP-induced p53 acetylation, we investigated
p53 acetylation in the presence of p300 siRNA or an inhibitor of
p300. As shown in Fig. 7B,
PTP-induced p53 acetylation was completely eliminated in
p300-depleted HT-29 cells. Also, PTP-induced p53 acetylation was
reduced in HT-29 cells treated with an inhibitor of p300, anacardic
acid (Fig. 7C). To determine if
the PTP-mediated p53 acetylation was p300-dependent, we
investigated the interaction between p53, p300, and SIRT1 or HDAC1
by in situ PLA. Fig. 7D
shows that p53–p300 interaction was induced in PTP-treated HT-29
cells by ~2.4-fold, based on the number of PLA foci per nucleus
compared to DMSO-treated cells. However, p53-SIRT1 and p53-HDAC1
interactions did not significantly change. These results suggest
that PTP-induced acetylation of p53 is mediated by p300, and not by
suppression of SIRT1- or HDAC1-dependent p53 deacetylation.
Discussion
Although there are recent studies that suggest that
p53 may act as a negative regulator of senescence (20), p53 is still considered a key
inducer of senescence (2). The
tumor suppressor p53 is known to promote arrest in G1-phase of the
cell cycle through the induction of p21 expression (21). It has also been reported that the
choice between p53-mediated quiescence and senescence is determined
by the mTOR pathway in a nutlin-3a-treated melanoma cell line and
mouse embryonic fibroblasts (22).
In the present study, we show that pyrimidine derivative PTP
effectively activates p53 in colorectal cancer cells, which induces
p53-dependent cell cycle arrest. Our study provides direct evidence
that PTP induced both cell cycle arrest and senescence, showing an
enlarged and flattened morphology with SA-β-gal expression, through
activation of p53. Even after removal of PTP, colorectal cancer
cells expressing wild-type p53 barely resume their proliferative
activity and almost completely lose their capacity to form colonies
(data not shown).
It is well established that many cellular stresses
lead to phosphorylation of p53 at multiple residues. DNA
damage-induced phosphorylation of p53 at Ser15 weakens the
association of p53 with MDM2 and leads to accumulation of p53 by
preventing MDM2-mediated degradation. We found no evidence that PTP
induces DNA damage or activates any canonical DNA damage signaling,
measured by phosphorylation of DNA damage signal molecules such as
ATM, Chk2 and H2AX (Fig. 5), which
raises the possibility that PTP induces p53 phosphorylation in
response to stress other than DNA damage. Recently, MAPKs p38 and
Erk1/2 have been implicated in phosphorylation of p53 at Ser15 in
response to UV irradiation and cisplatin treatment (23–25).
We have demonstrated that PTP induces phosphorylation of Erk1/2 in
a time-dependent manner and that the patterns of phosphorylation of
Erk1/2 are similar to those of p53 (Fig. 6A). Inhibition of Erk1/2 activation
(Fig. 6B) and suppression of
Erk1/2 expression with siRNA (Fig.
6C) reduces the levels of p53 phosphorylation. Phosphorylation
of p53 by PTP was significantly inhibited by knockdown of either
Erk1 or Erk2; however, the total p53 level was decreased by only a
small amount in Erk2-suppressed cells. This suggests that, whereas
Erk1 and 2 both participate in activation of p53 after exposure to
PTP, Erk1 appears to play a more important role than Erk2.
Acetylation of p53 is a powerful mechanism of p53
activation. It promotes p53 stabilization through inhibition of the
interaction of MDM2 and p53. It also prevents ubiquitination at the
same site and recruits cofactors for the promoter-specific
activator of p53 transcription activity (19,26,27).
Six lysine residues (Lys370, Lys372, Lys373, Lys381, Lys382 and
Lys386) in the C-terminal regulatory domain are acetylated by
p300/CBP or PCAF and ubiquitinated by MDM2. We observed that PTP
induces acetylation of p53 at Lys382 and of histone H3 at Lys14
(Fig. 7A). Also, inhibition of
p300 mostly reduces the level of PTP-induced p53 acetylation
(Fig. 7B and C). Although specific
phosphorylation of p53 at residue Ser15 could be one activation
step leading to p53–p300/CBP complex formation and subsequent p53
acetylation by p300/CBP, Ser15 phosphorylation is not the only
mechanism that can lead to p53 acetylation. Several studies
reported that actinomycin D is also a powerful reagent in inducing
p53 acetylation. However, it does not phosphorylate p53 at Ser15
(28,29). Kuroda et al (30) reported that, when rRNA
transcription was suppressed by nucleolar stress, MyBBP1A
translocated to the nucleoplasm and promoted p53–p300 interaction
to enhance acetylation of p53. However, the mechanism of p53
acetylation without Ser15 phosphorylation is not clear.
Histone deacetylases, HDAC1 and SIRT1 maintain
steady-state levels of p53 acetylation (31,32).
SIRT1 preferentially deacetylates p53 at Lys382 and reduces the
activity of p53 to induce the expression of target genes. Although
we did demonstrate that PTP induces the interaction between p53 and
p300, the interaction between p53 and HDAC1 or SIRT1 was not
changed by exposure to PTP in colorectal cancer cells (Fig. 7D). This shows that PTP-induced p53
acetylation is mediated by increased interaction of p300 with p53,
and that PTP may not affect the p53-deacetylating activity of SIRT1
and HDAC1.
In the present study, we have demonstrated that
inhibition of tumor proliferation and induction of cellular
senescence mediated by activation and accumulation of p53 may be
important aspects of the antitumor activity of pyrimidine
derivative PTP.
Acknowledgements
The present study was supported by the National
Nuclear R & D Program of the Ministry of Science, ICT and
Future Planning, Republic of Korea (50541-2013).
Abbreviations:
|
PTP
|
N-[2-(dimethylamino)ethyl]-2,3-dimethyl-4-oxo-4H-pyrido[1,2-a]thieno[2,3-d]pyrimidine-9-carboxamide
|
References
|
1
|
Vousden KH: p53: Death Star. Cell.
103:691–694. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Juven T, Barak Y, Zauberman A, George DL
and Oren M: Wild type p53 can mediate sequence-specific
transactivation of an internal promoter within the mdm2 gene.
Oncogene. 8:3411–3416. 1993.PubMed/NCBI
|
|
4
|
Barak Y, Juven T, Haffner R and Oren M:
mdm2 expression is induced by wild type p53 activity. EMBO J.
12:461–468. 1993.PubMed/NCBI
|
|
5
|
Miyashita T, Harigai M, Hanada M and Reed
JC: Identification of a p53-dependent negative response element in
the bcl-2 gene. Cancer Res. 54:3131–3135. 1994.PubMed/NCBI
|
|
6
|
Banin S, Moyal L, Shieh SY, Anderson CW,
Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y and Ziv Y:
Enhanced phosphorylation of p53 by ATM in response to DNA damage.
Science. 281:1674–1677. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tibbetts RS, Brumbaugh KM, Williams JM,
Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C and Abraham RT: A
role for ATR in the DNA damage-induced phosphorylation of p53.
Genes Dev. 13:152–157. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bykov VJ, Issaeva N, Shilov A, Hultcrantz
M, Pugacheva E, Chumakov P, Bergman J, Wiman KG and Selivanova G:
Restoration of the tumor suppressor function to mutant p53 by a
low-molecular-weight compound. Nat Med. 8:282–288. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miyachi M, Kakazu N, Yagyu S, Katsumi Y,
Tsubai-Shimizu S, Kikuchi K, Tsuchiya K, Iehara T and Hosoi H:
Restoration of p53 pathway by nutlin-3 induces cell cycle arrest
and apoptosis in human rhabdomyosarcoma cells. Clin Cancer Res.
15:4077–4084. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dai F, Chen Y, Song Y, Huang L, Zhai D,
Dong Y, Lai L, Zhang T, Li D, Pang X, Liu M and Yi Z: A natural
small molecule Harmine inhibits angiogenesis and suppresses tumour
growth through activation of p53 in endothelial cells. PLoS One.
7:e521622012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Q, Zeng SX, Zhang Y, Zhang Y, Ding
D, Ye Q, Meroueh SO and Lu H: A small molecule Inauhzin inhibits
SIRT1 activity and suppresses tumour growth through activation of
p53. EMBO Mol Med. 4:298–312. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jain KS, Chitre TS, Miniyar PB, Kathiravan
MK, Bendre VS, Veer VS, Shahane SR and Shishoo CJ: Biological and
medicinal significance of pyrimidines. Curr Sci. 90:793–803.
2006.
|
|
13
|
Zhang N, Ayral-Kaloustian S, Nguyen T,
Hernandez R and Beyer C: 2-Cyanoaminopyrimidines as a class of
antitumor agents that promote tubulin polymerization. Bioorg Med
Chem Lett. 17:3003–3005. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cocco MT, Congiu C, Lilliu V and Onnis V:
Synthesis and in vitro antitumoral activity of new
hyrazinopyriminide-5-carbonitrile derivatives. Bioorg Med Chem.
14:366–372. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cocco MT, Congiu C, Lilliu V and Onnis V:
Synthesis of new N-(2-(trifluoromethyl)pyridine-yl)anthranilic acid
derivatives and their evaluation as anticancer agents. Bioorg Med
Chem Lett. 14:5787–5791. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Longley DB, Harkin DP and Johnston PG:
5-Fluorouracil: mechanisms of action and clinical stratagies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lim MJ, Ahn JY, Han Y, Yu CH, Kim MH, Lee
SL, Lim DS and Song JY: Acriflavine enhances radiosensitivity of
colon cancer cells through endoplasmic reticulum stress-mediated
apoptosis. Int J Biochem Cell Biol. 44:1214–1222. 2012. View Article : Google Scholar
|
|
18
|
Harris SL and Levine AJ: The p53 pathway:
positive and negative feedback loops. Oncogene. 24:2899–2908. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Haupt Y, Maya R, Kazaz A and Oren M: Mdm2
promotes the rapid degradation of p53. Nature. 387:296–299. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Demidenko ZN, Korotchkina LG, Gudkov AV
and Blagosklonny MV: Paradoxical suppression of cellular senescence
by p53. Proc Natl Acd Sci USA. 107:9660–9664. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hofseth LJ, Hussain SP and Harris CC: p53:
25 years after its discovery. Trends Pharmacol. 25:177–181.
2004.
|
|
22
|
Korotchkina LG, Leontieva OV, Bukreeva EI,
Demidenko ZN, Gudkov AV and Blagosklonny MV: The choice between
p53-induced senescence and quiescence is determined in part by the
mTOR pathway. Aging (Albany, NY). 2:344–352. 2010.PubMed/NCBI
|
|
23
|
Melnikova VO, Santamaria AB, Bolshakov SV
and Ananthaswamy HN: Mutant p53 is constitutively phosphorylated at
Serine 15 in UV-induced mouse skin tumors: involvement of ERK1/2
MAP kinase. Oncogene. 22:5958–5966. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Persons DL, Yazlovitskaya EM and Pelling
JC: Effect of extra-cellular signal-regulated kinase on p53
accumulation in response to cisplatin. J Biol Chem.
275:35778–35785. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
She QB, Chen N and Dong Z: ERKs and p38
kinase phosphorylate p53 protein at serine 15 in response to UV
radiation. J Biol Chem. 275:20444–20449. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kubbutat MH, Jones SN and Vousden KH:
Regulation of p53 stability by Mdm2. Nature. 387:299–303. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Maltzman W and Czyzyk L: UV irradiation
stimulates levels of p53 cellular tumor antigen in nontransformed
mouse cells. Mol Cell Biol. 4:1689–1694. 1984.PubMed/NCBI
|
|
28
|
Ashcroft M, Taya Y and Vousden KH: Stress
signals utilize multiple pathways to stabilize p53. Mol Cell Biol.
20:3224–3233. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ito A, Lai CH, Zhao X, Saito S, Hamilton
MH, Appella E and Yao TP: p300/CBP-mediated p53 acetylation is
commonly induced by p53-activating agents and inhibited by MDM2.
EMBO J. 20:1331–1340. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kuroda T, Murayama A, Katagiri N, Ohta Y,
Fujita E, Masumoto H, Ema M, Takahashi S, Kimura K and Yanagisawa
J: RNA content in the nucleolus alters p53 acetylation via MYBBP1A.
EMBO J. 30:1054–1066. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Barneda-Zahonero B and Parra M: Histone
deacetylases and cancer. Mol Oncol. 6:579–589. 2012. View Article : Google Scholar
|
|
32
|
Vaziri H, Dessain SK, Eaton EN, Imai SI,
Frye RA, Pandita TK, Guarente L and Weinberg RA:
hSIR2SIRT1 functions as an NAD-dependent p53
deacetylation. Cell. 107:149–159. 2001. View Article : Google Scholar
|