Introduction
Osteosarcoma is the most common primary malignant
bone tumor in children and adolescents, with 70–75% of cases
occurring between the ages of 10 and 25 (1). Adult osteosarcoma is the second most
frequent sarcoma with a low rate of response to current therapy due
to inherent chemoresistance. The therapy of osteosarcoma has made
good progress with the development of surgery and screening
technologies and with the alliance of neoadjuvant chemotherapy,
radiotherapy, immunotherapy and thermotherapy. However, problems of
metastasis, recurrence and chemoresistance have not yet been
solved. Indeed, metastases occur in >80% of individuals that are
treated with surgery alone (2).
Moreover, despite aggressive treatment including adjuvant and
neoadjuvant chemotherapy, 30–40% of children die of osteosarcoma
(3,4). There is therefore an urgent need to
develop novel therapeutic agents.
Heat shock protein 90 (Hsp90) is an abundant
molecular chaperone which constitutes 1–2% of total cellular
protein. Hsp90 interacts with a variety of intracellular client
proteins involved in cell growth, differentiation and survival,
which facilitates their folding, activity, intracellular
localization and proteolytic turnover (5,6).
Hsp90 is abundantly expressed in eukaryotes and comprises >1% of
the eukaryote total cellular content (7,8).
However, Hsp90 is constitutively expressed at 2–10-fold higher
levels in tumor cells compared to normal cells, suggesting that it
may be critically important for tumor cell growth and survival
(9). These features make Hsp90 a
potential target for anticancer drug development. Geldanamycin (GA)
is a benzoquinone ansamycin antibiotic that interferes with the
action of Hsp90 leading to the degradation of Hsp90 client
proteins. Since many of these client proteins are oncogenic
proteins, GA inhibits the proliferation of cancer cells and shows
anticancer activity in experimental animals. Inhibition of Hsp90
activity not only results in rapid degradation of Hsp90 client
proteins but also induces apoptosis of various tumor cells
(10,11). Currently several drug candidates
that target Hsp90 are undergoing clinical trials for multiple
indications, either as a single agent or in combination therapy
(12). However, the molecular
mechanism of Hsp90 inhibitors in cancer cells needs to be further
elaborated.
Autophagy has recently gained attention because of
its paradoxical roles in cell survival and cell death, particularly
in the pathogenesis and treatment of cancer (13). Regulation of autophagy is highly
complex with inputs from the cellular environment through the
Akt/mTOR and MAPK/Erk1/2 signaling pathways (14). Hsp90 plays an important role in
autophagy (15). An Hsp90
inhibitor induces autophagy through inhibition of mTOR (16). Autophagy is activated during
starvation to provide an alternative energy source through
self-digestion. Autophagy thus serves as a temporary survival
mechanism. Autophagy is also important in the induction of tumor
cell death (17) and excessive
autophagy triggers autophagic cell death in tumors (18,19).
It is still under debate whether chemotherapy-induced autophagy in
tumor cells is a protective response or is invoked to promote cell
death (14). However, recent
studies have indicated that autophagy can function as a protective
mechanism in cells that are exposed to antitumor agents and that
blocking autophagy can trigger the activation of apoptosis
(20–22). Based on these findings, it has been
suggested that inhibitors of autophagy, such as the commonly used
inhibitor 3-methyladenine (3-MA), might be an effective treatment
for osteosarcoma because they activate apoptosis. Recent studies
demonstrate that 3-MA promotes chemotherapeutic drug-induced
apoptosis in cancer cells. Reportedly, 3-MA inhibits the activity
of PI3K and blocks the formation of pre-autophagosomes,
autophagosomes and autophagic vacuoles (23).
The aim of this study was to examine the effects of
the Hsp90 inhibitor GA, on osteosarcoma cells. We investigated
whether GA modulated the phosphorylation of proteins in the
Akt/mTOR signaling pathway and/or induced autophagy or apoptosis in
osteosarcoma cells. We further examined whether a combination of GA
and 3-MA enhanced GA-induced apoptosis in osteosarcoma cells.
Materials and methods
Chemical reagents
GA was purchased from StressMarq Biosciences, Inc.
(Victoria, BC, Canada), dissolved in dimethylsulfoxide (DMSO), and
stored at −20°C. The inhibitor of autophagy, 3-MA, was purchased
from Sigma Chemical Co. (St. Louis, MO, USA), dissolved in 1 mg/ml
dimethylformamide (DMF), and stored at room temperature.
Cell lines and cell culture
The KTHOS osteosarcoma cell line was used in this
study. This cell line was grown in culture medium consisting of
DMEM (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10%
fetal bovine serum (FBS) (Sigma-Aldrich) and 100 U/ml penicillin.
The cell line was routinely maintained at 37°C in a humidified 5%
CO2 atmosphere. Cells were divided into four treatment
groups; control (no inhibitor), GA, 3-MA and GA plus 3-MA (GA +
3-MA), for cell proliferation and the autophagy and apoptosis
assays. Cells were seeded onto culture dishes and were cultured in
growth medium for 48 h. The growth medium was then replaced with
fresh medium with or without inhibitors. In the experiments that
tested the combined effect of GA and 3-MA, cells were pre-treated
with 10 mM 3-MA for 1 h before GA was added to the culture medium,
and then cells were treated with 5 μM GA with 10 mM 3-MA for 24 h.
In experiments testing the effect of GA or 3-MA alone, cells were
treated with 5 μM GA or 10 mM 3-MA for 24 h.
In vitro cell proliferation assay
Cell proliferation was determined using the
CellTiter 96® AQueous One Solution Cell Proliferation
Assay (Promega Corporation, Madison, WI, USA). Cells were
trypsinized and seeded at a density of 1×104 cells/well
in 96-well cell culture plates with 200 μl culture medium
containing 10% FBS and were incubated for 48 h. Following this
initial incubation, the growth medium was replaced with medium
containing 10% FBS and GA at a concentration of 0, 0.01, 0.1, 1 or
10 μM. After 24 or 48 h, the medium was removed, the cells were
washed with phosphate-buffered saline (PBS), and fresh medium
containing the
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) reagent (100 μl medium plus 20 μl MTS reagent/well) was added
to each well. In the experiments that tested the combined effect of
GA and 3-MA, cells were pre-treated with 10 mM 3-MA for 1 h before
GA was added to the culture medium, and then cells were treated
with 5 μM GA with 10 mM 3-MA for 24 h. In experiments testing the
effect of GA or 3-MA, cells were treated with 5 μM GA or 10 mM 3-MA
for 24 h. Cells in the four treatment groups with/without GA
with/without 3-MA were also assayed for cell proliferation using
this MTS assay. The optical density was measured at 490 nm using an
automatic microplate reader after 2 h of further incubation
following the addition of the MTS reagent. Absorbance is directly
proportional to the number of living cells. Proliferation of each
well was calculated as a percent of control. At least three
independent experiments were performed for each assay.
Western blot analyses
Cells were trypsinized and seeded at a density of
6×105 cells/well in 6-well cell culture plates in 2 ml
culture medium with 10% FBS. After 48 h, cells were treated with
10% FBS containing GA for the indicated time and at the indicated
concentration. Cells of the four treatment groups were treated
with/without GA with/without 3-MA. In the experiments testing the
combined effect of GA and 3-MA, cells were pre-treated with 10 mM
3-MA for 1 h before GA was added to the culture medium, and then
cells were treated with 5 μM GA with 10 mM 3-MA for 24 h. In
experiments testing the effect of GA or 3-MA, cells were treated
with 5 μM GA or 10 mM 3-MA for 24 h. Following treatment, the
culture medium was replaced with lysis buffer (Cell Signaling
Technology, Inc., Beverly, MA, USA), and cells were lysed on ice
for 20 min. The cell lysates were spun at 15,000 × g using the Tomy
MTX-150 centrifuge (Tomy Seiko Co., Ltd., Fukuoka, Japan) at 4°C
for 30 min. The supernatant was then collected as the total
cellular protein extract. Protein concentration was determined
using the Protein Assay Bicinchoninate kit (Nacalai Tesque, Inc.,
Kyoto, Japan) and was standardized with bovine serum albumin. The
samples of total cellular protein were loaded onto an SDS
polyacrylamide gel (7.5, 10 or 12.5% commercial precast gel; Wako,
Tokyo, Japan), and the proteins were separated by SDS-PAGE under
reducing conditions. The mTOR, phospho-mTOR and cleaved PARP
proteins were separated on a 7.5% SDS gel; the Akt, phospho-Akt,
p70 ribosomal protein S6 kinase (p70S6K), phospho-p70S6K,
p62/SQSTM1 and α-tubulin proteins were separated on 10% SDS gel;
and 4E-binding protein 1 (4E-BP1), phospho-4E-BP1,
microtubule-associated protein light-chain 3 (LC-3), cleaved
caspase-9, and cleaved caspase-8 proteins were separated on a 12.5%
SDS gel. The separated proteins were electrophoretically
transferred to nitrocellose membranes (GE Healthcare Bio-Sciences
Corp., Piscataway, NJ, USA). The membranes were blocked for 90 min
in blocking buffer containing Tris-buffered saline-Tween-20 (TBS-T)
and 10% EZ block (Atto Co., Ltd., Tokyo, Japan). The membranes were
then incubated with primary antibodies, which were diluted in the
blocking buffer, overnight at 4°C. Antibodies against Akt and
phospho-Akt (Thr308) were purchased from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). Anti-mTOR and anti-phospho-mTOR
(Ser2448) antibodies were purchased from R&D Systems
(Minneapolis, MN, USA). Antibodies against 4E-BP1, phospho-4E-BP1
(Ser65), p70S6K, phospho-p70S6K (Thr421/Ser424), cleaved caspase-9
(Asp315), cleaved caspase-8 (Asp391) and cleaved PARP (Asp214) were
purchased from Cell Signaling Technology, Inc., and anti-α-tubulin
antibody was purchased from Sigma-Aldrich. Anti-LC-3 and
anti-p62/SQSTM1 antibodies were purchased from MBL Co., Ltd.
(Nagoya, Japan). These primary antibodies are listed and
characterized in Table I. The
specific HRP-conjugated secondary antibody incubations were
performed overnight at 4°C with gentle agitation. Bound antibodies
were detected using the ECL Plus Western Blotting Detection system
(GE Healthcare Bio-Sciences Corp.) and LAS-1000 Plus Image Analyzer
(Fujifilm Co., Tokyo, Japan). Specific signals were quantified by
densitometric analysis (Image J software).
 | Table IPrimary antibodies used in western
blot analysis. |
Table I
Primary antibodies used in western
blot analysis.
| Target | Source | Host | Dilution | Second antibody |
|---|
| LC-3 | MBL Co., Ltd. | Rabbit | 1:1,000 | Anti-rabbit |
| p62/SQSTM1 | MBL Co., Ltd. | Rabbit | 1:1,000 | Anti-rabbit |
| Akt | Santa Cruz
Biotechnology, Inc. | Rabbit | 1:1,000 | Anti-rabbit |
| Phospho-Akt | Santa Cruz
Biotechnology, Inc. | Rabbit | 1:1,000 | Anti-rabbit |
| mTOR | R&D Systems | Rabbit | 1:1,000 | Anti-rabbit |
| Phospho-mTOR | R&D Systems | Rabbit | 1:1,000 | Anti-rabbit |
| p70S6K | Cell Signaling
Technology, Inc. | Rabbit | 1:1,000 | Anti-rabbit |
| Phospho-p70S6K | Cell Signaling
Technology, Inc. | Rabbit | 1:1,000 | Anti-rabbit |
| 4E-BP1 | Cell Signaling
Technology, Inc. | Rabbit | 1:1,000 | Anti-rabbit |
| Phospho-4E-BP1 | Cell Signaling
Technology, Inc. | Rabbit | 1:1,000 | Anti-rabbit |
| Cleaved
caspase-9 | Cell Signaling
Technology, Inc. | Rabbit | 1:1,000 | Anti-rabbit |
| Cleaved
caspase-8 | Cell Signaling
Technology, Inc. | Rabbit | 1:1,000 | Anti-rabbit |
| Cleaved PARP | Cell Signaling
Technology, Inc. | Rabbit | 1:1,000 | Anti-rabbit |
| α-tubulin | Sigma-Aldrich | Mouse | 1:1,000 | Anti-mouse |
Flow cytometric TUNEL assay
TUNEL assay was performed using the MEBstain
Apoptosis TUNEL kit Direct (MBL Co., Ltd.) following the
manufacturer’s instructions. Cells were seeded at a density of
6×105 cells/well and were cultured for 48 h. Cells of
the four treatment groups were then treated with/without GA
with/without 3-MA. In the experiments testing the combined effect
of GA and 3-MA, cells were pre-treated with 10 mM 3-MA for 1 h
before GA was added to the culture medium, and then cells were
treated with 5 μM GA with 10 mM 3-MA for 24 h. In experiments
testing the effect of GA or 3-MA, cells were treated with 5 μM GA
or 10 mM 3-MA for 24 h. Cells were then washed gently three times
in PBS containing 0.2% BSA, fixed with 4% paraformaldehyde for 30
min at 4°C, and washed twice in PBS containing 0.2% BSA. Next, 200
μl of 70% ethanol were added to the sample, which was mixed gently
and then incubated for 30 min at −20°C. The samples were then
washed twice in PBS containing 0.2% BSA, 30 μl of TdT solution were
added and the samples were incubated for 1 h at 37°C. Next, the
samples were washed twice in PBS containing 0.2% BSA, suspended to
a final volume of 500 μl of PBS containing 0.2% BSA, and analyzed
using a flow cytometer (FC-500; Beckman Coulter, Inc., Brea, CA,
USA). Data are representative of three independent experiments.
Determination of apoptosis using Annexin
V-FITC and PI stain analysis
Cells were trypsinized and seeded at a density of
6×105 cells/well in 6-well cell culture plates in 2 ml
culture medium with 10% FBS and were then cultured for 48 h. Cells
of the four treatment groups were treated with/without GA
with/without 3-MA. In the experiments testing the combined effect
of GA and 3-MA, cells were pre-treated with 10 mM 3-MA for 1 h
before GA was added to the culture medium, and then cells were
treated with 5 μM GA with 10 mM 3-MA for 24 h. In experiments
testing the effect of GA or 3-MA, cells were treated with 5 μM GA
or 10 mM 3-MA for 24 h. Cells were then incubated for 15 min in a
dark room with Annexin V-FITC and PI using the Annexin V-FLUOS
Staining kit (Roche Applied Science, Penzberg, Germany) according
to the manufacturer’s recommendations. Stained cells were observed
under a fluorescence microscope (Keyence Co., Osaka, Japan)
equipped with a filter system (DAPI-BP for Annexin V: excitation
wavelength 377 nm and detection 447 nm, TRITC for PI: excitation
wavelength 543 nm and detection 593 nm). To quantify Annexin V and
PI incorporation, at least 100 cells from each treatment group were
examined under fluorescence microscopy, and the percentage of
Annexin V-positive or Annexin V-plus-PI positive cells were
calculated. The 100 cells sampled were chosen randomly to avoid
bias.
Detection of autophagic vacuoles using
monodansyl cadaverine
Autophagic vacuoles were detected using
mono-dansylcadaverine (MDC) by incubating cells with MDC solution
(1:1,000 in Cell-Based Assay Buffer, 50 μM) in PBS using the
Autophagy/Cytotoxicity Dual Staining kit (Cayman Chemical Co., Ann
Arbor, MI, USA). Cells were seeded at a density of 6×105
cells/well in 6-well cell culture plates in 2 ml culture medium
with 10% FBS and were then cultured for 48 h. In the experiments
testing the effect of GA, cells were treated with 5 μM GA for 24 h.
Cells were then incubated with MDC for 15 min and immediately
analyzed under a fluorescence microscope (DMI4000 B; Leica
Microsystems, Wetzlar, Germany) using a fluorescence microscope
equipped with a filter system (excitation wavelength of 460–500 nm,
emission wavelength of 512–542 nm). Bright-field and fluorescence
images were merged.
Statistical analysis
All data and results presented are representative
of, or calculated from, at least three independent experiments. For
the cell proliferation assay, TUNEL assay, and Annexin V-FITC/PI
stain analysis, differences between treatment groups were
determined using the GraphPad Prism 5 for Windows software package.
The data collected in three independent experiments for each group
are expressed as means ± standard deviation (SD) and were
statistically analyzed using ANOVA with Fisher’s PLSD post hoc
test. P<0.05 was regarded as statistically significant.
Results
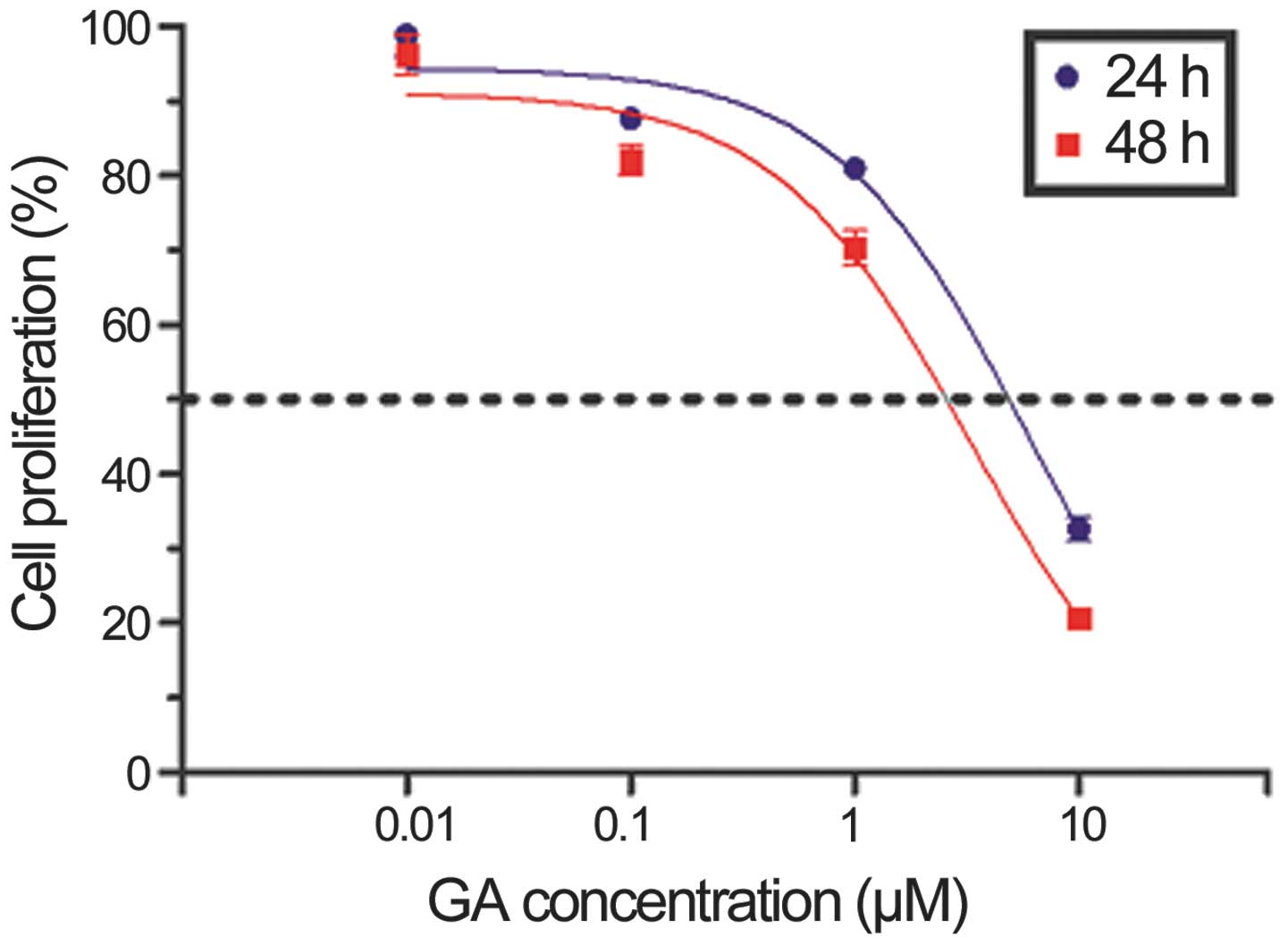
GA inhibits the proliferation of KTHOS
cells
Initially, we assessed the effects of GA on cellular
proliferation using the CellTiter 96® AQueous One
Solution Cell Proliferation Assay. KTHOS cells were cultured in the
presence of increasing doses of GA for 24 or 48 h. As shown in
Fig. 1, GA inhibited KTHOS
proliferation in a dose- and time-dependent manner. The
IC50 value of GA at 24 h was 5.974 μM.
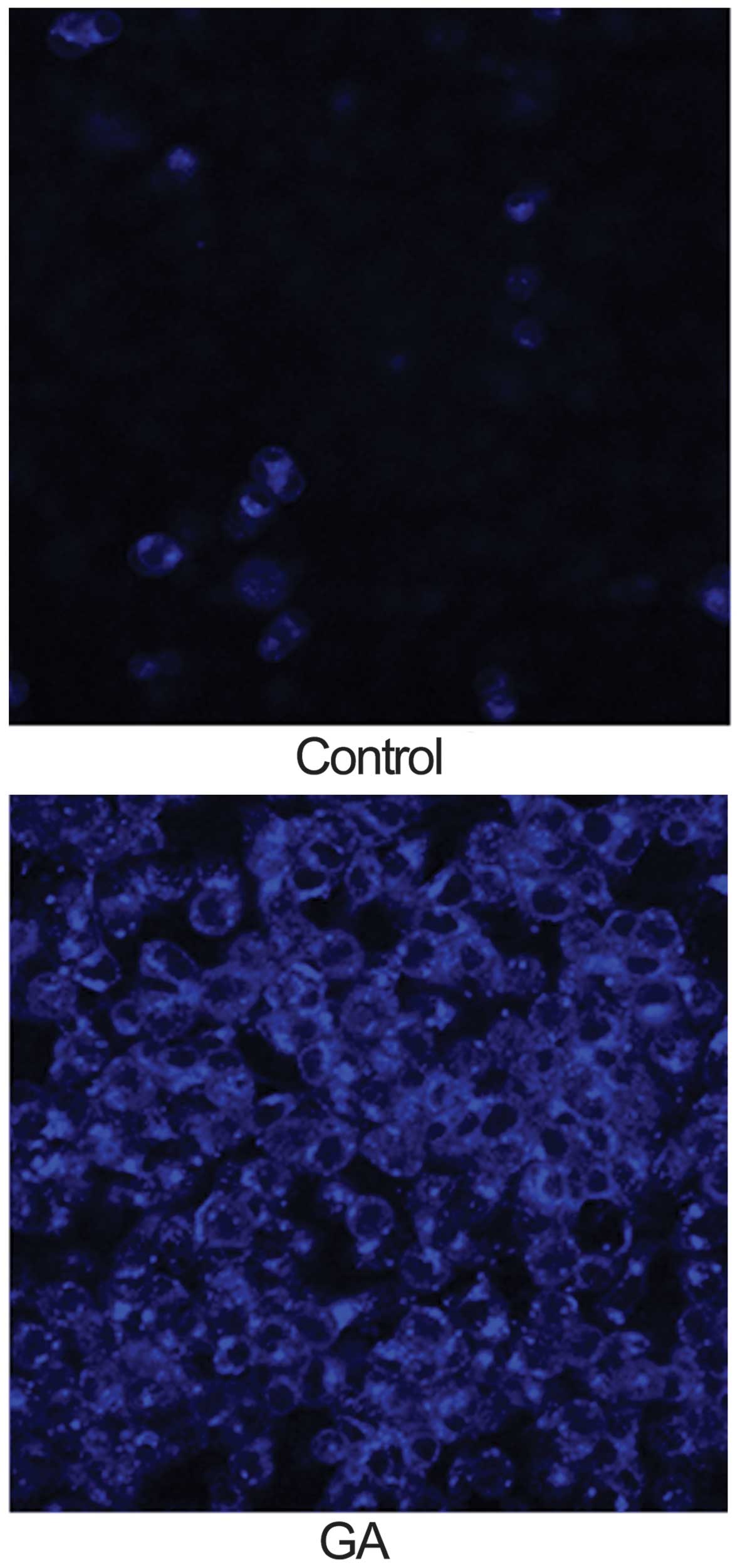
GA induces autophagy in KTHOS cells by
inhibiting Akt/mTOR/p70S6K signaling
We next investigated whether GA (5 μM for 24 h)
induced autophagy in KTHOS cells. The concentration of GA chosen
was based on the IC50 of GA after 24 h in the cell
proliferation assay. For analysis of autophagy, we used MDC
staining to detect autophagic vacuoles. MDC is an autofluorescent
dye that accumulates in mature autophagic vacuoles, such as
autophago-lysosomes, but not in the early endosome compartment; it
is therefore a specific marker for autophagic vacuoles. Cells were
incubated with MDC for 15 min after incubation with GA and were
then analyzed using a fluorescence microscope. MDC fluorescence was
observed in control and GA-treated KTHOS cells. However, GA-treated
KTHOS cells displayed higher and more frequent accumulation of MDC
accumulation than control cells (Fig.
2). These results suggest that GA treatment induces autophagy
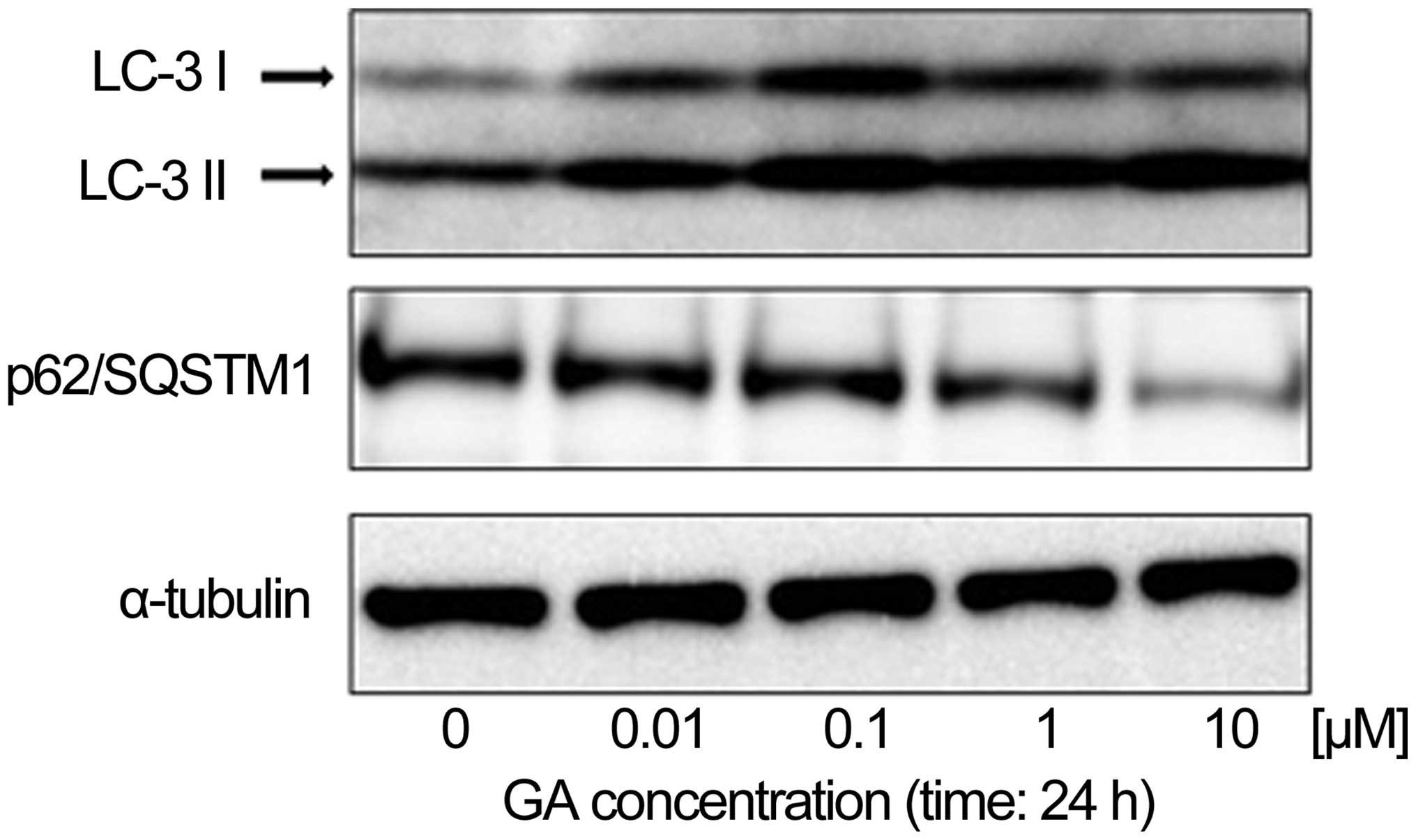
in KTHOS cells. To confirm that GA induced autophagy in these
cells, we first used western blot analysis to analyze the
expression of LC-3 and p62/SQSTM1 proteins, which are known to be
upregulated and downregulated respectively in autophagy, in KTHOS
cells exposed to concentrations of GA ranging from 0.01 to 10 μM,
for 24 h. Fig. 3 shows that GA
treatment induced a dose-dependent upregulation of LC3-II and
downregulation of the p62/SQSTM1 protein, which confirms induction
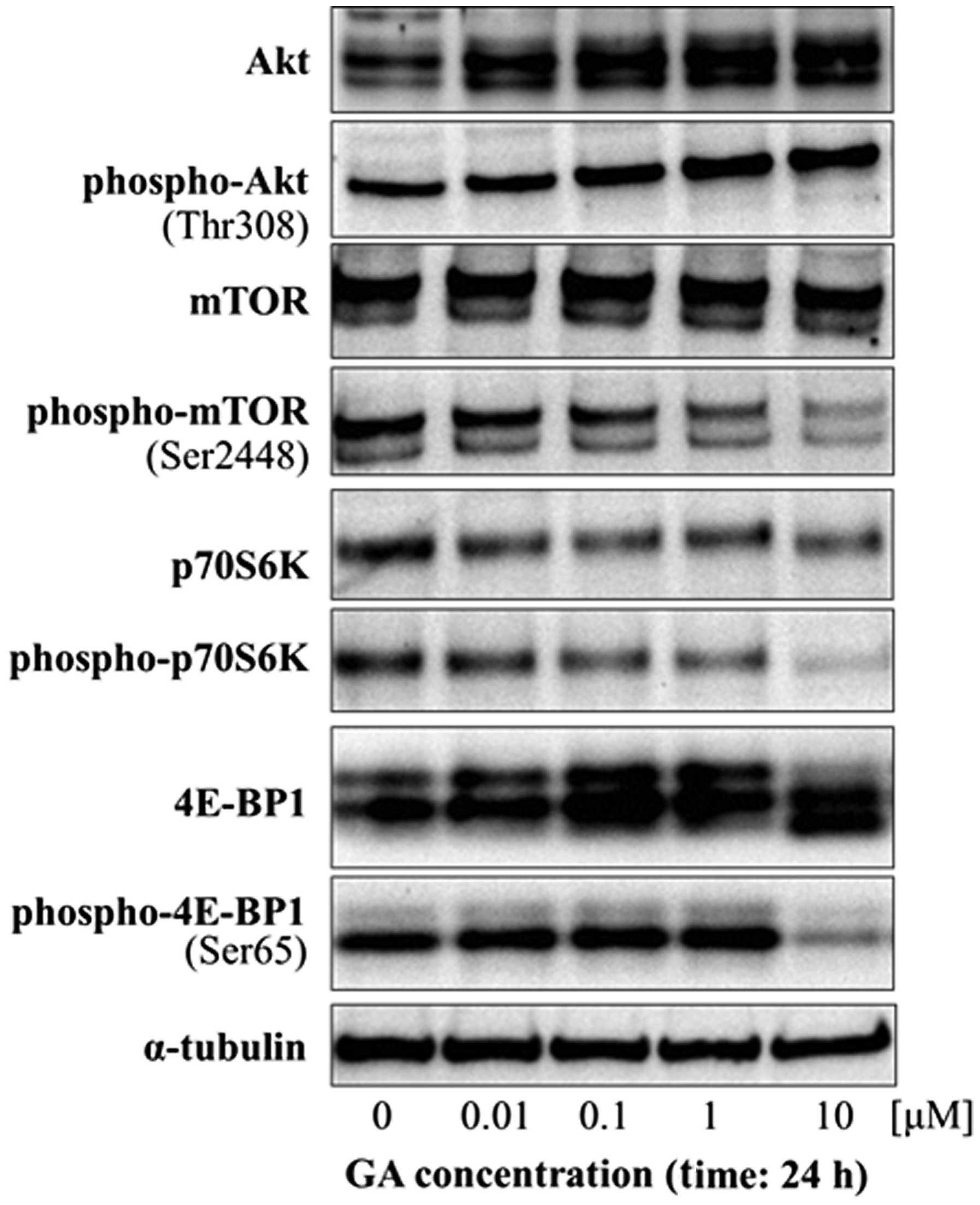
of autophagy in these cells. Activation of autophagy is associated
with the Akt/mTOR/p70S6K signaling pathway in mammalian cells;
Akt/mTOR/p70S6K signaling negatively regulates autophagy. We next
examined the potential role of Akt/mTOR/p70S6K signaling in
GA-induced autophagy by western blot analysis of the expression and
phosphorylation of Akt and mTOR and of the downstream effectors of
mTOR, p70S6K and 4E-BP1. GA did not cause any change in the levels
of phospho-Akt in KTHOS cells. However, GA treatment resulted in a
dose-dependent decrease in phospho-mTOR, phospho-p70S6K and
phospho-4E-BP1 (Fig. 4). These
findings indicate that GA affects the Akt/mTOR signaling pathway by
inhibiting the phosphorylation of downstream effectors of mTOR.
The combined results indicate that GA induces
autophagy by inhibition of the Akt/mTOR/p70S6K signaling
pathway.
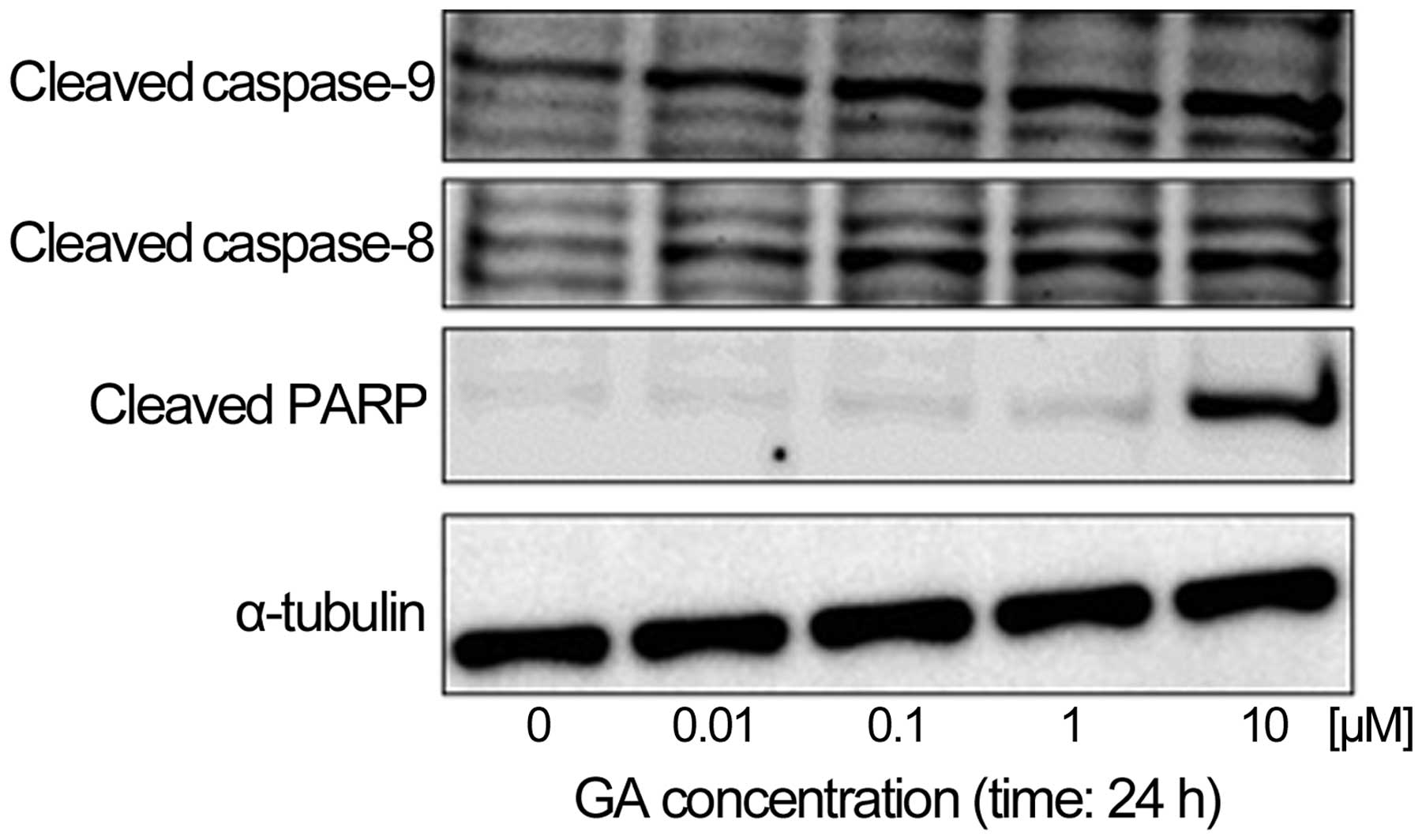
GA induces the caspase-dependent
apoptotic pathway in KTHOS cells
We next examined the effect of GA on caspase
activity to determine if GA induces caspase-dependent apoptosis in
KTHOS cells. Western blot analysis indicated that treatment of
KTHOS cells with concentrations of GA ranging from 0.01 to 10 μM
for 24 h resulted in dose-dependent cleavage of PARP, as well as
activation of caspase-8 and -9 (Fig.
5). These results suggest the ability of GA to induce apoptosis
in a caspase-dependent manner in KTHOS cells.
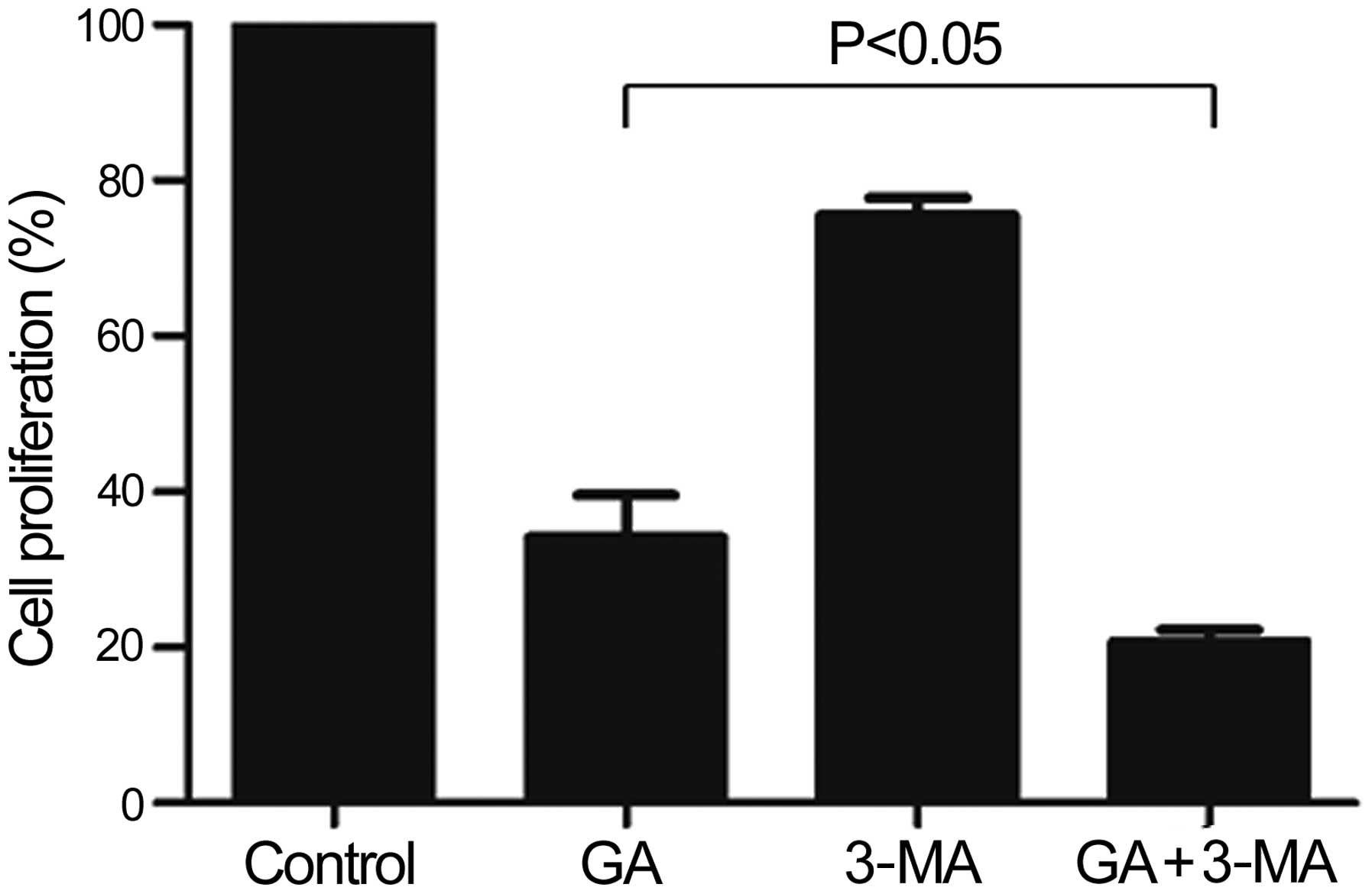
GA potently inhibits the proliferation of
KTHOS cells via induction of apoptosis following 3-MA
pre-treatment
We next determined whether GA-induced autophagy is a
protective or an apoptosis-promoting mechanism. For this purpose,
we assessed cellular proliferation following pre-treatment of KTHOS
cells with 10 mM 3-MA, which is commonly employed as a specific
inhibitor of autophagic sequestration, for 1 h prior to
administration of 5 μM GA for 24 h. As shown in Fig. 6, GA inhibition of KTHOS
proliferation following 3-MA pre-treatment was significantly higher
than that in the absence of 3-MA treatment (P<0.05). We next
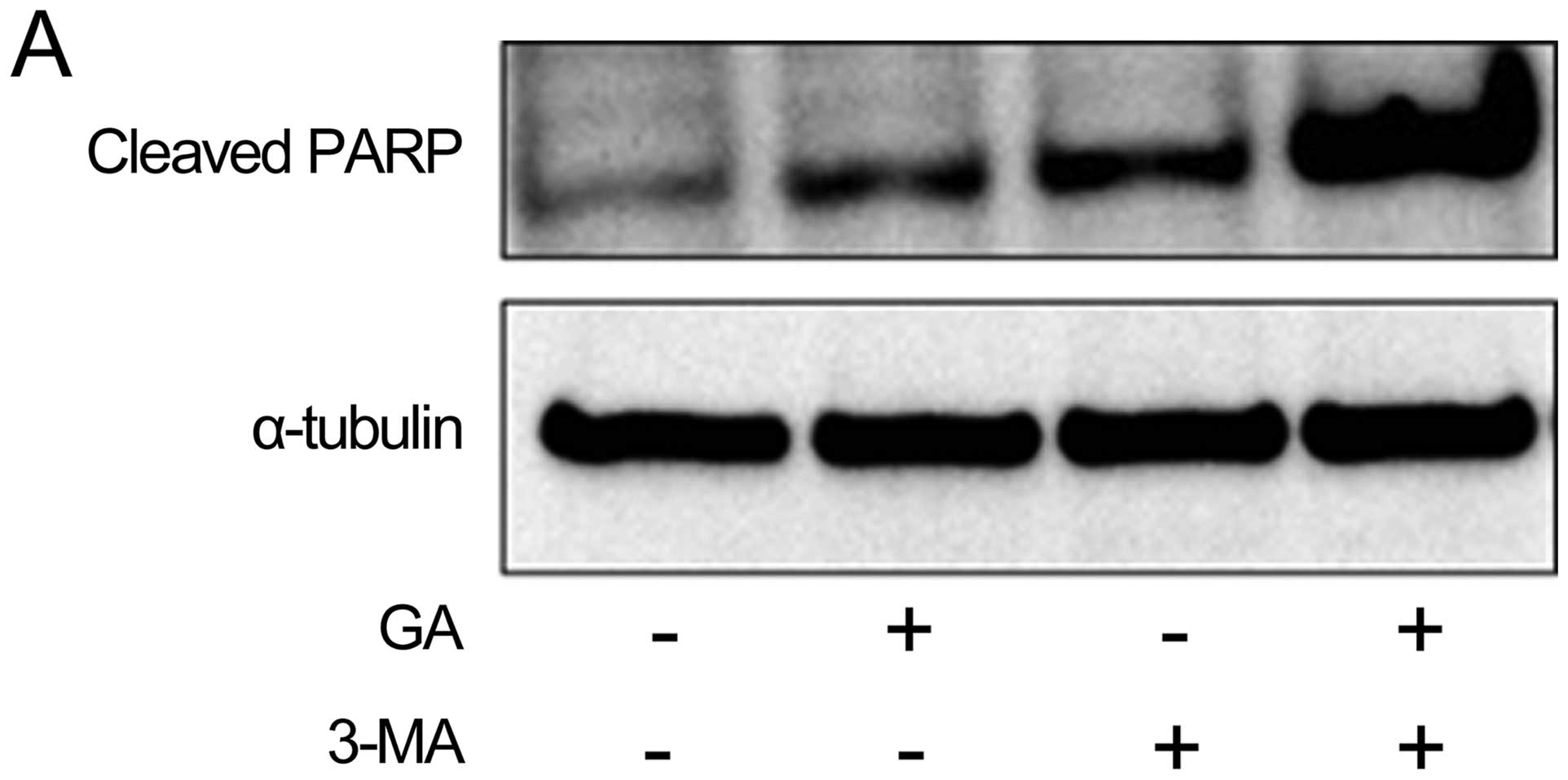
used western blot analysis to examine the effect of pre-treatment
with 10 mM 3-MA 1 h on the effect of GA treatment (5 μM for 24 h)
on protein expression of cleaved PARP, a marker of
caspase-dependent apoptosis. As shown in Fig. 7A, pre-treatment of cells with 3-MA
strongly increased the cleavage of PARP induced by GA. We next
examined induction of apoptotic cells by GA and the effect of 3-MA
pre-treatment on such induction. Apoptotic cells were assayed by
flow cytometry using the TUNEL assay. The number of apoptotic cells
induced by 24 h treatment with 5 μM GA was significantly increased
by 10 mM 3-MA pre-treatment (P<0.05) (Fig. 7B). We also assayed apoptotic cells
using Annexin V-FITC/PI staining and fluorescence microscopy.
Annexin V is a marker of early apoptosis, and PI is a marker of
late apoptosis and necrosis. As shown in Fig. 7C, the number of apoptotic cells as
measured by this assay that were induced by 24 h treatment with 5
μM GA was significantly increased by 10 mM 3-MA pre-treatment
(P<0.001). The combined results suggest that GA induces
autophagy as a protective mechanism in KTHOS cells. Furthermore, GA
potently inhibits the proliferation of KTHOS cells via induction of
apoptosis following 3-MA pre-treatment.
Discussion
Hsp90 is a molecular chaperone with several client
proteins that are known to contribute to tumorigenesis. Hsp90 has
recently been considered as a promising target for therapeutic
intervention in a variety of cancers. The biological activity of GA
and its semi-synthetic derivatives towards various hematopoietic
neoplasms and solid carcinomas has been demonstrated in
vitro and in murine xenograft models (24–27).
Several clinical trials evaluating both GA derivatives and other
novel Hsp90 inhibitors are ongoing. However, little is known
regarding the potential activity of Hsp90 inhibitors in sarcomas.
In this study, we demonstrate that GA inhibits the proliferation of
human osteosarcoma KTHOS cells via induction of apoptosis and also
induces autophagy. We further demonstrate that a combination of GA
and 3-MA potently inhibits the proliferation of KTHOS cells to a
greater extent than GA alone via induction of apoptosis.
We observed that GA induced time- and dose-dependent
inhibition of proliferation of KTHOS cells. GA also induced
apoptosis in KTHOS cells, resulting in altered cell morphology, DNA
fragmentation, multiple caspase activation and PARP cleavage.
Activation of caspase-8 indicated that the FasL/Fas pathway may be
involved in GA-induced apoptosis. GA also activated caspase-9,
which in turn, is known to activate the downstream effector
caspase-3 and lead to PARP cleavage. The combined results suggest
that GA-induced apoptosis is caspase-dependent.
Autophagy is a process in which subcellular
membranes undergo dynamic morphological change (autophagosomes form
and fuse with lysosomes) leading to the degradation of cellular
proteins and cytoplasmic organelles. Autophagy plays a protective
role when cells encounter environmental stresses such as starvation
or pathogen infection (28,29).
Autophagy also occurs under pathological conditions, such as in
neurodegenerative disease or hereditary myopathies. Recent
accumulating evidence indicates that autophagy often plays a role
in malignant diseases. Specifically, autophagy is believed to play
an important role in tumor development. During the early stages of
tumor formation, autophagy functions as a tumor suppressor, and
autophagic activity is often impaired in cancer cells. Many
anticancer drugs which lead to apoptosis can also induce
autophagy-related cell death in cancer cell lines (30,31).
In the present study autophagy was demonstrated in GA-treated cells
by MDC accumulation. GA treatment also induced dose-dependent
upregulation of expression of the autophagy marker LC3-II.
Inhibition of Hsp90 induces degradation of Hsp90 client proteins in
cancer cells, and it is widely thought to lead to reduced
proliferation. There are numerous Hsp90 client proteins. Akt is a
known Hsp90 client protein. Akt is a serine threonine kinase that
is downstream of PI3K and that has a large number of downstream
targets implicated in survival and cell cycle regulation (32). In the present study, GA inhibited
Akt/mTOR signaling, indicating that GA induces autophagy via
targeting of Akt/mTOR signaling. The combined results suggest that
GA-induced autophagy is associated with Akt protein degradation via
a mechanism that is dependent on Hsp90 inhibition and on inhibition
of Akt activation of mTOR.
3-MA is an inhibitor of autophagy. However, recent
reports indicate that when 3-MA is combined with chemotherapeutic
drugs it triggers apoptosis in some cancer cells (33). In the present study, we observed
that the use of a combination of GA and 3-MA induced more cell
death in KTHOS cells than the use of GA alone. We considered that
autophagy can function as a protective mechanism in KTHOS cells
that are subjected to GA and that blocking autophagy with 3-MA can
promote the activation of apoptosis. It therefore appears that the
combination of GA and 3-MA potently induced apoptotic cell death in
KTHOS cells by inhibition of autophagy.
In conclusion, GA had an inhibitory effect on cell
proliferation and inhibited the Akt/mTOR signaling pathway in KTHOS
cells. GA also induced autophagy in KTHOS cells. However, treatment
with a combination of GA and 3-MA suppressed autophagy and induced
much higher apoptosis in KTHOS cells than GA alone. We considered
that the autophagy inhibitor 3-MA suppressed a protective mechanism
induced by Hsp90 inhibitor in the tumor cells and induced
apoptosis. Therefore, the combination of an Hsp90 inhibitor and an
autophagy inhibitor may be an effective treatment for osteosarcoma
because this combination effectively induces apoptotic
pathways.
Acknowledgements
The authors thank Mr. Kouichi Yube (Division of
Research Instrument and Equipment, Kagawa University School of
Medicine, Kagawa, Japan) for his excellent technical assistance
with flow cytometry.
References
|
1
|
Mueller F, Fuchs B and Kaser-Hotz B:
Comparative biology of human and canine osteosarcoma. Anticancer
Res. 27:155–164. 2007.PubMed/NCBI
|
|
2
|
Withrow SJ, Powers BE, Straw RC and
Wilkins RM: Comparative aspects of osteosarcoma. Dog versus man.
Clin Orthop Relat Res. 270:159–168. 1991.PubMed/NCBI
|
|
3
|
Clark JC, Dass CR and Choong PF: A review
of clinical and molecular prognostic factors in osteosarcoma. J
Cancer Res Clin Oncol. 134:281–297. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Marina N, Gebhardt M, Teot L and Gorlick
R: Biology and therapeutic advances for pediatric osteosarcoma.
Oncologist. 9:422–441. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bishop SC, Burlison JA and Blagg BS:
Hsp90: a novel target for the disruption of multiple signaling
cascades. Curr Cancer Drug Targets. 7:369–388. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zuehlke A and Johnson JL: Hsp90 and
co-chaperones twist the functions of diverse client proteins.
Biopolymers. 93:211–217. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Solit DB and Chiosis G: Development and
application of Hsp90 inhibitors. Drug Discov Today. 13:38–43. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brown MA, Zhu L, Schmidt C and Tucker PW:
Hsp90 - from signal transduction to cell transformation. Biochem
Biophys Res Commun. 363:241–246. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Neckers L, Mimnaugh E and Schulte TW:
Hsp90 as an anti-cancer target. Drug Resist Updat. 2:165–172. 1999.
View Article : Google Scholar
|
|
10
|
Kamal A, Thao L, Sensintaffar J, et al: A
high-affinity conformation of Hsp90 confers tumor selectivity on
Hsp90 inhibitors. Nature. 425:407–410. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hertlein E, Wagner AJ, Jones J, et al:
17-DMAG targets the nuclear factor-kappaB family of proteins to
induce apoptosis in chronic lymphocytic leukemia: clinical
implications of HSP90 inhibition. Blood. 116:45–53. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Porter JR, Fritz CC and Depew KM:
Discovery and development of Hsp90 inhibitors: a promising pathway
for cancer therapy. Curr Opin Chem Biol. 14:412–420. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Amaravadi RK and Thompson CB: The roles of
therapy-induced autophagy and necrosis in cancer treatment. Clin
Cancer Res. 13:7271–7279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu C, Liu J, Hsu LC, Luo Y, Xiang R and
Chuang TH: Functional interaction of heat shock protein 90 and
Beclin 1 modulates Toll-like receptor-mediated autophagy. FASEB J.
25:2700–2710. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Palacios C, Martín-Pérez R, López-Pérez
AI, Pandiella A and López-Rivas A: Autophagy inhibition sensitizes
multiple myeloma cells to
17-dimethylaminoethylamino-17-demethoxy-geldanamycin-induced
apoptosis. Leuk Res. 34:1533–1538. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Roca H, Varsos Z and Pienta KJ: CCL2
protects prostate cancer PC3 cells from autophagic death via
phosphatidylinositol 3-kinase/AKT-dependent surviving
up-regulation. J Biol Chem. 283:25057–25073. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rami A and Kögel D: Apoptosis meets
autophagy-like cell death in the ischemic penumbra: two sides of
the same coin? Autophagy. 4:422–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu D, Yang Y, Liu Q and Wang J:
Inhibition of autophagy by 3-MA potentiates cisplatin-induced
apoptosis in esophageal squamous cell carcinoma cells. Med Oncol.
28:105–111. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kanematsu S, Uehara N, Miki H, et al:
Autophagy inhibition enhances sulforaphane-induced apoptosis in
human breast cancer cells. Anticancer Res. 30:3381–3390.
2010.PubMed/NCBI
|
|
22
|
Ren Y, Huang F, Liu Y, Yang Y, Jiang Q and
Xu C: Autophagy inhibition through PI3K/Akt increases apoptosis by
sodium selenite in NB4 cells. BMB Rep. 42:599–604. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Petiot A, Ogier-Denis E, Blommaart EF,
Meijer AJ and Codogno P: Distinct classes of phosphatidylinositol
3′-kinases are involved in signaling pathways that control
macroautophagy in HT-29 cells. J Biol Chem. 275:992–998. 2000.
View Article : Google Scholar
|
|
24
|
Al Shaer L, Walsby E, Gilkes A, et al:
Heat shock protein 90 inhibition is cytotoxic to primary AML cells
expressing mutant FLT3 and results in altered downstream
signalling. Br J Haematol. 141:483–493. 2008.PubMed/NCBI
|
|
25
|
Lang SA, Moser C, Gaumann A, et al:
Targeting heat shock protein 90 in pancreatic cancer impairs
insulin-like growth factor-I receptor signaling, disrupts an
interleukin-6/signal-transducer and activator of transcription
3/hypoxia-inducible factor-1α autocrine loop, and reduces
orthotopic tumor growth. Clin Cancer Res. 13:6459–6468. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Williams CR, Tabios R, Linehan WM and
Neckers L: Intratumor injection of the Hsp90 inhibitor 17AAG
decreases tumor growth and induces apoptosis in a prostate cancer
xenograft model. J Urol. 178:1528–1532. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yano M, Naito Z, Tanaka S and Asano G:
Expression and roles of heat shock proteins in human breast cancer.
Jpn J Cancer Res. 87:908–915. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Meijer AJ and Codogno P: Regulation and
role of autophagy in mammalian cells. Int J Biochem Cell Biol.
36:2445–2462. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu B, Cheng Y, Zhang B, Bian HJ and Bao
JK: Polygonatum cyrtonema lectin induces apoptosis and autophagy in
human melanoma A375 cells through a mitochondria-mediated
ROS-p38–p53 pathway. Cancer Lett. 275:54–60. 2009. View Article : Google Scholar
|
|
31
|
Wang Q, Chen Z, Diao X and Huang S:
Induction of autophagy-dependent apoptosis by the survivin
suppressant YM155 in prostate cancer cells. Cancer Lett. 302:29–36.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nishikawa T, Tsuno NH, Okaji Y, et al:
Inhibition of autophagy potentiates sulforaphane-induced apoptosis
in human colon cancer cells. Ann Surg Oncol. 17:592–602. 2010.
View Article : Google Scholar : PubMed/NCBI
|